
Zmeny názvoslovia, klasifikácie a diagnostických kritérií myeloproliferatívnych ochorení podľa WHO klasifikácie 2008
The changes in the nomenclature, classification and diagnostic criteria of myeloproliferative disorders according WHO classification 2008
The WHO classification of myeloid neoplasms published in 2008 (WHO-2008) is based on verified and accepted WHO classification 2001. It relies on tumor cell morphology evaluated in bone marrow (BM) biopsy and/or aspirate or in peripheral blood (PB) smears in correlation with clinical and laboratory (incl. genetic) data. The term “myeloproliferative disorders“ has been replaced by “myeloproliferative neoplasms“ to accent their malignant potential and the genetic approach has been introduced into MPN diagnostics. The myeloid neoplasms with eosinophilia and platelet derived - or fibroblast growth factor receptor 1 mutation are distinguished from chronic eosinophilic leukemia. The neoplastic mast cells proliferations are included in the MPN group, but their sub-classification remains unchanged. In the CML category the only change involves the need to reevaluate criteria of accelerated phase. The most important innovation is the implementation of specific Janus 2 tyrosine kinase mutation (JAK2 V617F) into the diagnostic criteria of chronic Ph1- MPN. Presence of this mutation or of activating JAK2 exon 12 mutations represents a typical finding in polycythemia vera (PV) patients, what together with typical clinical and laboratory data leads to PV diagnosis even in the absence of BM biopsy. WHO-2008 refers to initial stages of PV with borderline increase of hemoglobin and/or thrombocytosis in PB (clinically resembling essential thrombocythemia /ET/) and emphasizes the PV progress from initial to polycythemic phase with polyglobulia and final transformation to terminal “spent phase“ with distinct BM fibrosis and pancytopenia. Chronic idiopathic myelofibrosis is by WHO-2008 called “primary myelofibrosis“ (PMF). The key role in the PMF diagnosis plays BM biopsy (characteristic BM morphology) as well as demonstration of the clonality of the disease (JAK2V617F or thrombopoietin receptor mutations). WHO-2008 clearly defines the prefibrotic and fibrotic stages of PMF with significantly different patient’s survival rates. For ET diagnosis the arbitrary level of PB thrombocytes was lowered from previous 600 to 450x109/L. In contrast to previous classification requiring ET diagnosis per exclusion of other MPN, WHO-2008 introduces the positive diagnostic ET criteria (JAK2V617F or characteristic BM morphology).
Key words:
myeloproliferative disorders, myeloproliferative neoplasms, WHO classification, diagnostic criteria, chronic myeloid leukemia, polycythemia vera, primary myelofibrosis, esential thrombocythemia, JAK2
Autoři:
J. Marcinek; L. Plank; P. Szépe
Působiště autorů:
Komenského a Martinskej fakultnej nemocnice, Martin
; Ústav patologickej anatómie a Konzultačné centrum hematopatológie Jesseniovej lekárskej fakulty Univerzity
Vyšlo v časopise:
Transfuze Hematol. dnes,16, 2010, No. 1, p. 35-41.
Kategorie:
Souhrnné práce, původní práce, kazuistiky
Souhrn
Typizácia myeloidných neoplázií podľa WHO klasifikácie 2008 (WHO-2008) vychádza z osvedčenej a univerzálne akceptovanej WHO klasifikácie 2001 (WHO-2001). Je založená na korelácii morfológie nádorových buniek (biopsia a/alebo punktát kostnej drene – KD, resp. náter periférnej krvi - PK) s klinickými údajmi a výsledkami laboratórnych vyšetrení (obraz v PK, výsledky molekulárno-genetických analýz). Myeloproliferatívne ochorenia WHO-2008 nazýva myeloproliferatívnymi neopláziami (MPN), poukazujúc na ich malígny potenciál. Zdôrazňuje genetickú diagnostiku MPN, pričom oddeľuje skupinu MPN s eozinofíliou a mutáciami receptorov doštičkového (PDGFR) a fibroblastového rastového faktora 1 (FGFR1) od chronickej eozinofilnej leukémie. Nádorové ochorenia mastocytov boli zaradené do skupiny MPN, avšak ich bližšia klasifikácia ostáva identická s WHO-2001. Pri chronickej myeloidnej leukémii, s výnimkou potreby revízie kritérií akcelerovanej fázy, nedošlo k závažnejším zmenám. Najdôležitejšou zmenou je implementácia špecifickej bodovej mutácie Janus 2 tyrozínovej kinázy (JAK2 V617F) do diagnostických kritérií chronických, Filadelfia 1 chromozóm negatívnych (Ph1-) MPN. Prítomnosť tejto mutácie, resp. mutácie exónu 12 tohto génu je typickým znakom pravej polycytémie (PV) a v kontexte klinických a laboratórnych údajov potvrdzuje PV aj bez bioptického vyšetrenia KD. WHO-2008 poukazuje na iniciálne štádiá PV s hraničným zmnožením hemoglobínu a erytrocytov a/alebo trombocytózou v PK, klinicky imitujúce esenciálnu trombocytémiu (ET). Zároveň zdôrazňuje prechod PV z iniciálneho do polycytemického štádia so zreteľnou polyglobúliou až vznik terminálnej „spent fázy“ s rozvinutou fibrózou v KD a pancytopéniou v PK. Chronická idiopatická myelofibróza sa podľa WHO-2008 nazýva primárna myelofibróza (PMF), pričom kľúčovú úlohu v diagnostike ochorenia zohráva bioptické vyšetrenie KD (nález charakteristického morfologického obrazu) a potvrdenie klonality procesu (prítomnosť JAK2 V617F resp. mutácií trombopoetínového receptora). WHO-2008 jasne vymedzuje prefibrotické a fibrotické štádium PMF, líšiace sa dĺžkou prežívania pacientov. Pri ET došlo k zníženiu arbitrárne stanovenej hladiny trombocytov v PK potrebných na stanovenie diagnózy z pôvodných 600 na 450 x 109/L. Zatiaľ čo podľa WHO-2001 bola ET definovaná vylúčením iných typov MPN, WHO-2008 implementuje pozitívne diagnostické kritériá ET (prítomnosť JAK2V617F, alebo charakteristický morfologický obraz KD).
Kľúčové slová:
myeloproliferatívne ochorenia, myeloproliferatívne neoplázie, WHO klasifikácia, diagnostické kritériá, chronická myeloidná leukémia, pravá polycytémia, primárna myelofibróza, esenciálna trombocytémia, JAK2
Klasifikácia myeloidných neoplázií je dnes založená na ich medzinárodne akceptovanej a univerzálne použiteľnej WHO klasifikácii. Donedávna používané, tretie vydanie WHO klasifikácie myeloidných neoplázií z roku 2001 (WHO klasifikácia 2001-1) sa stretlo s veľkým úspechom nielen vo vedecko-výskumnej sfére, ale najmä u širokej odbornej verejnosti a zjednotilo tak rôzne klasifikačné schémy používané v Európe a Amerike, čo prinieslo benefit nielen pre ďalší výskum v tejto oblasti, ale najmä pre pacientov. Hematopatológia je dynamicky sa rozvíjajúci medicínsky odbor, v ktorom dochádza k odhaľovaniu stále nových genetických abnormalít a signálnych dráh vedúcich k vzniku ochorení. Tieto informácie umožňujú nielen precíznu diagnostiku ochorení, ale v ére špecifických inhibítorov tyrozínových kináz často identifikujú aj terapeutické ciele s následným zlepšením prognózy a kvality života pacientov. Preto bola v septembri 2008 zverejnená nová verzia WHO klasifikácie myeloidných neoplázií (WHO klasifikácia 2008-2), zohľadňujúca nové poznatky v oblasti etiopatogenézy, diagnostiky a terapie ochorení. Vzhľadom na jej objem a rozsiahlosť zmien sa v tejto práci budeme zaoberať len skupinou chronických myeloproliferatívnych ochorení (MPO). Cieľom tejto práce nie je kompletný prehľad diagnostiky MPO, ktorý je značne rozsiahly, ale zameranie sa na zmeny a prínos WHO klasifikácie 2008 v porovnaní s WHO klasifikáciou 2001.
Vo WHO klasifikácii 2008 v porovnaní s WHO klasifikáciou 2001 pozorujeme zmeny v nomenklatúre, diagnostickom prístupe a najmä v diagnostických kritériách ochorení, čo viedlo aj k založeniu nových klinicko-patologických jednotiek. MPO sú vo WHO klasifikácii 2008 označované ako myeloproliferatívne neoplázie (MPN) (tab. 1), pričom tento názov zdôrazňuje ich malígny potenciál. Aj keď v prípade adekvátnej terapie pacienti s MPN prežívajú relatívne dlhú dobu, v konečnom dôsledku tieto ochorenia spôsobujú svojimi komplikáciami smrť pacientov (3). Diagnostika MPN podľa WHO klasifikácie 2008 vychádza z pôvodnej WHO klasifikácie 2001 a je založená na multidisciplinárnej spolupráci viacerých medicínskych odborov pri korelácii morfológie neoplázií s klinickými informáciami a výsledkami laboratórnych a genetických vyšetrení.

Vo WHO klasifikácii 2008 sa stretávame s množstvom nových genetických abnormalít sprevádzajúcich myeloidné neoplázie. V prípadoch chronickej eozinofilnej leukémie (CEL), ako aj iných myeloidných (akútne myeloidné leukémie - AML, chronická myelomonocytová leukémia - CMMoL, iné MPN), menej často aj lymfoidných neoplázií (T-bunková akútna lymfoblastická leukémia) sprevádzanými eozinofíliou bola identifikovaná skupina mutácií postihujúcich receptor doštičkového rastového faktora alfa a beta (PDGFRA, PDGFRB) a fibroblastového rastového faktora 1 (FGFR1) (4, 5). Jednotlivé mutácie sa môžu morfologicky manifestovať širokým spektrom neoplázií, aj keď niektoré mutácie sa preferenčne spájajú s obrazom konkrétnej neoplázie. Úspešná terapia pacientov s mutáciami PDGFRA a PDGFRB inhibítormi tyrozínových kináz so zjavným zlepšením dĺžky prežívania a kvality ich života (6) viedla k vytvoreniu novej klinicko-patologickej jednotky s názvom: Myeloidné a lymfoidné neoplázie sprevádzané eozinofíliou a mutáciami PDGFRA, PDGFRB a FGFR1 (MaLNEaPDGFR/FGFR). WHO klasifikácia 2008 preto navrhuje nový algoritmus diferenciálnej diagnostiky eozinofílie v periférnej krvi (PK), kedy po vylúčení sekundárnych príčin eozinofílie a pri morfologickom podozrení na CEL (obr. 1) treba najskôr vylúčiť prítomnosť horeuvedených mutácií a až v prípade ich absencie môže byť stanovená diagnóza bližšie netypizovateľnej CEL (CEL - not otherwise specified - NOS), alebo inej myeloidnej neoplázie (schéma 1). U pacientov s CEL-NOS bez zjavného orgánového postihu alebo dysfunkcie WHO klasifikácia 2008 odporúča názov hypereozinofilný syndróm (HES), aj keď časť pacientov s HES sa neskôr transformuje do CEL-NOS. U pacientov s mutovaným FGFR1 zatiaľ nebol dokázaný benefit terapie dnešnými inhibítormi tyrozínových kináz, aj keď v blízkej budúcnosti sa objav takejto špecifickej terapie očakáva.


WHO klasifikácia 2008 s malými úpravami názvu naďalej zachováva aj ďalšie nozologické jednotky zo skupiny MPO podľa WHO klasifikácie 2001 (tab. 1), avšak skupina nádorových ochorení mastocytov (mastocytózy), ktorá bola vo WHO klasifikácii 2001 samostatnou kategóriu myeloidných neoplázií je vo WHO klasifikácii 2008 zaradená do skupiny MPN, keďže tieto ochorenia tiež vznikajú z kmeňovej bunky hemopoézy v KD (7). Bližšia typizácia mastocytóz však ostáva, až na malé zmeny, v zásade nezmenená (tab. 1) a pre jej rozsiahlosť od jej bližšieho opisu upúšťame.
Diagnostika chronickej myeloidnej leukémie (CML) ostáva založená na potvrdení BCR-ABL fúzneho génu, resp. jeho variant a biopsia KD nie je vyžadovaná pre diagnostiku ochorenia. Časť pacientov v skorých štádiách CML sa klinicky môže manifestovať trombocytózou v PK, bez ďalších sprievodných znakov CML, takže imitujú iné MPN, najmä esenciálnu trombocytémiu (ET) (8). Bioptické vyšetrenie KD umožňuje odlíšiť tieto prípady CML a zároveň sledovať rozvoj ochorenia, ktoré sa transformuje z počiatočnej chronickej fázy (CF) do akcelerovanej fázy (AF) (definovanej zmesou klinických, genetických a morfologických kritérií) až terminálnej blastickej fázy (BF), ktorá sa vyznačuje 20% podielom blastov PK a/alebo KD, resp. extramedulárnou proliferáciou blastov (1). Aj keď kritériá AF a BF CML sú v oboch WHO klasifikáciách identické, v ére terapie inhibítormi tyrozínových kináz sa po realizácií dostatočne veľkých klinických štúdií tieto kritériá môžu neskôr upravovať (9).
Najdôležitejším prínosom v skupine Ph1- MPN za posledné roky bol objav špecifickej bodovej mutácie Janus 2 tyrozínovej kinázy (JAK2 V617F), ktorá sa vyskytuje u všetkých Ph1- MPN (10, 11). Okrem MPN možno pozorovať túto mutáciu aj v prípadoch iných myeloidných neoplázií (cca 50 % pacientov s refraktérnou anémiou s prstencovými sideroblastami a trombocytózou (RARS-T), sporadicky aj v myelodysplastických syndrómoch (MDS), skupine MDS/MPN, akútnych myeloidných leukémiách). Naopak pacienti s BCR/ABL+ CML, lymfoidnými či epitelovými neopláziami, ako aj zdravá populácia sú negatívni (12, 13). Mutácia JAK2 V617F nie kauzálnou mutáciou spôsobujúcou vznik Ph1- MPN, avšak ovplyvňuje laboratórnu aj klinickú manifestáciu pacientov (10), aj keď jej konkrétny vplyv na dĺžku prežívania a prognózu pacientov je otázny. Prítomnosť JAK2 V617F potvrdzuje klonalitu ochorenia v zmysle myeloidnej neoplázie, a preto je vo WHO klasifikácii 2008 jedným z diagnostických kritérií všetkých Ph1- MPN (2), aj keď jej diferenciálno-diagnostický význam je limitovaný.
Hlavnú úlohu v diagnostike pravej polycytémie (PV) podľa WHO klasifikácie 2008 zohrávajú genetické vyšetrenia, najmä potvrdenie mutácie JAK2 V617F, ktorá sa vyskytuje u 90–95 % pacientov (11). V prípadoch PV bez JAK2 V617F pozorujeme skupinu bodových mutácií v exóne 12 JAK2, ktorých výskyt je pre PV patognostický (14). Prítomnosť týchto mutácií v kontexte polyglobúlie v PK pri splnení jedného „malého“ kritéria potvrdzuje diagnózu PV. PV možno diagnostikovať aj v zriedkavých prípadoch absencie oboch mutácií pri výskyte polyglobúlie a všetkých troch „malých“ diagnostických kritérií (tab. 2). WHO klasifikácia 2008 zdôrazňuje aj význam poznania dynamického rozvoja PV, keďže laboratórny a morfologický obraz, ako aj klinická manifestácia ochorenia sa postupne menia. Približne 10–15 % pacientov je diagnostikovaných v tzv. pre-polycytemickom štádiu, vyznačujúcom sa trombocytózou v PK a nižšou hladinou hemoglobínu, nedostatočnou pre potvrdenie PV (15). Toto štádium rýchlo progreduje do tzv. polyglobulického štádia so zreteľným zmnožením erytrocytov v PK a typickou klinickou aj laboratórnou manifestáciou. Keďže pacienti v pre-polycytemickom štádiu boli na základe klinických diagnostických kritérií v minulosti mylne diagnostikovaní ako ET a neskôr u nich vnikla polyglobúlia, začalo sa uvažovať o možnosti transformácie časti prípadov ET do PV. Pre-polycytemické aj polycytemické štádium PV sa však vyznačujú rovnakým morfologickým obrazom v KD, čo túto možnosť vylučuje (15). Vzhľadom na jednoduchú a šetrnú možnosť realizácie laboratórnych a genetických vyšetrení (potvrdenie „veľkých“ a dvoch „malých“ kritérií) nie je histologické vyšetrenie KD nevyhnutnou súčasťou diagnostiky PV. Napriek tomu WHO klasifikácia 2008 zdôrazňuje úlohu biopsie KD, umožňujúcu identifikáciu skorých štádií PV (klinickou a laboratórnou manifestáciou pripomínajúcimi iné MPN), ako aj odlíšenie reaktívnych zmien KD (16). Klasický morfologický obraz PV predstavuje hypercelulárna KD (vzhľadom na vek pacienta) a trilineárna myeloproliferácia (t.j. súčasné zmnoženie bieleho, červeného aj megakaryocytového radu krvotvorby). Dôležitým diagnostickým znakom je morfológia megakaryocytov (MGK), ktoré vykazujú značnú pleomorfiu (zmes malých, stredne veľkých až veľkých MGK), hyperlobulizované jadrá bez cytologických atypií (obr. 2). V interstíciu je normálne, či len minimálne zmnoženie retikulínových vlákien, bez prítomnosti makrofágov s obsahom železitého pigmentu (16, 17). V priebehu 10–15 rokov sa PV postupne transformuje do terminálneho fibrotického štádia (tzv. spent-fáza PV) s rozvinutou myelofibrózou v KD, pancytopéniou a leukoerytroblastovým obrazom v PK, ktoré je morfologicky a klinicky neodlíšiteľné od terminálnych štádií iných Ph1- PMN (16, 17).


Chronická idiopatická myelofibróza sa vo WHO klasifikácii 2008 nazýva primárna myelofibróza (PMF). Dôvodom zmeny názvu je definitívne objasnenie etiopatogenézy myelofibrózy v KD, ktorou je toto ochorenie príznačné. Proliferáciou bieleho a MGK radu a následným rozpadom dysplastických MGK pri PMF dochádza k uvoľňovaniu rôznych rastových faktorov (najdôležitejšími sú TGF-β a bFGF) stimulujúcich stromálne bunky KD (retikulárne bunky, fibroblasty, osteoblasty), ktoré následne produkujú väčšie množstvo retikulínových, neskôr aj kolagénových vlákien, spôsobujúcich fibrotizáciu KD (18). WHO klasifikácia 2008 taktiež značne upravuje diagnostické kritériá PMF (tab. 3), najmä v zmysle potvrdenia klonality ochorenia prítomnosťou mutácie JAK2 V617F, resp. mutácie trombopoetínového receptora (MPL W515L), vyskytujúcou sa cca v 10% PMF (19). Dôležitým prvkom je ustanovenie klasického morfologického obrazu ochorenia v KD ako „veľkého“ kritéria, potrebného pre potvrdenie tohto ochorenia. Preto bioptické vyšetrenie KD by malo byť zakaždým súčasťou diagnostiky PMF. Podobne ako WHO klasifikácia 2001, aj WHO klasifikácia 2008 zdôrazňuje dynamický rozvoj ochorenia, ktoré prebieha z počiatočného „prefibrotického“ štádia, bez výraznej fibrózy v KD, do „fibrotického“ štádia so zreteľnou myelofibrózou (1, 2). Toto rozdelenie má značný diagnostický a najmä prognostický význam. Prefibrotické štádium sa klinicky často manifestuje trombocytózou v PK s, alebo bez anémie a leukocytózy a svojou manifestáciou napodobuje ET (20). 30–40% pacientov diagnostikovaných v prefibrotickom štádium následne progreduje do fibrotického štádia s rozvinutou fibrózou v KD a typickou klinickou manifestáciou PMF (leukoerytroblastový krvný obraz s anémiou a extramedulárnou hemopoézou) (21). Zatiaľ čo medián prežívania sa v prefibrotickom štádiu pohybuje v rozmedzí 10–15 rokov, pacienti vo fibrotickom štádiu prežívajú len 3–7 rokov, a preto je dôležité odlíšiť skoré štádiá PMF od prípadov ET s podstatne lepšou prognózou (22). V tomto smere WHO klasifikácia 2008 aj podrobne opisuje morfologický obraz KD prefibrotického a fibrotického štádia PMF. V počiatočných štádiách ochorenia nachádzame hypercelulárnu KD s proliferáciou bieleho a MGK radu pri redukcii erytropoézy. MGK sú tvorené veľkými formami s hypolobulizovanými jadrami, ktoré nazývame tzv. obláčikovité jadrá a účasťou množstva dysplázií nielen MGK radu (obr. 3) (16, 17). Vo fibrotickom štádiu ochorenia pozorujeme taktiež hypercelulárnu KD s bilineárnou proliferáciou bieleho a MGK radu a „obláčikovité“ jadrá MGK, avšak prítomné sú aj početné malé a dysplastické formy MGK, často v intrasínusoidálnej lokalizácii (obr. 4). Terminálne môže byť celularita redukovaná rozsiahlou fibrotizáciou KD (16, 17). Nová klasifikácia striktne definuje množstvo fibrózy v KD v prefibrotickom štádiu PMF používaním Európskeho gradingu fibrózy v KD (23), pričom v tomto štádiu pozorujeme normálne množstvo fibrózy v KD (MF0), alebo len ľahkú retikulínovú fibrózu (MF1), zatiaľ čo fibrotické štádium sa vyznačuje rozvinutou retikulínovou (MF2) až kolagénovou (MF3) fibrózou s, alebo bez osteosklerotickej prestavby kostných trabekúl.



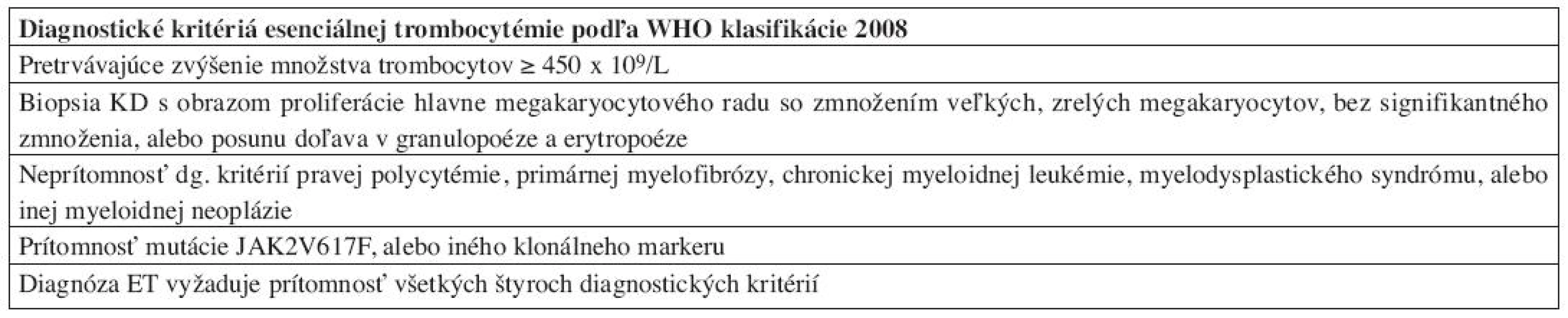
Najzásadnejšie zmeny v diagnostických kritériách WHO klasifikácie 2008 možno pozorovať u esenciálnej trombocytémie – ET. Arbitrárna hladina trombocytov v PK, potrebných pre stanovenie diagnózy ET, bola znížená na 450x109/L, keďže časť pacientov v skorých štádiách ochorenia nedosiahla vo WHO klasifikácii 2001 stanovené množstvo 600 x 109/L, a boli preto mylne diagnostikovaní ako reaktívne prípady trombocytózy (24). Rovnaké množstvo trombocytov v PK sa následne používa aj pre diagnostiku ochorení zo spektra MDS/MPN. Zatiaľ čo WHO klasifikácia 2001 diagnostikovala ET vylúčením iných príčin trombocytózy, WHO klasifikácia 2008 implementuje „pozitívne“ kritériá ochorenia (tab. 4), najmä v zmysle potvrdenia klonality ochorenia (mutácia JAK2 V617F) a klasického morfologického obrazu ET v KD. Preto je bioptické vyšetrenie KD štandardnou súčasťou diagnostiky ochorenia. ET sa vyznačuje prítomnosťou normocelulárnej, resp. len mierne hypercelulárnej KD s dominujúcou proliferáciou MGK. MGK sú tvorené veľkými formami s výrazne hyperlobulizovanými a hlboko lobulizovanými jadrami bez cytologických dysplázií, ktoré pripomínajú tzv. „jelenie parohy“ (obr. 5). V KD pozorujeme normálne množstvo, resp. len minimálne zmnoženie retikulínových vlákien (16, 17). Dôležitou skotočnosťou je, že trombocytóza v PK v kontexte pozitivity JAK2V617F nie je sama o sebe dostatočná pre potvrdenie diagnózy ET, keďže aj iné MPN sa môžu vyznačovať trombocytózou v PK (25). Rovnako je potrebné vylúčiť aj iné myeloidné neoplázie (vrátane CML, MDS/MPN, MDS) a v prípade absencie klonálneho markeru aj sekundárne príčiny trombocytózy. Z tohto hľadiska zohráva významnú úlohu bioptické vyšetrenie KD, umožňujúce odlíšenie reaktívnych zmien KD, ako aj iných myeloidných neoplázií, klinicky imitujúcich ET, najmä prefibrotické štádiá PMF (17, 20). ET naďalej ostáva stabilným ochorením s dobrou prognózou a dlhou dobou prežívania pri adekvátnej terapii (cca 15 rokov), pričom len minimálna časť pacientov sa transformuje do terminálneho štádia s myelofibrózou v KD (21).

Poslednou skupinou sú bližšie neklasifikovateľné MPN (MPN – NOS). Tu sa stretávame najmä s prípadmi skorých štádií MPN, kde najväčším problémom býva odlíšenie prefibrotických štádií PMF od ET, napriek odlišnosti bioptického obrazu ochorení v KD. Taktiež tu zaraďujeme pacientov diagnostikovaných až v terminálnych štádiách MPN s rozvinutou myelofibrózou, bez predchádzajúcej diagnózy MPN, keďže diferenciálna diagnostika MPN v terminálnych, fibrotických štádiách je problematická a v zásade nemožná (17). Treťou skupinou MPN-NOS sú prípady so závažnými sekundárnymi patologickými vplyvmi na KD, ktoré môžu natoľko alterovať morfológiu KD (najmä celularitu, množstvo fibrózy a morfológiu MGK) a morfologický obraz MPN, že histologická typizácia ochorenia je značne problematická. Keďže cytoredukčná terapia MPN značne ovplyvňuje spomínané morfologické črty KD (18), je potrebné biopsiu KD realizovať pred začatím terapie pacienta, resp. po odstránení, alebo minimalizovaní iných patologických faktorov ovplyvňujúcich morfológiu KD.
Základné princípy WHO klasifikácie 2008 ostávajú založené na WHO klasifikácii 2001, avšak nová WHO klasifikácia implementuje nové poznatky najmä v genetike MPN do diagnostických. kritérií ochorení, čím skvalitňuje diferenciálnu diagnostiku a vymedzuje aj osobitné nozologické jednotky s možnosťou účinnejšej liečby a lepšej prognózy. Zároveň zdôrazňuje význam bioptického vyšetrenia KD v diagnostike a najmä subtypizácií Ph1-MPN, ktoré umožňuje aj vylúčenie reaktívnych zmien KD a zároveň poskytuje informácie aj o niektorých prognosticky významných morfologických črtách KD (množstvo blastov a stupeň fibrózy). Dôležitou zmenou je aj zavedenie pozitívnych kritérií pre diagnostiku ET, ktorá už nie je „diagnosis per exclusionem“, ako aj zníženie kritéria hladiny trombocytov potrebných pre diagnózu ET. Vzhľadom na dynamický rozvoj poznatkov najmä v oblasti genetiky a špecifickej terapie MPN však možno v blízkej budúcnosti očakávať ďalšie návrhy na zmeny a zlepšenia WHO klasifikácie 2008.
Poďakovanie: Práca bola podporená Grantom UK 67/2009, Európskym sociálnym fondom a projektom „Centrum excelentnosti pre perinatologický výskum“, ktorý je spolufinancovaný zo zdrojov ES.
Zoznam skratiek
- AF – akcelerovaná fáza
- BF – blastická fáza
- bFGF – základný fibroblastový rastový faktor (basic fibroblast growth factor)
- CEL – chronická eozinofilná leukémia
- CF – chronická fáza
- CIMF – chronická idiopatická myelofibróza
- CML – chronická myeloidná leukémia
- CMMoL – chronická myelomonocytová leukémia
- CNL – chronická neutrofilná leukémia
- ET – esenciálna trombocytémia
- FGFR1 – receptor fibroblastového rastového faktoru 1
- HES – hypereozinofilný syndróm
- JAK2 – Janus 2 tyrozínová kináza
- KD – kostná dreň
- MaLNEaPDGFR/FGFR – myeloidné a lymfoidné neoplázie s eozinofíliou a mutáciou receptoru doštičkového rastového faktora alebo receptoru fibroblastového rastového faktora
- MDS – myelodysplastický syndróm
- MDS/MPN – myelodysplascký-myeloproliferatívny syndróm
- MF0 – normálne množstvo fibrózy v kostnej dreni
- MF1 – ľahká retikulínová fibróza
- MF2 – rozvinutá retikulínová fibróza
- MF3 – rozvinutá kolagénová fibróza
- MGK – megakaryocyty
- MPL – trombopoetínový receptor
- MPN – myeloproliferatívne neoplázie
- MPO – myeloproliferatívne ochorenia
- NOS – bližšie neklasifikovateľné (not othewise specified)
- PDGFRA – receptor doštičkového rastového faktora α
- PDGRFB – receptor doštičkového rastového faktora ß
- Ph1 – Filadelfia 1 chromozóm
- PK – periférna krv
- PMF – primárna myelofibróza
- PV – pravá polycytémia
- RARS-T – refraktérna anémia s prstencovými sideroblastami a trombocytózou
- TGF-ß – transformačný rastový faktor ß (transfoming frowth factor ß)
- WHO – Svetová zdravotnícka organizácia (World health organisation)
MUDr. Juraj Marcinek, PhD
Ústav
patologickej anatómie JLF UK a MFN
Kollárova
2
036
59 Martin
Slovensko
marcinek@jfmed.uniba.sk
Zdroje
1. Jaffe ES, Harris NL, Stein H, Vardiman JW. World Health Oraganization Classifikation of Tumours, Pathology & Genetics, Tumours of Haemopoietic and Lymphoid Tissues. IARC Press, Lyon, 2001; 351.
2. Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues. 4th ed. WHO Press, IARC, Lyon, 2008; 439.
3. Kvasnicka HM, Thiele J. The impact of clinicopathological studies on staging and survival in essential thrombocythemia, chronic idiopathic myelofibrosis, and polycythemia rubra vera. Semin Thromb Hemost 2006; 32 : 362–371.
4. Metzgeroth G, Walz C, Score J., et al. Recurrent finding of the FIP1L1-PDGFRA fusion gene in eosinophilia-associated acute myeloid leukemia and lymphoblastic T-cell lymphoma. Leukemia 2007; 21 : 1183–1188.
5. Bain BJ, Fletcher SH. Chronic eosinophilic leukemias and the myeloproliferative variant of the hypereosinophilic syndrome. Immunol Allergy Clin North Am 2007; 27 : 377–388.
6. Cools J, De Angelo DJ, Gotlieb J, et al. A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med 2003; 348 : 123–128.
7. Tefferi A., Pardanani A. Clinical, genetic, and therapeutic insights into systemic mast cell disease. Curr Opin Hematol 2004; 11 : 58–64.
8. Rice L, Popat U. Ever case of essential thrombocythemia should be tested for the Philadelphia chromosome. Am J Hematol 2005; 78 : 71–73.
9. Cortes J E, Talpaz M, OęBrien S, et al. Staging of chronic myeloid leukemia in the imatinib era: an evaluation of the World Health Organization proposal. Cancer 2006; 106 : 1306–1315.
10. Kralovics R, Passamonti F, Buser A S, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 2005; 332 : 1499–1502.
11. Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005; 365 : 1054–1061.
12. Schmitt-Graeff AH, Teo SS, Olschewski M, et al. JAK2V617F mutation status identifies subtypes of refractory anemia with ringed sideroblasts associated with marked thrombocytosis. Haematologica 2008; 93 : 4–6.
13. Jones AV, Kreil S, Zoi K, et al. Widespread occurence of the JAK2 V617F mutation in chronic myeloproliferative disorders. Blood 2005; 106 : 2162–2168.
14. Scott LM, Tong W, Levine RL, et al. JAK2 exon 12 mutations in Polycythemia vera and Idiopathic myelofibrosis. N Engl J Med 2007; 356 : 444–445.
15. Thiele J, Kvasnicka HM, Diehl V. Initial (latent) polycythemia vera with thrombocytosis mimicking essential thrombocythemia. Acta Haematol 2005; 113 : 213–219.
16. Thiele J, Kvasnicka HM, Orazi A. Bone marrow histopathology in myeloproliferative disorders – current diagnostic approach. Semin Hematol 2005; 42 : 184–195.
17. Thiele J, Kvasnicka HM, Vardiman J. Bone marrow histopathology in the diagnosis of chronic myeloproliferative disorders: a forgotten pearl. Best Pract Res Clin Haematol 2006; 19 : 413–437.
18. Tefferi A. Pathogenesis of myelofibrosis with myeloid metaplasia. J Clin Oncol 2005; 23 : 8520–8530.
19. Pikman Y, Lee BH, Mercher T, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PloS Med 2006; 3 : 1140–1151.
20. Thiele J, Kvasnicka HM. Chronic myeloproliferative disorders with thrombocythemia: a comparative study of two classification systems (PVSG, WHO) on 839 Patients. Ann Hematol 2003; 82 : 148–152.
21. Kreft A, Buche G, Ghalibafian M, Buhr T, Fischer T, Kirkpatrick C J. The incidence of myelofibrosis in Essential thrombocythemia, Polycythemia vera and Chronic idiopathic myelofibrosis: a retrospective evaluation of sequential bone marrow biopsies. Acta Haematol 2005; 113 : 137–143.
22. Tefferi A. Myelofibrosis with myeloid metaplasia. N Engl J Med 2000; 342 : 1255–1265.
23. Thiele J, Kvasnicka HM, Facchetti F, Franco V, Van der Walt J, Orazi A. European consensus on grading bone marrow fibrosis and assessment of cellularity. Haematologica 2005; 90 : 1128–1132.
24. Sacchi S, Vinci G, Gugliotta L, et al. Diagnosis of essential thrombocythemia at platelet counts between 400 and 600x10(9)/L. Gruppo Italiano Malattie Mieloproliferative Croniche (GIMMC). Haematologica 2000; 85 : 492–495.
25. Michiels JJ, Kvasnicka HM, Thiele J. Chronic Ph1-negative myeloproliferative disorders (MPDs). European MPD workshop on clinical and pathological features of Ph1 - MPDs Rotterdam 2004; 2–40.
26. Kreft A, Nolde C, Büsche G, Buhr T, Kreipe H, Georgii A. Polycythaemia vera: bone marrow histopathology under treatment with interferon, hydroxyurea and busulphan. Eur J Haematol 2000; 64 : 32–41.
Štítky
Hematologie a transfuzní lékařství Interní lékařství OnkologieČlánek vyšel v časopise
Transfuze a hematologie dnes

2010 Číslo 1
- Diosmin a hesperidin: Co ukazuje farmakokinetika?
- Evaluace skóre m7-FLIPI u pacientů s folikulárním lymfomem odhalila slibný prediktivní biomarker k určení vhodné chemoterapie
- Neuropatie výrazně zpomaluje proces hojení u povrchových ran
- Diagnostika von Willebrandovy choroby krok za krokem
- Těhotenství a porod u ženy s VWD − kazuistika
Nejčtenější v tomto čísle
- Neočekávané vedlejší nálezy při 18F-FDG-PET/CT hodnocení léčebné odpovědi či následném sledování pacientů léčených pro ne-hodgkinský lymfom
- Zmeny názvoslovia, klasifikácie a diagnostických kritérií myeloproliferatívnych ochorení podľa WHO klasifikácie 2008
- Nehodgkinovské lymfómy v detskom veku v Slovenskej republike – výskyt a výsledky liečby
- Novinky v klasifikaci MDS a stanovení prognózy dle WPSS
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy