
Neurogenetika a neurovývojová onemocnění
Autoři:
doc. MUDr. Dana Šafka Brožková, Ph.D.; doc. MUDr. Petra Laššuthová, Ph.D.; RNDr. Anna Uhrová Mészárosová, Ph.D.
Působiště autorů:
Neurogenetická laboratoř Kliniky dětské neurologie 2. LF UK a FN Motol, Praha
Vyšlo v časopise:
Svět praktické medicíny, 5, 2023, č. 4, s. 24-28
Kategorie:
Medicína v ČR: přehledový článek
Souhrn
Neurogenetické choroby představují početnou a různorodou skupinu chorob. I přes pokroky v genetické diagnostice je šance na objasnění genetické příčiny vzácných neurologických chorob kolem 30–40 %. Naštěstí dochází k neustálému rozšiřování diagnostických a výzkumných metod a šance na objasnění se stále zvyšuje. Na příkladu nemocí dědičná periferní neuropatie a spastická paraparéza si ukážeme, jak se tyto nemoci projevují a jak probíhá diagnostika vzácných neurogenetických chorob.
Neurogenetické choroby řadíme mezi vzácná onemocnění. Vyskytují se celosvětově, ale odhadem přes 8000 vzácných nemocí postihuje až 6 % populace v západních zemích. Téměř 80 % těchto nemocí je geneticky podmíněno a není na ně dostupná léčba. Vzácná onemocnění se většinou projeví v dětském věku, ale není to zdaleka pravidlo. Bohužel třetina dětí postižených vzácnou nemocí zemře před dosažením pátých narozenin.
Nalezení genetické příčiny nemoci znamená pro pacienta ukončení diagnostických vyšetření a upřesnění diagnózy. Pokud je již nemoc lépe známa, objasnění vede k upřesnění prognózy a v neposlední řadě může vést i k úpravě medikace, případně zahájení léčby formou genové tera pie.
Genetickým vyšetřením se podaří objasnit (najít) genetickou příčinu nemoci 30–40 % pacientů. Je to dáno tím, že naše DNA kóduje přes 20 000 genů, oblastí DNA, které jsou návodem pro vytvoření proteinu. Pokud je v samotném genu nějaká patogenní odchylka od normy (tzv. patogenní varianta), pak se protein vůbec nevytvoří nebo vytvoří, ale funguje špatně a daná varianta se projeví nemocí.
Dosud je ale s nějakou známou nemocí spojováno pouze 7385 genů z celkového počtu 20 000. Tedy patogenní varianta v těchto genech se projeví jako více či méně známá nemoc. Větší část z 20 000 genů stále čeká na to, až někdo prokáže a popíše jejich patogenní odchylku jako příčinu nějaké nemoci. U části pacientů tak neobjasníme genetickou diagnózu, protože ještě neznáme konkrétní gen, který bude u nemoci popsán teprve v budoucnu.
Do určité míry nás v počtu objasněných případů brzdí také diagnostické možnosti, i když se za posledních 10 let rozvíjejí v genetice závratným tempem. Dříve dostupné metody umožňovaly vyšetřovat jednotlivý gen po genu postupně, zdlouhavě a draze. V dnešní době je již běžné provedení sekvenování nové generace (NGS). Toto sekvenování umožňuje najednou vyšetřit několik desítek vybraných genů (NGS panel) nebo rovnou všechny známé geny, tedy celou kódující DNA s 20 000 geny (NGS exomové sekvenování). Z tohoto vyšetření je možné analyzovat odchylky v DNA malého rozsahu – varianty jednobodové a vícebodové (záměna jednoho/několika nukleotidů – A, C, T, G), případně i větší varianty, přestavby jako delece, duplikace, které pokrývají oblasti dlouhé několik stovek až tisíců nukleotidů. I toto vyšetření má své limity, které se mohou projevit nedostatečným pokrytím vyšetřovaných oblastí, případně při zpracování dat konkrétní varianta není vyhodnocena, a příčinu nemoci u pacienta tak neobjevíme.
Obecně je vyšší šance na objasnění u monogenních chorob, kdy je patogenní varianta pouze v jednom konkrétním genu. U těch platí mendelovský způsob dědičnosti. Nemoc se tak může dědit dominantně, recesivně nebo je vázána na pohlavní chromosom X. Neurologické choroby však mohou být i v důsledku komplexní dědičnosti, kdy se na vzniku nemoci podílí více genetických faktorů, a v tomto případě hraje nezanedbatelnou roli prostředí, šance na objasnění tohoto typu onemocnění je nižší.
Z důvodů výše uvedených je zřejmé, proč nelze genetickým vyšetřením podezření na určitou nemoc vyloučit. Nikdy totiž nevíme, zda nám genetická příčina nemoci zatím neuniká. Proto genetickým vyšetřením můžeme nemoc pouze potvrdit, ale nikdy vyvrátit. Situace je jiná v případě známé varianty, která způsobuje nemoc v rodině, a chceme vyšetřit, zda tato konkrétní varianta není u některého z příbuzných, tam lze tuto variantu vyloučit nebo potvrdit s jistotou.
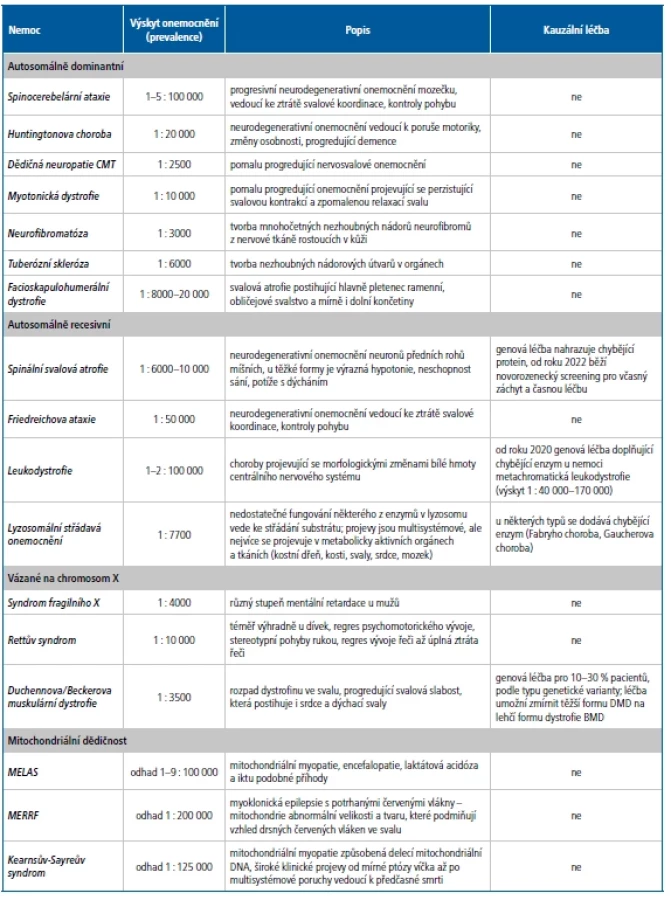
Skupina neurogenetických chorob představuje rozsáhlý seznam, pro představu zde uvádíme přehled některých častějších geneticky podmíněných neurologických nemocí i s krátkou charakteristikou (Tab. 1).
Pro konkrétní představu neurogenetických nemocí, jejich projevů a diagnostiky se zaměříme na tři skupiny nemocí, které vyšetřujeme v Neurogenetické laboratoři Kliniky dětské neurologie 2. LF UK a FN Motol. Jsou to dědičné neuropatie, spastické paraparézy a časné dětské epilepsie.
A. Dědičné neuropatie
Dědičné neuropatie CMT (neboli hereditární motorické a senzitivní neuropatie, HMSN) jsou nejčastější dědičná nervosvalová onemocnění, která postihují asi 5 tisíc osob v České republice. Název CMT pochází ze zkratky jmen vědců, kteří nemoc poprvé popsali, Charcot-Marie-Tooth. Dědičné neuropatie zahrnují velikou skupinu neuropatií, které mají podobné příznaky nemoci, ale jsou způsobeny patogenními variantami v různých genech. Podle toho, v jakém genu nalezneme patogenní variantu, lze zpřesnit prognózu nemoci u pacienta i samotný podtyp neuropatie.
Klinický obraz
U pacientů postižených CMT neuropatií se projevuje postižení motorických a senzitivních nervů. Postupně se zhoršuje síla a hybnost dolních a posléze i horních končetin jako následek poškození periferních motorických nervů. Nejčastěji si pacienti stěžují na zakopávání, nestabilitu kotníků a při postižení horních končetin neobratnost při otevírání lahví, zapínání knoflíků. Projevuje se také postižení senzitivních nervů ve smyslu zhoršeného čití, rozlišení teplé a studené, nejčastěji ponožkového rozsahu, různé parestezie, jako je brnění, mravenčení. Obecně jsou projevy nemoci lokalizované hlavně na prstech, chodidlech, lýtkách a obdobně při postižení horních končetin jsou postiženy drobné svaly ruky a části předloktí (Obr. 1). První projevy dědičné neuropatie se mohou vyskytnout v kterémkoliv věku, ale spíše očekáváme genetickou příčinu u neuropatie vzniklé v první a druhé dekádě, což platí i pro nejčastější formu dědičné neuropatie, typ CMT1A.
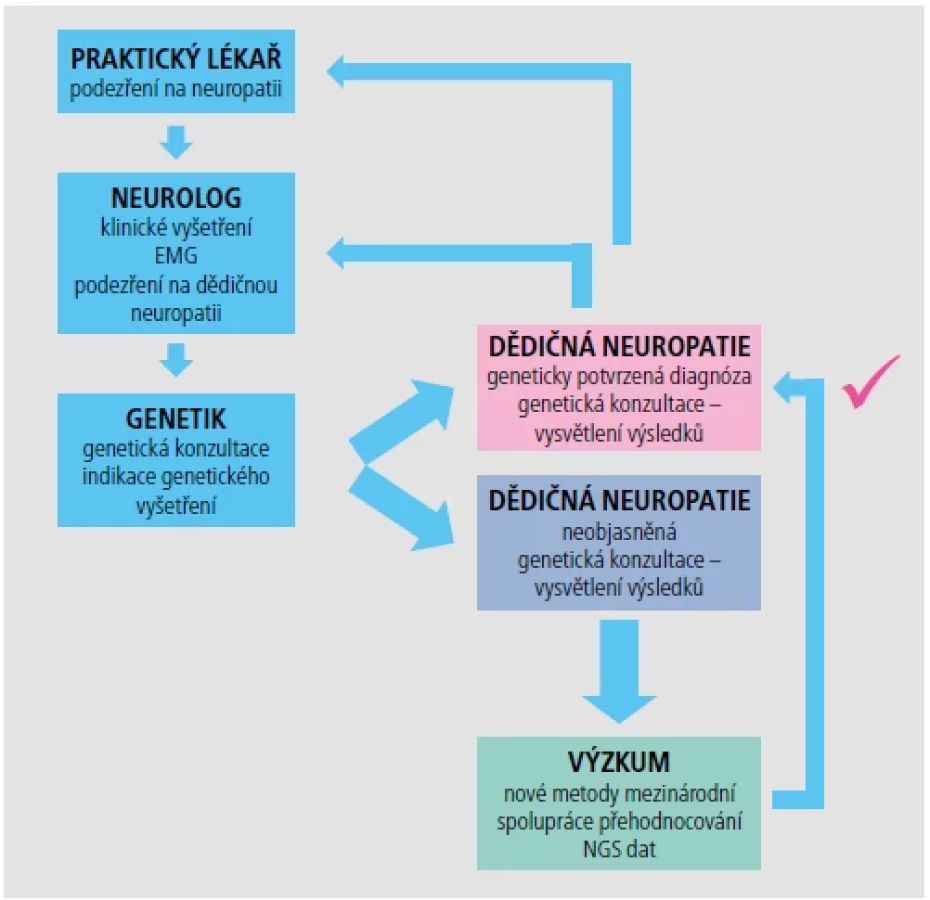
Klinické vyšetření neurologem doplněné o EMG vyšetření je dostačující k určení klinické diagnózy periferní neuropatie. Podle výsledku EMG se neuropatie dělí na dvě základní skupiny, a to demyelinizační, typ 1, se sníženou rychlostí vedení periferním nervem, a axonální typ 2, kde je rychlost vedení v normě, ale jsou snížené amplitudy.
CMT není smrtelné onemocnění a ve většině případů nezkracuje očekávanou délku života. Vede však často k tělesné invalidizaci a v těžkých případech jsou pacienti odkázáni na používání invalidního vozíku. Projevy CMT jsou velice variabilní, a to jak mezi jednotlivými typy neuropatií, tak i při stejném typu neuropatie v rámci jedné rodiny.
![Fotografie pacientů s dědičnou neuropatií. Atrofie lýtek a typická deformita nohy s vyklenutým nártem a kladívkovými prsty. Atrofie drobných svalů ruky.[Foto: archiv Neurogenetické laboratoře]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/5da96b06a8b8772e28db66c676baac82.png)
Genetika
Podle typu neuropatie se vyskytuje dědičnost dominantní, recesivní i vázaná na pohlavní chromosom X. Patogenní varianta, která způsobí neuropatii, se může vyskytnout v jednom z více než 100 genů. Každým rokem jsou patogenní varianty v dalších 1–2 genech nově popsány s dědičnou neuropatií.
Základní genetické vyšetření duplikace/delece pro typ neuropatie CMT1A nebo HNPP (dědičná neuropatie se sklonem k tlakovým parézám) objasní až 70 % pacientů s dědičnou demyelinizační neuropatií. Úspěšnost dalších vyšetření je již výrazně nižší a pouze u 30–40 % pacientů z těch, kteří mají negativní vyšetření CMT1A duplikace/HNPP delece, nalezneme příčinu dědičné neuropatie. Veškerá genetická vyšetření jsou poměrně finančně a časově náročná (Obr. 2). Proto například pokud je dospělý pacient zaslán k vyloučení genetické příčiny dědičné neuropatie a v diferenciální diagnostice je i CIDP (chronická zánětlivá demyelinizační neuropatie), přistupujeme pouze k základnímu vyšetření CMT1A duplikace/HNPP delece. Naopak u dětských pacientů s jasnou diagnózou periferní neuropatie přistupujeme hned k vyšetření NGS panelem.
Šance na objasnění příčiny dědičné neuropatie je také výrazně nižší u pacientů s pozdním nástupem (po 45. roce věku) a axonálním typem dědičné neuropatie. Obecně je šance na objasnění vyšší u dědičných neuropatií, kde je postiženo více členů rodiny a jedná se o typ demyelinizační.
Terapie
Slibně vypadá testování látky DTx-1252, která je určena k léčbě pacientů s nejčastějším typem neuropatie, typem CMT1A. Testování na myším modelu prokázalo výrazné zmírnění příznaků nemoci. Firma Novartis nedávno odkoupila práva na tuto látku a nyní plánuje začátek klinických studií. Principem léčby je snížení tvorby proteinu PMP22, který je v důsledku patogenní varianty u CMT1A pacientů v nadbytku. Dalším typem neuropatie, pro kterou bude dostupná léčba, je neuropatie způsobená patogenními variantami v genu SORD. U pacientů je v důsledku chybějícího enzymu SORD nedostatečně odbouráván sorbitol. Látka AT-007 sni žuje tvorbu sorbitolu a pro klinické studie užití látky AT-007 již probíhá nábor pacientů. Pro ostatní typy neuropatií kauzální terapie zatím není.
B. Spastické paraparézy
Klinický obraz
Hereditární spastické paraparézy/paraplegie (HSP nebo též SPG) jsou heterogenní skupinou dědičných onemocnění, která se klinicky projevují postupně se zhoršující oboustrannou spasticitou (ztuhlost, křeče) a slabostí dolních končetin. Onemocnění nezkracuje očekávanou délku života, ale může vést až ke ztrátě schopnosti samostatné chůze. Symptomy onemocnění se mohou začít projevovat v kterémkoli věku. U některých typů HSP, které označujeme jako komplikované, se mohou u pacientů postupně rozvinout další klinické znaky, např. atrofie částí mozku, atrofie optického nervu, kognitivní deficit, postižení horních končetin a další.
Genetika
Pro nemoc HSP platí také veliká genetická různorodost, proto v současné době při základní diagnostice vyšetřujeme přes 140 genů, ve kterých může být patogenní varianta zodpovědná za nemoc.
Terapie
V současné době kauzální terapie neexistuje. Proto je tak důležitá možnost genetického vyšetření a upřesnění rizika opakování nemoci v rodině, případně nabídnutí preimplantační diagnostiky s možností narození zdravého dítěte bez rodinné zátěže ve formě nemoci.
C. Časné dětské epilepsie
Závažné časné dětské epilepsie a epileptické encefalopatie (EE) jsou extrémně heterogenní skupinou onemocnění s časným nástupem epileptických záchvatů, obvykle farmakorezistentních (tzn. nereagujících na léčbu), provázených vývojovou stagnací či regresem. Objasnění genetických příčin tak heterogenní skupiny onemocnění umožnilo až sekvenování nové generace (NGS). Pravděpodobnost zjištění genetické příčiny se liší u různých typů epilepsie. U vzácných, závažných, sporadických epilepsií s nástupem v dětském věku bez patologického nálezu na magnetické rezonanci mozku je v současnosti až 50% pravděpodobnost, že se pomocí genetických metod podaří příčinu onemocnění objasnit. Ve většině případů (až 90 %) se jedná o patogenní variantu u pacienta nově vzniklou, s autosomálně dominantní dědičností. Tato varianta vznikla u jednoho z rodičů pouze v pohlavních buňkách. V takovém případě je riziko opakování nemoci u dalších potomků jen mírně zvýšené oproti riziku v běžné populaci. U běžných, byť často familiárních epilepsií je pravděpodobnost objasnění nesrovnatelně nižší.
Závěr
Spektrum neurogenetických chorob je velice široké. Neurogenetická laboratoř Kliniky dětské neurologie 2. LF UK a FN Motol se zabývá vyšetřováním příčin vzácných neurogenetických chorob v již popsaných genech v rámci rutinní diagnostiky. Pro vyšetření používáme široké spektrum metod (MLPA, klasické sekvenování jednotlivých genů, vyšetření NGS panelu genů, exomové i genomové sekvenování). Samozřejmostí je i hledání nových příčin těchto nemocí, a to v rámci výzkumu. V tomto případě je již nutná spolupráce se zahraničními týmy, tak abychom pomohli objasnit co nejvíce pacientů, pro které bude v budoucnu snad i kauzální léčba.
Podpořeno Islandem, Lichtenštejnskem a Norskem prostřednictvím Fondů EHP reg. č.: ZD-ZDOVA2-001.

Zdroje
1. An Online Catalog of Human Genes and Genetic Disorders, OMIM Gene map statistics [cit. 1.7.2023], dostupné z www.omim.org.
2. Cortese A, Zhu Y, Rebelo AP, et al. Biallelic mutations in SORD cause a common and potentially treatable hereditary neuropathy with implications for diabetes. Nat Genet 2020;52(5):473–81.
3. de Souza PVS, de Rezende Pinto WBV, de Rezende Batistella GN, et al. Hereditary spastic paraplegia: clinical and genetic hallmarks. Cerebellum 2017;16(2):525–51.
4. Eggermann K, Gess B, Hausler M, et al. Hereditary Neuropathies. Dtsch Arztebl Int. 2018;115(6):91–7.
5. Fridman V, Saporta MA. Mechanisms and treatments in demyelinating CMT. Neurotherapeutics 2021;18(4):2236–68.
6. Generation genome. Annual report of the chief medical offier. 2016 (https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/631043/CMO_annual_report_generation_genome.pdf).
7. Harding AE, Thomas PK. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain 1980;103(2):259–80.
8. Investigators GPP, Smedley D, Smith KR, Martin A, et al. 100,000 genomes pilot on rare-disease diagnosis in health care – preliminary report. N Engl J Med 2021;385(20):1868–80.
9. Knowles JK, Helbig I, Metcalf CS, et al. Precision medicine for genetic epilepsy on the horizon: Recent advances, present challenges, and suggestions for continued progress. Epilepsia 2022;63(10):2461–75.
10. Kramarz C, Rossor AM. Neurological update: hereditary neuropathies. J Neurol 2022;269(9):5187–91.
11. Lassuthova P, Safka Brozkova D, et al. Improving diagnosis of inherited peripheral neuropathies through gene panel analysis. Orphanet J Rare Dis 2016;11(1):118.
12. Lupski JR, de Oca-Luna RM, Slaugenhaupt S, et al. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell 1991;66(2):219–32.
13. Murala S, Nagarajan E, Bollu PC. Hereditary spastic paraplegia. Neurol Sci 2021;42(3):883–94.
14. Pipis M, Rossor AM, Laura M, et al. Next-generation sequencing in Charcot-Marie-Tooth disease: opportunities and challenges. Nat Rev Neurol 2019;15(11):644–56.
15. Portál pro vzácná onemocnění a léčivé přípravky pro vzácná onemocnění, [cit. 1.7.2023], dostupné z www.orpha.net.
16. Ruano L, Melo C, Silva MC, et al. The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies. Neuroepidemiology 2014;42(3):174–83.
17. Stanek D, Lassuthova P, Sterbova K, et al. Detection rate of causal variants in severe childhood epilepsy is highest in patients with seizure onset within the first four weeks of life. Orphanet J Rare Dis 2018;13(1):71.
Štítky
Praktické lékařství pro děti a dorost Praktické lékařství pro dospěléČlánek vyšel v časopise
Svět praktické medicíny

- I kondomový urinal může zabít
- Preskripce inkontinenčních pomůcek v praxi: pravidla platí i pro jejich kombinace
- Efektivita kartáčku Sonicare For Kids u dětí předškolního věku
- Metodika úhrady pomůcek pro inkontinentní pacienty
- Inkontinence jako důsledek operačního zákroku na prostatě
Nejčtenější v tomto čísle
- Zvýšené jaterní testy: jak postupovat v jejich vyhodnocování?
- Diferenciální diagnostika kulhání u dítěte
- Fixní kombinace ibuprofen/paracetamol
- Hematurie v pediatrii
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy