
Suppression of a Natural Killer Cell Response by Simian Immunodeficiency Virus Peptides
Natural killer (NK) cells recognize and kill infected cells without prior antigenic stimulation, and thus provide an important early defense against virus infection. NK cell responses in primates are regulated in part through interactions between two highly polymorphic molecules, the killer-cell immunoglobulin-like receptors (KIRs) on NK cells and their major histocompatibility complex (MHC) class I ligands on target cells. Inhibitory KIRs normally suppress NK cell responses through interactions with their MHC class I ligands on the surface of healthy cells. However, when these interactions are perturbed, this inhibition is lost resulting in NK cell activation and killing of the target cell. We investigated the functional implications of simian immunodeficiency virus (SIV) peptides bound by a common MHC class I molecule in the rhesus macaque that stabilize or disrupt binding to an inhibitory KIR. Whereas SIV peptides that stabilized KIR-MHC class I binding suppressed NK cell activation, peptides that disrupted this interaction did not and resulted in NK cell lysis. These findings demonstrate that viral peptides can modulate NK cell responses through KIR-MHC class I interactions, and are consistent with the possibility that human and simian immunodeficiency viruses may acquire changes in epitopes that increase the binding of MHC class I ligands to inhibitory KIRs as a mechanism to suppress NK cell responses.
Published in the journal:
. PLoS Pathog 11(9): e32767. doi:10.1371/journal.ppat.1005145
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1005145
Summary
Natural killer (NK) cells recognize and kill infected cells without prior antigenic stimulation, and thus provide an important early defense against virus infection. NK cell responses in primates are regulated in part through interactions between two highly polymorphic molecules, the killer-cell immunoglobulin-like receptors (KIRs) on NK cells and their major histocompatibility complex (MHC) class I ligands on target cells. Inhibitory KIRs normally suppress NK cell responses through interactions with their MHC class I ligands on the surface of healthy cells. However, when these interactions are perturbed, this inhibition is lost resulting in NK cell activation and killing of the target cell. We investigated the functional implications of simian immunodeficiency virus (SIV) peptides bound by a common MHC class I molecule in the rhesus macaque that stabilize or disrupt binding to an inhibitory KIR. Whereas SIV peptides that stabilized KIR-MHC class I binding suppressed NK cell activation, peptides that disrupted this interaction did not and resulted in NK cell lysis. These findings demonstrate that viral peptides can modulate NK cell responses through KIR-MHC class I interactions, and are consistent with the possibility that human and simian immunodeficiency viruses may acquire changes in epitopes that increase the binding of MHC class I ligands to inhibitory KIRs as a mechanism to suppress NK cell responses.
Introduction
By virtue of their ability to recognize and kill infected cells without prior exposure to antigen, natural killer (NK) cells provide an important innate defense against viral pathogens. NK cells differentiate virus-infected cells from healthy cells through the integration of complex signals from activating and inhibitory receptors, which in primates include the highly polymorphic killer-cell immunoglobulin-like receptors (KIRs). Whereas the molecular basis of ligand recognition for the activating KIRs is not fully understood, inhibitory KIRs selectively bind to subsets of major histocompatibility complex (MHC) class I molecules bearing particular sequence motifs in their α1-domains [1–3]. Inhibitory KIRs normally suppress NK cell activation through interactions with their MHC class I ligands on the surface of healthy cells. However, when these interactions are perturbed, for instance as a result of MHC class I downregulation by the human immunodeficiency virus (HIV)-1 Nef protein [4–6], this inhibition is lost resulting in NK cell degranulation and lysis of the infected cell.
Polymorphic differences in KIR and HLA class I genes can influence the course of HIV-1 infection [7–12], as well as the outcome of infection with other viral pathogens, including hepatitis C virus (HCV) [13], human papillomavirus (HPV) [14] and cytomegalovirus (CMV) [15]. In the case of HIV-1, activating and highly-expressed inhibitory alleles of KIR3DL1/S1, in combination with HLA-Bw4 alleles encoding isoleucine at position 80 (HLA-Bw4-80I), are associated with delayed progression to AIDS and greater suppression of viral replication in autologous CD4+ T cells by bulk NK cells [8,9,16]. In accordance with these genetic associations, NK cells expressing KIR3DS1 can suppress the in vitro replication of HIV-1 in HLA-Bw4-80I+ lymphocytes [17], and KIR3DS1+ and KIR3DL1+ NK cells preferentially expand during HIV-1 infection in HLA-Bw4-80I+ individuals [18]. KIR-expressing NK cells may also exert selective pressure on virus replication as demonstrated by HIV-1 polymorphisms associated with KIR2DL2 that confer resistance to NK cells expressing this KIR [19].
Consistent with three-dimensional structures of KIR-HLA class I complexes revealing that KIRs contact surfaces of their HLA class I ligands over C-terminal residues of the bound peptide [20–22], changes in C-terminal positions of the peptide can stabilize or disrupt interactions with inhibitory KIRs [23–26]. NK cells may therefore sense changes in the repertoire of peptides presented by the MHC class I ligands of inhibitory KIRs. Although this helps to explain how NK cells differentiate virus-infected cells from healthy cells, it also creates an opportunity for immune escape. By acquiring changes in epitopes that enhance MHC class I binding to inhibitory KIRs, viruses may prevent the elimination of infected cells by NK cells. Indeed, such a mechanism may account for the adaptation of HIV-1 for resistance to KIR2DL2+ NK cells [19].
We previously demonstrated that the binding of Mamu-A1*002, a common MHC class I molecule in the rhesus macaque, to Mamu-KIR3DL05 is stabilized by certain simian immunodeficiency virus (SIV) peptides, but not by others [27]. To investigate the functional implications of these interactions, we tested SIV peptides bound by Mamu-A1*002 for the ability to suppress the cytolytic activity of primary Mamu-KIR3DL05+ NK cells. Twenty-eight of the 75 peptides, including three immunodominant CD8+ T cell epitopes with the highest affinity for Mamu-A1*002, inhibited killing by Mamu-KIR3DL05+ NK cells. The inhibitory effects of these peptides were dominant over variants that did not suppress NK cell killing. Moreover, Mamu-KIR3DL05+ NK cells were impaired for the ability to suppress SIV replication in Mamu-A1*002+, but not in Mamu-A1*002-, lymphocytes. These observations demonstrate that viral peptides can modulate NK cell activity and are consistent with the possibility that HIV-1 and SIV may acquire changes in epitopes that increase the binding of MHC class I ligands to inhibitory KIRs to impair NK cell killing of virus-infected cells.
Results
Mamu-A1*002-SIV peptide complexes differentially inhibit Mamu-KIR3DL05+ NK cells
We previously reported that Mamu-A1*002 tetramers folded with some SIV peptides, but not others, bind to Mamu-KIR3DL05 [27]. To evaluate the functional implications of these interactions, we tested individual peptides for the ability to inhibit NK cell degranulation in cytotoxicity assays. Primary NK cells were expanded from rhesus macaque peripheral blood mononuclear cells (PBMCs) by stimulation with γ-irradiated K562 Clone9.mbIL21 cells [28] and sorted into Mamu-KIR3DL05+ and -KIR3DL05- subsets using Mamu-A1*002 Gag GY9 tetramers. These NK cells were then tested in cytoxicity assays for recognition of a Mamu-A1*002-expressing, transporter associated with antigen processing (TAP)-deficient 721.221 cell line (721.221-ICP47-A1*002) incubated with specific SIV peptides. Because TAP is necessary for the translocation of peptides from the cytoplasm into the endoplasmic reticulum, MHC class I molecules expressed in these cells cannot load peptides derived from endogenously synthesized proteins, and are rapidly internalized from the plasma membrane [29,30]. Thus, most of the Mamu-A1*002 molecules expressed by these cells are empty and can be loaded with exogenous peptides. Moreover, peptide binding to Mamu-A1*002 can be assessed by staining for an increase in steady-state levels of MHC class I expression on the cell surface.
Six CD8+ T cell epitopes of SIV bound by Mamu-A1*002 were initially tested for their ability to suppress the cytolytic activity of Mamu-KIR3DL05+ NK cells. These included Gag71-79 GY9, Nef159-167 YY9, Env788-795 RY8, Vif89-97 IW9, Env760-768 SY9, and Nef169-177 KL9 peptides (Table 1). Mamu-KIR3DL05- NK cells killed 721.221-ICP47-A1*002 cells incubated with each of these peptides to a similar extent. In contrast, the cytolytic activity of Mamu-KIR3DL05+ NK cells was selectively inhibited by Gag GY9, Nef YY9, and Env RY8, but not by Vif IW9, Env SY9, or Nef KL9 (Fig 1A). The same pattern of peptide-dependent inhibition was reproducible with Mamu-KIR3DL05+ NK cells from four different animals (Fig 1B), and was not the result of differences in MHC class I binding, as all of the peptides stabilized surface expression of Mamu-A1*002 on the target cells to similar levels (Fig 1C). These results are consistent with previously published data showing that Mamu-A1*002 tetramers folded with Gag GY9, Nef YY9 and Env RY8, but not Vif IW9, bind to Mamu-KIR3DL05 [27].
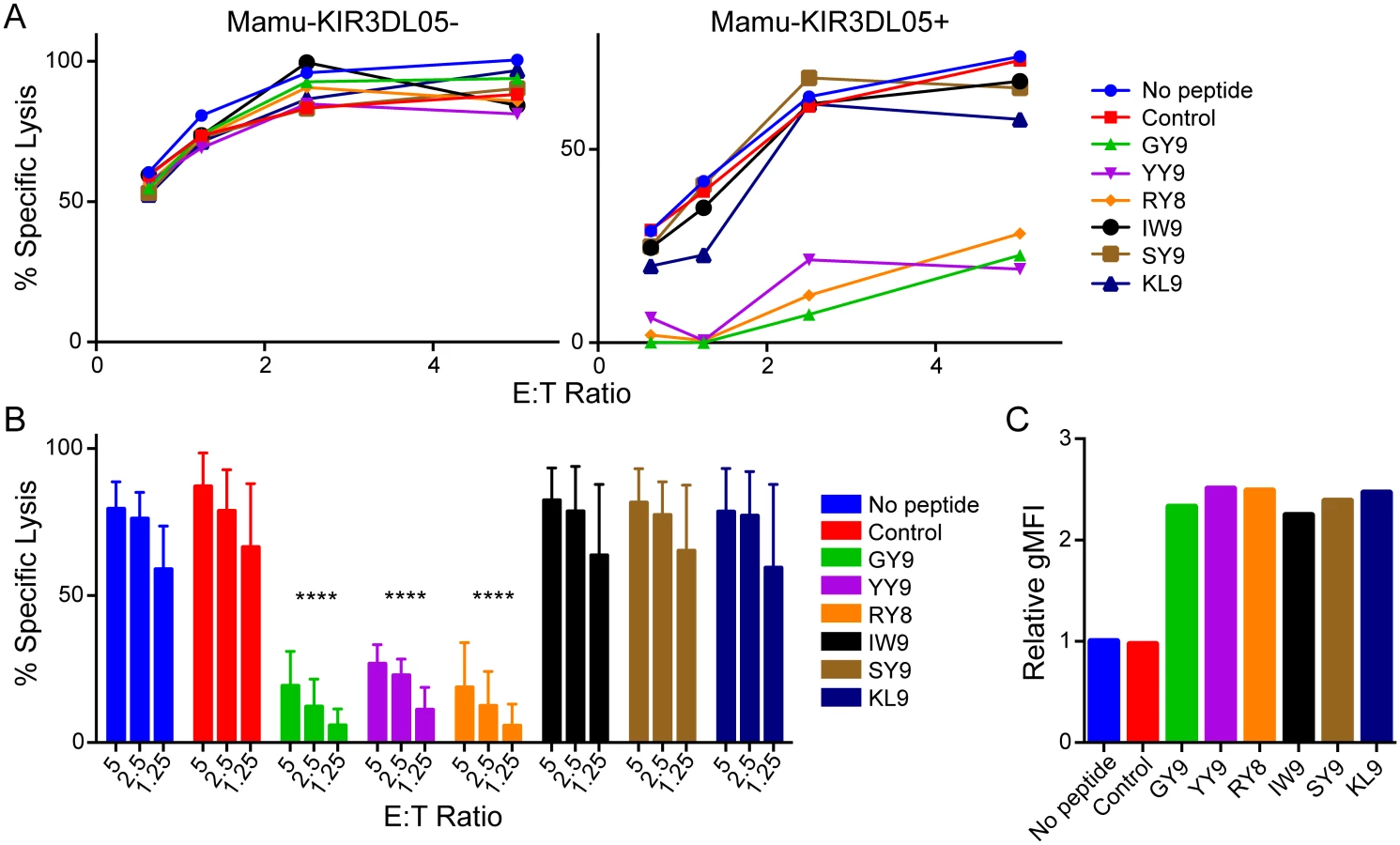

The same approach was used to screen 75 SIVmac239 peptides previously shown to bind to Mamu-A1*002 (Table 1) [31]. As expected, all of the peptides stabilized Mamu-A1*002 expression on the surface of 721.221-ICP47-A1*002 cells (S1 Fig) and had no effect on the cytolytic activity of Mamu-KIR3DL05- NK cells (S2 Fig). However, 28 of the 75 peptides significantly inhibited killing by Mamu-KIR3DL05+ NK cells (Fig 2). Ordered from highest to lowest binding affinity for Mamu-A1*002, these inhibitory peptides include seven previously defined CD8+ T cell epitopes known to be presented on the surface of SIV-infected cells [31]. Notably, the four peptides with highest affinity for Mamu-A1*002, including three immunodominant CD8+ T cell epitopes (Nef YY9, Env RY8, and Gag GY9; purple), strongly suppressed the cytolytic activity of Mamu-KIR3DL05+ NK cells (Fig 2). Because of their high affinity for Mamu-A1*002, these peptides may be presented at a greater density on the surface of SIV-infected cells than other viral peptides, and thus have a greater impact on Mamu-KIR3DL05+ NK cell responses.

Substitutions in C-terminal positions of Mamu-A1*002-bound peptides affect interactions with Mamu-KIR3DL05
To define the residues of Mamu-A1*002-bound peptides that differentially affect interactions with Mamu-KIR3DL05, variants of Gag GY9, Nef YY9, Env RY8 and Vif IW9 were screened for the ability to sensitize 721.221-ICP47-A1*002 cells to NK cell lysis. All of the variants tested bound to Mamu-A1*002 (S3A–S3D Fig) and did not affect the cytolytic activity of Mamu-KIR3DL05- NK cells (S3E–S3H Fig). However, several of the peptides did alter Mamu-KIR3DL05+ NK cell responses (Fig 3). In the case of Gag GY9, alanine or tryptophan substitutions at position 8 partially (GY9 L8A), or completely (GY9 L8W), abrogated Mamu-KIR3DL05+ NK cell inhibition (Fig 3A and 3B). Similar results were obtained for GY9 variants with tyrosine or phenylalanine at this position (S4 Fig), suggesting that the presence of a bulky aromatic side chain at position 8 may interfere with Mamu-A1*002 binding to Mamu-KIR3DL05. In the case of Nef YY9, opposite effects were observed for alanine versus tryptophan substitutions at position 8. Whereas replacement of arginine with alanine (YY9 R8A) further suppressed the cytolytic activity of Mamu-KIR3DL05+ NK cells compared to the wild-type peptide, a tryptophan substitution (YY9 R8W) allowed killing to a similar extent as the control peptide that does not bind to Mamu-A1*002 (Fig 3C and 3D). Substitutions at positions 6 and 7 also resulted in partial (YY9 G6A and G6W), or a nearly complete (YY9 I7W), loss of inhibition, suggesting that these residues may also contribute to interactions with Mamu-KIR3DL05. Similar to Gag GY9 and Nef YY9, a valine-to-tryptophan substitution in the penultimate position of Env RY8, in this case position 7 (RY8 V7W), completely abrogated inhibition (Fig 3E and 3F). A significant increase in cytolyic activity was also observed for a tryptophan substitution at position 6 (RY8 R6W) (Fig 3E and 3F). Yet neither alanine nor tryptophan substitutions at position 5 had a detectable effect on Mamu-KIR3DL05+ NK cell responses to any of these peptides (Fig 3A–3F). In the case of Vif IW9, single amino acid substitutions were not sufficient to change this disinhibitory peptide into an inhibitory variant. However, a combination of substitutions at positions 8 and 9 (IW9 F8A W9Y) resulted in significant inhibition of Mamu-KIR3DL05+ NK cells (Fig 3G and 3H), demonstrating that it is also possible to convert a peptide that permits a cytolytic response into one that suppresses this activity with only a couple of amino acid changes. Taken together, these results reveal a prominent role for residues at the penultimate position of Mamu-A1*002-bound peptides, with additional contributions of the two preceding residues, in modulating Mamu-A1*002 interactions with Mamu-KIR3DL05.

To confirm that these functional differences in NK cell responses reflect the effects of Mamu-A1*002-bound peptides on molecular interactions with Mamu-KIR3DL05, custom Mamu-A1*002 tetramers were synthesized with selected Gag GY9 and Nef YY9 variants and tested for their ability to bind to Mamu-KIR3DL05. Jurkat cells were electroporated with a plasmid DNA construct that expresses HA-tagged Mamu-KIR3DL05 and then stained with tetramer and an HA-specific monoclonal antibody (Fig 4A), as previously described [27]. The integrity of each of the tetramers was also verified by staining a cell line that stably expresses LILRB1, which binds to MHC class I molecules folded with β2-microglobulin independently of MHC-bound peptide (Fig 4B) [32–34]. A comparison of tetramer - versus HA-staining on transfected Jurkat cells revealed that the effects of peptides on Mamu-A1*002 binding to Mamu-KIR3DL05 directly mirrors their effects on cytolytic activity. Just as Gag GY9 L8A partially increases target cell lysis by Mamu-KIR3DL05+ NK cells (Fig 3A and 3B), this peptide diminishes Mamu-A1*002 binding to Mamu-KIR3DL05 (shifts tetramer staining to higher levels of Mamu-KIR3DL05/HA on the cell surface) (Fig 4A). Conversely, the Nef YY9 R8A variant, which enhances Mamu-KIR3DL05+ NK cell inhibition (Fig 3C and 3D), increases Mamu-A1*002 binding to Mamu-KIR3DL05 compared to the wild-type Nef YY9 peptide (shifts tetramer staining to slightly lower surface levels of Mamu-KIR3DL05/HA) (Fig 4A). Furthermore, the Gag GY9 L8W and Nef YY9 R8W variants, which allow full cytolytic activation of Mamu-KIR3DL05+ NK cells (Fig 3A–3D), completely abrogate Mamu-A1*002 binding to Mamu-KIR3DL05 (Fig 4A). These tryptophan substitutions do not interfere with the folding of Mamu-A1*002-peptide complexes, since Gag GY9 L8W and Nef YY9 R8W tetramers retain the ability to bind to LILRB1 (Fig 4B). Thus, the functional effects of amino acid changes at position 8 of Gag GY9 and Nef YY9 on Mamu-KIR3DL05+ NK cell responses correspond to their effects on Mamu-A1*002 binding to Mamu-KIR3DL05.

Structural modeling of amino acid replacements in SIV peptides bound by Mamu-A1*002
Three-dimensional crystal structures of Mamu-A1*002 in complex with Gag GY9 and Nef YY9 reveal how changes in C-terminal positions of these peptides may affect interactions with Mamu-KIR3DL05 [35,36]. These structures show that the side chains of the residues at position 8 of both peptides project over the α1-domain of Mamu-A1*002 in an orientation to interact with Mamu-KIR3DL05 [35,36]. In the case of Gag GY9 L8A, the methyl group of the alanine at this position should not interfere with Mamu-A1*002 binding to Mamu-KIR3DL05 (Fig 5A). However, this side chain is also unlikely to contribute to the stability of the interaction to the same extent as the longer aliphatic side chain of leucine, which probably explains the decrease in tetramer binding (Fig 4A) and the partial increase in cytolytic activity for this peptide (Fig 3A). In the case of Gag GY9 L8W, the bulky indole ring of tryptophan at position 8 projects out of the peptide-binding pocket in an orientation to directly interfere with binding to Mamu-KIR3DL05. Indeed, Mamu-A1*002 tetramer folded with this peptide failed to bind to Mamu-KIR3DL05 under conditions of protein over-expression in transfected cells (Fig 4A). Disruption of Mamu-KIR3DL05 binding to Mamu-A1*002 therefore explains the complete loss of NK cell inhibition and the strong cytolytic response against 721.221-ICP47-A1*002 cells pulsed with this peptide (Fig 3A). Similar modeling of Nef YY9 I7W and R8W in complex with Mamu-A1*002 reveals that the ring structures at positions 7 and 8 of these peptides protrude from the peptide-binding pocket (Fig 5B). Hence, these tryptophan substitutions may also hinder Mamu-KIR3DL05 binding to Mamu-A1*002, accounting for the lack of Mamu-A1*002-YY9 R8W tetramer binding to Mamu-KIR3DL05 (Fig 4A) and the robust cytolytic activity against cells pulsed with each of these peptide variants (Fig 3C).

Inhibitory peptides dominate over variants that do not suppress NK cell responses
MHC class I molecules present a diverse repertoire of peptides derived from viral and cellular antigens on the surface of virus-infected cells, of which some may stabilize and some may disrupt interactions with any given KIR. We therefore asked which signal dominates when a mixture of peptides that do or do not suppress NK cell activity is presented on the cell surface. 721.221-ICP47-A1*002 cells were pulsed with mixtures of Gag GY9 and GY9 L8W or Env RY8 and RY8 V7W at concentrations that did not over-saturate binding to Mamu-A1*002 (S5A Fig), and were tested for susceptibility to lysis by Mamu-KIR3DL05+ NK cells. Cell surface stabilization of Mamu-A1*002 was similar for GY9 versus GY9 L8W and RY8 versus RY8 V7W individually (S5A Fig), or in combination (S5B Fig), and none of the peptide mixtures affected Mamu-KIR3DL05- NK cell responses (S5C Fig). At 25% of the total peptide concentration, Gag GY9 and Env RY8 dominated over their respective disinhibitory variants to fully suppress the cytolytic activity of Mamu-KIR3DL05+ NK cells (Fig 6A–6D). To determine the threshold at which the inhibitory effects of these peptides are observed, increasing concentrations of Gag GY9 and Env RY8 were tested in the presence of a fixed, sub-saturating concentration of Gag GY9 L8W and Env RY8 V7W. Half-maximal inhibition of killing was achieved at approximately 5% of the total peptide concentration for both Gag GY9 and Env RY8 (Fig 6E and 6F). Thus, under conditions where inhibitory peptides and their corresponding disinhibitory variants are presented simultaneously by Mamu-A1*002, the inhibitory peptides dominate to suppress Mamu-KIR3DL05+ NK cell responses.

Mamu-KIR3DL05+ NK cells are impaired in their ability to suppress SIV replication in Mamu-A1*002+ lymphocytes
To determine if the presentation of viral epitopes by Mamu-A1*002 impairs the antiviral activity of Mamu-KIR3DL05+ NK cells, SIV replication was compared in Mamu-A1*002+ and -A1*002- CD4+ lymphocytes co-cultured with Mamu-KIR3DL05+ versus-KIR3DL05- NK cells from the same animal. Both NK cell subsets suppressed virus replication to a similar extent in CD4+ lymphocytes from Mamu-A1*002- animals (Fig 7A). However, virus replication was consistently higher in the presence of Mamu-KIR3DL05+ than-KIR3DL05- NK cells in Mamu-A1*002+ CD4+ lymphocytes (Fig 7A). A composite analysis of data obtained using cells derived from three different animals revealed significantly higher peak (p = 0.0370, paired t-test) and total (p = 0.0102, paired t-test) virus replication in Mamu-A1*002+ CD4+ lymphocytes cultured with Mamu-KIR3DL05+ NK cells than in those cultured with Mamu-KIR3DL05- NK cells (Fig 7B and 7C). Similar results were also obtained for a comparison of Mamu-KIR3DL05+ versus-KIR3DL05- NK cell lysis of SIV-infected 721.221 cell lines that do, or do not, express Mamu-A1*002. Whereas the killing of virus-infected Mamu-A1*002- cells was equivalent for both NK cell subsets, the lysis of SIV-infected Mamu-A1*002+ cells by Mamu-KIR3DL05+ NK cells was significantly inhibited compared to Mamu-KIR3DL05- NK cells (p = 0.0347, paired t-test) (Fig 7D). These differences in the ability of Mamu-KIR3DL05+ versus-KIR3DL05- NK cells to suppress SIV replication and to lyse virus-infected cells are therefore consistent with the specific impairment of Mamu-KIR3DL05+ NK cells by inhibitory viral peptides presented on the surface of SIV-infected cells by Mamu-A1*002.

Discussion
Genetic studies have associated certain combinations of KIR and HLA class I alleles with the control of HIV, HPV, and HCV infections, suggesting that KIR-MHC class I interactions play an important role in the immune response to these viruses [8,9,13,14]. However, less is known about whether viral peptides affect the interaction of KIR with their ligands during infection, despite abundant evidence demonstrating that KIR recognition is influenced by MHC class I-bound peptides [24–27,32,37], including variants of some HIV peptides [38–40]. In this study, we demonstrate that over a third of the SIV peptides bound by Mamu-A1*002, including the four peptides with the highest affinity for this MHC class I molecule, suppress the cytolytic activity of Mamu-KIR3DL05+ NK cells. These peptides therefore either stabilize, or do not interfere with, the recognition of Mamu-A1*002 by Mamu-KIR3DL05. We also found that variants of inhibitory peptides with substitutions at C-terminal positions, particularly the introduction of bulky aromatic residues at the penultimate position, no longer suppress Mamu-KIR3DL05+ NK cell lysis of Mamu-A1*002+ cells. When inhibitory and disinhibitory peptides are both presented on the surface of Mamu-A1*002+ cells, signals from inhibitory peptides overwhelmingly dominate to suppress Mamu-KIR3DL05+ NK cell responses. Accordingly, Mamu-KIR3DL05+ NK cells were less effective at suppressing viral replication in autologous Mamu-A1*002+ CD4+ lymphocytes and killing SIV-infected Mamu-A1*002+ cells than their Mamu-KIR3DL05- counterparts. These results suggest that viral peptides that promote the recognition of MHC class I ligands by inhibitory KIRs may reduce NK cell lysis of virus-infected cells as a mechanism of immune evasion.
We previously demonstrated that Mamu-A1*002 in complex with certain SIV peptides binds with unusually high avidity to Mamu-KIR3DL05 [27]. Direct staining of unstimulated primary macaque NK cells was observed for Mamu-A1*002 tetramers folded with either Gag GY9 or Env RY8 [27]. This contrasts with human KIRs for which tetramer staining is generally only detectable upon KIR overexpression in transfected cell lines or in activated NK cell clones [25,38,41]. The stabilization of Mamu-A1*002-KIR3DL05 interactions by these viral peptides may be important for the resistance of SIV-infected cells to Mamu-KIR3DL05+ NK cells under conditions of incomplete Mamu-A1*002 downregulation by Nef. Similar to the selective downregulation of HLA-A and -B molecules (but not HLA-C molecules) by HIV-1 Nef [4–6], all Mamu-A molecules, and some Mamu-B molecules, are downregulated by the SIV Nef protein [42]. Although MHC class I downmodulation increases the resistance of virus-infected cells to CD8+ T cells, decreased surface expression of ligands for inhibitory KIRs can increase their susceptibility to NK cells [6]. Thus, viral epitopes, such as Gag GY9 and Env RY8, that stabilize particularly high avidity MHC class I interactions with inhibitory KIRs may compensate for partial MHC class I downmodulation by Nef to protect virus-infected cells from elimination by NK cells.
The specific residues of SIV peptides bound by Mamu-A1*002 that modulate interactions with Mamu-KIR3DL05 were identified by testing peptide variants in cytotoxicity assays. Although changes at the penultimate position (position 7 of Env RY8 or position 8 of Gag GY9 and Nef YY9) had the most consistent impact on Mamu-KIR3DL05+ NK cell responses, changes at the preceding positions of Nef YY9 (positions 6 and 7) and Env RY8 (position 6) also affected cytolytic activity. Whereas partial effects were observed for alanine substitutions, tryptophan substitutions resulted in a complete, or nearly complete, loss of Mamu-KIR3DL05+ NK cell inhibition.
Three-dimensional crystal structures of Mamu-A1*002 in complex with Gag GY9 and Nef YY9 suggest possible explanations for these effects [35,36]. In the case of Gag GY9 L8A, the short methyl group of alanine at position 8 is unlikely to stabilize interactions with Mamu-KIR3DL05 as well as the longer aliphatic side chain of leucine, which may account for the partial increase in cytolytic activity against this peptide variant and the decrease in avidity of Mamu-KIR3DL05 for Mamu-A1*002 tetramer folded with this peptide. In contrast, the bulky indole ring of tryptophan at position 8 of Gag GY9 L8W, and at positions 7 and 8 of Nef YY9 I7W and R8W, projects out of the peptide-binding pocket in an orientation predicted to interfere with engagement by Mamu-KIR3DL05. In each case, disruption of Mamu-A1*002 binding to Mamu-KIR3DL05 may satisfy “missing self” through the loss of inhibitory signals from this ligand resulting in full cytolytic activity against these peptide variants and absence of binding of Mamu-KIR3DL05 by Mamu-A1*002 tetramers folded with GY9 L8W and YY9 R8W. A similar mechanism probably explains the inability of Vif IW9 to inhibit Mamu-KIR3DL05+ NK cell responses. Vif IW9 naturally contains a bulky phenylalanine residue at position 8, and consistent with the inability of Mamu-A1*002 tetramers folded with this peptide to bind to Mamu-KIR3DL05, even under conditions of KIR overexpression in transfected cells [27], Vif IW9 resulted in robust KIR3DL05+ NK cell activity against Mamu-A1*002+ target cells. However, replacement of phenylalanine with alanine at position 8, in combination with a tryptophan-to-tyrosine substitution at position 9 (an alternative anchor residue for Mamu-A1*002) [31], resulted in a variant (IW9 F8A W9Y) that partially suppresses Mamu-KIR3DL05+ NK activation. Together, these observations illustrate how amino acid changes at C-terminal positions of MHC class I-bound peptides that participate in interactions with inhibitory KIR can modulate NK cell responses, and provide a glimpse into the underlying molecular mechanisms by which these receptors contribute to protective immunity versus immune evasion.
Assays with mixtures of Mamu-A1*002-bound peptides that do or do not inhibit Mamu-KIR3DL05+ NK cell responses revealed that signals from inhibitory peptides overwhelmingly dominate to suppress NK cell activation. When present at a concentration of just 5% of a mixture with their respective disinhibitory variants, Gag GY9 and Env RY8 exhibited approximately 50% of their maximal capacity to inhibit the lysis of Mamu-A1*002+ target cells. These observations are consistent with the need to prevent NK cell lysis of normal healthy cells presenting diverse self-peptides in complex with MHC class I ligands, and imply that for a viral peptide to cause NK cell degranulation by preventing engagement of an inhibitory KIR, it would need to be presented on the cell surface at a sufficiently high density to overcome negative signals from the same MHC class I ligand loaded with other peptides. In this respect, our results contrast with previous studies, which found that low concentrations of ‘antagonistic’ peptides were sufficient to overcome inhibition by peptides that stabilize HLA-Cw*0102 binding to KIR2DL2 and KIR2DL3 [26,43]. A possible explanation for the difference in our results may be the nature of the peptides examined. Whereas antagonistic peptides are distinguished by weak KIR binding that does not induce NK cell inhibition [26], the peptide variants used in our assays contain tryptophan substitutions with bulky aromatic side chains predicted to abrogate KIR binding. Furthermore, because antagonism is less efficient for high avidity HLA-KIR interactions [26], any antagonistic effects of our variants may have been overcome by the unusually high avidity of Mamu-A1*002-Gag GY9 and -Env RY8 complexes for Mamu-KIR3DL05 [27]. Alternatively, differences in our results may reflect fundamental differences in ligand recognition for KIRs with two versus three immunoglobulin-like domains. In the absence of a D0 domain to stabilize MHC class I binding, 2D KIRs may be more sensitive than 3D KIRs to MHC class I-bound peptides. In support of this possibility, changes at position 8 of HLA class I-bound peptides have been shown to have a more dramatic effect on the Kd of HLA-Cw3 binding to KIR2DL2 than on that of HLA-B*57 binding to KIR3DL1 [20,21].
While inhibitory peptide dominance is compatible with the need to prevent NK cell lysis of healthy cells, it also provides an avenue for viruses to gain resistance to NK cells by acquiring changes in epitopes that increase the binding of MHC class I ligands to inhibitory KIRs. Indeed, this may explain the selective pressure for HIV-1 to acquire polymorphisms that increase the binding of KIR2DL2 to virus-infected cells and impair virus suppression by NK cells from KIR2DL2+ individuals in vitro [19]. Therefore, it is perhaps not a coincidence that the four SIV peptides bound with the highest affinity by Mamu-A1*002, including three immunodominant CD8+ T cell epitopes known to be processed and presented on the surface of virus-infected cells (Nef YY9, Env RY8 and Gag GY9), all suppress Mamu-KIR3DL05+ NK cell responses. Although multiple factors determine the efficiency of MHC class I presentation, the high affinity of Mamu-A1*002 for these peptides suggests that they may be presented in abundance on the surface of SIV-infected cells expressing this MHC class I molecule. Given that Mamu-KIR3DL05 and -A1*002 are commonly expressed in Indian origin rhesus macaques [27], it is therefore conceivable that SIV may have acquired changes in one or more of these epitopes to stabilize this KIR-MHC class I interaction as a mechanism of NK cell evasion.
In accordance with this possibility, Mamu-KIR3DL05+ NK cells were less effective than Mamu-KIR3DL05- NK cells at suppressing SIV replication in autologous CD4+ lymphocytes from Mamu-A1*002+ macaques. Moreover, the lysis of SIV-infected cells expressing Mamu-A1*002 was also less efficient for Mamu-KIR3DL05+ NK cells than for Mamu-KIR3DL05- NK cells. As these differences were not observed using CD4+ lymphocytes from Mamu-A1*002- animals, or target cells that lack Mamu-A1*002, these results do not reflect inherent differences in Mamu-KIR3DL05+ versus-KIR3DL05- NK cell function. Thus, under physiological conditions of virus infection in primary CD4+ lymphocytes and in a stable cell line expressing Mamu-A1*002, Mamu-KIR3DL05+ NK cells are specifically impaired in their ability to contain SIV replication.
Similar observations in humans suggest that NK cells exert selective pressure on HIV-1 replication in certain individuals. KIR2DL2-associated HIV-1 polymorphisms were identified in chronically infected individuals that increase the binding of this inhibitory KIR to virus-infected cells and impair the ability of NK cells from KIR2DL2+ individuals to suppress virus replication in vitro [19]. Although the HLA class I ligands and specific viral peptides associated with these polymorphisms were not identified, subsequent studies defined a few HIV-1 peptides that could enhance the binding of KIR2DL2 to two HLA-C1 allotypes [39,40]. These observations are analogous to the identification of SIV peptides that inhibit Mamu-KIR3DL05+ NK cell responses. However, because rhesus macaques do not express KIR2D receptors of the D1-D2 configuration or their HLA-C ligands [44–47], the mechanisms of NK cell inhibition in humans and macaques may differ. Whereas HLA-C is not downregulated by HIV-1 Nef [5,6], Mamu-A1*002 is subject to downregulation by the SIV Nef protein [42]. Thus, it is conceivable that amino acid changes in HIV-1 epitopes that merely eliminate interference with HLA-C binding to KIR2DL2 would be sufficient to inhibit KIR2DL2+ NK cells, but substitutions in SIV epitopes that increase the stability of Mamu-KIR3DL05 binding to Mamu-A1*002 may be necessary to overcome incomplete downregulation of this ligand by Nef to adequately suppress KIR3DL05+ NK cells.
Although HLA class I-bound peptides also influence interactions with human KIR3DL1 [38,41], functional parallels with Mamu-KIR3DL05 are limited. Whereas Mamu-KIR3DL05+ NK cells exhibit impaired NK cell suppression of SIV replication in autologous Mamu-A1*002+ lymphocytes, human NK cells from individuals possessing highly-expressed alleles of KIR3DL1 are better able to suppress HIV-1 replication in autologous HLA-B*57+ lymphocytes [16]. Moreover, unlike Mamu-KIR3DL05, which appears to be associated with elevated viral loads in SIV-infected animals [48], certain highly expressed alleles of KIR3DL1, in combination with their HLA-Bw4 ligands, are associated with lower viral loads and slower progression to AIDS in HIV-1 infected individuals [9]. These contrary observations may reflect fundamental differences in the molecular interactions of Mamu-KIR3DL05 versus KIR3DL1 with their ligands, as these receptors represent highly divergent products of non-orthologous genes [27]. However, from our limited knowledge of the MHC class I ligands of rhesus macaque KIRs and the effects of viral peptides on KIR-MHC class I interactions in either humans or macaques, it is difficult to generalize these findings to other KIR-MHC class I interactions. Indeed, it is possible that Mamu-KIR3DL05+ NK cells may suppress SIV replication in the context of MHC class I ligands other than Mamu-A1*002 that have yet to be defined, or that the presentation of HIV-1 epitopes by some HLA-Bw4 molecules may suppress KIR3DL1+ NK cell responses.
In summary, we show that 28 of 75 SIV peptides bound by Mamu-A1*002 inhibit Mamu-KIR3DL05+ NK cell responses, that changes at C-terminal positions of these peptides differentially affect NK cell activity, and that signals from inhibitory peptides dominate over disinhibitory variants to suppress NK cell lysis. Insights into the molecular basis for these peptide-dependent effects on NK cell responses are provided by modeling amino acid replacements into available three-dimensional structures of SIV Gag GY9 and Nef YY9 in complex with Mamu-A1*002. To our knowledge, this represents the most comprehensive analysis to date of the functional effects of viral peptides on a specific KIR-MHC class I interaction. The identification of multiple viral peptides bound with high affinity by Mamu-A1*002 that enhance binding to Mamu-KIR3DL05 and suppress NK cell activation, together with a deficit in the ability of Mamu-KIR3DL05+ NK cells to suppress SIV replication in Mamu-A1*002+ lymphocytes, supports a mechanism by which immunodeficiency viruses may reduce their susceptibility to NK cells by acquiring changes in epitopes that stabilize MHC class I interactions with inhibitory KIRs.
Materials and Methods
Ethics statement
The Indian origin rhesus macaques (Macaca mulatta) used in this study were housed and cared for in an indoor ABSL2+ facility at the New England Primate Research Center (NEPRC) in accordance with standards of the Association for Assessment and Accreditation of Laboratory Animal Care and the Harvard Medical School Animal Care and Use Committee. Animal experiments were approved by the Harvard Medical Area Standing Committee on Animals under protocol 04873 and conducted according to the principles described in the Guide for the Care and Use of Laboratory Animals [49]. Steps taken to improve animal welfare included social housing and environmental enrichment such as foraging opportunities and manipulable devices. Water was supplied ad libitum and animals were provided commercial monkey chow twice daily and fresh produce at least three times per week. Animals were monitored twice daily by animal care and veterinary staff, and suffering due to experimental procedures was minimized by sedation with ketamine HCl prior to blood collection.
Stable cell lines
To produce 721.221-ICP47-A1*002 cells, rhesus macaque Mamu-A1*002 cDNA was cloned into the pQCXIP retroviral vector (Clontech). This vector was cotransfected with pVSV-G (Clontech) into GP2-293 cells and supernatant was harvested two days post-transfection. The supernatant was centrifuged in Ultracel 50k filter centrifuge tubes (Millipore) to yield concentrated VSV-G pseudotyped MLV-based particles. 721.221-ICP47 cells, a derivative of the MHC class I-deficient 721.221 cell line [50] that expresses HSV-1 ICP47 to inhibit the transporter associated with antigen processing (TAP) complex [29,30], were transduced by incubation with concentrated virus for 3 hours at 37°C. Three days later, cells were placed under selection with 0.4 μg/mL puromycin (Invitrogen). 721.221-CD4-A1*002-LTRluc cells were produced by sequential transduction of 721.221 cells with pQCXIP expressing rhesus macaque CD4, a pLNSX-derived retroviral vector containing the firefly luciferase gene under the control of the SIV LTR, and pQCXIH (Clontech) expressing Mamu-A1*002 using a similar procedure. Cells were placed under selection with 0.4 μg/mL puromycin, 500 μg/mL of G418, and 100 μg/mL of hygromycin additively after each transduction.
NK cell expansion and cell culture
PBMC (5 × 106 cells) were stimulated with 1 × 107 γ-irradiated K562 Clone 9.mbIL21 cells [28] in a volume of 40 mL of NKEM: RPMI 1640 (Invitrogen) supplemented with 10% FBS (Invitrogen), glutamine (Invitrogen), Primocin (InvivoGen), and 50 U/mL IL-2 (NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH, contributed by Dr. Maurice Gately, Hoffmann—La Roche Inc.) [51]. On days 3 and 5 after culture, cells were resuspended in fresh medium. The expanded NK cells were re-stimulated on day 7, and weekly thereafter, with additional γ-irradiated K562 Clone 9.mbIL21 cells at a 1 : 1 ratio. From day 7 onward, expanded cells were resuspended in fresh medium at 4 × 105 cells/mL 2–3 times weekly.
NK cell sorting
Expanded NK cell cultures were incubated with anti-CD3 Ab (clone 6G12) and T cells were depleted using pan-mouse IgG Dynabeads (Dynal Biotech). Mamu-KIR3DL05+ and -KIR3DL05- subsets were separated by FACS using a Mamu-A1*002 tetramer folded with Gag71-79 GY9 that binds Mamu-KIR3DL05 [27]. NK cells were stained with PE-conjugated Mamu-A1*002-GY9 tetramer for 30 minutes at 37°C followed by staining with anti-CD3-Pacific Blue (clone SP34-2; BD Biosciences) and anti-NKG2A-APC (clone Z199; Beckman Coulter) or anti-NKG2A-Pacific Blue and anti-CD3-FITC (clone SP34; BD Biosciences) for 20 minutes at 25°C. Tetramer+CD3-NKG2A+ and Tetramer-CD3-NKG2A+ subsets were sorted using a FACSAria II (BD Biosciences). After sorting, these NK cells subsets were stimulated with γ-irradiated K562 Clone 9.mbIL21 cells and maintained as described above. Mamu-KIR3DL05+ and -KIR3DL05- NK cell subsets were sorted from PBMC by the same procedure for immediate use in viral suppression assays.
Calcein acetoxymethyl ester (CAM) cytotoxicity assay
721.221-ICP47-A1*002 cells were incubated overnight at 26°C with SIV peptides (GenScript and Mimotopes) in Hybridoma-Serum Free Medium (Invitrogen) to stabilize cell surface A1*002-peptide complexes. A GY9 variant with S2A and Y9G substitutions at anchor residues was included as a non-A1*002-binding peptide control. An aliquot of 2 × 105 peptide-pulsed cells were then stained with a PE-conjugated pan-MHC class I specific antibody (clone W6/32; Dako) to verify the surface stabilization of MHC class I molecules. The remaining peptide-pulsed cells were stained with CAM (Invitrogen) at a 1 : 100 dilution for 1 hour at 26°C. CAM-stained cells were washed and then incubated with Mamu-KIR3DL05+ or -KIR3DL05- NK cells for 4 hours at E:T ratios between 0.5 : 1 and 10 : 1 in NKEM at 26°C. The release of CAM into the supernatant was measured using a fluorescent plate reader (excitation 485 nm, absorption 530 nm). Percent specific lysis was calculated as (test release—spontaneous release) / (maximum release—spontaneous release).
Tetramer staining
Ten million Jurkat cells were electroporated with 25 μg of pCGCG-KIR3DL05*008-HA construct or empty pCGCG (250 V, 950 μF) in 400 μl RPMI in a 4 mm cuvette (BioRad) then cultured in 9 ml RPMI supplemented with 10% FBS for 22 hours. One million cells were stained with APC-conjugated Mamu-A1*00201 tetramers (NIH Tetramer Core Facility) folded with either GY9 (GSENLKSLY), GY9 L8A (GSENLKSAY), GY9 L8W (GSENLKSWY), YY9 (YTSGPGIRY), YY9 R8A (YTSGPGIAY), or YY9 R8W (YTSGPGIWY) peptide (Genscript; 30 minutes, 37°C) followed by staining with anti-HA-PE (Miltenyi) for 10 minutes at 4°C. Cells were washed in PBS and fixed in 4% paraformaldehyde. To verify the proper folding of tetramers with peptide variants, 1 × 106 Ba/F3 cells expressing LILRB1 (kindly provided by Simon Kollnberger, University of Oxford) were stained with each tetramer as previously described [32]. All data was acquired using a LSR II flow cytometer (BD Biosciences) and analyzed using FlowJo 8.8.6.
Structural modeling
Molecular graphics and analyses were performed with the UCSF Chimera package developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311). Modeling of amino acid substitutions in the structures of Mamu-A1*002 in complex with GY9 (PDB 3JTS) [35] or YY9 (PDB 3JTT) [36] were carried out using the swapaa function in which rotamer position is determined by minimal clash score, maximal hydrogen bonding, and probability according to the Dunbrack rotamer library [52].
Viral suppression assay
The target cells were prepared by depletion of CD8+ cells from PBMC by treatment with CD8-Dynabeads (Dynal Biotech) followed by ConA-activation (5 μg/ml) in RPMI 1640 supplemented with 10% FBS, glutamine, HEPES (Invitrogen), and Primocin (R10) for 4 days. On the day of the assay, 2 × 106 target cells were infected with 100 ng p27 SIVmac239 in minimal volume for 3 hours at 37°C, then washed. Mamu-KIR3DL05+ and -KIR3DL05- NK cell subsets were sorted as described above and were incubated with 2 × 104 SIV-infected autologous CD4+ target cells in R10 + 20 U/mL IL-2 in duplicate wells at a 5 : 1 or 3 : 1 E:T ratio. On days 3, 5, 7, and 9/10, 50 uL of supernatant was sampled with replacement. SIV titer was determined by SIV p27 Antigen Capture Assay (Advanced Bioscience Laboratories). Area under the curve was calculated using Prism 6 (GraphPad).
Luciferase lysis assay
Four days prior to the assay, 721.221-CD4-A1*002-LTRluc cells, which express luciferase when infected with SIV, were infected by spinoculation [53] in the presence of 4 μg/mL of Polybrene (Sigma-Aldrich). On the day of the assay, infected cells were washed three times and 104 infected cells were plated with between 1.5 x 104 and 105 effector cells in 200 μL of NKEM and incubated for 8 hours at 37°C. Luciferase activity was measured using BriteLite Plus (Perkin Elmer). Control wells contained uninfected 721.221-CD4-A1*002-LTRluc cells. % RLU was defined as (RLU infected with NK cells—RLU uninfected with NK cells) / (RLU infected without NK cells—RLU uninfected without NK cells)*100.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Gumperz JE, Litwin V, Phillips JH, Lanier LL, Parham P. The Bw4 public epitope of HLA-B molecules confers reactivity with natural killer cell clones that express NKB1, a putative HLA receptor. J Exp Med. 1995;181(3):1133–44. Epub 1995/03/01. 7532677; PubMed Central PMCID: PMC2191933.
2. Biassoni R, Falco M, Cambiaggi A, Costa P, Verdiani S, Pende D, et al. Amino acid substitutions can influence the natural killer (NK)-mediated recognition of HLA-C molecules. Role of serine-77 and lysine-80 in the target cell protection from lysis mediated by "group 2" or "group 1" NK clones. J Exp Med. 1995;182(2):605–9. Epub 1995/08/01. 7629517; PubMed Central PMCID: PMC2192139.
3. Schafer JL, Colantonio AD, Neidermyer WJ, Dudley DM, Connole M, O'Connor DH, et al. KIR3DL01 Recognition of Bw4 Ligands in the Rhesus Macaque: Maintenance of Bw4 Specificity since the Divergence of Apes and Old World Monkeys. J Immunol. 2014. Epub 2014/01/24. doi: 10.4049/jimmunol.1302883 24453246.
4. Collins KL, Chen BK, Kalams SA, Walker BD, Baltimore D. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature. 1998;391(6665):397–401. Epub 1998/02/05. doi: 10.1038/34929 9450757.
5. Schwartz O, Marechal V, Le Gall S, Lemonnier F, Heard JM. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat Med. 1996;2(3):338–42. Epub 1996/03/01. 8612235.
6. Cohen GB, Gandhi RT, Davis DM, Mandelboim O, Chen BK, Strominger JL, et al. The selective downregulation of class I major histocompatibility complex proteins by HIV-1 protects HIV-infected cells from NK cells. Immunity. 1999;10(6):661–71. Epub 1999/07/14. doi: 10.1016/S1074-7613(00)80065-5 10403641.
7. Carrington M, Martin MP, van Bergen J. KIR-HLA intercourse in HIV disease. Trends Microbiol. 2008;16(12):620–7. Epub 2008/11/04. doi: 10.1016/j.tim.2008.09.002 18976921.
8. Martin MP, Gao X, Lee JH, Nelson GW, Detels R, Goedert JJ, et al. Epistatic interaction between KIR3DS1 and HLA-B delays the progression to AIDS. Nat Genet. 2002;31(4):429–34. Epub 2002/07/23. doi: 10.1038/ng934 12134147.
9. Martin MP, Qi Y, Gao X, Yamada E, Martin JN, Pereyra F, et al. Innate partnership of HLA-B and KIR3DL1 subtypes against HIV-1. Nat Genet. 2007;39(6):733–40. Epub 2007/05/15. doi: 10.1038/ng2035 17496894.
10. Qi Y, Martin MP, Gao X, Jacobson L, Goedert JJ, Buchbinder S, et al. KIR/HLA pleiotropism: protection against both HIV and opportunistic infections. PLoS Pathog. 2006;2(8):e79. Epub 2006/08/29. doi: 10.1371/journal.ppat.0020079 16933987; PubMed Central PMCID: PMC1550271.
11. Gaudieri S, DeSantis D, McKinnon E, Moore C, Nolan D, Witt CS, et al. Killer immunoglobulin-like receptors and HLA act both independently and synergistically to modify HIV disease progression. Genes Immun. 2005;6(8):683–90. Epub 2005/08/27. doi: 10.1038/sj.gene.6364256 16121209.
12. Barbour JD, Sriram U, Caillier SJ, Levy JA, Hecht FM, Oksenberg JR. Synergy or independence? Deciphering the interaction of HLA Class I and NK cell KIR alleles in early HIV-1 disease progression. PLoS Pathog. 2007;3(4):e43. Epub 2007/04/24. doi: 10.1371/journal.ppat.0030043 17447840; PubMed Central PMCID: PMC1853116.
13. Khakoo SI, Thio CL, Martin MP, Brooks CR, Gao X, Astemborski J, et al. HLA and NK cell inhibitory receptor genes in resolving hepatitis C virus infection. Science. 2004;305(5685):872–4. Epub 2004/08/07. doi: 10.1126/science.1097670 15297676.
14. Carrington M, Wang S, Martin MP, Gao X, Schiffman M, Cheng J, et al. Hierarchy of resistance to cervical neoplasia mediated by combinations of killer immunoglobulin-like receptor and human leukocyte antigen loci. J Exp Med. 2005;201(7):1069–75. Epub 2005/04/06. 15809352; PubMed Central PMCID: PMC2213116.
15. Chen C, Busson M, Rocha V, Appert ML, Lepage V, Dulphy N, et al. Activating KIR genes are associated with CMV reactivation and survival after non-T-cell depleted HLA-identical sibling bone marrow transplantation for malignant disorders. Bone Marrow Transplant. 2006;38(6):437–44. Epub 2006/08/08. doi: 10.1038/sj.bmt.1705468 16892071.
16. Song R, Lisovsky I, Lebouche B, Routy JP, Bruneau J, Bernard NF. HIV Protective KIR3DL1/S1-HLA-B Genotypes Influence NK Cell-Mediated Inhibition of HIV Replication in Autologous CD4 Targets. PLoS Pathog. 2014;10(1):e1003867. Epub 2014/01/24. doi: 10.1371/journal.ppat.1003867 24453969; PubMed Central PMCID: PMC3894215.
17. Alter G, Martin MP, Teigen N, Carr WH, Suscovich TJ, Schneidewind A, et al. Differential natural killer cell-mediated inhibition of HIV-1 replication based on distinct KIR/HLA subtypes. J Exp Med. 2007;204(12):3027–36. Epub 2007/11/21. 18025129; PubMed Central PMCID: PMC2118524.
18. Alter G, Rihn S, Walter K, Nolting A, Martin M, Rosenberg ES, et al. HLA class I subtype-dependent expansion of KIR3DS1+ and KIR3DL1+ NK cells during acute human immunodeficiency virus type 1 infection. J Virol. 2009;83(13):6798–805. Epub 2009/04/24. 19386717; PubMed Central PMCID: PMC2698561.
19. Alter G, Heckerman D, Schneidewind A, Fadda L, Kadie CM, Carlson JM, et al. HIV-1 adaptation to NK-cell-mediated immune pressure. Nature. 2011;476(7358):96–100. Epub 2011/08/05. doi: 10.1038/nature10237 21814282.
20. Vivian JP, Duncan RC, Berry R, O'Connor GM, Reid HH, Beddoe T, et al. Killer cell immunoglobulin-like receptor 3DL1-mediated recognition of human leukocyte antigen B. Nature. 2011;479(7373):401–5. Epub 2011/10/25. doi: 10.1038/nature10517 22020283.
21. Boyington JC, Motyka SA, Schuck P, Brooks AG, Sun PD. Crystal structure of an NK cell immunoglobulin-like receptor in complex with its class I MHC ligand. Nature. 2000;405(6786):537–43. Epub 2000/06/13. doi: 10.1038/35014520 10850706.
22. Fan QR, Long EO, Wiley DC. Crystal structure of the human natural killer cell inhibitory receptor KIR2DL1-HLA-Cw4 complex. Nat Immunol. 2001;2(5):452–60. Epub 2001/04/27. doi: 10.1038/87766 11323700.
23. Rajagopalan S, Long EO. The direct binding of a p58 killer cell inhibitory receptor to human histocompatibility leukocyte antigen (HLA)-Cw4 exhibits peptide selectivity. J Exp Med. 1997;185(8):1523–8. Epub 1997/04/21. 9126935; PubMed Central PMCID: PMC2196286.
24. Mandelboim O, Wilson SB, Vales-Gomez M, Reyburn HT, Strominger JL. Self and viral peptides can initiate lysis by autologous natural killer cells. Proc Natl Acad Sci U S A. 1997;94(9):4604–9. Epub 1997/04/29. 9114037; PubMed Central PMCID: PMC20770.
25. Hansasuta P, Dong T, Thananchai H, Weekes M, Willberg C, Aldemir H, et al. Recognition of HLA-A3 and HLA-A11 by KIR3DL2 is peptide-specific. Eur J Immunol. 2004;34(6):1673–9. Epub 2004/05/27. doi: 10.1002/eji.200425089 15162437.
26. Fadda L, Borhis G, Ahmed P, Cheent K, Pageon SV, Cazaly A, et al. Peptide antagonism as a mechanism for NK cell activation. Proc Natl Acad Sci U S A. 2010;107(22):10160–5. Epub 2010/05/05. doi: 10.1073/pnas.0913745107 20439706; PubMed Central PMCID: PMC2890497.
27. Colantonio AD, Bimber BN, Neidermyer WJ Jr., Reeves RK, Alter G, Altfeld M, et al. KIR polymorphisms modulate peptide-specific binding to an MHC class I ligand with a Bw6 motif. PLoS Pathog. 2011;7(3):e1001316. Epub 2011/03/23. doi: 10.1371/journal.ppat.1001316 21423672.
28. Somanchi SS, Senyukov VV, Denman CJ, Lee DA. Expansion, purification, and functional assessment of human peripheral blood NK cells. J Vis Exp. 2011;e2540(48). Epub 2011/02/23. doi: 10.3791/2540 21339714.
29. Hill A, Jugovic P, York I, Russ G, Bennink J, Yewdell J, et al. Herpes simplex virus turns off the TAP to evade host immunity. Nature. 1995;375(6530):411–5. Epub 1995/06/01. doi: 10.1038/375411a0 7760935.
30. Fruh K, Ahn K, Djaballah H, Sempe P, van Endert PM, Tampe R, et al. A viral inhibitor of peptide transporters for antigen presentation. Nature. 1995;375(6530):415–8. Epub 1995/06/01. doi: 10.1038/375415a0 7760936.
31. Loffredo JT, Sidney J, Wojewoda C, Dodds E, Reynolds MR, Napoe G, et al. Identification of seventeen new simian immunodeficiency virus-derived CD8+ T cell epitopes restricted by the high frequency molecule, Mamu-A*02, and potential escape from CTL recognition. J Immunol. 2004;173(8):5064–76. Epub 2004/10/08. doi: 10.4049/jimmunol.173.8.5064 15470050.
32. O'Connor GM, Vivian JP, Widjaja JM, Bridgeman JS, Gostick E, Lafont BA, et al. Mutational and structural analysis of KIR3DL1 reveals a lineage-defining allotypic dimorphism that impacts both HLA and peptide sensitivity. J Immunol. 2014;192(6):2875–84. Epub 2014/02/25. doi: 10.4049/jimmunol.1303142 24563253; PubMed Central PMCID: PMC3948114.
33. Jones DC, Kosmoliaptsis V, Apps R, Lapaque N, Smith I, Kono A, et al. HLA class I allelic sequence and conformation regulate leukocyte Ig-like receptor binding. J Immunol. 2011;186(5):2990–7. Epub 2011/01/29. doi: 10.4049/jimmunol.1003078 21270408.
34. Willcox BE, Thomas LM, Bjorkman PJ. Crystal structure of HLA-A2 bound to LIR-1, a host and viral major histocompatibility complex receptor. Nat Immunol. 2003;4(9):913–9. Epub 2003/08/05. doi: 10.1038/ni961 12897781.
35. Feng Y, Qi J, Zhang H, Wang J, Liu J, Jiang F, et al. X-ray crystallographic characterization of rhesus macaque MHC Mamu-A*02 complexed with an immunodominant SIV-Gag nonapeptide. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2006;62(Pt 1):13–5. Epub 2006/03/03. doi: 10.1107/S1744309105038704 16511250; PubMed Central PMCID: PMC2150927.
36. Liu J, Dai L, Qi J, Gao F, Feng Y, Liu W, et al. Diverse peptide presentation of rhesus macaque major histocompatibility complex class I Mamu-A 02 revealed by two peptide complex structures and insights into immune escape of simian immunodeficiency virus. J Virol. 2011;85(14):7372–83. Epub 2011/05/13. 21561910; PubMed Central PMCID: PMC3126565.
37. Peruzzi M, Wagtmann N, Long EO. A p70 killer cell inhibitory receptor specific for several HLA-B allotypes discriminates among peptides bound to HLA-B*2705. J Exp Med. 1996;184(4):1585–90. Epub 1996/10/01. 8879234; PubMed Central PMCID: PMC2192820.
38. Fadda L, O'Connor GM, Kumar S, Piechocka-Trocha A, Gardiner CM, Carrington M, et al. Common HIV-1 peptide variants mediate differential binding of KIR3DL1 to HLA-Bw4 molecules. J Virol. 2011;85(12):5970–4. Epub 2011/04/08. 21471246; PubMed Central PMCID: PMC3126328.
39. Fadda L, Korner C, Kumar S, van Teijlingen NH, Piechocka-Trocha A, Carrington M, et al. HLA-Cw*0102-restricted HIV-1 p24 epitope variants can modulate the binding of the inhibitory KIR2DL2 receptor and primary NK cell function. PLoS Pathog. 2012;8(7):e1002805. Epub 2012/07/19. doi: 10.1371/journal.ppat.1002805 22807681; PubMed Central PMCID: PMC3395618.
40. van Teijlingen NH, Holzemer A, Korner C, Garcia-Beltran WF, Schafer JL, Fadda L, et al. Sequence variations in HIV-1 p24 Gag-derived epitopes can alter binding of KIR2DL2 to HLA-C*03 : 04 and modulate primary natural killer cell function. AIDS. 2014;28(10):1399–408. Epub 2014/05/03. doi: 10.1097/QAD.0000000000000284 24785948.
41. Thananchai H, Gillespie G, Martin MP, Bashirova A, Yawata N, Yawata M, et al. Cutting Edge: Allele-specific and peptide-dependent interactions between KIR3DL1 and HLA-A and HLA-B. J Immunol. 2007;178(1):33–7. Epub 2006/12/22. doi: 10.4049/jimmunol.178.1.33 17182537.
42. DeGottardi MQ, Specht A, Metcalf B, Kaur A, Kirchhoff F, Evans DT. Selective downregulation of rhesus macaque and sooty mangabey major histocompatibility complex class I molecules by Nef alleles of simian immunodeficiency virus and human immunodeficiency virus type 2. J Virol. 2008;82(6):3139–46. Epub 2008/01/18. 18199657; PubMed Central PMCID: PMC2258998.
43. Cassidy S, Mukherjee S, Myint TM, Mbribindi B, North H, Traherne J, et al. Peptide selectivity discriminates NK cells from KIR2DL2 - and KIR2DL3-positive individuals. Eur J Immunol. 2014. Epub 2014/11/02. doi: 10.1002/eji.201444613 25359276.
44. Sambrook JG, Bashirova A, Palmer S, Sims S, Trowsdale J, Abi-Rached L, et al. Single haplotype analysis demonstrates rapid evolution of the killer immunoglobulin-like receptor (KIR) loci in primates. Genome Res. 2005;15(1):25–35. Epub 2005/01/06. doi: 10.1101/gr.2381205 15632087; PubMed Central PMCID: PMC540275.
45. Hershberger KL, Shyam R, Miura A, Letvin NL. Diversity of the killer cell Ig-like receptors of rhesus monkeys. J Immunol. 2001;166(7):4380–90. Epub 2001/03/20. 11254692.
46. Blokhuis JH, van der Wiel MK, Doxiadis GG, Bontrop RE. The mosaic of KIR haplotypes in rhesus macaques. Immunogenetics. 2010;62(5):295–306. Epub 2010/03/06. doi: 10.1007/s00251-010-0434-3 20204612; PubMed Central PMCID: PMC2858804.
47. Boyson JE, Shufflebotham C, Cadavid LF, Urvater JA, Knapp LA, Hughes AL, et al. The MHC class I genes of the rhesus monkey. Different evolutionary histories of MHC class I and II genes in primates. J Immunol. 1996;156(12):4656–65. Epub 1996/06/15. 8648109.
48. Bostik P, Kobkitjaroen J, Tang W, Villinger F, Pereira LE, Little DM, et al. Decreased NK cell frequency and function is associated with increased risk of KIR3DL allele polymorphism in simian immunodeficiency virus-infected rhesus macaques with high viral loads. J Immunol. 2009;182(6):3638–49. Epub 2009/03/07. doi: 10.4049/jimmunol.0803580 19265142.
49. National Research Council (U.S.). Committee for the Update of the Guide for the Care and Use of Laboratory Animals, Institute for Laboratory Animal Research (U.S.), National Academies Press (U.S.). Guide for the care and use of laboratory animals. 8th ed. Washington, D.C.: National Academies Press. xxv, 220 p. p.
50. Shimizu Y, Geraghty DE, Koller BH, Orr HT, DeMars R. Transfer and expression of three cloned human non-HLA-A,B,C class I major histocompatibility complex genes in mutant lymphoblastoid cells. Proc Natl Acad Sci U S A. 1988;85(1):227–31. Epub 1988/01/01. 3257565; PubMed Central PMCID: PMC279517.
51. Lahm HW, Stein S. Characterization of recombinant human interleukin-2 with micromethods. J Chromatogr. 1985;326 : 357–61. Epub 1985/06/19. 3875623.
52. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–12. Epub 2004/07/21. doi: 10.1002/jcc.20084 15264254.
53. O'Doherty U, Swiggard WJ, Malim MH. Human immunodeficiency virus type 1 spinoculation enhances infection through virus binding. J Virol. 2000;74(21):10074–80. Epub 2000/10/12. 11024136; PubMed Central PMCID: PMC102046.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 9
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Epicellular Apicomplexans: Parasites “On the Way In”
- Fiat Luc: Bioluminescence Imaging Reveals In Vivo Viral Replication Dynamics
- Knocking on Closed Doors: Host Interferons Dynamically Regulate Blood-Brain Barrier Function during Viral Infections of the Central Nervous System
- A KSHV microRNA Directly Targets G Protein-Coupled Receptor Kinase 2 to Promote the Migration and Invasion of Endothelial Cells by Inducing CXCR2 and Activating AKT Signaling
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy