
Přehled maligních chorob odvozených od histiocytárních a dendritických buněk
Overview of malignant disorders derived from the histiocytic and dendritic cells
Histiocytic and dendritic cell disorders are very rare disease. Langerhans cell histiocytic is more frequent, that any other of this group. The characteristics of these diseases are summarized in this overview with respect of the recent WHO classification, published 2017 and Histocyte Society classification published 2016.
Keywords:
histiocytic and dendritic cell neoplasms
Autoři:
Z. Král; Z. Adam 1; Marta Ježová 2; J. Neubeuer 3; Z. Řehák 4; L. Pour 1
Působiště autorů:
Interní hematoonkologická klinika LF MU a FN Brno
1; Patologický ústav LF MU a FN BRNO
2; Radiologická klinika LF MU a FN Brno
3; PET CT Oddělení Masarykův onkologická ústav
4
Vyšlo v časopise:
Transfuze Hematol. dnes,25, 2019, No. 2, p. 131-146.
Kategorie:
Souhrnné/edukační práce
Souhrn
Neoplazie z histiocytárních a dendritických buněk jsou velmi raritní a tedy obtížně diagnostikovalné nemoci. Nejčastější z nich je histiocytóza z Langerhansových buněk, incidence ostatních nemocí je ještě menší. V následujícím přehledu je shrnuta jejich charakteristika a uvedeny recentní klasifikace, standardně používaná WHO klasifikace (poslední verze publikovaná v roce 2017) a paralelně s ní existující klasifikace, vytvořená společností Histiocyte Society (poslední verze publikovaná 2016).
Klíčová slova:
neoplazie odvozené od histiocytárních a dendritických buněk
ÚVOD
Histocytózy, neboli nemoci odvozené od histiocytárních a dendritických buněk, se vyskytují velmi vzácně, podstatně vzácněji než nemoci odvozené od buněk linie myeloidní nebo linie lymfocytární, či nemoci odvozené od buněk plazmocytárních. Jejich výskyt je natolik vzácný, že hematolog se za svůj pracovní život ani se všemi nesetká. Nejčastější nemocí z této skupiny je histiocytóza z Langerhansových buněk. Této nemoci se budeme věnovat podrobněji v dalším článku (v příštím čísle Transfuze a hematologie dnes, pozn. redakce). V rámci tohoto textu uvedeme přehled všech nemocí, které jsou řazeny v poslední WHO klasifikaci krevních chorob z roku 2017 [1] a klasifikaci Histiocyte Society z roku 2016 [2] do této kategorie nemocí.
Dendritické buňky, monocyty a makrofágy jsou součástí mononukleárního fagocytujícího systému.
Historickým termínem histiocyt označujeme makrofágy usazené ve tkáních. Monocyty vznikají v kostní dřeni z myeloidní kmenové buňky. Po průniku do tkání vyzrávají v makrofágy nadané schopností fagocytózy – odstraňují apoptotické buňky, cizorodý materiál a patogeny. Jsou to velké ovoidní buňky s excentrickým oválným nebo ledvinovitým jádrem. Mohou střádat lipidy (xantomové buňky) a splývat v buňky mnohojaderné. Při imunohistochemickém vyšetření jsou cytoplazmaticky pozitivní s CD68 a lysozymem, cytoplazmaticky nebo membránově s CD163.
Dendritické buňky nemají jednotný původ. Většina je stejně jako monocyty odvozována z myeloidní kmenové buňky kostní dřeně. Úlohou dendritických buněk je prezentovat antigen a komplex histokompatibilních molekul a aktivovat naivní T-buňky. Dendritické buňky se dělí na myeloidní a plazmocytoidní. Plazmocytoidní dendritické buňky cirkulují v periferní krvi, tvoří speciální kategorii, a budou proto z dalšího pojednání vyjmuty. V lidském organismu jsou hlavními typy dendritických buněk Langerhansovy buňky a dermální dendritické buňky (syn. intersticiální dendritické buňky). Langerhansovy buňky nacházíme v kůži a na sliznicích. Charakterizuje je exprese antigenu CD1a, langerinu (CD 207), S100 a ultrastrukturálně zvláštní Birbeckova granula ve tvaru tenisových raket. Při aktivaci migrují do drénujících lymfatických uzlin, kde se pravděpodobně mění na interdigitující dendritické buňky. Ty ztrácejí většinu antigenů s výjimkou S100. Indeterminované buňky jsou považovány za prekurzory Langerhansových buněk, alternativně za zralé buňky ve stadiu migrace. Nemají Birbeckova granula, a proto nereagují s langerinem, exprese CD1a je zachována. Dermální dendritické buňky se nalézají v kožní škáře, hlubokých měkkých tkáních i vnitřních orgánech. Imunohistochemický profil je relativně nespecifický, pozitivní bývá faktor XIIIa, CD68; CD1a je negativní a S100 variabilní, spíše negativní.
Kromě toho jsou známy nejméně dvě další skupiny dendritických buněk, které nepocházejí z krvetvorné linie, ale z nediferencované mezenchymové buňky (stromální kmenové buňky). Mají vřetenitý tvar a podobu fibroblastů či myofibroblastů. Folikulární dendritické buňky jsou v lymfatických uzlinách. Nemigrují, tvoří stabilní síť v zárodečných centrech lymfatických foliklů osídlenou B lymfocyty. Exprimují unikátní znaky CD21 a CD23. Fibroblastické retikulární buňky jsou přítomny v uzlinách podél postkapilárních venul. Ve slezině a kostní dřeni tvoří opěrnou kostru. Reagují pozitivně s protilátkou proti hladkosvalovému aktinu [1, 2].
Z tohoto morfologického podkladu se odvíjejí klasifikace. Poslední tři desetiletí jsme svědky existence dvou klasifikačních systémů vedle sebe. V roce 2017 vyšla v pořadí již čtvrtá WHO klasifikace krevních chorob a v této světově platné klasifikaci je také kapitola nazvaná Histocytic and dedritic cell neplasms. A pararelně s touto klasifikací vychází klasifikace tvořená skupinou „Working Group of the Histiocyte Society“. Klasifikace tvořená Histiocyte Society obsahuje více klinických pohledů a zaměřuje se na ty četnější, ale méně agresivní nemoci a obsahuje definice některých jednotek, které ve WHO klasifikaci nejsou, neboť jsou spíše reaktivní než neoplastické etiologie, například hemofagocytující lymfohistiocytóza, Rosaiova-Dorfmanova choroba a skupina mukokutánních histiocytárních proliferací. Jednotlivá onemocnění se nyní odvozují od koncových zralých buněk a mají tomu odpovídající imunofenotyp. Nejnovější revize zohledňují i molekulárně genetické poznatky. Obě klasifikace nejsou ve vzájemném rozporu, pouze se liší členěním na některé podjednotky [1, 2].
První klasifikace histiocytóz, zvěřejněná v roce 1987 skupinou Working Group of the Histiocyte Society, definovala tři zásadní kategorie:
- Langerhans cell histiocytosis
- non-Langerhans cell histiocytoses a
- maligní histiocytózy.
V roce 2016 navrhla Working Group of the Histiocyte Society novou klasifikaci. Tato nová klasifikace dělí histiocytární choroby do celkem 5 velkých skupin. Do první skupiny řadí histiocytózu z Langerhansových buněk. Druhou skupinu pak tvoří kožní a mukokutánní histiocytózy, třetí skupinu maligní histiocytózy, čtvrtou skupinu Rosaiova-Dorfmanova nemoc a pátou pak hemofagocytující lymfohistiocytóza a syndrom aktivace makrofágů [2].
Překvapivé je, že autoři klasifikace Histiocyte Society do první skupiny k histiocytózám z Langerhansových buněk přiřadili i Erdheimovu-Chesterovu nemoc, kterou WHO klasifikace krevních chorob řadí pod kapitolu juvenilní xantogranulom. Autoři Histiocyte Society uvádějí, že v histologických vzorcích se často podařilo indentikovat současně obě nemoci a že obě nemoci mají některé společné vlastnosti: postihují stopky hypofýzy a způsobují diabetes insipidus. Při dlouhém trvání obou těchto nemocí se jako pozdní komplikace vyskytuje neurodegenerativní onemocnění mozku [2]. Tyto přesuny jen ilustrují, jak málo o této skupině nemocí víme.
Obě klasifikace v přehledu uvádíme v tabulce 1 a v dalším textu pak stručně budeme charakterizovat tyto nemoci, u těch, s nimiž jsme se v praxi setkali, přidáme ilustrační fotografie.

STRUČNÁ CHARAKTERISTIKA JEDNOTLIVÝCH JEDNOTEK
V dalším popisu se budeme držet poslední WHO klasifikace krevních chorob, kde to budeme považovat za užitečné, zmíníme i pojetí klasifikace druhé.
1. Histiocytóza z Langerhansových buněk (LCH)
Diagnózu histiocytózy z Langerhansových buněk lze stanovit pouze histologickým vyšetřením. Mikroskopicky vidíme infiltráty z velkých oválných buněk se světle eozinofilní cytoplazmou a ledvinovitými nebo zprohýbanými jádry se zářezy. Počet mitóz je různý, ale stanovení proliferačního indexu Ki67 nemá prognostický význam. V pozadí je nenádorová příměs, v níž převládají eozinofily. Diagnostické buňky dávají difuzně pozitivní reakci s CD1a a S100. Náročná detekce Birbeckových granul, pro což je nutná elektronová mikroskopie, byla nahrazena průkazem znaku CD207 (langerinu), což je proveditelné i na fixovaných vzorcích [1, 2, 3].
Nemoc zvaná histiocytóza z indeterminovaných buněk (ICH) nemá přítomný znak CD207 a tím ji lze odlišit. V případě Rosaiovy-Dorfmanovy nemoci jsou S100+ histiocyty často vícejaderné a mají prokazatelný jev zvaný emperipolesis a neexprimují ani CD1a a ani CD207 [1, 2, 3]. Emperipolesis je jev podobný fagocytóze, má s ní společné to, že se jedna buňka dostane do nitra druhé buňky, ale liší se tím, že v případě jevu jménem emperipolesis nedochází k destrukci buňky, zatímco v případě fagocytózy dochází k destrukci fagocytované buňky.
Uvádí se, že histiocytóza z Langerhasových buněk může provázet jiné krevní nemoci, ale tento jev jsme u našich pacientů zatím nepozorovali. Autoři z Histiocyte Society uvádějí, že klasické dělení histiocytóz na Langerhansovu histiocytózu a non-Langerhansovy histiocytózy, kam patří Erheimova-Chesterova nemoc mírně ztrácí na své důležitosti, protože 20 % pacientů s Erdheimovou-Chesterovou nemocí má také ložiska LCH [2, 4].
Working Group of Histiocyte Society proto doporučeje zahrnout histiocytózu z Langerhanosových buněk, Erdheimovu-Chesterovu nemoc a extrakutánní juvenilní xantogranulom do jedné skupiny nemocí. Obě nemoci mají klonální mutace, postihující geny MAPK signální cesty, a to více než v 80 % případů. Monocyty z periferní krve přitom mívají tu samou mutaci, jaká se popisuje v patologických buňkách [2, 4].
Přelom v léčbě této nemoci přinesla detekce mutace V600E genu BRAF. Tato mutace má za následek konstitutivní aktivaci MAPK signální cesty, které způsobuje malignizace u více typů tumorů. Přítomna je přibližně u poloviny případů LCH. Průkaz BRAF mutace u CD34+ buněk kostní dřeně u některých pacientů s vysoce rizikovou formou nemoci signalizuje, že LCH může být odvozena od hemopoetických progenitorových buněk. Dále asi u 19 % případů byly detekovány mutace MAP2K1 nebo MEK1 kinázy, která také souvisí s MAPK signální cestou. V posledních letech byly popsány mutace i v dalších signálních cestách. Molekulárně biologickou charakteristiku podrobně rozebírá recentní publikace zveřejněná v časopise Klinická onkologie 2018 [5, 6].
Po stanovení diagnózy je vždy třeba stanovit rozsah nemoci a podle toho pak zvolit vhodnou léčbu. Pro dětské pacienty byla publikována mezinárodní doporučení jak pro vyšetření rozsahu nemoci, tak pro léčbu [7, 8]. Pro pacienty s prokázanou mutací BRAF lze po domluvě s pojišťovnou použít vemurafenib, excelentní výsledky podání vemurafenibu popsali i slovenští autoři [9]
Vzácnější než LCH je sarkom z Langerhansových buněk. Je tvořen Langerhansovými buňkami s cytologickými znaky malignity a nezvykle vysokou mitotickou aktivitou včetně mitóz atypických. Eozinofily na pozadí mizí. Imunofenotyp je shodný. Nádor se chová zhoubně [10].
Protože se LCH vyskytuje nejčastěji ze všech histiocytóz, budeme se jí podrobněji věnovat v samostatném článku (v příštím čísle Transfuze a hematologie dnes, pozn. redakce). Na našem pracovišti za posledních 28 let evidujeme přes 40 osob s diagnózou LCH.
2. Indeterminate-cell histiocytosis
Je choroba morfologicky podobná LCH, odlišit ji lze na základě imunofenotypu. Je podstatně vzácnější než LCH a postihuje dominantně kůži ve formě makul a papul [11, 12]. Za posledních 28 let jsme se na našem pracovišti setkali s touto diagnózou jen jedenkrát [13]. Kožní postižení u našeho pacienta ilustrují obr. 1–3. Histologické a klinické znaky odlišující LCH a indeterminate cell histiocytosis uvádíme v popisu případu [13]



3. Erdheimova-Chesterova choroba
Erdheimova-Chesterova choroba (Erheim-Chester disease) je histocytární onemocnění, patřící do skupiny juvenilního xantogranulomu. Choroba se projevuje symetrickou osteosklerózou, postihující diafýzu i metafýzu dlouhých kostí, šetřící epifýzy. Radiologický nález je pro tuto nemoc patognomický. Nicméně 5–8 % pacientů může mít také postiženy ploché kosti. Erdheimova-Chesterova choroba představuje vlastně systémovou formu xantogranulomatózního onemocnění.
Patologické infiltráty tvoří pěnité histiocyty, histiocyty s eozinofilní cytoplazmou a příměs malých reaktivních lymfocytů, plazmocytů a neutrofilů. Jsou i případy, kdy dominuje neurčitá fibróza. Nález připomíná zánět nebo reparativní změny. Diagnosticky cenné jsou vícejaderné buňky Toutonova typu s věnečkem jader kolem eozinofilního středu a lemem pěnité cytoplazmy na periferii. Histiocyty jsou pozitivní s markery CD68, CD163 a faktorem XIIIa, negativní s S100, CD1a a langerinem. Diferenciální diagnostika je široká a zahrnuje histiocytózu z Langerhansových buněk, obrovskobuněčné kostní léze, chronické sklerotizující a hnisavé záněty, xantomy a specifické záněty (lepru, mykobakteriózu). Malá biopsie může být nevýtěžná. Patolog musí být klinikem podrobně informován o podezření na toto vzácné onemocnění včetně radiologického nálezu. Nemoc je považována za blízkou formu juvenilního xantogranulomu, histologicky jsou obě jednotky identické. V předcházející WHO klasfikaci byla proto zařazena do kapitoly juvenilní xantogranulom [14].
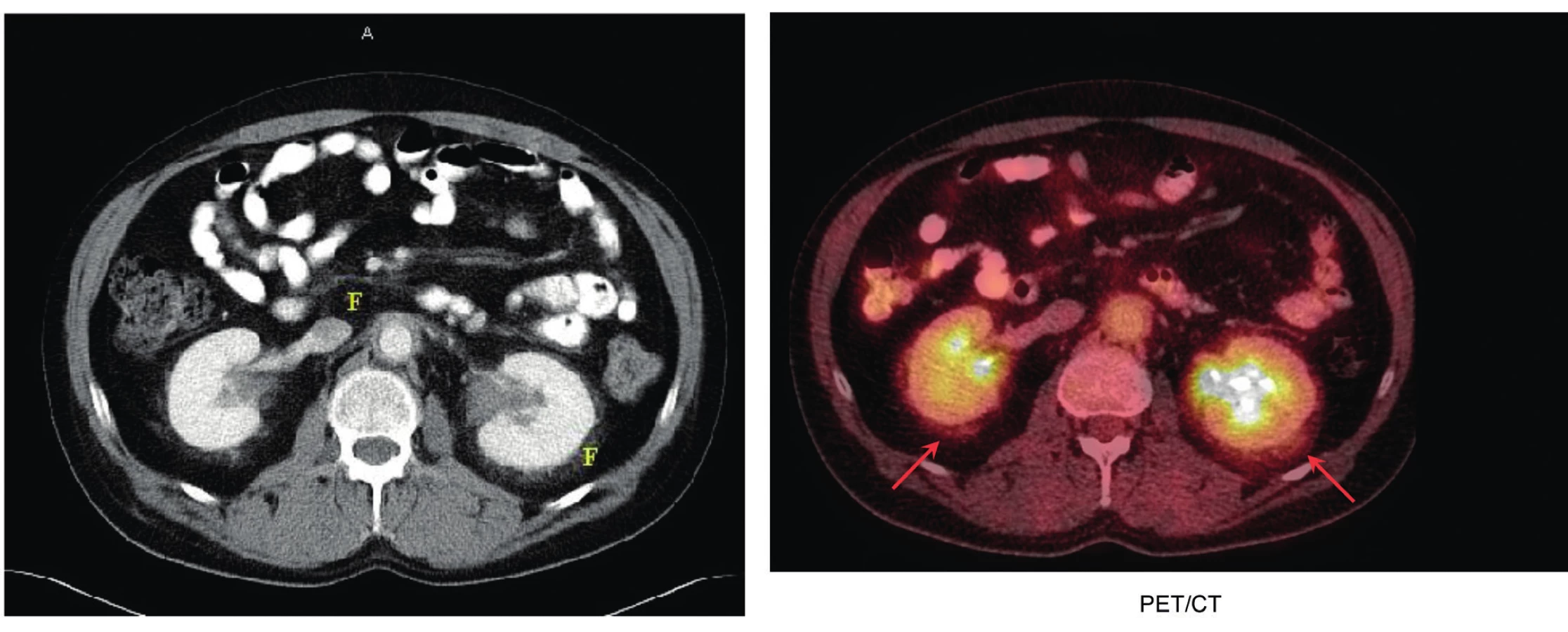
Mimokostní postižení je u této nemoci popisováno v 50 % případů. Byly popsány následující komplikace: postižení hypotalamu s následným diabetem insipidem a hypopituarismem, retroperitonální infiltrace s postižením ledvin s perirenální fibrózou, případy s ložisky na očních víčkách vzhledu xantomů, exoftalmus, a také postižení plic a velkých cév se zesílením cévní stěny. Plicní fibróza s dušností a srdeční selhání jsou nejčastější příčiny úmrtí. Neurologické postižení může způsobovat ataxii či parézy. Charakteristické nálezy na zobrazovacích vyšetřeních ilustrují obr. 4–7.
Velmi dobře ji znázorňuje scintigrafie kostí (radiofarmakum technecium
pyrofosfát), ale je velmi dobře prokazatelné pomocí FDG-PET/CT
zobrazení, které zobrazí zvýšenou akumulaci fluorodeoxyglukózy ve
stejných lokalizacích. Na rentgenovém či CT zobrazení těchto lokalit se
zvýšenou akumulací Tc-pyrofosfátu a fluorodeoxyglukózy je pak dobře
patrná osteoskleróza.


Vlevo na klasickém CT snímku jsou místa s fibrotickými změnami označena písmenem F, vpravo na FDG-PET/CT zobrazení jsou místa perirenální
fibrózy označená šipkami. Perirenální fibróza je pro Erdheimovu-Chesterovu chorobu typická.
Ilustruje to zobrazení aorty se zesílenou cévní stěnou.

Šipky ukazují zesílení stopky hypofýzy u pacienta s Erdheimovou-Chesterovou nemocí, tedy stejné postižení k jakému dochází u pacientů
s histiocytózou z Langerhansových buněk. Tato infiltrace má za následek diabetes insipidus.

Xantogranulomatózní proces při Edheimově--Chesterově nemoci může mimo viscerální orgány či kosti postihovat také kůži, dutinu orbity či paranazální dutiny.
Klinicky se nemoc projevuje bolestmi končetin a může způsobit klasické zánětlivé projevy zvané v tomto případně „B-symptomy“, úbytek hmotnosti, subfebrilie či febrilie, noční pocení, patologickou únavu. Laboratorně tomu odpovídají vysoké hodnoty CRP obvykle s normální hodnotou prokalcitoninu.
Průběh nemoci je velmi individuální a odpovídá stupni poškození organismu, nezřídka byl popsán fatální konec [15]. Někteří autoři popisují současný výskyt Erdheimovy-Chesterovy nemoci s myeloidními neoplaziemi [2, 16].
V léčbě této nemoci byly testovány všechny dostupné léky, nejvíce zřejmě interferon alfa a nyní v případně prokázané mutace BRAF pak Vemurafenib [17, 18].
Za posledních 28 let jsme se touto diagnózou setkali jen 2krát.
V jednom případně léčba založená na kladribinu vedla k dlouholeté kompletní remisi, v druhém případě pouze k parciální remisi a tento pacient je na udržovací léčbě preparátem anakinra (preparát Kineret) [19–22].
4. Kožní a mukokutánní histiocytózy
Working Party of Histocyte Society dále definuje kožní a mukokutánní formy a vytváří označení kožní formy xantogranulomových chorob. U dětí tyto formy spontálně mizí. Pro jednotlivé klinické formy byla vyvinuta speciální označení, která uvádíme v přehledu v tabulce 2. Při výjimečnosti těchto chorob je těžké se v nich orientovat a zřejmě častěji než hematologové s nimi přicházejí do kontaktu kožní specialisté a v případě periorbitální lokalizace oční lékaři.
![Non-LCH histiocytózy kůže a sliznic podle klasifikace Histiocyte Society [2]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/b2a6bcfe3c12481f1e5527bbda382a5f.png)
Juvenilní xantogranulom (synonymum névoxantoendoteliom) je častý kožní tumorek u malých dětí. Vypadá jako solitární červenožlutý nebo žlutý uzel na kůži hlavy, krku a trupu. Histologicky je pod epidermis neostře ohraničené ložisko smíšené stavby, z jednojaderných okrouhlých histiocytů s eozinofilní, vakuolizovanou i pěnitou cytoplazmou, vřetenitých histiocytů a vícejaderných Toutonových buněk. Na pozadí jsou malé lymfocyty a neutrofily. Mnohočetné, gigantické, hluboké, viscerální a diseminované formy jsou mikroskopicky podobné, ale mnohem vzácnější. Podtypem je pak nekrobiotický xantogranulom, který je velmi často spojen s přítomností monoklonálního imunoglobulinu.
Nemoc probíhá indolentně, ale setkali jsme se s pacientkou, u níž postižení víček a snaha o operační řešení způsobily lagoftalmus a oboustrannou slepotu. Někdy se pro ploché morfy užívá termínu xanthoma planum, pro indurované morfy pak xanthogranuloma [23–34].
Za posledních 28 let jsme se setkali s periorbitálními xantogranulomy celkem 3krát a v jednom případě šlo o velké ploché plochy xantogranulomu na trupu v souvislosti s monoklonální gamapatií, která trvá bez vývoje již druhé desetiletí [35, 36] – viz obr. 8–11.




Koncentrace cholesterolu a triglyceridů byly při dolní hranici fyziologického rozmezí.
5. Rosaiova-Dorfmanova nemoc
Pro podrobnosti odkazujeme na náš recentní přehledný článek [38]. Formy Rosaiovy-Dorfmanovy nemoci uvádí tabulka 3.
![Formy Rosaiovy-Dorfmanovy nemoci (Rosai-Dorfman Disease – RDD) podle klasifikace Histocyte Society [2]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/89c1c1f2322b8e9fa738475d1b4a9a67.png)
6. Maligní histiocytární choroby
Do skupiny maligních chorob, odvozených od histiocytů podle WHO patří:
- histiocytární sarkom,
- sarkom z folikulárních dendritických buněk,
- sarkom z interdigitujících dendritických buněk,
- fibroblastický retikulární tumor.
Před zavedením imunofenotypizace byla diagnóza maligní histiocytózy stanovena mnohem častěji, protože četné B - i T-buněčné lymfoproliferace byly považovány za histiocytární malignity. V současné době jsou patology diagnostikovány velice zřídka.
Přibližně třetina histiocytárních sarkomů se manifestuje lokalizovanou lymfadenopatií, třetina se manifestuje kožními ložisky (solitární či mnohočetná) a poslední třetina vzniká extranodálně, často v oblasti zažívacího traktu.
Někteří nemocní mají systémové postižení s mnohočetnými ložisky, jehož popis se může shodovat s dřívějšími popisy maligní histiocytózy. WHO klasifikace by nyní pro tento stav použila termínu generalizovaná či diseminovaná forma histiocytárního sarkomu.
6. 1. Lokalizovaný histiocytární sarkom
Tato jednotka je odvozena od fagocytujících mononukleárních buněk ve stadiu tkáňové fixace a diferen-ciace, tj. zralých makrofágů. Může vzniknout jak v kůži, tak zažívacím traktu či v kostech. Za posledních 28 let jsme se setkali s 2 případy lokalizovaného histiocytárního sarkomu, kdy po radikálním operačním odstranění a lokální radioterapii se podařilo onemocnění eliminovat.
6. 2. Diseminovaný histiocytární sarkom, synonymem maligní histiocytóza
Někteří pacienti s histiocytárním sarkomem mají mnohočetné postižení včetně hepatomegalie a splenomegalie, což odpovídá staršímu popisu maligní histiocytózy. Tento termín se dnes již nepoužívá a místo něj se používá termín diseminovaný histiocytární sarkom [39–41].
Tato diseminovaná forma histiocytárního sarkomu je velmi agresivně probíhající nemoc. Klinické příznaky se podobají projevům lymfoblastické leukémie s generalizovaným postižením orgánů. Maligní histiocytózu velmi často provází vysoké horečky nad 39 °C, splenomegalie (100 %), lymfadenopatie (92 %), hepatomegalie (67 %). Mohou však být infiltrovány i jiné orgány, například plíce, mozek, kůže, což k výše uvedeným příznakům může přidat dušnost či bolesti hlavy. Někdy způsobuje osteolýzu a s ní spojené bolesti kostí. Kožní manifestace může nabývat různých podob, od benigně vyhlížejícího exantému až po četné kožní tumory trupu a končetin. Postižení střeva se často projeví obstrukčními příznaky.
Nemoc charakterizují následující laboratorní změny: trombocytopenie (92 %), anémie (92 %), leukocytopenie (67 %). V biochemickém vyšetření se u těchto pacientů velmi často detekují vysoké hodnoty LDH a bilirubinu, přičemž jaterní enzymy a renální funkce bývají jen nepatrně zhoršené. Nepravidelně se vyskytuje zvýšení ACE-inhibitoru (Angiotensin Converting Enzym Inhibitor) a TNF (Tumor Necrosis Factor).
Při postižení CNS lze často nalézt v mozkomíšním moku patologické fagocytující neoplastické histiocyty [42].
Vyšetření kostní dřeně metodou trepanobiopsie je nejpřístupnější cestou ke zjištění diagnózy. Je však nutno upozornit na skutečnost, že první vzorky mohou být hodnoceny jako negativní a teprve při výraznější infiltraci se podaří identifikovat proliferující anaplastické histiocyty.
Nádorovou populaci tvoří velké atypické buňky rostoucí v plochách. V uzlinách se mohou šířit sinusy. Buňky jsou oválné, nepravidelné nebo vřetenité s velkými hyperchromními jádry. Cytoplazma je eozinofilní i slabě vakuolizovaná, hemofagocytóza není typická. Některé buňky jsou vícejaderné nebo vyloženě bizarní. Nenádorovou příměs reprezentují malé lymfocyty, neutrofily, eozinofily a blandní histiocyty. V některých případech převládají a vlastní nádorové buňky překryjí. Histologická diagnostika je velmi obtížná. Nádor se totiž v základním barvení podobá jak velkobuněčným lymfomům (ALCL, DLBCL aj.), tak nediferencovanému karcinomu, melanomu, vřetenobuněčnému nebo pleomorfnímu sarkomu. Jedině široký panel protilátek dokáže tyto jednotky vyloučit. Musí být pozitivní alespoň některé histiocytární znaky (CD68, CD163, lysozym, CD4). Komplikované je také odlišení myelosarkomu, v čemž může pomoci klinická anamnéza. Klinicky se tyto histocytární sarkomy chovají velmi agresivně, asi v 70 % je nemoc rozpoznána v generalizovaném stadiu (III a IV), a proto asi 60 % nemocných zemře v průběhu léčby na progresi nemoci.
Pro léčbu diseminonvané formy histiocytárního sarkomu (postaru maligní histiocytózy) se používají stejná cytostatická schémata jako pro léčbu agresivních lymfomů.
Také u maligní histiocytózy či histiocytárního lymfomu lze použít k léčbě cladribin neboli 2-chlorodeoxy-adenosin [43–45]. Ojedinělé případy maligní histiocytózy diagnostikové na našem pracovišti měly vždy velmi agresivní průběh a malou léčebnou odpověď [46].
6. 3. Sarkom z folikulárních dendritických buněk
Sarkom z folikulárních dendritických buněk často (asi ve dvou třetinách případů) tvoří lokalizovanou lymfadenopatii, která má tendenci k lokálním recidivám po léčbě. Méně často vzniká primárně extranodálně, a to v jakékoliv lokalizaci, například v tonzile. Tendence k diseminaci není velká.
Někteří autoři uvádějí, že v 10–20 % je tento typ tumoru asociován s hyalinně-vaskulárním typem Castlemanovy nemoci. Pro sarkom z folikulárních dendritických buněk je typická pomalu rostoucí nádorová masa bez přítomnosti systémových příznaků. Sarkom z folikulárních dendritických buněk se chová indolentně, jako low grade sarkom [47–49].
Nádor je tvořen protáhlými nebo ovoidními buňkami, které rostou ve svazcích, vírech nebo se rohožovitě proplétají. Jádra jsou oválná a chromatin bledý. Buňky nemusí být výrazně atypické, počet mitóz je také variabilní. Mezi vřetenitými buňkami jsou roztroušeny malé lymfocyty. Buňky sarkomu lze při imunohistochemickém vyšetření potvrdit podle markerů folikulárních dendritických buněk (CD21, CD23 a CD35).
Většina nemocných je léčena i vyléčena kompletní chirurgickou resekcí s nebo bez adjuvantní chemoterapie a radioterapie. Lokální recidivy se vyskytují asi v 50 % případů a metastázy asi u 25 % případů. Asi 10–20 % nemocných s tímto typem tumoru mu po delším boji (léčbě) podlehne [50–52].
6. 4. Sarkom z interdigitujících dendritických buněk
Sarkom z interdigitujících dendritických buněk je velmi vzácné onemocnění. Může vznikout primárně v uzlině, ale i kůži a v měkkých tkáních. Byly také popsány různé formy viscerálního postižení. Nemoc se většinou projeví symptomatickou nádorovou masou, klasické B-symptomy jsou popisovány spíše výjimečně [53, 54].
Nádorové buňky jsou vřetenité, s popraškem malých T lymfocytů a plazmocytů. Bez imunohistochemie jej nelze rozpoznat od sarkomu z folikulárních dendritických buněk, CD21 i CD23 jsou však negativní. Pozitivní bývá vimentin, S100 a slabě CD68.
Zásadní pro léčbu je možnost provedení totální resekce. Pokud to není možné, používají se stejné chemoterapeutické režimy jako pro léčbu nehodgkinských lymfomů. Uvádí se, že efekt samotné chemoterapie není tak dobrý, jako je u maligních lymfomů. Transplantace kostní dřeně je proto vždy ke zvážení, pokud není možná radikální operace a odstranění patologické masy. Prognóza této nemoci je v případě nemožnosti radikální operace nepříznivá [55, 56].
Na našem pracovišti jsme léčili mladého muže s interdigitujícím dendritickým sarkomem dolní končetiny, který byl rezistentní ke klasické i vysokodávkované chemoteapii BEAM s podporou autologní transplantace krvetvorných buněk a svému onemocnění nakonec podlehl [57, 58].
7. Hemofagocytární lymfohistiocytóza
7. 1. Patofyziologie hemofagocytárních lymfohistiocytóz
Hemofagocytární lymfohistiocytóza představuje reaktivní zmnožení lymfocytů a histocytů s probíhají hemofagocytózou. Existuje familiární forma této nemoci s prokázanou mutací více než dvou genů, z nichž každý narušuje cytotoxickou funkci NK - a T-buněk. Defekt NK - a T-buněk má klíčovou roli pro uvedenou poruchu. A dále byla definována podobná jednotka – syndrom aktivace makrofágů, včetně diferenciálně diagnostických postupů [59].
Získané formy se mohou vyskytnout u osoby s vrozenou nebo získanou poruchou imunity. Vyvolávající stimulem pak může být infekce. Podmínkou, aby tato získaná forma mohla vzniknout, je však výrazný defekt NK - a T-buněčné imunity.
Hereditární i získané formy se klinicky velmi podobají, a proto se pro ně používá společný termín hemofagocytární lymfohistiocytóza. Společným jmenovatelem je narušená cytotoxická funkce lymfocytů, vedoucí k přetrvávající aktivitě imunitního systému, tedy k proliferaci a akumulaci lymfocytů a histiocytů v postižených orgánech.
Familiární hemofagocytující lymfohistiocytóza je jednotka, u níž byla prokázána mutace různých genů, které jsou důležité pro cytotoxickou funkci T - a NK-buněk.
První byla popsána mutace genu pro perforin, další pak byla mutace genu Munc 13-4, která způsobuje defektní fúzi cytoplazmatických granulí. Následovalo odhalení dalších genů, unc13d, syntaxin 11. Také vrozené defekty imunity predisponují pro tuto nemoc (Chédiak Higashi syndrom, Griscelli syndrom 2 a na X chromozom vázaný lymfoproliferativní syndrom) [60].
V případě získaného hemofagocytárního syndromu byla prokázána excesivní tvorba cytokinů normálními nebo maligními T-lymfocyty. Kontinuálně zvýšená produkce určitých cytokinů pak indukuje hemofagocytání syndrom. Důkazem excesivní imunitní stimulace je zvýšená hladina solubilního receptoru IL-2 u pacientů s aktivní nemocí [61].
Příznaky a jejich příčiny shrnuje tabulka 4.

Charakteristickým nálezem v biopsii lymfatických uzlin je deplece lymfocytů a široké, jakoby prázdné sinusy s velkými makrofágy. Ty při podrobném cytologickém hodnocení vykazují známky aktivace, je zvýšeno množství cytoplazmy a je zřetelná fagocytóza erytrocytů, leukocytů, krevních destiček a jejich framentů. V postižených orgánech je vždy smíšená lymfo-histiocytární infiltrace. Výrazná histiocytární proliferace je zřetelná v celém retikuloendoteliálním systému, nejvíce je postižena kostní dřeň, červená pulpa sleziny, jaterní sinusy a lymfatické uzliny. Infiltrace kostní dřeně je vždy zřetelná u hemofagocytující lymfohistiocytózy spojené s infekcí, ale může být opožděná v případě familiární formy, kdy iniciální histologie kostní dřeně může prokázat hyperplazii červené krvetvorby bez hemofagocytózy. Je proto vhodné bioptovat i jiné tkáně a orgány.
Musíme upozornit, že hemofagocytóza není nález zcela specifický pro HLH. Může doprovázet i jiné stavy s aktivací retikuloendoteliálního systému tj. sepsi, hemolytické anémie, podání krevní transfuze a reakci štěpu vůči hostiteli (GvHD). Diagnóza HLH se musí zakládat na korelaci projevů klinických, biochemických, imunologických a morfologických. V doporučeních je požadováno splnění 5 z 8 vyjmenovaných diagnostických kritérií (viz tabulka 5), hemofagocytóza je pouze jedním z nich.
![Diagnostická kritéria fagocytární lymfohistiocytózy publikovaná 2007 [73]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/f3bdc2425e2f00dd0df4b73c6f478d8c.png)
Podle souvislosti lze hemofagocytární lymfohistiocytózu dělit do tří až čtyř skupin:
- Familiární erytrofagocytární lymfohistiocytóza
- Hemofagocytární lymfohistiocytóza asociovaná s infekcí
- Hemofagocytární lymfohistiocytóza asociovaná s maligní neoplazií
- Hemofagocytární lymfohistiocytóza asociovaná s neznámým vyvolávajícím činitelem [62–64].
7. 2. Familiární erytrofagocytární lymfohistiocytóza (FEL)
FEL je vzácné, často fatální multiorgánové onemocnění, postihující játra, slezinu, lymfatické uzliny a centrální nervový systém. Rodinná anamnéza může být pozitivní, choroba má autozomálně recesivní způsob dědičnosti. Choroba se manifestuje u kojenců a batolat. Projevuje se horečkou nejasného původu, úbytkem na hmotnosti, cytopenií a hepatosplenomegalií. Někdy lze detekovat makulopapulární exantem červeno-fialového zbarvení (eflorescence u LCH bývají žluto-hnědé). FEL se obtížně diagnostikuje, neboť první bioptické vyšetření kostní dřeně nezachytí hemofagocytózu. Ta je přítomna až po delším průběhu nemoci, kdy se objevuje také pancytopenie a žloutenka. Diagnóza nemoci je podpořena průkazem laboratorních změn, vysoké hladiny ferritinu, což je vedle trigliceridů velmi jednoduchý a všude dostupný parametr, lymfohistiocytárních infiltrátů a přítomnosti erytrocytofagocytózy ve vzorcích z lymfatických uzlin, sleziny, jater, kostní dřeně, nebo plic a průkaz familární formy pak na molekulárně genetickém vyšetření [65, 66]. Diagnostická kritéria z roku 2007 uvádí tabulka 5.
Porucha se projeví náhle vzniklými horečkami a postižením výše uvedených orgánů. Možné je i postižení CNS, dezorientace, křeče, porucha vědomí, kóma. Laboratorní vyšetření mohou odhalit hyperlipidemii, hypofibrinogenemii a poruchu buněčné imunity – (snížení aktivity cytotoxicity). Onemocnění je autozomálně recesivně vázané. Průkaz pozitivní rodinné anamnézy může při stanovení diagnózy napomoci. Průběh nemoci je rychlý a velmi často fatální. Brněnští autoři popisují kazuistiku rodiny, v níž první dítě zemřelo v novorozeneckém věku na familiární HLH pod obrazem fulminantního selhání jater. Posmrtně nalezená příčinná mutace genu pro perforin umožnila úspěšné provedení prenatální diagnostiky v dalším těhotenství, z něhož se narodil zdravý sourozenec [67].
Klasickým lékem je etoposid, dále kortikosteroidy, vinblastin a další formy imunosuprese. Při postižení CNS se intratekálně aplikují steroidy a metotrexát. Novým a velmi účinným preparátem, podobně jako u Langerhansovy histiocytózy, je cladribin. S terapeutickým cílem byla u těchto dětí dělána také splenektomie.
Klasickým léčebným protokolem pro hemofagocytující lymfohistiocytózu, který se používá jak u familiární, tak u nefamiliární formy, je trojkombinace složená z etoposidu, dexametazonu a cyklosporinu. Léčebný protokol: www.histio.org/society/protocols se všemi podrobnostmi. U méně rozvinuté dětské formy je možné použít jen kortikoidy a imunoglobuliny. Alogenní transplantace krvetvorné tkáně je indikována v případě selhání léčby 1. linie.
Formou kazuistik byly popsány případy, kdy pomohl daclizumab, nebo alemtuzumab, či etanercept. Dokonce i aplikace anakinry je zmiňována [68–77].
Uvedená cytostatická léčba má potenciál dosáhnout u dětí zpomalení průběhu, nicméně zastavení procesu a vyléčení se uvedenou chemoterapií nepodaří vždy dosáhnout. Jediným zásadním léčebným postupem je alogenní transplantace. Ta je považována za léčbu volby, pokud je vhodný dárce.
7. 3. Sekundární hemofagocytují lymfohistiocytóza
Sekundární hemofagocytující lymfohistiocytóza, asociovaná s infekcí, byla poprvé popsána při virové infekci u imunokompromitovaného pacienta, později i u řady jiných virových, bakteriálních, mykotických a parazitárních onemocnění, ale i maligních onemocnění [78, 79].
Podmínkou vzniku byl stav imunodeficience, a to buď vrozeného, získaného či iatrogenního původu. Sekundární hemofagocytující lymfohistiocytózu lze rovněž pozorovat v souvislosti s některými T-lymfoidními malignitami. Klinická symptomatologie je obdobná jako u familiární erytrofagocytující lymfohistiocytóze. Dominuje horečka, hepatopatie, anémie a koagulopatie. Příčina koagulopatie je zřejmě v infiltraci jater.
Základem pro stanovení diagnózy je biopsie kostní dřeně, v níž jsou benigně vyhlížející histiocytární buňky obsahující fagocytované erytrocyty a další krvinky. Fenotyp a cytochemická charakteristika je shodná s fyziologickými histiocyty a odlišná od maligních histiocytů. Podobný obraz lze nalézt i v uzlinách. V kostní dřeni může být přitom zřetelné zmnožení tvorby jak erytrocytů, tak trombocytů, přičemž v periferní krvi je jich nedostatek a nejsou přítomny specifické protilátky, které by způsobily jejich zánik na autoimunitním podkladě [80]. Velmi podrobný popis případu a hluboký pohled do patofyziologie nemoci uvedli autoři z hematologického pracoviště v Plzni v časopise Vnitřní lékařství 2018 [81].
Sekundární hemofagocytární syndrom tedy není pravou maligní histiocytózou (histiocytárním sarkomem), od které musí být naopak bezpečně odlišen. Uvádíme to proto, že agresivní nástup nemoci může vést ke zmýlení s maligní histiocytózou (genaralizovným histiocytárním sarkomem) [80, 81].
Léčba se zaměřuje na zvládnutí souběžně probíhající infekce, imunodeficitního stavu, případně vyvolávajícího maligního onemocnění. Pokud nemoc vznikne u pacientů na imunosupresi, je to indikací k přerušení imunosuprese.
V případech asociovaných s maligní nemocí je třeba paralelně s intenzivní symptomatickou léčbou HLH léčit i základní maligní onemocnění [82–86].
8. Kikuchi Fujimoto histiocytární nekrotizující lymfadenitida
Tato nemoc není uvedena ve výčtu histocytárních onemocnění ani ve WHO klasifikaci ani v klasifikaci Histiocyte Society. Jde o reaktivní změny. Tato nemoc do kapitoly histiocytárních onemocnění nepatří, ale protože ve svém názvu nese přídavné jmého „histiocytární“, tak ji stručně zmíníme.
Kikuchi Fujimoto histiocytární nekrotizující lymfadenitida je termín pro self limiting cervikální lymfadenopatii nejasného původu. Předpokládá se, že se jedná o postvirální hyperimunní reakci. Je zde možné spojení s lupus erythematodes a s nespefickými hyperimunitními reakcemi na různé vyvolávající příčiny. Klinicky se projeví jako zvětšené uzliny, nejčastěji v oblasti krku, případně spojené s horečkou nejasného původu.
Diagnózu lze stanovit pouze histologicky z exstirpované uzliny. V uzlině jsou přítomny ložiska nekrózy, které mohou splývat, agretáty histiocytů a aktivované lymfocyty.
Léčba této nemoci se odvíjí od tíže příznaků. Lehčí příznaky by měla zvládnout nesteroidní antiflogistika, závažnější průběh s horečkami pak léčba glukokortikoidy. Vzhledem k tomu, že podobný obraz může mít i lupusová lymfadenitida, doporučuje se vždy vyšetření cílené na průkaz systémových chorob pojiva [87–90].
ZÁVĚR
Stručně jsme charakterizovali jednotlivé choroby, které současná WHO klasifikace krevních chorob z roku 2017 řadí do skupiny histiocytárních chorob. Popis jednotlivých z nich je uveden v citované literatuře.
Podíl autorů na přípravě rukopisu
ZA a LP – odpovídají za úhel pohledu klinického lékaře
MJ – odpovídá za úhel pohledu patologa
JN – odpovídá za uvedené zobrazovací vyšetření CT a MR
ZŘ – odpová za uvedené zobrazovací vyšetření pomocí radioizotopů
Čestné prohlášení
Autoři práce prohlašují, že v souvislosti s tématem, vznikem a publikací tohoto článku nejsou ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou.
Doručeno do redakce dne 30. 4. 2018.
Přijato po recenzi dne 12. 11. 2018.
Doc. MUDr. Luděk Pour, Ph.D.
Interní hematologická a onkologická klinika
Fakultní nemocnice Brno
Jihlavská 340/20
625 00 Brno
e-mail: pour.ludek@fnbrno.cz
Zdroje
1. Pileri SA, Jaffe R, Facchetti F, Jones DM, Jaffe ES. Histiocytic and dendritic cell neoplasms. In: Swerdlow SH, Campo E, Harris NL, et al. WHO classification of the tumours of haematopoietic and lymphoid dissue. Revised 4th edition Lyon, International Agency for Research on Cancer 2017 : 465–482.
2. Emile JF, Abla O, Fraitag S, et al. Histiocyte Society. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood 2016;127(22):2672–2681.
3. Vaiselbuh SR, Bryceson YT, Allen CE, et al. Updates on histiocytic disorders. Pediatr Blood Cancer 2014;61(7):1329–1335.
4. Chen M, Ding C, Lu T, et al. Langerhans cell histiocytosis and Erdheim--Chester disease overlap syndrome with bone marrow involvement and type 2 diabetes mellitus. Ann Hematol 2018;97(1):189–192.
5. Novosad O, Skrypets T, Pastushenko Y, et al. MAPK/ERK signal pathway alterations in patients with Langerhans cell histiocytosis. Klin Onkol 2018;31(2):130–136.
6. Diamond EL, Subbiah V, Lockhart AC, et al. Vemurafenib for BRAF V600-Mutant Erdheim-Chester disease and Langerhans cell histiocytosis: Analysis of data from the histology-independent, phase 2, open-label VE-BASKET study. JAMA Oncol 2018;4(3):384–388.
7. Girschikofsky M, Arico M, Castillo D, et al. Management of adult patients with Langerhans cell histiocytosis: recommendations from an expert panel on behalf of Euro-Histio-Net. Orphanet J Rare Dis 2013;8 : 72.
8. Haupt R, Minkov M, Astigarraga I, et al. Euro Histio Network. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer 2013;60(2):175–184.
9. Kolenová A, Bubanská E, Špotová A, et al. Cielená liečba závažnej multisystémovej histiocytózy z Langerhansových buniek. Pediatr Prax 2018;19(1):27–31.
10. Nakamine H, Yamakawa M, Yoshino T, et al. Langerhans cell histiocytosis and Langerhans cell sarcoma: current understanding and differential diagnosis. J Clin Exp Hematop 2016;56(2):109–118.
11. Xu XL, Bu WB, Zong WK, Sun JF. Indeterminate cell histiocytosis: a case series and review of the literature. Eur J Dermatol 2017;27(5):559–561.
12. Rezk SA, Spagnolo DV, Brynes RK, et al. Indeterminate cell tumor: a rare dendritic neoplasm. Am J Surg Pathol 2008;32(12):1868–1876.
13. Adam Z, Ježová M, Šlampa P, et al. Histiocytóza z indeterminovaných buněk – vymizení kožní infiltrace po ozáření elektronovým svazkem a aplikace 2-chlorodeoxyadenozinu: kazuistika. Vnitř Lék 2017;63(4):284–288.
14. Cohen-Aubart F, Emile JF, Carrat F, et al. Phenotypes and survival in Erdheim-Chester disease: Results from a 165-patient cohort. Am J Hematol; publikováno elektronicky 2. února 2018; DOI: 10.1002/ajh.25055.
15. Suzuki H, Wanibuchi M, Komatsu K, et al. Erdheim-Chester disease involving the central nervous system with the unique appearance of a coated vertebral artery. NMC Case Rep J 2016;3(4):125–128.
16. Papo M, Diamond EL, Cohen-Aubart F, et al. High prevalence of myeloid neoplasms in adults with non-Langerhans cell histiocytosis. Blood 2017;130(8):1007–1013.
17. Goyal G, Shah MV, Call TG, Litzow MR, et al. Efficacy of biological agents in the treatment of Erdheim-Chester disease. Br J Haematol; publikováno elektronicky 30. října 2017; DOI:10.1111/bjh.14997.
18. Cohen Aubart F, Emile JF, et al. Targeted therapies in 54 patients with Erdheim-Chester disease, including follow-up after interruption (the LOVE study). Blood 2017;130(11):1377–1380.
19. Adam Z, Petrášová H, Řehák Z, et al. Hodnocení pěti let léčby Erdheimovy-Chesterovy nemoci anakinrou. Vnitř Lék 2016;62(10): 820–832.
20. Adam Z, Řehák Z, Koukalová R, et al. PET-CT dokumentovaná kompletní remise Erdheimovy-Chesterovy nemoci, trvající více než 4 roky od zahájení léčby kladribinem. Vnitř Lék 2014;60(5-6):501–511.
21. Szturz P, Adam Z, Řehák Z, et al. Xanthelasma palpebrarum responding to interleukin-1 blockade. Intern Med J 2014;44(6):617–618.
22. Adam Z, Szturz P, Pour L, et al. Kladribin je vysoce účinný v léčbě histiocytózy z Langerhanosových buněk a vzácných chorob ze skupiny juvelního xantogranulomu. Vnitř Lék 2012;58(6):455–465.
23. Luder CM, Nordmann TM, et al. Histiocytosis – cutaneous manifestations of hematopoietic neoplasm and non-neoplastic histiocytic proliferations. J Eur Acad Dermatol Venereol; publikováno elektronicky 16. ledna 2018; DOI: 10.1111/jdv.14794.
24. Mahendran P, Wee J, Chong H, Natkunarajah J. Necrobiotic xanthogranuloma treated with lenalidomide. Clin Exp Dermatol 2018;43(3):345–347.
25. Sagiv O, Thakar SD, Morrell G, et al. Rituximab monotherapy is effective in treating orbital necrobiotic xanthogranuloma. Ophthal Plast Reconstr Surg 2018;34(1):e24–e27.
26. Techavichit P, Sosothikul D, Chaichana T, et al. BRAF V600E mutation in pediatric intracranial and cranial juvenile xanthogranuloma. Hum Pathol 2017;69 : 118–122.
27. Fölster-Holst R. Severe systemic juvenile xanthogranuloma is an indication for systemic therapy. Br J Dermatol 2017;176(2):302–304.
28. Wruhs M, Feldmann R, Sawetz I, et al. Necrobiotic xanthogranuloma in a patient with multiple myeloma. Case Rep Dermatol 2016;8(3):350–353.
29. Kusumgar P, Vijaya PH, Monappa V. Adult-onset asthma and periocular xanthogranuloma: A rare case report. Can J Ophthalmol 2016;51(6):e168–e171.
30. Klingner M, Hansel G, Schönlebe J, et al. Disseminated necrobiotic xanthogranuloma. Hautarzt 2016;67(11):902–906.
31. Honda Y, Nakamizo S, Dainichi T, et al. Adult-onset asthma and periocular xanthogranuloma associated with IgG4-related disease with infiltration of regulatory T cells. J Eur Acad Dermatol Venereol 2017;31(2):e124–e125.
32. Miguel D, Lukacs J, Illing T, et al. Treatment of necrobiotic xanthogranuloma - a systematic review. J Eur Acad Dermatol Venereol 2017;31(2):221–235.
33. Maintz L, Wenzel J, Irnich M, et al. Successful treatment of systemic juvenile xanthogranulomatosis with cytarabine and 2-chlorodeoxyadenosine: case report and review of the literature. Br J Dermatol 2017;176(2):481–487.
34. Higgins LS, Go RS, Dingli D, et al. Clinical features and treatment outcomes of patients with necrobiotic xanthogranuloma associated with monoclonal gammopathies. Clin Lymphoma Myeloma Leuk 2016;16(8):447–452.
35. Adam Z, Veselý K, Motyčková I, et al. Oční víčka se žlutými granulomy a kašel. Periokulární xanthogranulom asociovaný s adult-onset astmatem. Popis případu a přehled klinických forem juvenilního xantogranulomu. Vnitř Lék 2012;58(5):365–377.
36. Adam Z, Zahradová L, Krejčí M, et al. Difúzní ploché normolipemické xanthomatomy a nekrobiotický xantogranulom asociovaný s monoklonální gamapatií. Vnitř Lék 2010;56(11):1158–1168.
37. Ranganathan S. Histiocytic proliferations. Semin Diagn Pathol 2016;33(6):396–409.
38. Adam Z, Koukalová R, Řehák Z, Čermák A, Krejčí M, Pour L. Neinfekční nemaligní lymfadenopatie – sinusová histiocytóza s masivní lymfadenopatií, nemoc Rosaiova-Dorfmanova. Transfuze Hematol dnes 2018;24(3):166–173.
39. Huhn D. Therapy of malignant histiocytosis. Haematol Blood Transfus 1981;27 : 211–216.
40. Kommalapati A, Tella SH, Durkin M, et al. Histiocytic sarcoma: a population-based analysis of incidence, demographic disparities, and long-term outcomes. Blood 2018;131(2):265–268.
41. Jiang M, Bennani NN, Feldman AL. Lymphoma classification update: T-cell lymphomas, Hodgkin lymphomas, and histiocytic/dendritic cell neoplasms. Expert Rev Hematol 2017;10(3):239–249.
42. Vokuhl C, Oschlies I, Klapper W, et al. Histiocytic diseases in child-hood and adolescence. Pathologe 2015;36(5):443–450.
43. Magro CM, Kazi N, Sisinger AE. Primary cutaneous histiocytic sarcoma: A report of five cases with primary cutaneous involvement and review of the literature. Ann Diagn Pathol 2018;32 : 56–62.
44. Voruz S, Cairoli A, Naveiras O, et al. Response to MEK inhibition with trametinib and tyrosine kinase inhibition with imatinib in multifocal histiocytic sarcoma. Haematologica 2018;103(1):e39–e41.
45. Iwabuchi H, Kawashima H, Umezu H, et al. Successful treatment of histiocytic sarcoma with cladribine and high-dose cytosine arabinoside in a child. Int J Hematol 2017;106(2):299–303.
46. Létalová E, Moulis M, Klincová M, et al. Histiocytární sarkom. Vnitř Lék 2013;59(12):1117–1122.
47. Chen T, Gopal P. Follicular dendritic cell sarcoma. Arch Pathol Lab Med 2017;141(4):596–599.
48. Facchetti F, Lorenzi L. Follicular dendritic cells and related sarcoma. Semin Diagn Pathol 2016;33(5):262–276.
49. Li J, Zhou ML, Zhou SH. Clinical and pathological features of head and neck follicular dendritic cell sarcoma. Hematology 2015;20(10):571–583.
50. Jain P, Milgrom SA, Patel KP, et al. Characteristics, management, and outcomes of patients with follicular dendritic cell sarcoma. Br J Haematol 2017;178(3):403–412.
51. Sasaki M, Izumi H, Yokoyama T, et al. Follicular dendritic cell sarcoma treated with a variety of chemotherapy. Hematol Oncol 2017;35(4): 905–908.
52. Purkait S, Mallick S, Joshi PP, et al. Retroperitoneal and mediastinal follicular dendritic cell sarcoma: report of 3 cases with review of literature. Hematol Oncol 2017;35(3):374–379.
53. Ninkovic S, Cole-Sinclair MF. Interdigitating dendritic cell sarcoma: diagnostic pitfalls, treatment challenges and role of transdifferentation in pathogenesis. Pathology 2017;49(6):643–646.
54. Nguyen CM, Cassarino D. Primary cutaneous interdigitating dendritic cell sarcoma: a case report and review of the literature. Am J Dermatopathol 2016;38(8):628–631.
55. Pokuri VK, Merzianu M, Gandhi S, et al. Interdigitating dendritic cell sarcoma. J Natl Compr Canc Netw 2015;13(2):128–132.
56. Di Liso E, Pennelli N, Lodovichetti G, et al. Braf mutation in interdigitating dendritic cell sarcoma: a case report and review of the literature. Cancer Biol Ther 2015;16(8):1128–1135.
57. Adam Z, Pour L, Veselý K, et al. Interdigitating dendritic cell sarcoma of the leg. Onkologie 2009;32(6):364–365.
58. Adam Z, Veselý K, Krejčí M, et al. Interdigitující dendritický sarkom dolní končetiny rezistentní k vysokodávkované chemoterapii BEAM s transplantací autologních hemopoetických kmenových buněk. Vnitř Lék 2009;55(2):147–157.
59. Minoia F, Bovis F, Davì S, et al. Development and initial validation of the macrophage activation syndrome/primary hemophagocytic lymphohistiocytosis score, a diagnostic tool that differentiates primary hemophagocytic lymphohistiocytosis from macrophage activation syndrome. J Pediatr 2017;189 : 72–78.
60. Janka GE, Lehmberg K. Hemophagocytic syndromes – an update. Blood Rev 2014;28(4):135–142.
61. Lehmberg K, Sprekels B, Nichols KE, et al. Malignancy-associated haemophagocytic lymphohistiocytosis in children and adolescents. Br J Haematol 2015;170(4):539–549.
62. Ruscitti P, Rago C, Breda L, et al. Macrophage activation syndrome in Still‘s disease: analysis of clinical characteristics and survival in paediatric and adult patients. Clin Rheumatol 2017;36(12):2839–2845.
63. Grom AA. Primary hemophagocytic lymphohistiocytosis and macrophage activation syndrome: the importance of timely clinical differentiation. J Pediatr 2017;189 : 19–21.
64. Otrock ZK, Daver N, Kantarjian HM, Eby CS. Diagnostic challenges of hemophagocytic lymphohistiocytosis. Clin Lymphoma Myeloma Leuk 2017;17S:S105–S110.
65. Ammann S, Lehmberg K, Zur Stadt U, et al. Effective immunological guidance of genetic analysesi exome sequencing in patients evaluated for hemophagocytic lymphohistiocytosis. J Clin Immunol 2017;37(8):770–780.
66. Al-Samkari H, Berliner N. Hemophagocytic lymphohistiocytosis. Annu Rev Pathol 2018;13 : 27–49.
67. Henter JI, Horne A, Aricó M, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007;48(2):124–131.
68. Saarela M, Senthil K, Jones J, et al. Hemophagocytic lymphohistiocytosis in 2 patients with multiple sclerosis treated with alemtuzumab. Neurology 2018;90(18):849–851.
69. Marsh RA, Allen CE, McClain KL, et al. Salvage therapy of refractory hemophagocytic lymphohistiocytosis with alemtuzumab. Pediatr Blood Cancer 2013;60(1):101–109.
70. Machaczka M, Vaktnäs J, Chiang SC, et al. Alemtuzumab treatment for hemophagocytic lymphohistiocytosis. Nat Rev Clin Oncol 2010;7(10):596.
71. Strout MP, Seropian S, Berliner N. Alemtuzumab as a bridge to allogeneic SCT in atypical hemophagocytic lymphohistiocytosis. Nat Rev Clin Oncol 2010;7(7):415–420.
72. Olin RL, Nichols KE, Naghashpour M, et al. Successful use of the anti-CD25 antibody daclizumab in an adult patient with hemophagocytic lymphohistiocytosis. Am J Hematol 2008;83(9):747–749.
73. Makay B, Yilmaz S, Türkyilmaz Z, et al. Etanercept for therapy-resistant macrophage activation syndrome. Pediatr Blood Cancer 2008;50(2):419 – 421.
74. Takahashi N, Naniwa T, Banno S. Successful use of etanercept in the treatment of acute lupus hemophagocytic syndrome. Mod Rheumatol 2008;18(1):72–75.
75. Kikuchi H, Yamamoto T, Asako K, et al. Etanercept for the treatment of intractable hemophagocytic syndrome with systemic lupus erythematosus. Mod Rheumatol 2012;22(2):308–311.
76. Ramanan AV, Schneider R. Macrophage activation syndrome fol-lowing initiation of etanercept in a child with systemic onset juvenile rheumatoid arthritis. J Rheumatol 2003;30(2):401–403.
77. Divithotawela C, Garrett P, Westall G, et al. Successful treatment of cytomegalovirus associated hemophagocytic lymphohistiocytosis with the interleukin 1 inhibitor – anakinra. Respirol Case Rep 2015;4(1):4–6.
78. Ježová M, Gaillyová R. Familiární hemofagocytující lymfohistiocytóza: od autopsie k prenatální diagnóze. Kazuistika. Cesk Patol 2017;53(1):29–34.
79. Wang H, Xiong L, Tang W, Zhou Y, Li F. A systematic review of malignancy-associated hemophagocytic lymphohistiocytosis that needs more attentions. Oncotarget 2017;8(35):59977–59985.
80. Daver N, McClain K, Allen CE, et al. A consensus review on malignancy-associated hemophagocytic lymphohistiocytosis in adults. Cancer 2017;123(17):3229–3240.
81. Šrámek J, Karvunidis T, Lysák D, Harazim M, Karas M, Jindra P. Hemofagocytární lymfohistiocytóza u dospělých – review a kazuistika. Vnitřní Lék 2018;64(3):300–307.
82. Saevels K, Robert D, Van den Broeck S, et al. EBV-associated hemophagocytic lymphohistiocytosis complicated by severe coagulation disorders and opportunistic infections: case report of a survivor. Clin Case Rep 2017;6(1):115–118.
83. Bergsten E, Horne A, Aricó M, et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long-term results of the cooperative HLH-2004 study. Blood 2017;130(25):2728–2738.
84. Zandvakili I, Conboy CB, Ayed AO, et al. Ruxolitinib as first-line treatment in secondary hemophagocytic lymphohistiocytosis: A second experience. Am J Hematol; publikováno elektronicky 8. února 2018; DOI: 10.1002/ajh.25063.
85. Broglie L, Pommert L, Rao S, et al. Ruxolitinib for treatment of refractory hemophagocytic lymphohistiocytosis. Blood Adv 2017;1(19):1533–1536.
86. Lorenz F, Klimkowska M, Pawłowicz E, et al. Clinical characteristics, therapy response, and outcome of 51 adult patients with hematological malignancy-associated hemophagocytic lymphohistiocytosis: a single institution experience. Leuk Lymphoma 2018;3 : 1–11.
87. Pepe F, Disma S, Teodoro C, et al. Kikuchi-Fujimoto disease: a clinicopathologic update. Pathologica 2016;108(3):120–129.
88. Mathew LM, Kapila R, Schwartz RA. Kikuchi-Fujimoto disease: a diagnostic dilemma. Int J Dermatol 2016;55(10):1069–1075.
89. Cuglievan B, Miranda RN. Kikuchi-Fujimoto disease. Blood 2017;129(7):917–918.
90. Szturz P, Adam Z, Chovancová J, et al. Cytokine analysis in a patient with relapsing Kikuchi-Fujimoto disease. Leuk Lymphoma 2012;53(4):743–745.
Štítky
Hematologie a transfuzní lékařství Interní lékařství OnkologieČlánek vyšel v časopise
Transfuze a hematologie dnes

2019 Číslo 2
- Profesionální expozice azbestu a riziko vzniku karcinomu plic
- Aktuální možnosti léčby mnohočetného myelomu
- Proces hojení ran krok za krokem a co ho může zkomplikovat
- Mutace BRCA1/2 nezvyšuje vliv perorální antikoncepce na riziko karcinomu prsa a ovarií
- Limity glykovaného hemoglobinu a význam dalších glykovaných proteinů
Nejčtenější v tomto čísle
- Transplantácia krvotvorných buniek u pacientov s myelodysplastickým syndrómom – skúsenosti jedného centra
- Přehled maligních chorob odvozených od histiocytárních a dendritických buněk
- Cytológia imprintov lymfatických uzlín: jedenásťročné skúsenosti
- Analýza fluktuace dárců krve mezi státním zařízením transfuzní služby a soukromým plazmaferetickým centrem
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy