
Esenciální trombocytemie v dětském věku
Essential thrombocythaemia in children
Essential thrombocythaemia (ET), a Philadelphia chromosome negative chronic myeloproliferative disorder, is usually a disease of middle age and it is extremely rare in paediatric population. In this report we have analysed 15 children (9 girls and 6 boys) diagnosed with ET in the Czech paediatric population from 1987 to 2007. There is a growing incidence of ET in children: 0.1/1 million children/year in years 1987–1996 but 0.6/1 million children/year in years 1997–2006. Platelet count at the time of diagnosis was 681–2428x10⁹/l (median 1720x10⁹/l), Hb and leukocyte counts were within normal range. Splenomegaly was found in 8 children. The median follow-up was 19 months. The clinical picture in our paediatric patients was milder than in adults. The diagnosis was made only during routine blood analysis in more than half of the cases. No thrombosis or major bleeding was observed at the time of diagnosis, although symptoms of microvascular obstruction were present in some patients. Erythropoietin (Epo) hypersensitivity of haematopoietic progenitors in vitro was found in the majority of the patients (11/13), with EEC formation in 9/11 patients. These results are indicative of the diagnosis of myeloproliferative disease. We detected JAK2 V617F mutation in peripheral blood leukocytes or in separated platelets only in one female patient with borderline level of haemoglobin. Monoclonal haematopoiesis was noted only in one another female patient. During the EEC analysis we found some colonies bearing heterozygous or homozygous V617F mutation in 3/5 examined patients. Our data suggests that childhood ET patients could bear minor JAK2 V617F-positive subclones. These patients did not show any phenotypic differences from the cohort. Treatment strategies for paediatric ET have not yet been fully defined. The treatment approach to our paediatric patients was variable. As the majority of paediatric haematologists prefer anagrelide to hydroxyurea, 6 patients were treated with anagrelide, 5 patients with acetylosalicylic acid and in 4 patients the „wait and see“ strategy was used. United approach towards the diagnosis and treatment of childhood ET is still to be established. The paediatric patients have to be closely monitored in case of possible disease progression to clonal haematopoiesis, of appearance of JAK2 V617F mutation, or of evolvement into clinical picture of polycythaemia vera. Finally, the question if adult and paediatric ET are different stages of the same disease or whether they represent separate disorders is still to be answered possibly by international cooperation in large multicentral studies and by long-term monitoring of paediatric patients.
Key words:
essential thrombocythaemia, children, clonality, erythroid colonies, hypersensitivity to erythropoietin, JAK2 V617F mutation
Autoři:
D. Pospíšilová 1; J. Veselovská 2; M. Horváthová 2; R. Solná 2; J. Kučerová 2; J. Čmejlová 3; R. Čmejla 3; M. Beličková 3; S. Peková 4; K. Petrtýlová 5; V. Mihál 1; T. Votava 6; J. Hak 7; O. Zapletal 8; J. Starý 5; V. Divoký 2
Působiště autorů:
Dětská klinika LF UP a FN v Olomouci, 2Ústav biologie LF UP v Olomouci, 3Ústav hematologie a krevní transfúze
Oddělení buněčné fyziologie, Praha, 4Oddělení klinické biochemie, hematologie a imunologie, Nemocnice na Homolce
Praha 5Klinika dětské hematologi
; Dětská klinika LF UK a FN Hradec Králové, 8Oddělení klinické hematologie FN Brno
7
Vyšlo v časopise:
Transfuze Hematol. dnes,14, 2008, No. 2, p. 63-70.
Kategorie:
Souhrnné práce, původní práce, kazuistiky
Souhrn
Esenciální trombocytemie (ET) je Ph₁ negativní myeloproliferativní onemocnění charakterizované zvýšenou proliferací megakaryocytů se zvýšenou produkcí trombocytů, trvalou trombocytózou a zvýšeným rizikem trombotických a krvácivých epizod. Je považována za onemocnění středního věku, u dětí se vyskytuje velmi vzácně s incidencí kolem 1/10 milionů dětí za rok. U většiny pacientů byla prokázána snížená exprese receptoru pro trombopoetin na trombocytech. V roce 2005 byla u 23–57 % dospělých s ET popsána mutace genu JAK2 Val617Phe. Role této mutace v patogenezi ET dosud nebyla přesně objasněna. V letech 1987–2007 bylo v České republice diagnostikováno 15 dětí s ET (9 dívek a 6 chlapců ve věku 6–17 let). Incidence ET v dětské populaci má narůstající tendenci, v letech v letech 1987–1996 byla 0,1/1 milion dětí/rok, v letech 1997–2006 0,6/1 milion dětí/rok. Onemocnění bylo u 8 dětí zjištěno náhodně, u žádného z pacientů se neobjevily závažné trombotické a krvácivé komplikace. U 5 dětí byly přítomny klinické známky mikrovaskulární obstrukce, u dvou dětí přechodná nezávažná krvácení. Monoklonální hematopoéza byla přítomna jen u jedné nemocné. Většina (11/13) dětí vykazovala hypersenzitivitu hematopoetických progenitorových buněk na erytropoetin v in vitro kulturách. Tzv. endogenní erytroidní kolonie, tj. kolonie rostoucí i v médiu bez přidání exogenního erytropoetinu, byly detekovány u 11 dětí. Mutace genu JAK2 Val617Phe na úrovni leukocytů z periferní krve detekována jen u jednoho pacienta. Při analýze hematopoetických kolonií na přítomnost mutace genu JAK2 Val617Phe byly nalezeny ojedinělé homozygotní a heterozygotní kolonie nesoucí mutaci u 3/5 vyšetřených dětí, přestože u nich mutace nebyla detekována na úrovni periferní krve. Naše výsledky ukazují, že i u dětských esenciálních trombocytemií se mohou velmi vzácně vyskytovat subklony nesoucí tuto mutaci. Nález subklonů nebyl asociován se závažnějším klinickým fenotypem. Šest pacientů bylo léčeno anagrelidem, pět pacientů kyselinou acetylosalicylovou, čtyři pacienti byli pouze sledováni. Dosud nebyl stanoven jednotný diagnostický a léčený přístup u dětí s ET. Dětské pacienty je nutno pečlivě sledovat vzhledem k možné progresi onemocnění s vývojem klonální hematopoézy, získání mutace genu JAK2 Val617Phe, popřípadě rozvoji klinického obrazu polycytemia vera. Zda se v případě ET u dětí a dospělých jedná o biologicky odlišné onemocnění nebo zda jde o různé vývojové fáze téhož onemocnění, mohou ukázat pouze velké mezinárodní studie a především dlouhodobé sledování dětských pacientů.
Klíčová slova:
esenciální trombocytemie, dětský věk, klonalita, erytroidní kolonie, hypersenzitivita na erytropoetin, mutace JAK2 Val617Phe
Úvod
Esenciální trombocytemie (ET) patří spolu s pravou polycytemií a idiopatickou myelofibrózou mezi Ph1 negativní chronická myeloproliferativní onemocnění (MPD). ET je charakterizovaná zvýšenou proliferací megakaryocytů s nekontrolovanou produkcí trombocytů, trvalou trombocytózou, zvýšeným rizikem trombotických a krvácivých epizod. Může progredovat do myelodysplastického syndromu nebo leukemie. Je považována za onemocnění středního věku, diagnóza je nejčastěji stanovena mezi 50.–60. rokem. Asi 20 % případů je diagnostikováno ve věku pod 40 let. Incidence v dospělém věku je 1–5/100 000 osob/rok (1, 2).
Etiologie ET dosud nebyla plně objasněna. U většiny pacientů byla prokázána snížená exprese receptoru pro trombopoetin (MPL) na povrchu progenitorových buněk pro megakaryocyty a na povrchu trombocytů. U části pacientů s familiárním výskytem trombocytemie byly nalezeny mutace genu pro trombopoetin (THPO) a genu pro jeho receptor (MPL) (3, 4).
V roce 2005 byla identifikována mutace v JAK2 tyrozinové kináze vedoucí k aminokyselinové záměně valinu za fenylalanin v pozici 617 kinázové domény (JAK2 V617F) jako častá genetická abnormalita u různých typů MPD včetně ET. Mutace JAK2 V617F je popisována u > 90 % dospělých nemocných s pravou polycytemií, u 23–57 % dospělých s ET a u > 50 % nemocných s idiopatickou myelofibrózou (5–8). Jedná se o aktivační mutaci signální dráhy receptorů pro růstové faktory. Přesný etiologický význam této mutace u ET dosud nebyl objasněn. Dle posledních prací zvyšuje přítomnost této mutace riziko trombóz, především arteriálních (9, 10), nemá však vliv na celkové přežití pacientů ani na transformaci do leukemie (11). Poslední studie potvrzují, že přinejmenším u části dospělých pacientů s ET není JAK2 V617F mutace kauzální a objevuje se později v průběhu onemocnění. Pouze část populace klonálních buněk nese totiž tuto mutaci (12).
V dětském věku se ET vyskytuje vzácně, je uváděna incidence 1 na 10 milionů dětí/rok (13). K dnešnímu dni bylo v odborném písemnictví popsáno asi 100 případů dětských pacientů s ET (14). Malý počet pacientů a variabilita vyšetřených a srovnávaných parametrů nedává zatím prostor k žádným definitivním závěrům v této oblasti. Panel vyšetření požadovaných pro přesnou diagnostiku dětské ET dosud nebyl přesně definován, byly publikovány pouze ojedinělé zkušenosti s léčbou dětských pacientů. Indikace léčby proto nejsou jednotné a jsou obvykle odvozovány z výsledků získaných ze studií provedených u dospělých pacientů. Dvě dosud publikované studie dětských pacientů ukázaly, že většina dětí s ET má polyklonální onemocnění a že mutace JAK2 V617F se u dětí s ET na rozdíl od dospělých pacientů nachází pouze ojediněle (15, 16).
Soubor pacientů
Cílem této multicentrické studie je zhodnocení klinických a laboratorních nálezů u dětských pacientů s ET, jejichž diagnóza byla stanovena v jednotlivých centrech dětské hematologie-onkologie v České republice v letech 1987–2007. V uvedeném časovém intervalu bylo v České republice diagnostikováno 15 dětí s ET. Jednalo se o 9 dívek a 6 chlapců. Věk při stanovení dg. byl 6–17 let (medián 10,5 let). Všichni pacienti splňovali diagnostická kritéria PVSG (17). U všech pacientů bylo provedeno retrospektivní zhodnocení anamnestických a klinických údajů a výsledků vyšetření provedených při diagnóze.
U všech pacientů byla dále provedena následující vyšetření:
- hladiny erytropoetinu (Epo) a trombopoetinu (Tpo);
- analýza genů pro trombopoetin (THPO) a jeho receptor (MPL);
- stanovení in vitro senzitivity erytroidních progenitorových buněk k Epo a přítomnost spontánního růstu erytroidních kolonií („endogenous erythroid colonies“ – EEC);
- klonální krvetvorba u dívek;
- analýza JAK2 V617F mutace v leukocytech, v trombocytech a v koloniích EEC.
Etická komise Univerzity Palackého a Fakultní nemocnice Olomouc tuto studii schválila. Všichni pacienti nebo zákonní zástupci podepsali informovaný souhlas.
Metody
Měření sérových hladin Epo a Tpo
Koncentrace Epo byla měřena RIA metodou a vyjádřena v IU/l. K měření hladin sérového Tpo byl použit komerční kit (Quantikine, Elisa, RD system Europe), hladiny byly vyjádřeny v pg/ml a vztaženy k normám pro příslušný věk dítěte (18).
Analýza genů THPO a MPL
Pro identifikaci mutací byla použita metoda polymerázové řetězové reakce (PCR) a následné sekvenování PCR fragmentů. Primery pro amplifikaci z genomické DNA byly navrženy tak, aby bylo možné u obou genů zanalyzovat všechny exony s přilehlými oblastmi. PCR produkty byly elektroforeticky separovány a purifikovány z gelu pomocí QIAquick Gel Extraction Kit (Qiagen) a použity jako templát pro sekvenační reakci. Sekvence byly analyzovány na přístroji ABI3130 Genetic Analyzer (Applied Biosystems). Sekvence primerů a přesné podmínky amplifikace byly publikovány (19).
Analýza klonální krvetvorby
Z periferní krve 9 pacientek byla získána mononukleární frakce (MNC) a granulocyty separací na gradientu Ficol-Histopaque. Z mononukleární frakce byly izolovány CD3+ a CD14+ buňky pomocí magnetické separace (MiniMACS, Miltenyi Biotec). 0,5 μg genomické DNA bylo štěpeno po dobu nejméně 16 hodin při 37 ľC za užití reakční směsi obsahující 25 jednotek metylsenzitivní endonukleázy HpaII. Pro kontrolu štěpení bylo prováděno souběžné štěpení mužské genomické DNA se stejným množstvím restrikčního enzymu. Naštěpená DNA byla amplifikována za podmínek dříve publikovaných (20). PCR produkt byl analyzován v kapilární elektroforéze ABI PRISM 310 (Applied Biosystems) s použitím GeneScan softwaru (Applied Biosystems). Procentuální zastoupení alel bylo vypočítáno jako podíl alely s větší délkou ku alele s menší délkou násobený 100 %. Za mono-klonální vzorky byly považovány výsledky v intervalu 90–100 % nebo 0–10 %, za polyklonální v rozsahu 20–80 %. Ostatní výsledky byly označeny za oligoklonální.
Hypersenzitivita na Epo – klonální test hematopoetických progenitorů
Tato metoda využívá proliferační a diferenciační kapacitu hematopoetických progenitorů in vitro tvořit kolonie krevních buněk za definovaných podmínek (21). MPD na rozdíl od sekundárních stavů se vyznačují hypersenzitivitou hematopoetických progenitorů na různé růstové faktory např. na Epo.
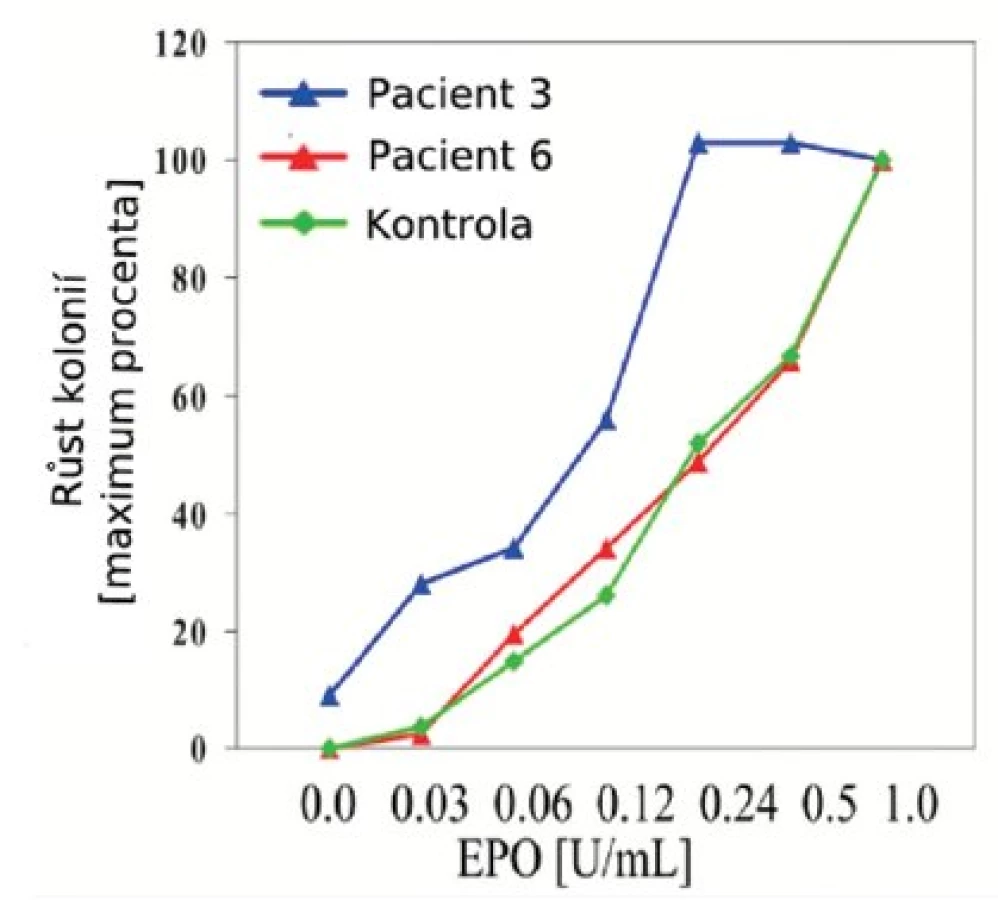
Mononukleární frakce buněk obsahující progenitory byla izolována z heparinizované periferní krve centrifugací na hustotním gradientu (Histopaque 1077 Hybri-Max, Sigma-Aldrich). 2,5 ml polotekutého média obsahujícího metylcelulózu (MethoCult H4531, StemCell Technologies) bylo připraveno do 35mm Petriho misek (1,1 ml na jednu Petriho misku) se 100 μl suspenze mononukleárních buněk (2,5x105 buněk na misku) a s přídavkem Epo (Epogen Epoetin Alfa, Amgen). Standardní koncentrace Epo v tomto systému je 1 U/ml média, pro test hypersenzitivity bylo využito koncentrací 0,03; 0,06; 0,12; 0,24; 0,5 U/ml média. Kultury byly inkubovány v atmosféře s 5 % CO2, 21 % O2, 70 % vlhkostí, při 37 °C. 14. den kultury byl hodnocen počet a morfologie kolonií (erytroidní kolonie BFU-E). BFU-E rostoucí v kulturách bez přidání Epo byly hodnoceny jako tzv. endogenní erytroidní kolonie (EEC) – (obr. 1).

Detekce mutace JAK2 V617F
Mutace JAK2 V617F byla detekována na úrovni genomické DNA izolované z leukocytů z periferní krve pomocí alelově-specifické (AS) PCR a pomocí restrikční analýzy BsaXI (6), a dále na úrovni RNA izolované z trombocytů z periferní krve pomocí reverzní transkripce s následnou alelově-specifickou PCR (AS-RT-PCR) podle protokolu dle Campbella et al. (22).
Detekce JAK2 V617F na úrovni jednotlivých myeloidních hematopoetických kolonií
Detekce mutace JAK2 V617F byla založena na metodě real-time alelické diskriminace na přístroji RotorGene 300 (Corbett Research) nebo na přístroji LightCycler 480 (Roche Applied Science). Jednotlivé kolonie byly přeneseny pod mikroskopem do 500 μl Trizol Reagent (Invitrogen). DNA se vyprecipitovala přes noc přidáním 300 μl 96% etanolu. Vysrážená DNA byla stočena při 14 000 g 30 min., a dvakrát promyta v 10% etanolu v 1x PBS, usušena a rozpuštěna v 10 μl deionizované, autoklávované vody. Vlastní metoda využívá jeden pár primerů k amplifikaci odpovídajícího fragmentu JAK2 genu pomocí PCR reakce a dvou fluorescenčně značených LNA-modifikovaných sond (Exiqon, Dánsko), které dokáží rozlišit mezi oběma JAK2 genotypy (mezi V617F mutací a nemutovaným – “wild-type”) (15).
Výsledky
Základní charakteristika souboru pacientů s výsledky retrospektivních analýz jsou shrnuty v tabulce 1.

Při analýze anamnestických údajů byly zjištěny klinické příznaky, které by mohly být dávány do souvislosti s trombocytemií, pouze u 7 dětí. Ve třech případech šlo o bolesti hlavy, které mohly být známkou mikrovaskulární obstrukce, v jednom případě o opakované synkopy, pravděpodobně v důsledku přechodné ischemie mozkových cév. Pacientka s pozitivním nálezem mutace JAK2 V617F byla vyšetřena pro intermitentní bolesti na hrudníku. U dvou pacientů byly při diagnóze přítomny krvácivé projevy: v jednom případě přechodné krvácení z rekta bez zjevné zevní příčiny a ve druhém krvácení do kolenního kloubu provokované úrazem. U osmi dětí byla diagnóza zjištěna náhodně při vyšetření krevního obrazu indikovaném z jiných příčin, většinou v rámci předoperačního vyšetření. U žádného z dětí nebyla v době diagnózy ani v průběhu sledování zaznamenána trombóza, ani závažné krvácení. Počet trombocytů v době diagnózy byl 681–2428x109/l (medián 1720x109/l). Hladiny hemoglobinu a počet leukocytů v době diagnózy byly v mezích normy pro daný věk. Doba sledování pacientů v době vyšetření se pohybovala od 5 do 192 měsíců s mediánem 19 měsíců. Tři pacienti dosáhli v době námi prováděných analýz již dospělého věku.
U pěti pacientů byla při prvním klinickém vyšetření přítomna splenomegalie, u dalších dvou byla splenomegalie zjištěna při ultrazvukovém vyšetření břicha.
Hladiny Epo byly normální u 13 pacientů, u dvou pacientů byly hladiny lehce snížené. Hladiny Tpo byly u 13 pacientů normální, u dvou lehce zvýšené. Morfologie megakaryocytů v kostní dřeni ukázala dominantní proliferaci megakaryocytů se zvýšením počtu zralých polyploidních megakaryocytů. Nebyly popsány patologické změny erytropoézy ani granulopoézy. Cytogenetická analýza aspirátu kostní dřeně provedená u všech pacientů vyloučila patologické nálezy.
Čtyři pacienti nebyli léčeni, pouze sledováni. Pěti pacientům byla podávána kyselina acetylosalicylová, v jednom případě po přechodnou dobu s interferonem-α (IFN-α). Jedna pacientka byla v minulosti přechodně léčena hydroxyureou a posléze IFN-α. Šest pacientů bylo léčeno anagrelidem (Thromboreductin). Pacienti léčení anagrelidem udávali minimální vedlejší účinky léčby. V jednom případě šlo o přechodné bolesti břicha, ve druhém o intermitentní mírné bolesti v epigastriu.
Přehled výsledků současných analýz je uveden
v tabulce 2
U žádného z pacientů nebyly nalezeny mutace genu THPO ani MPL. Hypersenzitivita hematopoetických progenitorů na Epo in vitro byla prokázána u 11/13 vyšetřených pacientů (tab. 1, obr. 1). U dvou pacientů jsme konstatovali nulový nebo slabý růst kolonií, který znemožnil závěrečné hodnocení. U dvou dětí s ET odpovídal výsledek křivce růstu kolonií u zdravých kontrol (obr. 2). U 9 pacientů s prokázanou hypersenzitivitou byl také popsán růst kolonií EEC (tab. 2, obr. 1).
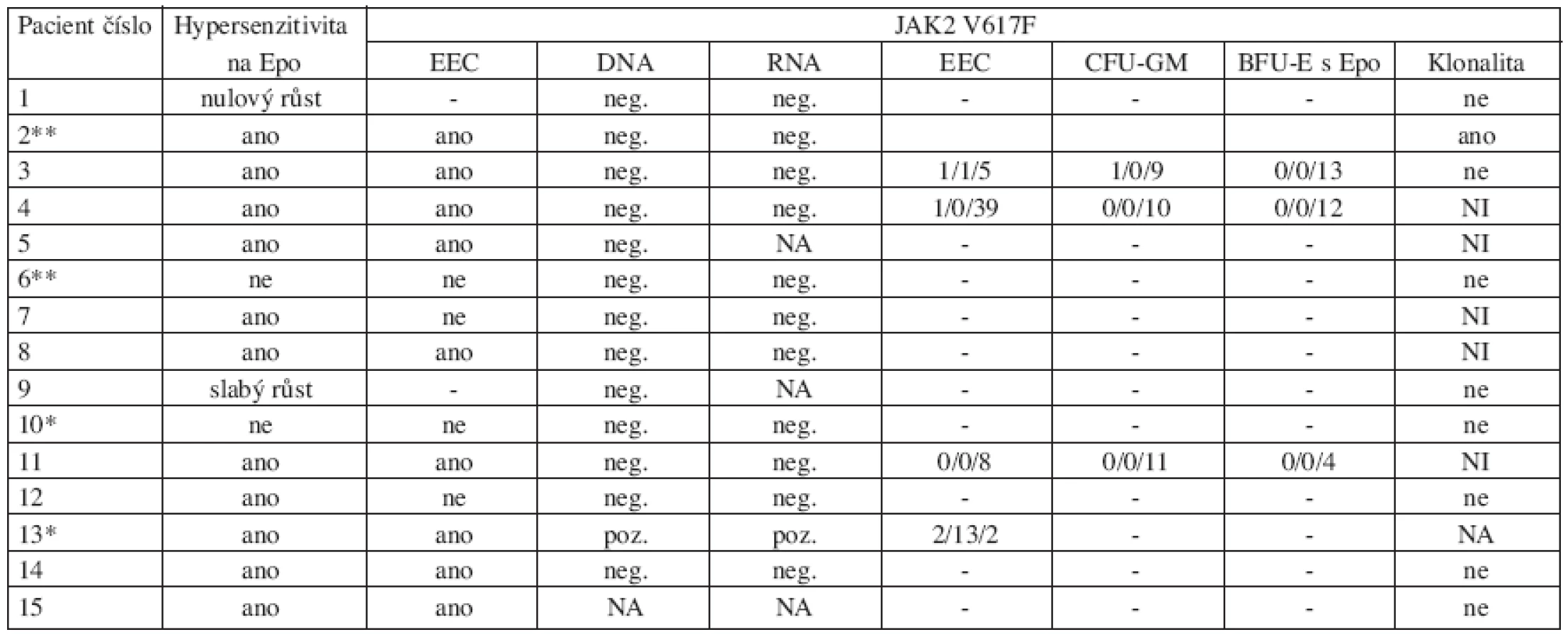
U 14 pacientů nebyla prokázána JAK2 V617F mutace na DNA úrovni v leukocytech periferní krve ani na RNA úrovni v purifikovaných trombocytech a granulocytech. Pouze u pacientky č. 13 byl nález JAK2 V617F mutace na úrovni DNA i RNA pozitivní. Klonální krvetvorba byla zjištěna pouze u pacientky č. 2. U pacientky s pozitivním nálezem JAK2 V617F mutace i u pacientky s klonálním onemocněním byla prokázána hypersenzitivita progenitorových buněk k Epo i růst EEC.
Další analýza ukázala přítomnost JAK2 V617F mutace u ojedinělých heterozygotních a dokonce i homozygotních kolonií u 3 z 5 vyšetřených dětí (tab. 2).
Diskuse
Trombocytemie se v dětském věku vyskytuje relativně často (až u 15 % hospitalizovaných dětí). V naprosté většině případů se jedná o sekundární zvýšení trombocytů, které provází celou řadu onemocnění, jako jsou akutní a chronické infekce, sideropenická anémie, onemocnění pojiva, nádorová onemocnění, hemolytické anémie, stavy po splenektomii a jiné. Většinou nepřináší žádné komplikace a nevyžaduje léčbu (23). Primární – esenciální trombocytemie je naproti tomu u dětí velmi vzácná. Incidence ET v dětské populaci má narůstající tendenci, v letech v letech 1987–1996 byla 0,1/1 milion dětí/rok, v letech 1997–2006 0,6/1 milion dětí/rok. U většiny pacientů souboru (11/15) byla diagnóza stanovena po roce 2000. Nedomníváme se, že je to v důsledku zvyšující se incidence choroby, ale v důsledku skutečnosti, že je diagnóza v dnešní době obvykle stanovena dříve než v minulosti a často náhodně. U většiny dětí souboru (8/15) bylo onemocnění v době diagnózy asymptomatické a diagnóza byla u dětí stanovena náhodně při vyšetření krevního obrazu indikovaného z jiných důvodů. Při používání automatických analyzátorů a s častějším vyšetření krevního obrazu u dětí lze očekávat časnější záchyt onemocnění v asymptomatické fázi.
Klinický obraz ET u pediatrických pacientů je tedy mírnější než u dospělých nemocných, u kterých je popisován výskyt trombotických komplikací až ve 29 % případů (10). V době diagnózy ani v průběhu sledování jsme nezaznamenali u žádného pacienta z našeho souboru trombózu nebo závažnější krvácení. Nejčastěji se zde vyskytovaly projevy mikrovaskulární obstrukce (bolesti hlavy, synkopy, bolest na hrudníku). Pouze u dvou pacientů byly pozorovány mírné krvácivé projevy, a to přechodné krvácení z rekta a krvácení do kolenního kloubu. Pacientka s pozitivním nálezem JAK2 mutace udávala v době diagnózy pouze bolesti na hrudníku.
Mutace genu THPO jsou popisovány vzácně u dětí s familiárním výskytem ET. Mutace genu MPL (receptoru pro Tpo) je v souborech dospělých pacientů nalézána s frekvencí 1 % (24). V našem souboru nebyly mutace žádného z obou genů nalezeny, dokonce ani u pacienta s familiárním výskytem trombocytózy. U většiny dětí s ET tedy tyto mutace pravděpodobně nehrají etiologickou roli.
U většiny vyšetřených dětí byla prokázána hypersenzitivita hematopoetických progenitorů na Epo v in vitro kulturách. Erytroidní kolonie rostoucí i v médiu bez přidání exogenního Epo (EEC) byly detekovány u 9 dětí. Pozitivní nálezy hypersenzitivity na Epo a průkaz EEC jsou charakteristickým rysem myeloproliferativních onemocnění a považujeme je za významný diagnostický znak MPD. Nepřítomnost těchto znaků však nevylučuje diagnózu ET. Pouze u jedné pacientky s prokázanou hypersenzitivitou na Epo a s růstem EEC byla prokázána mutace JAK2 V617F. Tento nález potvrzuje výsledky studií dospělých pacientů, které popisují nález EEC jak u JAK2 V617F pozitivních, tak i negativních pacientů s ET (25).
Pouze u jediné pacientky byla prokázána klonální krvetvorba. Diagnóza u této pacientky byla stanovena již ve věku 9 let, a to náhodně při předoperačním vyšetření. V době námi prováděných analýz byla tato pacientka již dospělá. Pozitivní nález klonální krvetvorby nekoreloval s nálezem mutace genu pro JAK2 kinázu, pouze s pozitivním nálezem hypersenzitivity na Epo a s průkazem EEC. Většina dětských pacientů klasifikovaných jako ET tedy nemá klonální onemocnění. Otázkou zůstává, zda a jak rychle se z monoklonálního onemocnění může časem vyvinout onemocnění klonální.
Mutace JAK2 V617F byla prokázána v periferní krvi pouze u jediné pacientky našeho souboru (6,5 %). Kromě námi publikované studie (15) byla dosud publikována jediná práce o výskytu JAK2 V617F mutace u dětských pacientů (16), která rovněž poukazuje na skutečnost, že u většiny dětských pacientů s ET tuto mutaci v periferní krvi nelze detekovat. Autoři prokázali mutaci u 2 z 20 pacientů (10 %). Položili jsme si otázku, zda mutace může být přítomna v minoritní subpopulaci hematopoetických progenitorů, což by bylo pod hranicí citlivosti detekce mutace na úrovni DNA nebo RNA z periferní krve. V in vitro kulturách jsou hematopoetické progenitory stimulovány k diferenciaci a vytvářejí kolonii. Na přítomnost JAK2 V617F mutace byly tedy analyzovány jednotlivé myeloidní kolonie (BFU-E a CFU-GM). Přesto, že jsme nedetekovali JAK2 V617F v mononukleárních buňkách periferní krve u 14/15 pacientů, ani v granulocytární nebo v destičkové RNA při použití standardních PCR protokolů, u tří z pěti vyšetřených dětí byly nalezeny ojedinělé JAK2 V617F pozitivní erytroidní a granulocytární kolonie homozygotní a heterozygotní pro tuto mutaci (tab. 2). Pozitivní nálezy nebyly asociovány se závažnějším klinickým fenotypem ani se specifickými laboratorními znaky. Naše výsledky ukazují, že i u dětských ET se mohou velmi vzácně vyskytovat JAK2 V617F pozitivní subklony. Je evidentní, že typ vyšetřovaných buněk a senzitivita použitých technik jsou pro detekci JAK2 V617F mutace velmi důležité. V kontrastu s publikovanými pracemi jsme prokázali, že homozygotní klony u ET existují (26). Raritní výskyt JAK2 V617F homozygotních progenitorových buněk však provokuje k úvaze, že mutantní subklony mohou být u ET buďto potlačeny, jak předpokládá Lippert et al (8), nebo mohou reprezentovat časnější událost ve vývoji choroby, než se dříve předpokládalo (12). Naše výsledky zatím nedovolují vyvozovat, zda JAK2 V617F pozitivní subklony mají u dětských pacientů s ET prognostický význam. Nenalezli jsme asociaci klinických ani laboratorních nálezů (vyšší výskyt trombotických nebo krvácivých příhod nebo hladina hemoglobinu, počet leukocytů a trombocytů) s pozitivitou mutace v koloniích. Z tohoto pohledu zajímavým nálezem byly nápadně vyšší hladiny hemoglobinu u jediné pacientky s pozitivitou nálezu JAK2 V617F mutace v periferní krvi. Hodnota však ještě nepřekročila horní hranici normy pro věk pacientky. V době diagnózy měla pacientka také nižší hodnoty počtu trombocytů ve srovnání s ostatními pacienty – (tab. 1). Tyto nálezy jsou v souladu s názory některých autorů (27), podle kterých je u pacientů s ET s pozitivním nálezem JAK2 V617F mutace vyšší riziko přechodu do pravé polycytemie. Proto bude velmi důležité sledovat dětské pacienty s ET vzhledem k možné progresi onemocnění zahrnující rozvoj klonální hematopoézy a JAK2 V617F mutace, popřípadě vývoj onemocnění do klinického obrazu polycytemia vera.
Vzhledem k nízkému výskytu závažných komplikací u dětí s ET by bylo možno klasifikovat toto onemocnění u dětí jako méně rizikové než je tomu u dospělých pacientů. Individuální klinický průběh je však variabilní a i u některých dětských pacientů se mohou objevit život ohrožující komplikace (23, 28, 29). Vzhledem k předpokladu dlouhého přežití pacientů, u kterých byla diagnóza ET stanovena již v dětství, je jasným imperativem poskytnout dětským pacientům dobře tolerovanou, minimálně toxickou a přitom účinnou léčbu. Podobně jako u dospělých je léčba považována za kompromis mezi riziky potenciální lékové toxicity na straně jedné a nezbytností prevence trombotických a hemoragických komplikací na straně druhé. V dnešní době dosud není k dispozici dostatek klinických studií hodnotících léčbu dětských pacientů s ET. Byla publikována pouze kazuistická sdělení nebo soubory s velmi malým počtem pacientů. Proto zatím neexistují obecně platná a jasná doporučení léčebných postupů.
Pro asymptomatické pacienty, u kterých není přítomno žádné přídatné riziko trombózy nebo krvácení, doporučuje většina autorů taktiku pečlivého monitorování vývoje nemoci bez léčby („watch and wait“). U dětí s projevy mikrovaskulární obstrukce, jejichž příznakem mohou být bolesti hlavy až charakteru migrény, erytromelalgie a přechodné projevy mozkové nebo kardiální ischemie, je doporučováno podání malé dávky kyseliny acetylosalicylové. Jednoznačné indikace a taktika cytoreduktivní léčby dosud nebyly stanoveny. Je bezpochyby opodstatněná u pacientů, kteří již prodělali trombotickou komplikaci, případně u pacientů s pozitivním nálezem JAK2 V617F mutace, u které se předpokládá vyšší riziko vzniku trombóz. Při indikaci cytoreduktivní terapie je možno zvolit jeden ze základních tří léků: hydroxyureu, IFN-α nebo anagrelid. Hydroxyurea je u dospělých pacientů většinou dobře tolerována. Rozsáhlé diskuse o jejím možném vlivu na indukci vzniku leukemie, které zatím nebyly ukončeny, vedou řadu dětských hematologů k názoru, že by lékem první volby pro dětské pacienty s ET měl být anagrelid, případně IFN-α. Hydroxyurea je tedy většinou dětských hematologů doporučována jako lék druhé volby (23, 27, 29).
IFN-α je používán u dětí s ET jen vzácně pro nepřijatelné vedlejší účinky (23). Anagrelid, imidazochinazolinový derivát, způsobuje selektivní supresi megakaryocytů bez ovlivnění erytropoézy nebo granulopoézy. Redukuje masu megakaryocytů a jejich ploidii a snižuje obrat produkce a tím počet trombocytů. Více než 90% dospělých odpovídá na anagrelid dobře bez ohledu na předchozí léčbu, a je lépe snášen než IFN-α (28). Jeho podání může však doprovázet celá řada vedlejších účinků jako jsou bolesti hlavy, retence tekutin, tachykardie, arytmie, anémie, které vedly k přerušení léčby u 16–20 % mladých dospělých pacientů (30). Zkušenosti s jeho podáním u dětí jsou malé, byly publikovány ve formě kazuistik nebo hodnocení malého souboru pacientů (31–34). Anagrelid byl úspěšně použit i u 6 dětí z našeho souboru. Potíže pacientů léčených anagrelidem byly zcela minimální a lék byl velmi dobře snášen.
Závěr
Hodnocení našeho souboru pacientů a rozbor již publikovaných dat o ET v dětském věku otevírá celou řadu otázek, na které je potřeba v budoucnu odpovědět. ET je v dětském věku vzácné onemocnění s heterogenními laboratorními i klinickými příznaky. Jeho patogeneze může být v některých aspektech odlišná od patogeneze ET u dospělých. Zda se jedná o biologicky odlišné onemocnění nebo zda se jedná o různé vývojové fáze téhož onemocnění mohou ukázat pouze velké studie na velkém počtu pacientů a především dlouhodobé sledování dětských pacientů. Totéž platí o stanovení racionálního diagnostického přístupu. Diferenciální diagnóza trombocytemie u dětí je velmi složitá a odlišení ET od sekundární trombocytemie je základní podmínkou volby správného léčebného přístupu. Velmi důležité je hodnocení morfologie megakaryocytů v kostní dřeni zkušeným cytopatologem, které však u dětí s trombocytemií může přinést rozpaky. Jak jsme ukázali v našem souboru v souhlase s dalšími autory, při diagnostice ET u dětí se nelze spoléhat na vyšetření klonality ani na analýzu JAK2 V617F mutace. Otázkou zůstává význam vyšetření mutace v erytroidních nebo granulocytárních koloniích. V našich rukách se jako nejspolehlivější test jeví vyšetření hypersenzitivity na Epo a detekce růstu kolonií EEC (15). Jako nejpřesnější a nejvíce specifické se ukazuje vyšetření růstu endogenních megakaryocytárních kolonií in vitro (8). Ke sjednocení diagnostického a léčebného přístupu jsou však zcela nezbytné studie na větším počtu pacientů, které vzhledem ke vzácnosti ET budou bezesporu vyžadovat mezinárodní spolupráci.
Došlo do redakce: 7. 3. 2008
Přijato: 18. 4. 2008
Práce byla podporována grantem IGA MZ: NR 9471-3.
Doc. MUDr. Dagmar Pospíšilová, Ph.D.
Březinova 5
772 00 Olomouc
Zdroje
1. Jensen MK, de Nully Brown P, Nielsen OJ, Hasselbach HC. Incidence, clinical features and outcome of essential thrombocythemia in a well defined geographical area. Eur J Haematol 2000; 65 : 132–139.
2. Mesa RA, Silverstein MN, Jacobsen SJ, Wollan PC, Tefferi A. Population-based incidence and survival figures in essential thrombocythemia and agnogenic myeloid metaplasia: an Olmsted County Study, 1975–1995. Am J Hematol 1999; 61 : 10–15.
3. Ghilardi N, Skoda RC. A single base deletion in the thrombopoietin (TPO) gene causes familial essential thrombocythemia through a mechanism of more efficient translation of TPO mRNA. Blood 1999; 94 : 1480–1482.
4. Ding J, Komatsu H, Wakita A, et al. Familial essential thrombocythemia associated with a dominant-positive activating mutation of the c-Mpl gene, which encodes for the receptor for thrombopoietin. Blood 2004; 103 : 4198–4200.
5. Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 2005; 352 : 1779–1790.
6. Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005; 365 : 1054–1061.
7. Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005; 7 : 387–397.
8. Lippert E, Boissinot M, Kralovics R, et al. The JAK2-V617F mutation is frequently present at diagnosis in patients with essential thrombocythemia and polycythemia vera. Blood 2006; 108 : 1865–1867.
9. Finazzi G, Rambaldi A, Guerini V, Carobbo A, Barbui T. Risk of thrombosis in patients with essential thrombocythemia and polycythemia vera according to JAK2 V617F mutation status. Haematologica 2007; 92 : 135–136.
10. Tefferri A, Elliott M. Thrombosis in myeloproliferative disorders: prevalence, prognostic factors, and the role of leukocytes and JAK2V617F. Semin Tromb Hemost 2007; 33 : 313 : 320.
11. Gangat N, Wolanskyj AP, McClure RF, Li CY, Schwager S, Wu W, Tefferi A. Risk stratification for survival and leukemic transformation in essential thrombocythemia: a single institutional study of 605 patients. Leukemia 2007; 21 : 270–276.
12. Kralovics R, Teo SS, Li S, et al. Acquisition of the V617F mutation of JAK2 is a late genetic event in a subset of patients with myeloproliferative disorders. Blood 2006; 108 : 1377–1380.
13. Hasle H. Incidence of essential thrombocythaemia in children. Br J Haematol 2000; 110 : 751.
14. Dame Ch, Sutor AH. Primary and secondary thrombocytosis in childhood. Br J Haematol 2005; 129 : 165–177.
15. Veselovska J, Pospisilova D, Pekova S, et al. Most pediatric patients with essential thrombocythemia show hypersensitivity to erythropoietin in vitro, with rare JAK2 V617F-positive erythroid colonies. Leuk Res 2008; 32 : 369–377.
16. Randi ML, Putti MC, Scapin M, et al. Pediatric patients with essential thrombocythemia are mostly polyclonal and V617FJAK2 negative. Blood 2006; 108 : 3600–3602.
17. Murphy S, Peterson P, Iland H, Laszlo J. Experience of the Polycythemia vera Study Group with essential thrombocythemia: a final report of diagnostic criteria, survival and leukemic transition by treatment. Semin Hematol 1997; 34 : 29–39.
18. Ishiguro A, Nakahata T, Matsubara K, Hayashi Y, Kato T, Suzuki Y. Age-related changes in thrombopoietin in children: reference interval for serum thrombopoietin levels. Br J Haematol 1999; 106 : 884–888.
19. Pospisilova D, Cmejlova J, Slavik L, Cmejla R. Elevated thrombopoietin levels and alterations in the sequence of its receptor c-Mpl in patients with Diamond-Blackfan anaemia. Haematologica 2004; 89 : 1382–1383.
20. Cermak J, Belickova M, Krejcova H, et al. The presence of clonal cell subpopulations in peripheral blood and bone marrow of patients with refractory cytopenia with multilineage dysplasia but not in patients with refractory anemia may reflect a multistep pathogenesis of myelodysplasia. Leukemia Research 2005; 29 : 371–379.
21. Prchal JT, Sokol L. “Benign erythrocytosis” and other familial and congenital polycythaemias. Eur J Haematol 1996; 57 : 263–268.
22. Campbell PJ, Scott LM, Buck G, et al. United Kingdom Myeloproliferative Disorders Study Group; Medical Research Council Adult Leukaemia Working Party; Australasian Leukaemia and Lymphoma Group. Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2 V617F mutation status: a prospective study. Lancet 2005; 366 : 1945–1953.
23. Randi ML, Putti MC. Essential thrombocythaemia in children: is treatment needed? Expert Opin Pharmacother 2004; 5 : 1009–1014.
24. Pardanani AD, Levine RL, Lasho T, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood 2006; 108 : 3472–6.
25. Pardanani A, Lasho TL, Finke C, Mesa RA, Hogan WJ, Ketterling RP, Gilliland DG, Tefferi A. Extending Jak2V617F and MplW515 mutation analysis to single hematopoietic colonies and B and T lymphocytes. Stem Cells 2007; 25 : 2358–2362.
26. Scott LM, Scott MA, Campbell PJ, Green AR. Progenitors homozygous for the V617F mutation occur in most patients with polycythemia vera, but not essential thrombocythemia. Blood 2006; 108 : 2435–2437.
27. Rumi E, Passamonti F, Pietra D, Della Porta MG, Arcaini L, Boggi S, et al. JAK2 (V617F) as an acquired somatic mutation and a secondary genetic event associated with disease progression in familial myeloproliferative disorders. Cancer 2006; 107 : 2206–2211.
28. Michiels JJ, van Genderen PJ. Essential Thrombocythemia in Childhood. Seminars Thromb Hemost 1997; 23 : 295–301.
29. Alvarez-Larrán A, Cervantes F, Bellosillo B, Giralt M, Juliá A, Hernández-Boluda JC, Bosch A, Hernández-Nieto L, Clapés V, Burgaleta C, Salvador C, Arellano-Rodrigo E, Colomer D, Besses C. Essential thrombocythemia in young individuals: frequency and risk factors for vascular events and evolution to myelofibrosis in 126 patients. Leukemia 2007; 21(6): 1218–1223.
30. Scherer S, Ferrari R, Rister M, Treatment of essential thrombocythemia in childhood. Pediatr Hematol Oncol 2003; 20 : 361–365.
31. Storren E, Tefferi A. Long term use of anagrelide in young patients with essential thrombocythemia. Blood 2001; 97 : 863–866.
32. Hermann J, Fuchs D, Sauerbrey A, Hemple L, Zintl F. Successful treatment of essential thrombocythemia with anagrelide in a child. Med Pediatr Oncol 1998; 30 : 367–371.
33. El-Moneim AA, Kratz CP, Böll S, Rister M, Pahl HL, Niemeyer CM. Essential versus reactive thrombocythemia in children: retrospective analyses of 12 cases. Pediatr Blood Cancer 2007; 49(1): 52–55.
34. Lackner H, Urban C, Benesch M, Moser A, Sovinz P, Schwinger W, Dornbusch HJ. Long-term use of anagrelide in the treatment of children with essential thrombocythemia. Eur J Haematol 2006; 77(4): 358–359.
Štítky
Hematologie a transfuzní lékařství Interní lékařství OnkologieČlánek vyšel v časopise
Transfuze a hematologie dnes

2008 Číslo 2
- Diosmin a hesperidin: Co ukazuje farmakokinetika?
- Evaluace skóre m7-FLIPI u pacientů s folikulárním lymfomem odhalila slibný prediktivní biomarker k určení vhodné chemoterapie
- Neuropatie výrazně zpomaluje proces hojení u povrchových ran
- Diagnostika von Willebrandovy choroby krok za krokem
- Těhotenství a porod u ženy s VWD − kazuistika
Nejčtenější v tomto čísle
- Esenciální trombocytemie v dětském věku
- Transientní antigenemie HBsAg po očkování vakcínou proti hepatitidě B u dárkyně krve
- Kontroly správné výrobní praxe v krevních bankách
- Pokroky v léčbě Ph pozitivní akutní lymfoblastické leukemie dospělých
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy