
Klinický význam génových mutácií u akútnych myeloidných leukémií s normálnym karyotypom
Clinical relevance of gene mutations in acute myeloid leukemia with normal karyotype
Acute myeloid leukemia (AML) is characterized by distinct clinical and genetic heterogeneity. Chromosomal aberrations are present in about 55% of patients at the time of diagnosis however a large group of AML (about 45%) comprise patients with normal karyotype (NK-AML). In the latter group, gene mutations and changes in expression profiles enable us to allocate subsets with different prognosis. In NK-AML specific mutation profiles are associated with worse prognosis (FLT3-ITD, MLL-PTD mutations) or mark for better prognosis (isolated NPM1 and CEBPA mutations, NPM1mutated AML without FLT3-ITD). Treatment decisions based on molecular stratification however, remain controversial. This review assesses the current insight on the prognostic and therapeutic significance of gene mutations in NK-AML.
Key words:
AML, normal karyotype, mutation, molecular markers, FLT3, NPM1, CEBPA, WT1, IDH1/2
Autoři:
B. Katrincsáková; T. Szotkowski; M. Divoká; K. Indrák; M. Jarošová
Působiště autorů:
Hemato-onkologická klinika Fakultní nemocnice Olomouc
Vyšlo v časopise:
Transfuze Hematol. dnes,17, 2011, No. 2, p. 72-80.
Kategorie:
Souhrnné práce, původní práce, kazuistiky
Souhrn
Akútne myeloidné leukémie (AML) sa vyznačujú výraznou klinickou i genetickou heterogenitou. Asi 55 % AML charakterizujú v dobe diagnózy chromozomálne aberácie, ale obsiahlu skupinu (asi 45 %) predstavujú pacienti s normálnym karyotypom (NK). Na základe génových mutácií a zmien expresného profilu je dnes možné vyčleniť v rámci AML s NK podskupiny pacientov s odlišnou prognózou. Špecifické mutačné profily sú u AML s NK spojené s horšou prognózou (mutácie FLT3-ITD, MLL-PTD) alebo sú naopak známkou lepšej prognózy (izolované mutácie NPM1 a CEBPA, NPM1mutované AML bez FLT3-ITD). Liečba na základe molekulárnej stratifikácie však ostáva zatiaľ kontroverzná. Tento prehľad hodnotí súčasný pohľad na prognostický a terapeutický význam génových mutácií u AML s NK.
Kľúčové slová:
AML, normálny karyotyp, mutácie, molekulárne markery, FLT3, NPM1, CEBPA, WT1, IDH1/2
Úvod
Akútne myeloidné leukémie (AML) predstavujú klinicky, morfologicky, imunologicky a geneticky heterogénnu skupinu hematologických malignít, ktoré charakterizuje klonálna proliferácia nezrelých krvotvorných buniek. Napriek významným pokrokom v diagnostike a liečbe patria AML, s výnimkou akútnych promyelocytárnych leukémií (APL), stále k prognosticky najmenej priaznivým hematologickým diagnózam. Dlhodobo prežíva asi 40 % mladších (< 60 rokov) a iba 10 % starších (≥ 60 rokov) pacientov (1). Vyšší vek (> 60–65 rokov) je u AML obecne známkou horšej prognózy, poznanie ďalších prognosticky významných faktorov je však v prípade tejto závažnej skupiny onkologických ochorení nevyhnutné. Diagnostika a následná klasifikácia si v prípade AML vyžaduje komplexné zhodnotenie morfológie, imunofenotypu, cytogenetiky a molekulárnej genetiky leukemických buniek (2).
Nenáhodné, klonálne chromozomálne zmeny sa vyskytujú asi u 55 % AML dospelého veku. Pacienti sa v závislosti na prítomnosti či neprítomnosti cytogenetických abnormalít radia do priaznivej, nepriaznivej a intermediárnej rizikovej skupiny. Priaznivou prognózou sa vyznačujú AML s cytogenetickým nálezom t(15;17)(q22;q12)/PML-RARA, t(8;21)(q22;q22)/RUNX1-RUNX1T1 a inv(16)(p13.1q22) alebo t(16;16)(p13.1;q22)/CBFB-MYH11 (2). Pacienti s t(15;17), ktoré sú špecificky prítomné u APL, bývajú vďaka odlišným liečebným výsledkom dokonca vyčleňovaní zo skupiny s dobrou prognózou do samostatnej skupiny s veľmi priaznivou prognózou. Nález iných numerických a štrukturálnych cytogenetických abnormalít, vrátane monozómií chromozómov 5q, 7q, delécie celého chromozómu 7, prestavieb MLL/11q23 (s výnimkou t(9;11)(p22;q23)/ /MLL-AF9) a komplexný karyotyp (nález ≥ 3 klonálnych cytogenetických abnormalít) býva u AML známkou nepriaznivej prognózy. Mimoriadne početnú cytogenetickú skupinu AML (asi 45 %) predstavujú pacienti, u ktorých metódy klasickej cytogenetiky neumožňujú identifikovať žiadne zmeny karyotypu. Cytogenetický nález je v prípade AML s normálnym karyotypom (NK) prognosticky nevýznamný a radí týchto pacientov uniformne do kategórie so strednou prognózou.
Pohľad na prognózu strednej cytogenetickej skupiny AML sa pritom v poslednej dobe významne prehodnotil. Podstatne tomu napomohli pomerne nedávno identifikované molekulárne genetické zmeny u AML s NK, ktoré dnes predstavujú molekulárne najlepšie charakterizovanú skupinu AML. Poznáme somatické mutácie viacerých génov, ktoré pomáhajú odhaľovať dosiaľ nepoznané molekulárne profily predovšetkým v rámci AML s NK (3). Tieto poznatky, ktoré rozširujú naše chápanie leukemogenézy, postupne prenikajú do klasifikácie AML a mali by nám pomáhať pri identifikácii skupín AML s priaznivým a menej priaznivým rizikom. S pribúdaním molekulárnych dát sa však pomerne zložito hodnotí otázka konkurencie viacerých mutácií u jedného pacienta, ktoré v rámci AML s NK pozorujeme čoraz častejšie. Génové mutácie neustále pribúdajú, ale význam komplexných mutačných profilov ostáva v klinickej praxi zatiaľ často kontroverzný. I napriek mnohým otázkam sme však nedávno zaznamenali významný pokrok v klasifikácii AML. Najnovšie kritériá definované WHO klasifikáciou nádorov krvotvorby a lymfoidných tkanív z roku 2008 začali u AML významne zohľadňovať nielen diagnostické cytogenetické zmeny, ale dokonca i vybrané génové mutácie. Nová WHO klasifikácia rozlišuje v rámci primárnej AML dve predbežné molekulárne podjednotky – AML s mutovaným NPM1 a AML s mutovaným CEBPA (4). Ďalší prínos poznania detailného molekulárneho profilu AML spočíva v možnosti sledovania diagnostických génových mutácií v post-terapeutickom období pri monitorovaní minimálnej reziduálnej choroby (MRD), čo má obrovský význam práve u AML s NK.
Cieľom tohto prehľadu je zhrnúť aktuálne poznatky o význame génových mutácií u AML s NK a zhodnotiť ich i) prognostický význam, ii) možnosti uplatnenia na úrovni monitorovania minimálnej reziduálnej choroby a iii) význam pri voľbe liečebného postupu, vrátane cielenej terapie.
Patogenéza génových mutácií u AML s normálnym karyotypom
Somatické mutácie nachádzame u AML s NK najčastejšie v génoch FLT3 (Fms-related tyrosine kinase 3), NPM1 (nucleophosmin), CEBPA (CCAAT/enhancer binding protein alpha), MLL (mixed-lineage leukemia), RUNX1 (runt-related transcription factor 1), NRAS (neuroblastoma RAS viral oncogene) a WT1 (Wilmęs tumor 1) (5). Akumulácia získaných somatických mutácií v krvotvorných bunkách je u AML dlhodobo známa. Mnohostupňový proces leukemogenézy sprevádzajú spravidla 2 typy génových mutácií. Z nich prvý typ (typ I) zahŕňa mutácie, ktoré aktivujú signálne dráhy, čím sa leukemické bunky selekčne zvýhodňujú a následne sú schopné efektívnejšej proliferácie, či prežívania. Mutácie typu I sa na procese leukemogenézy podieľajú spoločne s mutáciami v transkripčných faktoroch a zložkách transkripčného ko-aktivačného komplexu (mutácie typu II). Mutácie typu II zodpovedajú za nevyhnutnú poruchu v diferenciácii krvotvorných buniek, čo je primárny znak AML. Hoci sa všetky hore uvedené mutácie vyskytujú prednostne u AML s NK, objavujú sa i u AML s cytogenetickými abnormalitami.
Mutácie v géne FLT3 (Fms-related tyrosine kinase 3, 13q12.2)
Úloha receptorovej tyrozínkinázy FLT3 v patogenéze AML sa diskutovala v tomto časopise pomerne nedávno (6). Ide o najčastejšie mutovaný gén u AML s výskytom mutácií asi u 40–45 % AML s NK. Mutácie FLT3 sa prednostne sústreďujú v 2 funkčných doménach enzýmu, juxtamembránovej (JMD) a tyrozínkinázovej (TKD), zodpovedajú za konštitutívne aktívnu signálnu dráhu a prejavujú sa nekontrolovanou proliferáciou krvotvorných buniek (obr. 1A). Z nich interná tandemová duplikácia génu FLT3 (FLT3-ITD) v oblasti JMD je prítomná u významnej skupiny (~ 30–40 %) AML s NK. Duplikácie postihujú prevažne oblasť 14. exónu FLT3, miesto inzercie a dĺžka duplikovanej génovej oblasti sa však významne líšia medzi pacientmi, u ktorých sa nezriedka pozoruje i prítomnosť viacpočetných mutovaných alel vo variabilnom kvantitatívnom zastúpení (obr. 1 B, C). Biologická podstata vysokých diagnostických hladín FLT3-ITD u časti pacientov sa objasňuje mechanizmom uniparentálnej dizómie chromozómu 13, ktorá môže už v počiatočnom štádiu leukemogenézy viesť k vzniku leukemického klonu s prevahou mutovaného FLT3. V priebehu progresie AML dokáže identický efekt navodiť mechanizmus mitotickej rekombinácie a vysvetliť homozygotný mutačný status u pacienta, ktorý sa pôvodne vyznačoval heterozygotným zastúpením FLT3-ITD.
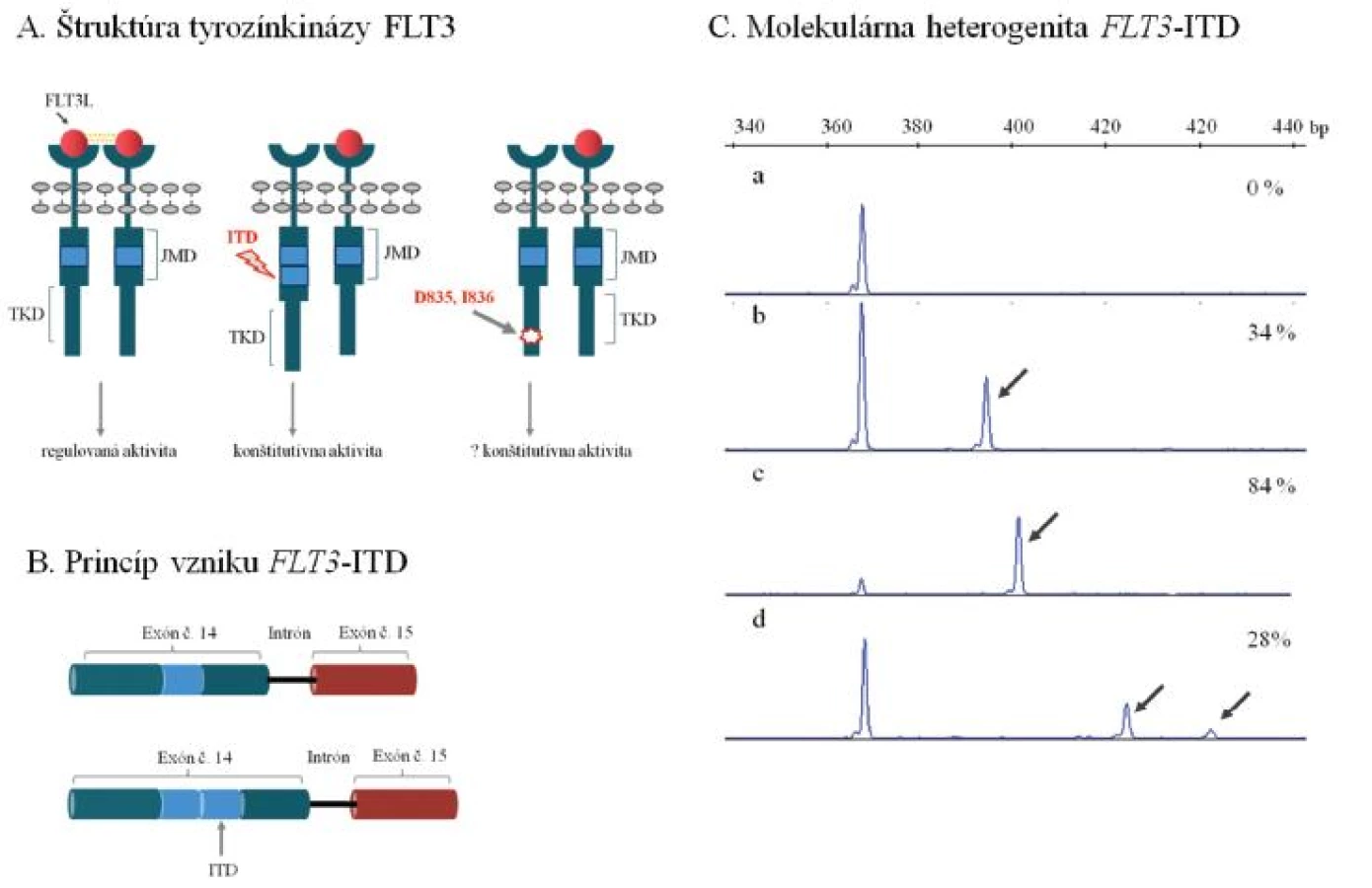
Klinický význam FLT3-ITD, vrátane štúdií detailne zohľadňujúcich jeho mimoriadnu genetickú variabilitu bol dosiaľ predmetom početných štúdií na rôzne obsiahlych súboroch zahŕňajúcich i AML s NK. Nález FLT3-ITD je u AML obecne uznávaným nezávislým nepriaznivým ukazovateľom prežitia bez príznakov ochorenia a celkového prežitia, spája sa s chemorezistenciou a zvýšeným rizikom relapsu (7, 8).
Zatiaľ sa jednotne neuzavrel klinický význam inter-individuálnej variability v mieste inzercie, dĺžke alebo celkovom počte duplikovaných mutovaných alel. Z biologického hľadiska je zaujímavé, že veľkosť tandemovo duplikovanej oblasti by mohla významne ovplyvňovať interakciu medzi juxtamembránovou doménou a aktivačnou slučkou tyrozínkinázy FLT3 tým, že od seba rôzne vzďaľuje tieto 2 kľúčové domény enzýmu a znemožňuje mu tým nadobudnúť inaktívnu konformáciu. Za predpokladu, že úmerne veľkosti ITD by dochádzalo (iba) k čiastočnej až absolútnej konštitutívnej aktivácii tyrozínkinázy, pacienti s dlhším ITD by mohli predstavovať klinicky vysoko rizikovú skupinu (9, obr. 1 A, C).
Pacienti s homozygotným mutačným statusom sa u AML s FLT3-ITD vyznačujú najhoršími klinickými výsledkami (9, 10). Horším prežitím bez príznakov ochorenia a celkovo horším prežitím sa v rámci tejto skupiny AML obecne vyznačujú pacienti s vysokým zastúpením FLT3-ITD alely (prevažne > 50 % z celkovej distribúcie FLT3 alel) (8, 9).
Klinický význam nízkych diagnostických hladín FLT3-ITD však už nie je jednotne stanovený. Vzhľadom na lepšiu prognózu navrhli Thiede a kolektív odlišovať týchto pacientov od skupiny s vysokým zastúpením FLT3-ITD (7). V silnom protipóle tomuto názoru stoja dôkazy o potenciálnej chemorezistencii FLT3-ITD pozitívnych leukemických buniek (7, 8). Táto selekčná výhoda môže pri konvenčnej (chemo)terapii zvyšovať riziko relapsu, ktorý by sa prejavil nárastom počtu mutovaných buniek, nezávisle na minoritnom zastúpení buniek s ITD pri diagnóze. Tento trend potvrdili nezávislé štúdie, ktoré sledovali FLT3-ITD v post-terapeutickom období a paralelne analyzovali vzorky pacientov z doby diagnózy a relapsu (11, 12). Minoritné/nízke diagnostické zastúpenie ITD (1–24 %) by sa na podklade týchto pozorovaní nemalo vylučovať z prognostickej stratifikácie AML (9). Na druhej strane, zmieňovaná evolúcia FLT3-ITD v priebehu progresie AML radí FLT3-ITD medzi nestabilné faktory klinického stavu, čo je nutné dôkladne zvažovať v prípade sledovania tejto mutácie ako markeru MRD v post-terapeutickom období. U významnej skupiny pacientov (4 až 27 %) nie je možné potvrdiť diagnostický nález FLT3-ITD pri relapse AML, prípadne sa FLT3-ITD vyskytuje v podobe, ktorá sa líši od diagnostického nálezu (11, 12, 13).
Ďalší typ známych mutácií FLT3 predstavujú u AML bodové mutácie v aktivačnej slučke druhej tyrozínkinázovej domény (tzv. mutácie FLT3-TKD), ktoré sa vyskytujú asi u 5–14 % AML s NK (3, obr. 1A). Hoci tieto mutácie taktiež aberantne ovplyvňujú aktivitu enzýmu, ich klinický význam je v porovnaní s FLT3-ITD kontroverzný a významne závislý na ďalších kooperujúcich mutáciách (14, 15).
Naproti tomu vysoké riziko relapsu (> 80 %) pri štandardnej terapii je u AML s FLT3-ITD obecne známe a predurčuje pacientov s FLT3-ITD za kandidátov na intenzívnu liečbu, ktorá zahŕňa v prípade dostupnosti vhodného darcu alogénnu transplantáciu krvotvorných kmeňových buniek (alloHSCT), prípadne klinické štúdie smerované k experimentálnejšej terapii, vrátane inhibície enzymatickej aktivity FLT3 (16). Vieme, že alloHSCT významne zlepšuje prežívanie u AML, nevynímajúc AML s FLT3-ITD. Dosiaľ sa však nepodarilo potvrdiť, že alloHSCT v 1. kompletnej remisii dokáže prekonať nepriaznivú prognózu, ktorá je spájaná s diagnostickým nálezom FLT3-ITD (17, 18). Dôkazy v tomto smere nepriniesla ani nedávna štúdia, ktorá hodnotila skupinu 94 mladších (≤ 60 rokov) pacientov s NK, ktorí prekonali relaps AML po indukčnej terapii. Diagnostický nález FLT3-ITD a vyšší vek si v tejto štúdii zachovali nepriaznivý vplyv na prežitie i po intenzívnej salvážovej terapii, ktorá po relapse AML zahŕňala re-indukčnú liečbu alebo alloHSCT (19).
V posledných rokoch sa sústredila značná pozornosť na vývoj FLT3-tyrozínkinázových inhibítorov (FLT3I), ktoré by dokázali regulovať aberantný enzým a zabrániť permanentnej aktivite signálnej dráhy. Na základe dostupných výsledkov klinických štúdií však krátkodobé remisie navodené FLT3I rozhodne nesplnili úvodné očakávania. Napriek obmedzenému klinickému efektu, ktorý boli schopné navodiť na úrovni monoterapie sa však ďalej uvažuje o aplikácii FLT3I v kombinácii s bežnými chemoterapeutikami, čím by sa mohla dosiahnuť lepšia efektivita FLT3I pri liečbe AML (20).
Mutácie v géne NPM1 (Nucleophosmin, 5q35.1)
Mutácie génu NPM1 (NPM1m) predstavujú jednu z najčastejších molekulárnych zmien u AML dospelého veku. Vyskytujú sa u 55 % AML s NK (21). Dosiaľ boli popísané rôzne, prevažne 4-nukleotidové posunové mutácie, ktoré sa sústreďujú výlučne v 12. exóne NPM1 (22, obr. 2 A, B, C). Mutácie vedú k abnormálnej lokalizácii proteínu NPM1 do cytoplazmy buniek, čo je dôsledok straty jadrového lokalizačného signálu, prípadne vzniku nového jadrového exportného signálu (21). Vďaka unikátnemu klinickému a molekulárnemu profilu zohľadňuje nová WHO klasifikácia NPM1m AML v samostatnej, provizórnej kategórii (4). Na priaznivý prognostický význam NPM1m pôvodne upozornili Schnittger a kol. na súbore 401 AML s NK, ktorý zahŕňal pacientov s primárnou i sekundárnou AML bez vekovej hranice (23). Ich závery potvrdili nezávislé štúdie na súboroch AML s NK vo vybranej skupine mladších (≤ 60 rokov) pacientov a celkom nedávno sa poukázalo na menej výrazný, ale stále priaznivý prognostický význam izolovaných NPM1m u AML staršieho (> 60 rokov) veku (21, 22, 24).

Asi u 40 % NPM1m AML sa pozorujú paralelné mutácie v géne FLT3, predovšetkým FLT3-ITD.Prítomnosť FLT3-ITDvýznamne zhoršuje priaznivú prognózu NPM1m AML (16).Naproti tomu Döhner a kolektív poukázali u pacientov s NPM1m AML bez FLT3-ITD na 60% pravdepodobnosť prežitia 5 rokov od diagnózy, čo je porovnateľné s odhadovaným prežitím CEBPA mutovaných AML s NK a dokonca s prežitím „core binding factor“ (CBF) leukémií, u ktorých je priaznivá prognóza dlhodobo známa (25). Začalo sa preto dokonca zvažovať preradenie AML s izolovanými NPM1m do priaznivej rizikovej skupiny AML (2, 16).Pilotné štúdie zamerané na NPM1m AML bez FLT3-ITD(25) ukázali, že alloHSCT v 1. línii liečby neprináša u týchto pacientov zlepšenie liečebných výsledkov v porovnaní s pacientmi, ktorí podstúpili konvenčnú chemoterapiu bez alloHSCT. Ďalšie štúdie na súbore mladších (≤ 60 rokov) AML taktiež nepotvrdili prínos alloHSCT v skupine AML s izolovaným NPM1m genotypom (tj. bez FLT3-ITD a bez MLL-PTD) (16).
Vzhľadom na častý výskyt a predovšetkým stabilitu sa NPM1m radia medzi perspektívne markery MRD, čo predstavuje prínos u významnej skupiny AML s NK (21). Zastúpenie NPM1m reziduálnych buniek sa úspešne monitoruje na báze kvantitatívnych techník založených na metóde polymerázovej reťazovej reakcie (26).
V súvislosti s terapiou NPM1m AML sa uvažuje o cielenom zásahu priamo voči narušenému mechanizmu transportu mutovaného proteínu (21). Úvodné sľubné účinky kombinácie indukčnej terapie s kyselinou all-trans retinovou (ATRA) u NPM1mAML (27) sa nepodarilo potvrdiť v nezávislých štúdiách, ktoré prebehlina obsiahlejších súboroch(28), hoci sa nedá vylúčiť, že tieto odporujúce výsledky sú dôsledkom analýzy súboru mladších (do 60 rokov) pacientov, ktorí sa vyznačujú vyššími hodnotami dosiahnutých kompletných remisií nezávisle na efekte ATRA.
Mutácie v géne CEBPA (CCAAT/enhancer binding protein alpha, 19q13.11)
Transkripčný faktor CEBPA predstavuje ďalší významný proteín, ktorý je v rámci regulácie krvotvorby zodpovedný za líniovú špecifikáciu a diferenciáciu multipotentných myeloidných progenitorov v zrelé granulocyty (29). Deficit CEBPA vedie u myší k diferenciačnému bloku na úrovni nezrelých granulocytov, čo je špecifický znak AML (30, 31). Proteín (izoforma p42) pozostáva z C-terminálnej DNA-väzobnej a dimerizačnej (bZIP) domény a 2 ďalších N-terminálnych trans-aktivačných domén (TAD1 a TAD2). Kódujúcu oblasť génu predstavuje jediný exón s prevahou GC bohatých (> 70 %) oblastí (32). Mutácie CEBPA sa pozorujú asi u 10 % AML s NK (33). Vyskytujú sa po celej kódujúcej oblasti génu a rozlišujú sa na N-terminálne, posunové mutácie, ktoré vedú k tvorbe skrátenej proteínovej izoformy (p30) a na C-terminálne mutácie, ktoré v dôsledku delécií či inzercií porušujú homo - a heterodimerizáciu, blokujú väzbu na DNA a v konečnom dôsledku zabraňujú aktivácii granulocytovo-špecifických cieľových génov (32). Je známe, že významná časť pacientov nesie dokonca 2 rôzne mutácie CEBPA (biCEBPAm). Pomerne často sa v týchto prípadoch popisujú kombinácieN-terminálnych mutácií na jednej alele s C-terminálnymi mutáciami na ďalšej alele génu.
Vo WHO klasifikácii predstavuje mutačný profil CEBPA jednu z dvoch nových samostatných molekulárnych podskupín AML (4). V rozpore s úvodnými výsledkami, ktoré poukazovali na celkovo priaznivý prognostický význam mutovaného profilu CEBPA u AML s NK, recentné štúdie na obsiahlych súboroch zhodne potvrdzujú priaznivý efekt CEBPA mutácií, ale výlučne v prípade biCEBPAm AML (34, 35). Na základe štúdií expresného profilu formujú tieto AML unikátnu molekulárnu podskupinu so špecifickým profilom génovej expresie (36). V porovnaní so skupinou AML s jednou mutáciou CEBPA (CEBPAm) sú biCEBPAm AML charakteristické menej častým výskytom ďalších, konkurentných mutácií. Podľa dostupných výsledkov multi-variabilných analýz nevykazuje nález CEBPAm/biCEBPAm v skupine NPM1m AML ďalší benefit pre prognózu tejto (priaznivej rizikovej) skupiny AML. Podobne ako v skupine NPM1m AML sa však i u CEBPAm/biCEBPAm AML poukazuje na význam FLT3-ITD. Nepriaznivý účinok FLT3-ITD na prognózu CEBPAm/biCEBPAm AML však zatiaľ nie je jednotne určený (16, 35, 36, 37). Aktuálne poznatky o klinickom význame konkurentných mutácií u CEBPAm/biCEBPAm AML pritom dovoľujú opatrne zvažovať limitáciu mutačného skríningu CEBPA na skupinu AML bez nálezu NPM1m a bez FLT3-ITD. V tejto molekulárnej skupine AML by mala predstavovať prítomnosť biCEBPAm priaznivý prognostický faktor.
Pacienti s izolovanými CEBPAm sa pokladajú popri AML s izolovanými NPM1m za ďalšiu priaznivú molekulárnu skupinu AML, v ktorej sa intenzívne zvažujú liečebné možnosti, predovšetkým prínos alloHSCT v 1. línii liečby. Záverečné zhodnotenie významu alloHSCT v tejto skupine AML vyžaduje objasnenie dosiaľ neúplne pochopených funkčných súvislostí medzi jednotlivými mutačnými profilmi CEBPA (predovšetkým CEBPAm v porovnaní s biCEBPAm, prípadne N-terminálnych CEBPAm v porovnaní s C-terminálnymi CEBPAm).
Parciálne tandemové duplikácie v géne MLL (Mixed-Lineage Leukemia, 11q23.3)
Gén MLL kóduje histón metyltransferázu významnú v regulácii krvotvorby a expresie Hox génov. Na rozdiel od obecne známych onkogénnych prestavieb MLL (38) sa asi u 8 % AML s NK vyskytujú špecifické mutácie, ktoré vedú k čiastočnej (parciálnej) tandemovej duplikácii MLL (MLL-PTD) (3). Od prvej zmienky o klinickom význame MLL-PTD u AML pred takmer 20 rokmi sa zhodnotil prognostický význam tejto mutácie na niekoľkých obsiahlych súboroch zahŕňajúcich i AML s NK, u ktorých je nález MLL-PTD rizikovým faktorom skorého relapsu (39). Nedávna analýza vybranej skupiny mladších (< 60 rokov) pacientov s AML a NK však prekvapivo nepotvrdila v tejto skupine klinicky významné rozdiely v závislosti na prítomnosti MLL-PTD (40). Nález MLL-PTD sa u AML s NK natrvalo považuje za rizikový faktor skorého relapsu, zohľadňuje sa však podiel ďalších, kooperujúcich (nezávislých negatívnych) markerov, predovšetkým prítomnosť konkurentných FLT3-ITD, ktoré sa vyskytujú asi u 30 % AML s nálezom MLL-PTD (41).
Napriek kontroverznému pohľadu na leukemogénny potenciál MLL-PTD je z hľadiska možností cielenej terapie významné, že inhibícia aktivity DNA metyltransferázy a histón deacetylázy dokáže reverzibilne navrátiť expresiu štandardnej alely MLL, ktorá je v MLL-PTD pozitívnych leukemických blastoch potlačená (42).
Ďalšie génové mutácie u AML s NK
Kombinované mutačné analýzy zamerané na skríning NPM1m, CEBPAm a FLT3m, prípadne MLL-PTD umožňujú identifikovať aspoň jednu molekulárnu zmenu asi u 70-80 % AML s NK (2). V zmysle molekulárnej stratifikácie AML sa radia pacienti s nálezom NPM1m bez FLT3-ITD do priaznivej molekulárnej skupiny. Naproti tomu AML bez NPM1m, ale s nálezom FLT3-ITD, rovnako ako AML bez NPM1m a súčasne bez FLT3-ITD predstavujú vysoko rizikovú molekulárnu skupinu AML. Prítomnosť mutácií CEBPA radí pacientov za predpokladu, že u nich nie je prítomná FLT3-ITD, taktiež do priaznivej molekulárnej skupiny.
Uvedenú molekulárnu stratifikáciu však stále nemôžeme uvažovať u významnej skupiny (asi 20–30 %) AML s NK, u ktorých sa tieto mutácie jednoducho nevyskytujú. Na patogenéze AML sa u týchto pacientov určite podieľajú ďalšie molekulárne mechanizmy, ktoré zahŕňajú i ďalšie génové mutácie. Aktivácia signálnej dráhy RAS, či abnormálna funkcia transkripčného faktoru CBF ako dôsledok mutácií v géne NRAS, resp. mutácií v géne RUNX1 sa predpokladá asi u 10 % AML s NK (3). Jednotný prognostický význam však zatiaľ nie je v prípade ani jedného z týchto 2 typov génových mutácií u AML s NK stanovený. Naproti tomu, medzi intenzívne diskutované (nové) mutácie, ktorých prognostický význam sa zatiaľ hodnotí sa zaraďujú mutácie v géne WT1 a najnovšie i mutácie v génoch IDH1, resp. IDH2.
Mutácie v géne WT1 (Wilmęs tumor 1, 11p13)
Narušená expresia WT1 je u leukémií, nevynímajúc AML dlhodobo známa a i niektoré tuzemské pracoviská využívajú hladiny expresie tohto génu na sledovanie MRD (43). Napriek dôkazom o význame WT1 v regulácii prežívania, proliferácie, či diferenciácie krvotvorných buniek ostáva presná úloha tohto funkčne duálneho transkripčného faktoru v normálnej i leukemickej krvotvorbe neobjasnená. Celkom nedávno sa opakovane upozornilo na výskyt mutácií WT1 (WT1m) približne u 10 % AML s NK (44). Mutácie sa prevažne koncentrujú v 2 mutačných hot-spot oblastiach WT1 (exóny 7 a 9), hoci niektoré detailné štúdie poukazujú na sporadické mutácie i v exónoch 1, 2, 3 a 8 (45).Posunové mutácie v exóne 7 vedú k skráteniu proteínu WT1, ktorý v tejto podobe nie je schopný regulácie transkripcie. Predpokladá sa tiež strata jadrových lokalizačných signálov ako aj strata schopnosti viazať domény ďalších interagujúcich proteínov, vrátane tumor supresorového génu p53 a jeho homológu p73. Naproti tomu posunové mutácie v exóne 9 génu WT1 sú jednak u AML menej časté a navyše výsledné proteíny by sa mali vyznačovať odlišnými funkčnými dôsledkami v porovnaní s proteínmi s mutáciami v exóne 7. V exóne 9 WT1 sa častejšie popisujú mutácie meniace zmysel. Tieto mutácie primárne zasahujú do aminokyselinového zloženia výsledného proteínu a podľa dostupných informácií vedú k destabilizácii štruktúry zinkového prstu a k narušeniu DNA väzobnej kapacity (44).
Mutácie WT1 hodnotí niekoľko pracovných skupín ako nepriaznivý ukazovateľ prognózy AML dospelého veku. V dostupných štúdiách majú WT1m nepriaznivý vplyv na výsledky indukčnej liečby a dosiahnutie kompletných remisií, sú spájané s častejšou kumulatívnou incidenciou relapsov a horším celkovým prežitím. Tieto štúdie aktuálne zhrnul Owen a kolektív (44). Významná nemecko/rakúska skupina poukázala na negatívny vplyv WT1m jedine u pacientov s koexistujúcimi WT1m a FLT3-ITD. V kontraste s ďalšími prácami tak nepotvrdila nezávislý prognostický význam WT1m u AML s NK (45). Diskusia o príčinách nejednotných výsledkov upozorňuje i na odlišné liečebné protokoly porovnávaných nezávislých štúdií, ktoré sa líšia v celkovej kumulatívnej dávke cytarabínu (44). Pokiaľ by sa podarilo potvrdiť, že intenzívnejšia liečba dokáže zabrániť negatívnemu účinku, ktorými sa WT1m u AML vyznačujú, potom by predstavoval mutačný status WT1 ďalší významný prognostický ukazovateľ u AML s NK, rovnako ako dôležitý marker MRD.
Mutácie v génoch IDH1 (Isocitrate Dehydrogenase 1, 2q34) a IDH2 (Isocitrate Dehydrogenase 2, 15q26.1)
Na gén izocitrátdehydrogenázy 1 (IDH1) upozornili celkom nedávno výsledky sekvenovania kompletného genómu u pacienta s AML a NK (46). V krátkom čase bol výskyt mutácií IDH1 (IDH1m) potvrdený u 5,5 % primárnych AML a asi u 11 % AML s NK (47). Frekvencia výskytu IDH1m u AML s NK však nie je zatiaľ jednotne stanovená. Vieme, že IDH1msa u AML sústreďujú prevažne v kodóne 132, ktorý kóduje evolučne konzervovanú aminokyselinu arginín (R132). Je ďalej známe, že popri mutáciám v IDH1 sa u AML vyskytujú nukleotidové substitúcie i v géne IDH2 (IDH2m),ktorý kóduje mitochondriálnu izoformu cytoplazmatickej izocitrátdehydrogenázy IDH1 (48). V géne IDH2 postihujú mutácie prednostne kodóny 140 a 172 a zodpovedajú v týchto pozíciách taktiež za zámenu aminokyseliny arginín (mutácie R140, resp R172) (49).
Na základe dostupných funkčných štúdií sa dá predpokladať onkogénny efekt IDH1/2m v patogenéze AML (47). Je známe, že izocitrátdehydrogenáza dokáže v prítomnosti IDH1/2m premieňať α-ketoglutarát na 2-hydroxyglutarát (47,48). Ten sa ako metabolit hromadí v mutovaných leukemických bunkách a pravdepodobne blokuje aktivitu enzýmov, ktoré využívajú α-ketoglutarátako svoj substrát. Medzi takéto enzýmy sa zaraďujú i vybrané metyltransferázy a demetyltransferázy, ktorých aktivita ovplyvňuje hladiny metylácie DNA v bunkách. Je významné, že na molekulárnej úrovni reguluje metylačný profil DNA a histónov nielen mieru génovej expresie, ale i bunkovú diferenciáciu. Výsledky mutačných a epigenetických štúdií poukázali u AML s IDH1/2m na unikátny epigenetický profil, ktorý charakterizuje globálna hypermetylácia DNA (50). V súlade s uvedenými sa predpokladá, že zmena metylačného profilu môže u IDH1/2m AML viesť k narušenej expresii onkogénov a tumor supresorových génov a tým aberantne ovplyvňovať funkciu kľúčových signálnych a metabolických dráh (50). Figueroa a kolektív preto navrhujú zaradiť IDH1/2m medzi tzv. epigenetické regulátory, ktoré by mohli predstavovať v patogenéze AML novú kategóriu génových mutácií. Súčasne podávajú predbežné dôkazy pre zváženie klasifikácie IDH1/2m AML v rámci samostatného podtypu AML (50).
Pilotné štúdie poukázali prevažne na nepriaznivý prognostický efekt IDH1/2m u AML (47). Niektoré štúdie hodnotia IDH1m ako nepriaznivý ukazovateľ v skupine NPM1m AML (51), prípadne diskutujú súvislosť IDH1m s chemorezistenciou leukemických buniek (52). Záverečné zhodnotenie prognostického významu IDH1/2m u AML však vyžaduje ďalšie analýzy. V tejto súvislosti, testovanie klinického významu IDH1m bude pri multivariabilnom hodnotení mutačných profilov NPM1, FLT3 a CEBPA vyžadovať precízne navrhnuté štúdie na dostatočne rozsiahlych, uniformne liečených súboroch. Mimoriadne prínosné bude záverečné zhodnotenie významu IDH1m u NPM1m AML. Zatiaľ sa predpokladá, že IDH1m by mohli predstavovať popri FLT3-ITD ďalší kandidátny marker, ktorý prednostne asociuje s NPM1m a mohol by ovplyvňovať priebeh ochorenia v tejto prognosticky priaznivej molekulárnej skupine AML (51).
Na základe analýz párovaných vzoriek z doby diagnózy a relapsu je možné predbežne usudzovať, že IDH1m sa objavujú v skorých štádiách leukemogenézy, sú stabilné v priebehu progresie ochorenia a tým predurčené za vhodného kandidáta na marker MRD u AML (53).
Z terapeutického hľadiska sú zatiaľ v teoretickej línii zaujímavé úvahy o možnosti blokovania funkčných dôsledkov akumulácie 2-hydroxyglutarátu v leukemických bunkách cestou cielenej inhibície enzymatickej aktivity mutovaných izocitrátdehydrogenáz u pacientov s IDH1/2m (47).
Záver
Intenzívna pozornosť venovaná AML s NK predovšetkým v posledných asi 5 až 6 rokoch a uplatnenie nových techník molekulárnej genetiky pri analýze genómu leukemických buniek priniesli výrazný pokrok v pochopení biologickej podstaty AML. Poznáme nové, klinicky významné (molekulárne) markery, ktoré ovplyvňujú klasifikáciu, odpoveď na liečbu, prežitie a uplatňujú sa ako markery MRD, obzvlášť v skupine AML s NK. Na podklade mutačného profilu génov CEBPA a NPM1 sme schopní identifikovať nové, zatiaľ provizórne podskupiny AML a uvažujeme možnosti uplatnenia našich vedomostí o biológii AML pri optimalizácii liečebnej stratégie, od indikácie alloHSCT po cielenú molekulárnu terapiu. Prognóza AML s NK sa vo svetle pribúdajúcich informácií o vzájomnom vzťahu kooperujúcich mutácií a funkčných následkov podrobne charakterizovaných mutačných profilov neustále upravuje. Napriek mnohým otázkam, ktoré stále čakajú na objasnenie, sa v dnešnej klinickej praxi jednotne odporúča hodnotiť mutačný profil génov NPM1, FLT3 a CEBPA aspoň u AML s NK. V nasledujúcich rokoch by mali práve tieto mutácie predstavovať rozhodujúce markery pri voľbe liečebného postupu, v ktorom sa perspektívne otvoria možnosti pre významnejšie zohľadňovanie molekulárneho profilu AML. Po dôkladnom preskúmaní úlohy ďalších kandidátnych génov (WT1, IDH1/2 a iné) v procese leukemogenézy AML si budú i tieto pravdepodobne nachádzať svoje miesto v klasifikácii AML a my budeme schopní pochopiť súvislosť medzi biologickou podstatou AML a klinickou heterogenitou, ktorou sa AML, obzvlášť AML s NK vyznačujú.
Zoznam použitých skratiek
- DNA – deoxyribonukleotidová kyselina (z angl. deoxyribonucleic acid)
- MRD – minimálna reziduálna choroba (z angl. minimal residual disease)
- WHO – svetová zdravotnícka organizácia (z angl. world health organisation)
Poďakovanie
Laboratórne
výsledky zmienené v tejto práci boli podporené grantom MSM
6198959205 a LF-2010-004.
Mgr. Beáta Katrincsáková
Laboratoř
molekulární biologie
Hemato-onkologická
klinika
Fakultní
nemocnice Olomouc
I. P. Pavlova
6
775
20 Olomouc
Doručeno
do redakce: 14. 2. 2011
Přijato
po recenzi: 9. 3. 2011
Zdroje
1. Estey E, Döhner H. Acute myeloid leukaemia. Lancet 2006 Nov 25; 368 (9550): 1894-907.
2. Döhner H, Estey EH, Amadori S, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European Leukemia Net. Blood 2010 Jan 21; 115(3): 453-74.
3. Mrózek K, Marcucci G, Paschka P, Whitman SP, Bloomfield CD. Clinical relevance of mutations and gene-expression changes in adult acute myeloid leukemia with normal cytogenetics: are we ready for a prognostically prioritized molecular classification? Blood 2007 Jan 15; 109(2): 431-48.
4. Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC Press; 2008.
5. Döhner K, Döhner H. Molecular characterization of acute myeloid leukemia. Haematologica 2008 Jul; 93(7): 976-82.
6. Gazdová J, Dvořáková D, Ježíšková I, Rázga F, Jurček T, Mayer J. Úloha FLT3 mutací v patogenezi akutní myeloidní leukemie. Transfuze Hematol dnes 2009 Prosinec; 15 : 229-236.
7. Thiede C, Steudel C, Mohr B, et al Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood 2002 Jun 15; 99(12): 4326-35.
8. Yanada M, Matsuo K, Suzuki T, Kiyoi H, Naoe T. Prognostic significance of FLT3 internal tandem duplication and tyrosine kinase domain mutations for acute myeloid leukemia: a meta-analysis. Leukemia 2005 Aug; 19(8): 1345-9.
9. Gale RE, Green C, Allen C, et al. The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood 2008; 111(5): 2776–84.
10. Whitman SP, Archer KJ, Feng L, et al. Absence of the wild-type allele predicts poor prognosis in adult de novo acute myeloid leukemia with normal cytogenetics and the internal tandem duplication of FLT3: a Cancer and Leukemia Group B study. Cancer Res 2001; 61(19): 7233–9.
11. Kottaridis PD, Gale RE, Langabeer SE , et al. Studies of FLT3 mutations in paired presentation and relapse samples from patients with acute myeloid leukemia: implications for the role of FLT3 mutations in leukemogenesis, minimal residual disease detection, and possible therapy with FLT3 inhibitors. Blood 2002 Oct 1; 100(7): 2393-8.
12. Schnittger S, Schoch C, Kern W, et al. FLT3 length mutations as marker for follow-up studies in acute myeloid leukaemia. Acta Haematol 2004; 112(1-2): 68-78.
13. Beretta C, Gaipa G, Rossi V, et al. Development of a quantitative-PCR method for specific FLT3/ITD monitoring in acute myeloid leukemia. Leukemia 2004 Aug; 18(8): 1441-4.
14. Whitman SP, Ruppert AS, Radmacher MD, et al. FLT3 D835/I836 mutations are associated with poor disease-free survival and a distinct gene-expression signature among younger adults with de novo cytogenetically normal acute myeloid leukemia lacking FLT3 internal tandem duplications. Blood 2008 Feb 1; 111(3): 1552-9.
15. Bacher U, Haferlach C, Kern W, et al. Prognostic relevance of FLT3-TKD mutations in AML: the combination matters-an analysis of 3082 patients. Blood 2008 Mar 1; 111(5): 2527-37.
16. Schlenk RF, Döhner K, Krauter J, et al Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med 2008 May 1; 358(18): 1909-18.
17. Gale RE, Hills R, Kottaridis PD, et al. No evidence that FLT3 status should be considered as an indicator for transplantation in acute myeloid leukemia (AML): an analysis of 1135 patients, excluding acute promyelocytic leukemia, from the UK MRC AML10 and 12 trials. Blood 2005 Nov 15; 106(10): 3658-65.
18. Doubek M, Muzík J, Szotkowski T, et al. Is FLT3 internal tandem duplication significant indicator for allogeneic transplantation in acute myeloid leukemia? An analysis of patients from the Czech Acute Leukemia Clinical Register (ALERT). Neoplasma 2007; 54(1): 89-94.
19. Wagner K, Damm F, Thol F, et al. FLT3-internal tandem duplication and age are the major prognostic factors in relapsed acute myeloid leukemia with normal karyotype. Haematologica 2011 Jan 17 (Epub ahead of print).
20. Weisberg E, Barrett R, Liu Q, Stone R, Gray N, Griffin JD. FLT3 inhibition and mechanisms of drug resistance in mutant FLT3-positive AML. Drug Resist Updat 2009 Jun; 12(3): 81-9.
21. Falini B, Sportoletti P, Martelli MP. Acute myeloid leukemia with mutated NPM1: diagnosis, prognosis and therapeutic perspectives. Curr Opin Oncol 2009; 21 : 573–581.
22. Rau R, Brown P. Nucleophosmin (NPM1) mutations in adult and childhood acute myeloid leukaemia: towards definition of a new leukaemia entity. Hematol Oncol 2009 Dec; 27(4): 171-81.
23. Schnittger S, Schoch C, Kern W, et al Nucleophosmin gene mutations are predictors of favorable prognosis in acute myelogenous leukemia with a normal karyotype. Blood 2005 Dec 1; 106(12): 3733-9.
24. Becker H, Marcucci G, Maharry K, et al. Favorable prognostic impact of NPM1 mutations in older patients with cytogenetically normal de novo acute myeloid leukemia and associated gene - and microRNA-expression signatures: a Cancer and Leukemia Group B study. J Clin Oncol 2010 Feb 1; 28(4): 596-604.
25. Döhner K, Schlenk RF, Habdank M, et al. Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics:interaction with other gene mutations. Blood 2005 Dec 1; 106(12): 3740-6.
26. Schnittger S, Kern W, Tschulik C, et al. Minimal residual disease levels assessed by NPM1 mutation-specific RQ-PCR provide important prognostic information in AML. Blood 2009 Sep 10; 114(11): 2220-31.
27. Schlenk RF, Döhner K, Kneba M, et al. Gene mutations and response to treatment with all-trans retinoic acid in elderly patients with acute myeloid leukemia: results from the AMLSG Trial AML HD98B. Haematologica 2009; 94 : 54-60.
28. Burnett AK, Hills RK, Green C, et al. The impact on outcome of the addition of all-trans retinoic acid to intensive chemotherapy in younger patients with nonacute promyelocytic acute myeloid leukemia: overall results and results in genotypic subgroups defined by mutations in NPM1, FLT3, and CEBPA. Blood 2010 Feb 4; 115(5): 948-56.
29. Pabst T, Mueller BU, Zhang P, et al. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-alpha (C/EBPalpha), in acute myeloid leukemia. Nat Genet 2001; 27 : 263–270.
30. Zhang DE, Zhang P, Wang ND, Hetherington CJ, Darlington GJ, Tenen DG. Absence of granulocyte colony-stimulating factor signaling and neutrophil development in CCAAT enhancer binding protein alpha-deficient mice. Proc Natl Acad Sci USA 1997; 94(2): 569-574.
31. Rosenbauer F, Tenen DG. Transcription factors in myeloid development: balancing differentiation with transformation. Nat Rev Immunol 2007; 7(2): 105-117.
32. Reckzeh K, Cammenga J. Molecular mechanisms underlying deregulation of C/EBPalpha in acute myeloid leukemia. Int J Hematol 2010 May; 91(4): 557-68.
33. Fröhling S, Schlenk RF, Stolze I, et al. CEBPA mutations in younger adults with acute myeloid leukemia and normal cytogenetics: prognostic relevance and analysis of cooperating mutations. J Clin Oncol 2004 Feb 15; 22(4): 624-33.
34. Taskesen E, Bullinger L, Corbacioglu A. et al Prognostic impact, concurrent genetic mutations and gene expression features of AML with CEBPA mutations in a cohort of 1182 cytogenetically normal AML: further evidence for CEBPA double mutant AML as a distinctive disease entity. Blood. 2010 Dec 21 (Epub ahead of print).
35. Green CL, Koo KK, Hills RK, Burnett AK, Linch DC, Gale RE. Prognostic significance of CEBPA mutations in a large cohort of younger adult patients with acute myeloid leukemia: impact of double CEBPA mutations and the interaction with FLT3 and NPM1 mutations. J Clin Oncol 2010 Jun 1; 28(16): 2739-47.
36. Wouters BJ, Lowenberg B, Erpelinck-Verschueren CA, van Putten WL, Valk PJ, Delwel R. Double CEBPA mutations, but not single CEBPA mutations, define a subgroup of acute myeloid leukemia with a distinctive gene expression profile that is uniquely associated with a favorable outcome. Blood 2009; 113(13): 3088-3091.
37. Renneville A, Boissel N, Gachard N, et al. The favorable impact of CEBPA mutations in patients with acute myeloid leukemia is only observed in the absence of associated cytogenetic abnormalities and FLT3 internal duplication. Blood 2009 May 21; 113(21): 5090-3.
38. Slany RK. The molecular biology of mixed lineage leukemia. Haematologica 2009 Jul; 94(7): 984-93.
39. Basecke J, Whelan JT, Griesinger F, Bertrand FE. The MLL partial tandem duplication in acute myeloid leukaemia. Br J Haematol 2006 Nov; 135(4): 438-49.
40. Whitman SP, Ruppert AS, Marcucci G, et al. Long-term disease-free survivors with cytogenetically normal acute myeloid leukemia and MLL partial tandem duplication: a Cancer and Leukemia Group B study. Blood 2007 Jun 15; 109(12): 5164-7.
41. Olesen LH, Nyvold CG, Aggerholm A, NŅrgaard JM, Guldberg P, Hokland P. Delineation and molecular characterization of acute myeloid leukemia patients with coduplication of FLT3 and MLL. Eur J Haematol 2005 Sep; 75(3): 185-92.
42. Whitman SP, Liu S, Vukosavljevic T, et al. The MLL partial tandem duplication: evidence for recessive gain-of-function in acute myeloid leukemia identifies a novel patient subgroup for molecular-targeted therapy. Blood 2005 Jul 1; 106(1): 345-52.
43. Polák J, Marková J, Schwarz J, et al. The use of quantitative assessment of Wilms tumour gene 1 for monitoring of residual disease in acute myeloid leukemia patients. Cas Lek Cesk 2006; 145(1): 36-42.
44. Owen C, Fitzgibbon J, Paschka P. The clinical relevance of Wilms Tumour 1 (WT1) gene mutations in acute leukaemia. Hematol Oncol 2010 Mar; 28(1): 13-9.
45. Gaidzik VI, Schlenk RF, Moschny S, et al. German-Austrian AML Study Group. Prognostic impact of WT1 mutations in cytogenetically normal acute myeloid leukemia: a study of the German-Austrian AML Study Group. Blood 2009 May 7; 113(19): 4505-11.
46. Mardis ER, Ding L, Dooling DJ, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 2009 Sep 10; 361(11): 1058-66.
47. Dang L, Jin S, Su SM. IDH mutations in glioma and acute myeloid leukemia. Trends Mol Med 2010 Sep; 16(9): 387-97.
48. Gross S, Cairns RA, Minden MD, et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med. 2010 Feb 15; 207(2): 339-44.
49. Marcucci G, Maharry K,Wu YZ, et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B Study. J Clin Oncol 2010; 28(14): 2348–55.
50. Figueroa ME, Abdel-Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010 Dec 14; 18(6): 553-67.
51. Boissel N, Nibourel O, Renneville A, et al. Prognostic impact of isocitrate dehydrogenase enzyme isoforms 1 and 2 mutations in acute myeloid leukemia: a study by the Acute Leukemia French Association group. J Clin Oncol 2010 Aug 10; 28(23): 3717-23.
52. Green CL, Evans CM, Hills RK, Burnett AK, Linch DC, Gale RE. The prognostic significance of IDH1 mutations in younger adult patients with acute myeloid leukemia is dependent on FLT3/ITD status. Blood 2010 Oct 14; 116(15): 2779-82.
53. Chou WC, Hou HA, Chen CY, et al. Distinct clinical and biological characteristics in adult acute myeloid leukemia bearing isocitrate dehydrogenase 1 (IDH1) mutation. Blood 2010; 115 : 2749–2754.
Štítky
Hematologie a transfuzní lékařství Interní lékařství OnkologieČlánek vyšel v časopise
Transfuze a hematologie dnes

2011 Číslo 2
- Srovnání vlivu omeprazolu a pantoprazolu na antiagregační účinek klopidogrelu
- Fixní kombinace kandesartan/amlodipin v terapii arteriální hypertenze
- Současné postavení a přínos sartanů v klinické praxi
- Prognostický význam hladiny natriuretických peptidů při léčbě empagliflozinem
- Hypertrofická obstrukční kardiomyopatie ve světle moderní farmakoterapie – kazuistika
Nejčtenější v tomto čísle
-
Chronická myeloidní leukemie
Doporučení pro diagnostiku, monitorování a léčbu CML u dospělých, aktualizovaná verze 2011 (střed časopisu) - Kazuistika: sekundární trombocytopenie a diseminovaná intravaskulární koagulace u pacienta s generalizovaným adenokarcinomem colon descendens
- Klinický význam génových mutácií u akútnych myeloidných leukémií s normálnym karyotypom
- „Stanovení rezistence na clopidogrel pomocí vícenásobné impedanční a optické transmisní agregometrie“
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy