
Varicella Viruses Inhibit Interferon-Stimulated JAK-STAT Signaling through Multiple Mechanisms
In this manuscript we demonstrate that the immediate early protein ORF63 encoded by varicella zoster virus (VZV) and simian varicella virus (SVV) interferes with interferon type I-mediated activation of JAK-STAT signaling and thereby inhibits the expression of interferon stimulated genes. ORF63 blocks this pathway by degrading IRF9, which plays a central role in JAK-STAT signaling. In addition, both viruses code for immune evasion mechanisms affecting the JAK-STAT pathway upstream of IRF9, which results in the inhibition of STAT2 phosphorylation. By fusing a degradation domain derived from dihydrofolate reductase (DHFR) to ORF63 we further demonstrate that this protein is essential for SVV growth and gene expression, indicating that ORF63 also affects IFN-signaling indirectly by regulating the expression of other immune evasion genes.
Published in the journal:
. PLoS Pathog 11(5): e32767. doi:10.1371/journal.ppat.1004901
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004901
Summary
In this manuscript we demonstrate that the immediate early protein ORF63 encoded by varicella zoster virus (VZV) and simian varicella virus (SVV) interferes with interferon type I-mediated activation of JAK-STAT signaling and thereby inhibits the expression of interferon stimulated genes. ORF63 blocks this pathway by degrading IRF9, which plays a central role in JAK-STAT signaling. In addition, both viruses code for immune evasion mechanisms affecting the JAK-STAT pathway upstream of IRF9, which results in the inhibition of STAT2 phosphorylation. By fusing a degradation domain derived from dihydrofolate reductase (DHFR) to ORF63 we further demonstrate that this protein is essential for SVV growth and gene expression, indicating that ORF63 also affects IFN-signaling indirectly by regulating the expression of other immune evasion genes.
Introduction
The alphaherpesvirus varicella zoster virus (VZV) is the causative agent of chickenpox. After primary infection, VZV establishes latency in sensory ganglia. Reactivation from latency, which typically occurs in elderly individuals, can cause shingles or herpes zoster that is associated with a number of debilitating complications, including postherpetic neuralgia [1]. In vivo research on VZV is limited because the virus does not produce varicella or zoster in animals [2, 3]. Simian varicella virus (SVV) is closely related to VZV sharing about 75% DNA homology and exhibiting a highly similar genome organization [4]. Inoculation of nonhuman primates, including African green monkeys and Cynomolgus macaques, results in a persistent viremia [4]. In contrast, infection of rhesus macaques (RM) with SVV results in a primary infection followed by latency that is similar to VZV infection in humans. SVV-induced skin lesions are resolved by 21 days post infection which correlates with the absence of virus DNA in blood. Latent SVV can be detected in ganglia of infected RM [5]. Infection of RM with SVV thus represents a robust animal model that recapitulates most hallmarks of a primary human VZV infection.
The innate host immune response to viral infection is dominated by interferons (IFNs) that are subdivided in three families, namely types I, II and III. In particular, several subtypes of IFNα and IFNβ that represent type I IFNs are key players in the anti-viral innate immune response [6]. Transcription of IFN is initiated by pattern recognition receptors (PRRs) engaging pathogen associated molecular patterns (PAMPs) such as double-stranded RNA, lipopolysaccharide and cytosolic DNA. Downstream signaling pathways lead to the activation of transcription factors such as IFN regulatory factor (IRF) 3 and nuclear factor κB (NFκB) that induce the transcription of IFNβ. Secreted IFNβ can signal in an autocrine and paracrine fashion by interacting with the type I IFN receptor complex (consisting of IFNAR1 and IFNAR2) both on infected and neighboring uninfected cells [7]. Receptor binding activates the JAK-STAT signaling pathway, which results in the expression of hundreds of IFN-stimulated genes (ISG) and corresponding proteins that inhibit virus growth by counteracting multiple molecular steps of the replication cycle and by signaling to innate immune cells including natural killer cells [8, 9]. In addition, type I IFNs have been shown to be involved in dendritic cell maturation and antigen presentation thereby stimulating the development of virus-specific adaptive immune responses [10, 11].
The ability of VZV and SVV to spread and establish latency in the presence of these immediate immune responses implies that both viruses display evasion strategies that circumvent or counteract the induction or function of IFNs and ISGs. VZV infection of human skin xenografts in severe combined immunodeficiency (SCIDhu) mice showed that VZV-infected cells do not express type I IFN, while uninfected bystander cells stained positive for the cytokine [12]. Several reports have shown that the effect of IFN is counteracted by at least four different VZV-encoded proteins: both IE62 and ORF47 alter the phosphorylation of IRF3 and prevent gene activation [13, 14], whereas ORF61 inhibits pathogen-induced cytokine expression by degrading IRF3 [15] and by blocking the activation of NFκB via inhibiting IκBα [16]. In addition, Cohen et al. showed the deletion of ORF63 severely attenuates the growth of the virus in the presence of IFNα but not IFNγ, suggesting a possible involvement of ORF63 in regulating JAK-STAT signaling [17].
Expression of IFNα and IFNβ and subsequent binding to their receptor triggers the dimerization of IFNAR1 and IFNAR2, which activates the Janus kinases JAK1 and TYK2 that are constitutively associated with the receptor. The kinases phosphorylate the receptor creating a docking site for the transcription factors STAT1 and STAT2. Subsequent phosphorylation of the STAT proteins by the JAKs leads to conformational changes within the molecules that allow the formation of stable STAT1, STAT2 and IFN regulatory factor 9 (IRF9) complex termed ISGF3. ISGF3 then shuttles to the nucleus where it activates the transcription of type I IFNs and ISGs [18]. VZV-infected cells in skin xenografts in the SCIDhu mice did not show nuclear localization or phosphorylation of STAT1, in contrast to uninfected bystander cells [12]. Since IFNα produced by bystander cells can induce JAK-STAT signaling in VZV-infected cells, the absence of pSTAT1 in infected cells suggests that VZV interferes not only with IRF3-mediated activation of IFN transcription but also with JAK/STAT signaling.
The VZV ORF63 protein is a 30 kDa immediate early protein that is phosphorylated by host and viral kinases [19, 20]. The protein is abundantly present during lytic infection and its expression has also been observed in latently infected ganglia [21, 22]. A duplicate of the ORF63 gene, designated ORF70, is found in the terminal repeat region [19]. VZV ORF63/70 regulates viral gene expression and impairs the expression of certain cellular genes [23–26]. In addition, VZV ORF63/70 inhibits apoptosis in cultured primary human neurons [27]. SVV encodes both ORF63/70 orthologous proteins that share 52% amino acid identity with their VZV homologs [28]. These gene products are required for replication of SVV in cell culture [29]. In vivo studies using SVV-infected RM and African green monkeys confirmed expression of the ORF63 protein in ganglia during latent infection [5].
In this report, we show that both SVV and VZV interfere with type I IFN signaling. SVV inhibits type I IFN-induced ISG expression by downregulating the expression of IRF9 and prevents phosphorylation of STAT2 upon IFN stimulation. We also observed a minor decrease in STAT2 levels in SVV-infected cells. SVV ORF63 was found to be responsible for the reduction in IRF9 expression, but was not directly involved in downregulation of STAT2 expression or inhibition of STAT2 phosphorylation. These data suggest that multiple SVV proteins counteract type I IFN signaling. Similarly, we observed reduced levels of STAT2 and IRF9 proteins in VZV-infected cells and the cells were refractory to IFN-induced STAT2 phosphorylation. Ectopic expression of VZV ORF63 affected steady state levels of IRF9, but did not block STAT2 expression or phosphorylation. Thus, the multi-level inhibition of JAK-STAT signaling seen in SVV-infected cells is conserved in VZV-infected cells.
Materials and Methods
Cell lines and recombinant viruses
Telomerized rhesus fibroblasts (TRFs), TRF-ISRE cells [30], the African green monkey kidney epithelial cell line Vero, Vero-CRE (kindly provided by Dr. Linda van Dyk, University of Colorado, Denver) [31], telomerized human fibroblasts (THF)-ISRE [32], human embryonic kidney (HEK) 293T cells (ATCC), and the human fibroblast cell line MRC-5 (ATCC) were maintained in DMEM supplemented with 10% heat-inactivated fetal bovine serum (FBS), 140 IU of penicillin/ml and 140 μg of streptomycin/ml.
TRFs and Vero cells were infected with a recombinant SVV Delta strain, in which eGFP was inserted between US2 and US3 through homologous recombination [33]. A confluent monolayer of TRF or Vero cells was infected by cocultivation of SVV.eGFP-infected cells with uninfected cells at the indicated ratios. Complete infection was confirmed by visualizing eGFP using fluorescence microscopy. Infected cells were maintained in DMEM supplemented with 2% FBS and harvested at 48 hours p.i. For VZV infections, we used the recombinant VZV Oka strain, in which eGFP was fused to the N-terminus of ORF66 (generously provided by P.R. Kinchington, University of Pittsburgh, Pennsylvania) [34]. VZV.eGFP-infected MRC-5 cells were cocultivated with uninfected MRC-5 cells at a 1 : 5 ratio.
Reagents and antibodies
Rhesus IFNα2 was obtained from R&D systems. Human IFNα and universal type I IFN (uIFN) were obtained from PBL Assay Science. MG132 (Fisher Scientific) was dissolved in DMSO and used at the indicated concentrations for 16 hours. Control wells were treated with same concentration of DMSO without MG132. Trimethoprim (TMP; Sigma-Aldrich) was dissolved in DMSO and used 10 μM or less where indicated. Viral cultures were supplemented with fresh TMP every 24 hours. The following antibodies were used for detection of endogenous and viral proteins in western blot: anti-ISG15 F-9 (Santa Cruz), anti-ISG54/IFIT2 (Abcam), anti-Mx-1 (GeneTex), anti-STAT1 M22 (Santa Cruz), anti-phosphorylated STAT1 Tyr701 (Santa Cruz), anti-STAT2 C20 (Santa Cruz), anti-phosphorylated STAT2 Tyr690 (Cell Signaling Technology), anti-IRF9/ISGF3γ clone 6 (BD Biosciences), anti-GAPDH 6C5 (Santa Cruz), anti-p84 5E10 (GeneTex), anti-IRF1 H-205 (Santa Cruz), anti-IRF3 (Santa Cruz) and anti-FLAG M2 (Sigma-Aldrich). The monoclonal antibodies specific for SVV and VZV ORF63 (clone 63_6), ORF62 (clone 62_6) and ORF31 (clone 31C_8) have been previously described [35]. STAT2 C20 (Santa Cruz) was also used for immunofluorescence microscopy.
Luciferase reporter assays
TRFs were transduced with a replication-defective lentivirus encoding firefly luciferase downstream of an IFN-stimulated response element (ISRE) and a lentivirus constitutively expressing renilla luciferase driven by a CMV promotor (Qiagen). Transduced TRFs (TRF-ISRE cells) were selected by culturing in the presence of 4 μg/ml puromycin. TRF-ISRE cells were infected with SVV.eGFP as described above and 24 hours p.i. the cells were seeded in a black 96 well plate (Corning Incorporated). At 42 hours p.i., the cells were stimulated with rhesus or human IFNα to induce expression of ISRE-driven firefly luciferase. After 6 hours, expression of firefly and renilla luciferase was measured using the Dual-Glo luciferase assay system (Promega). Luminescence was measured on a Veritas microplate luminometer (Promega). Data are presented as the ratio between firefly luciferase expression and renilla luciferase expression. For the experiment described in Fig 1B the cells were sorted for high GFP expression at 40 hours p.i. using a FACS Aria II cytometer. We seeded 2000 of the sorted and mock-infected cells cells in black 96 well plate and stimulated with uIFN for 6 hours, after which luciferase expression was measured using the ONE-Glo luciferase assay system (Promega).

HEK 293T cells were co-transfected with an ISRE-firefly luciferase reporter plasmid (pGL3-ISRE-Luc) and the pcDNA3.1 expression vectors (described below). At 24 and 42 hours post transfection, the cells were treated with 5000 U/ml uIFN for 6 hours. Cells were transferred to a black 96 wells plate before or right after treatment with uIFN treatment. Expression of firefly luciferase was measured using the ONE-Glo luciferase assay system (Promega).
Nuclear extractions and Western blotting
To generate nuclear and cytoplasmic fractions, cells were resuspended in dounce buffer (100 mM KCl, 20 mM Hepes [pH 7.4], 0.1 mM EDTA, 3% sucrose) supplemented with HALT protease and phosphatase inhibitors (Thermo Scientific). After 15 minutes 10% Nonidet P-40 was added to the lysate in a 1 : 20 ratio and lysates were vortexed for 10 seconds. The cytoplasmic fraction was removed immediately and nuclei were washed twice with D-PBS and lysed in RIPA buffer (10 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1% Sodium Deoxycholate, 0.1% SDS) supplemented with protease and phosphatase inhibitors. For all other experiments, cells were lysed directly in Laemmli sample buffer (100 mM Tris-HCL [pH 8.0], 4% SDS, 20% glycerol, 10% 2-mercaptoethanol, Bromophenol blue). Proteins were separated by SDS-page and transferred to polyvinylidene difluoride membranes (Thermo Scientific). Membranes were first incubated with the indicated antibodies, which was followed by incubation with horseradish peroxidase (HRP)-conjugated secondary antibodies specific for mouse (Santa Cruz) or rabbit (Thermo Scientific) IgG. Binding of secondary antibodies to the membranes was visualized by using Pierce ECL 2 (Thermo Scientific).
Immunofluorescence microscopy
Mock - or SVV-infected TRFs were grown on cover slips, washed twice with PBS and fixed with 3.7% formaldehyde (Fisher Scientific) at room temperature (RT) for 40 minutes. After washing with PBS, residual formaldehyde was quenched with 50 mM Ammonium Chloride for 10 minutes and the cells were permeabilized with 0.1% Triton for 5 to 7 minutes. Non-specific protein binding sites were blocked with 2% bovine serum albumin (BSA) (Fisher Scientific) and cells were incubated with STAT2-specific antibody in 2% BSA for 1 hour at 37°C. Cells were washed with 2% BSA and incubated with the secondary antibody Alexa Fluor 594 Goat anti-Rabbit (Life Technologies). Cells were then washed with 2% BSA, followed by one PBS wash. Cover slips were mounted on glass slides using Prolong Gold Anti-fade reagent (Cell Signaling). Staining was visualized on a Zeiss Axioskop 2 Plus fluorescence microscope, and images were taken using AxioVision v4.6 software (Zeiss).
Adenovirus production and infection
The recombinant adenoviruses expressing SVV ORF63 (AdORF63) and VZV AdORF63 were produced as previously described [36]. The vector contains a tetracycline-responsive promoter and requires the addition of a tetracycline-regulated transactivator (tTA) [37], which was provided by co-infecting with AdtTA. TRFs cultured in six-well clusters were co-transduced with the purified ORF63 adenoviruses and AdtTA at the indicated MOI in 0.5 ml of serum-free DMEM. After 1.5 hour of incubation at 37°C, 1.5 ml of DMEM supplemented with 10% FBS was added and incubation continued for a total of 48 hours. Where indicated, doxycycline was added to the infections to regulate tTA-dependent gene expression.
RNA isolation and semiquantative PCR
Total RNA was extractedand treated with DNase using the NucleoSpin RNA isolation kit (Machery Nagel) according to the manufacturer's protocol. The concentration of the RNA samples was measured using the NanoDrop 1000 Spectrophotometer (Thermo Scientific). Single-stranded cDNA was made from total RNA using random hexamers (TaKaRa) to prime first-strand synthesis by Maxima Reverse Transcriptase (Thermo Scientific) as recommended by the manufacturer. The induction of ISG54 and Mx-1 mRNA expression upon uIFN stimulation was determined using SYBR green-based semiquantitative real-time RT-PCR (qPCR) using the following primers: ISG54 Fw: 5’-gttactggaactaataggacac-3’, ISG54 Rev: 5’-tggcaagaatggaaca-3’, Mx-1 Fw: 5’-atgatcgtcaagtgccg-3’, Mx-1 Rev: 5’-gccttgccttcctcca-3’. SVV ORF63 expression was confirmed using the primers ORF63 Fw: 5’-CAGCGTCCTACAGTGAC-3’ and ORF63 Rev: 5’-GTTGCTGGTAGCATCATC-3’. Levels of IRF9 mRNA were determined using the primers IRF9 Fw: 5’ - TACCCGAAAACTCCGGAAC-3’ and IRF9 Rev: 5’-AAGAAGGCAGCATCCTGG-3’. Levels of STAT2 mRNA were determined using the primers STAT2 Fw: 5’ - ATGGCGCAGTGGGAAATG-3’ and STAT2 Rev: 5’-ctgccagttctggtcttc-3’. Reactions were performed using SYBR green PCR core reagents and Platinum Taq DNA Polymerase (Invitrogen). Relative expression of ISG54, Mx-1, and ORF63 was calculated using the method described by Livak and Schmittgen [38]. GAPDH was used as a housekeeping gene to establish a baseline against which target genes were compared (Fw: 5’-GCACCACCAACTGCTTAGCAC-3’, Rev: 5’ - TCTTCTGGGTGGCAGTGATG-3’). For IRF9 and STAT2 mRNA expression we calculated delta cycle threshold (ΔCt) by subtracting background Ct (GAPDH) from the Ct value for IRF9/STAT2.
Plasmids and transient transfections
Purified DNA from TRFs infected with SVV.eGFP was used as a template for PCR amplification of SVV ORF63. PCR was performed using AccuPrime Taq DNA polymerase High Fidelity (Life Technologies) using the primers 5’-AATAAAGGATCCGCCACCATGCAGGCGCCCCGAG-3’ (Fw) and 5’-AATAAAGAATTCTTATGTATTGTGTACAGACTCTCGTAACTCCGTG-3’ (Rev) to amplify the coding sequence of the SVV ORF63 gene. The PCR-generated product was inserted into pcDNA3.1-IRES-nlsGFP using BamHI/EcoRI sites, creating pcDNA3.1 ORF63. To create pcDNA3.1 FLAG-ORF32 we amplified ORF32 from the same template with the primers 5'-(AATAAAGGATCCGCCACCATGGCATCATCTAATACTTGCGAAGAACAAAATAATTCTA)-3' (Fw) and 5'-(AATAAAGAATTCTTActtatcgtcgtcatccttgtagtcATCCGTTTCGCTCTCGCTAGATGAAGGTTG)-3' (Rev) using the Expand High Fidelity PCR system (Roche). The PCR-generated product was inserted into the pcDNA3.1-IRES-nlsGFP vector using BamHI/EcoRI sites. VZV ORF63 was amplified from DNA extracted from VZV.eGFP-infected MRC-5 cells using the Expand Expand High Fidelity PCR system (Roche) and the following primers: 5'-(AATAAAGGATCCGCCACCATGTTTTGCACCTCACCGGC)-3' (Fw) and 5'-(AATAAAGAATTCCTACACGCCATGGGGGGGCGGTATATC)-3' (Rev). The resulting insert was cloned into pcDNA3.1-IRES-nlsGFP using BamHI/EcoRI sites. Rhesus IRF9 was synthesized and codon-optimized for expression in rhesus cell lines by GenScript. The insert was cloned from the pUC57 plasmid into pcDNA3.1-IRES-nlsGFP using BamHI/EcoRI restriction sites, creating pcDNA3.1 IRF9. All ligations were performed using the Rapid DNA Dephos and Ligation kit (Roche). The DNA sequences of all expression plasmids were verified.
HEK 293T cells were transfected with the indicated plasmids and pGL3-ISRE-Luc (a kind gift from Dr. John Hiscott, Vaccine and Gene Therapy Institute, Florida) using the Lipofectamine 2000 reagent (Life Technologies) using the manufacturers protocol. For the IRF9 overexpression experiment we used 1 μg pGL3-ISRE-Luc and the indicated amounts pcDNA3.1 (p)ORF63 and pcDNA3.1 (p)IRF9. Control pcDNA3.1 or pRetro-E2 expressing GFP was used to equalize all transfection samples to a total of 6 μg DNA. Transfection efficiency was confirmed measuring GFP expression in all samples using Synergy HTX Multi-Mode Reader (Bio-Tek) or by staining for specific proteins in western blot. To test whether VZV ORF63 inhibits IFN-signaling, we used 1 μg of pGL3-ISRE-Luc and 3 μg of the other indicated plasmids. 48 hours post transfection IFN-signaling was assessed using a luciferase assay, described below.
Lentivirus construction and infection
GIPZ lentivirus constructs expressing shRNA specific for human IRF9 were obtained from Open Biosystems/GE Healthcare. The constructs used are V3LHS-322329 (shRNA-1), V3LHS-322332 (shRNA-2), and V2LHS-69847 (shRNA-3). Replication deficient lentiviruses were produced by transfecting the shRNA vectors into HEK 293T cells and providing the vesicular stomatitis virus G (pMD2.G VSV-G, Addgene) protein and the packaging plasmid psPAX2 (Addgene) in trans. The plasmids were transfected using Lipofectamine LTX (Life Technologies). 48 hours post transfection the supernatant containing lentivirus was harvested and transferred to target cells, which were transduced in the presence of 5 μg/ml Polybrene (Hexadimethrine bromide; Sigma-Aldrich). After 24 hours, this process was repeated. The resulting cell lines were grown in the presence of 3 μg/ml Puromycin to select for shRNA expressing cells.
Statistical analysis
P-values were determined using unpaired Student’s t-test.
SVV BAC mutagenesis
To prepare the SVV ORF63/70 mutant, we used an SVV BAC containing the complete SVV genome and eGFP driven by the CMV immediate-early promoter [39]. To introduce mutations into SVV ORF63/70, we used the two-step red-mediated mutagenesis protocol [40]. Mutagenesis of SVV BAC using this protocol has been previously described [29]. Briefly, we used a recombinant plasmid encoding red fluorescent protein (RFP) interrupted by the kanamycin gene (kindly provided by Dr. Benedikt Kaufer, Freie Universität Berlin, Germany). Using oligonucleotide primers specific for regions flanking SVV ORF63/70 at the 5’-end and RFP-specific sequences at the 3’-end, we amplified a 1748 bp DNA fragment containing RFP/kanamycin (ORF63 mRFP Fw: TACCATCTGAATGTTACGTACATAAATAAAACGCTTCTCAATGGCCTCCTCCGAGGACG, ORF63 mRFP Rev: GACAGGGGTAACATGTTAGCGGCTCCCTATTGGGTAAGGGACTACAAGGCGCCGGTGGAG). The DNA fragment was used to transform E. coli GS1783 containing wild-type SVV BAC. We selected kanamycin-resistant colonies and extracted recombinant BAC DNA and confirmed recombination using Hind III digestion and agarose gel electrophoresis. We identified the recombinant BAC clones that contained RFP/kanamycin in place of SVV ORF63 and eliminated the kanamycin cassette. Complete replacement of SVV ORF 63 sequences by RFP was confirmed by sequence analysis.
DHFR domains were introduced at the C-terminus of ORF63/70 using a plasmid containing the destabilization domain dihydrofolate reductase (DHFR) derived from E. coli (kindly provided by Dr. Thomas Wandless, Stanford University, California). We introduced the kanamycin-cassette at a unique restriction site (PmeI) within the sequences encoding DHFR. We amplified DHFR/kanamycin with primers specific for SVV ORF63/70 (ORF70 DHFR Fw: CCATCTGAATGTTACGTACATAAATAAAACGCTTCTCAATGATCAGTCTGATTGCGGCGTTAGCGGT, ORF70 DHFR Rev: CCATCTGAATGTTACGTACATAAATAAAACGCTTCTCAATGATCAGTCTGATTGCGGCGTTAGCGGT) by PCR and transformed of E. coli GS1783 containing mutant SVV BAC in which SVV ORF63 was replaced with RFP. After elimination of the kanamycin cassette, mutant SVV BAC in which DHFR was fused at the amino terminus of SVV ORF70 was identified by HindIII digestion and gel electrophoresis. Proper fusion of DHFR to SVV ORF70 was confirmed by sequence analysis. The recombinant BAC was purified and used to transfect Vero cells. Infected cells were grown in the presence of 10 μM trimethoprim (TMP) to stabilize the ORF70-DHFR fusion protein. SVV plaques expressing eGFP and RFP were identified and isolated using a fluorescent microscope. Sequentially, mutant SVV was passaged four to five times in Vero-CRE cells. Passaging the virus allowed recombination of ORF70-DHFR to ORF63 location, which lead to the loss of RFP. In addition, BAC vector and eGFP sequences within the virus are flanked by loxP sites, thus passing the virus in Vero cells stably expressing cre recombinase resulted in the elimination of these non-viral sequences [39]. SVV plaques that were negative for both eGFP and RFP were purified and transferred from Vero-CRE cells to TRFs via serial passage. DNA extracted from Vero cells infected with SVV mutant was used for sequence analysis to confirm proper fusion of DHFR to the C-termini of both ORF63 and ORF70.
Wild type and mutant SVV growth curves
In vitro growth curves for wild type and mutant SVV were generated as described [41]. Briefly, a monolayer of uninfected Vero cells in 25 cm2 tissue culture flasks were infected with approximately 5X102 Vero cells previously infected with either wild type or ORF63-DHFR SVV. At 3, 24, 48, 72, 96, 120, 144 and 168 hours p.i. cells were trypsinized, diluted and seeded on triplicate dishes containing uninfected Vero cells. After approximately one week, infected cells were stained with crystal violet and infectious plaques were counted.
Accession numbers
SVV Delta, ORF63/70-DHFR SVV, SVV ORF63 gene, and SVV ORF32 gene: GenBank NC_002686. VZV pOka and VZV ORF63 gene: GenBank AB097933.
Results
SVV inhibits type I IFN-induced ISG expression
To determine if SVV interferes with type I IFN-mediated responses, we studied IFN-stimulated response element (ISRE)-dependent transcription in SVV-infected luciferase reporter cells. We used telomerized rhesus fibroblasts (TRFs) stably expressing firefly luciferase under the control of the ISRE as well as constitutively expressing renilla luciferase to control for differences in cell viability between the samples. TRF-ISRE cells were infected with SVV.eGFP at a ratio of 5 : 1 (uninfected to SVV-infected cells) and, after 42 hours, incubated with rhesus or human IFNα for 6 hours. Productive virus infection was confirmed by visualizing eGFP expression using immunofluorescence microscopy. The firefly and renilla signal was measured and the ratio of these values reflected ISRE activity. In mock-infected cells, incubation with increasing concentrations of rhesus IFNα corresponded with increased ISRE activity. However, only a minimal response to IFNα was observed in SVV-infected cells (Fig 1A, left panel). The rhesus reporter cells were also activated by human IFNα and a comparable reduction in ISRE activity was observed in the SVV-infected cells (Fig 1A, right panel). A dose-dependent increase of luciferase activity in SVV-infected cells was not due to the presence of uninfected cells since this was also observed when the infected cells were sorted for high GFP expression by flow cytometry before IFN-treatment (Fig 1B), suggesting that high concentrations of IFN can partially overcome the inhibition by SVV. The ISRE element drives the expression of interferon stimulated genes (ISG). To study if SVV inhibits IFNα-induced ISG-expression, we infected TRFs with SVV.eGFP for 40 hours and incubated with increasing concentrations of recombinant universal type I IFN (uIFN) for 8 hours. Productive SVV infection was confirmed by the detection of SVV ORF63 expression (Fig 1C). Expression of ISG15, ISG54 and Mx-1 was observed in all mock-infected IFNα-stimulated samples, but was absent in SVV-infected cells (Fig 1C). These data show that SVV inhibits IFNα-mediated activation of ISRE-dependent reporter gene expression and ISG protein expression.
SVV blocks IFN-mediated nuclear translocation of STAT by preventing STAT2 phosphorylation
The engagement of IFN with the IFN-receptor results in the activation of the JAK-STAT signal transduction pathway. The resulting phosphorylation of STAT1 and STAT2 allows their heterodimerization and association with IRF9, forming the ISGF3 complex that subsequently shuttles to the nucleus to initiate ISRE-dependent transcription [42]. The nuclear translocation of the ISGF3 complex is thus essential for ISRE activation. We analyzed IFN-induced nuclear localization of STAT in SVV-infected cells. TRFs were infected at a 10 : 1 ratio with SVV.eGFP and incubated with uIFN for 40 minutes at 48 hours post infection (p.i.). In uninfected cells, STAT2 was found predominantly in the cytosol in the absence of IFN and in the nucleus upon IFN-treatment (Fig 2A). In contrast, STAT2 was not translocated to the nucleus in SVV-infected cells (green/eGFP) upon IFN-treatment (Fig 2A). In addition, we isolated cytoplasmic and nuclear fractions of SVV.eGFP-infected Vero cells (ratio 5 : 1) and determined the cellular localization of STAT2 by western blot. Separation of cytosol and nuclei was confirmed using GAPDH and the nuclear matrix protein p84 (Fig 2B). In uninfected cells, STAT2 was found in the cytosolic fraction in the absence of IFN-treatment, whereas stimulation with uIFN led to the redistribution of STAT2 to both cytoplasmic and nuclear fractions. In contrast, STAT2 remained predominantly cytosolic in SVV-infected cells even upon IFN-treatment (Fig 2B). The SVV ORF62 protein was found in both cytoplasmic and nuclear fractions. This distribution is consistent with reports for the homologous VZV protein that, while primarily nuclear during early times of infection, localizes to the cytoplasm at later times of infections as a results of phosphorylation by ORF66 [43, 44]. Thus, in an asynchronous infection one would expect both nuclear and cytoplasmic expression of ORF62. Taken together, these data suggest that SVV inhibits IFN-dependent ISG-induction by abrogating the IFN-associated translocation of STAT2.

Next we examined whether the inhibition of STAT2 nuclear translocation correlated with a SVV-mediated reduction in steady state levels of ISGF3 members or impaired STAT1/STAT2 phosphorylation. TRFs infected with SVV.eGFP for 48 hours were stimulated with IFN for 20 minutes. Steady state levels and IFN-induced phosphorylation of STAT1 were comparable between SVV - and mock-infected cells (Fig 3A). In contrast, IFN-induced STAT2 phosphorylation was absent in SVV-infected cells and steady state levels of the protein also appeared to be reduced (Fig 3A). Densitometric analysis of STAT2 protein using four independent experiments confirmed an approximately 25% decrease in STAT2 levels (Fig 3B). However, this decrease was not statistically significant. In contrast, we observed a significant decrease of more than 50% in IRF9 levels by SVV (Fig 3A and 3B). Interestingly, the reductions in STAT2 and IRF9 protein levels were observed regardless of IFN stimulation (Fig 3B). Since IRF9 drives the nuclear translocation and retention of phosphorylated STAT1 and STAT2 [42], we studied the localization of residual IRF9 in IFN-stimulated SVV-infected Vero cells by analyzing isolated cytoplasmic and nuclear fractions in western blots. In control cells, IFN-stimulation triggered the increased translocation of IRF9 from the cytosol to the nucleus. However, this increased nuclear translocation of IRF9 was not observed in SVV-infected cells (Fig 3C). Furthermore, this experiment confirmed the SVV-mediated reduction in IRF9 expression levels. These data suggest that SVV abrogates JAK-STAT signaling by both preventing the phosphorylation of STAT2 and reducing STAT2 and IRF9 protein levels.
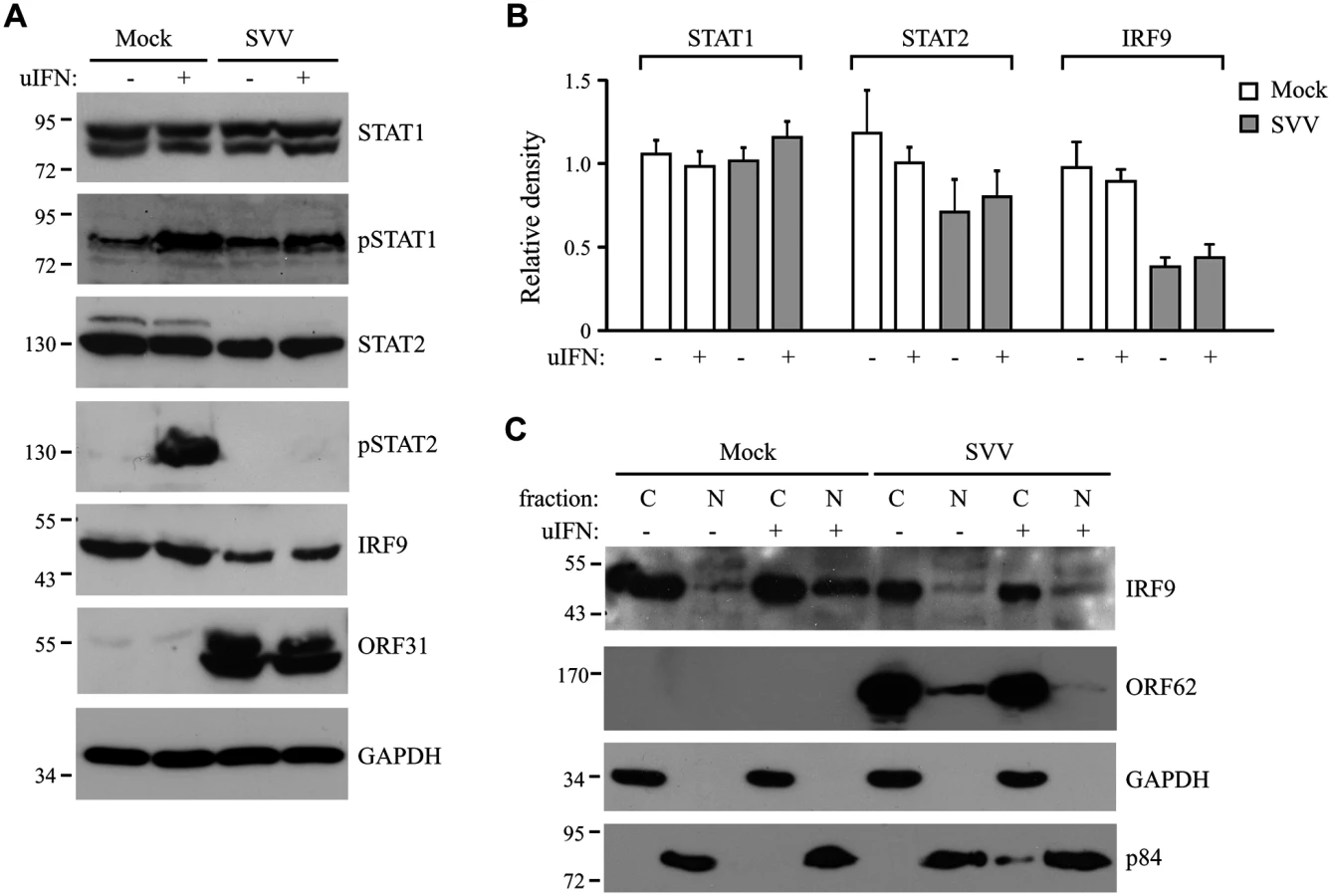
SVV ORF63 inhibits IFN-mediated JAK-STAT signaling
Ambagala et al. showed that wild type VZV can replicate in cells preincubated with IFNα, but an ORF63 deletion mutant could not [17]. This observation suggested that VZV ORF63 might be involved in the ability of VZV to evade IFN responses. VZV and SVV ORF63 share 52% overall amino acid homology [4, 45]. To determine if SVV ORF63 plays a role in the inhibition of IFN-stimulated responses observed in SVV-infected cells, we constructed a recombinant adenovirus expressing SVV ORF63 under the control of a tetracycline-responsive promotor (AdORF63). ORF63 expression is induced by co-infection with a recombinant adenovirus expressing the tetracycline-regulated transactivator (AdTA) [37]. These adenoviruses lack the E1-region [36], and therefore unable to interfere with IFN-signaling [46]. TRFs were co-transduced with a multiplicity of infection (MOI) of 10 of AdTA and with increasing MOIs of AdORF63. The expression of ORF63 was monitored at 48 hours p.i. (S1A Fig). Increasing MOI correlated with increasing ORF63 expression levels as expected. However, higher ORF63 levels also resulted in decreased GAPDH expression, suggesting that high expression levels of ORF63 may be cytotoxic. The transactivator expressed by AdTA can be inactivated by the tetracycline derivative doxycycline (Dox). To fine-tune ORF63 expression levels, we transduced TRFs with AdORF63 (MOI 20) and AdTA (MOI 10) in the presence of decreasing amounts of Dox. We detected robust ORF63 expression in cells that were incubated with 1 ng/ml Dox (S1B Fig) and these expression levels were comparable to ORF63 expression in cells infected with SVV (S1C Fig). At 1 ng Dox, GAPDH levels were not affected whereas lower Dox concentrations resulted in decreased GAPDH levels reflecting reduced cell viability (S1B Fig). To examine whether ORF63 inhibits IFN signaling we treated TRFs with IFN for up to 16 hours in the presence of 1ng Dox and studied ISG expression using qPCR (Fig 4A). In addition we used 1000 ng/ml Dox to inhibit ORF63 expression and under these conditions we observed increased expression of Mx-1 and ISG54 mRNA, reaching peak expression at 8 and 4 hours of IFN stimulation, respectively. In ORF63 expressing cells, however, Mx-1 and ISG54 mRNA levels were severely reduced at all time points (Fig 4A). ORF63 expression was confirmed by qPCR (Fig 4A, lower right panel). We also examined ISG protein expression by western bloting in the absence or presence of ORF63: after 8 hours of stimulation with IFN, high levels of ISG15, ISG54 and Mx-1 were detected in mock-transduced cells and in AdORF63-transduced cells treated with 1000 ng of Dox, whereas expression was absent or barely detectable in AdORF63-transduced cells treated with 1 ng Dox (Fig 4B). Taken together, these data indicate that SVV ORF63 inhibits type I IFN-induced gene expression.

SVV ORF63 induces IRF9 degradation in a proteasome-dependent manner
The inhibition of IFN-induced ISG expression by ORF63 correlated with our observations in SVV-infected cells. Since reduced STAT2 phosphorylation as well as decreased amounts of STAT2 and IRF9 proteins were observed in SVV-infected cells (Fig 3A and 3B), we examined whether expression of ORF63 leads to the inhibition of IFN-induced STAT2 phosphorylation and reduced steady state levels of STAT2 and IRF9. Expression and phosphorylation status of members of the JAK-STAT pathway were examined in AdORF63/AdTA-transduced TRFs stimulated with uIFN for 20 minutes or 8 hours. Despite an inhibition of IFN-induced ISG expression in ORF63-expressing cells (Fig 5A, lower panel) the expression levels and phosphorylation status of STAT1 and STAT2 were unchanged (Fig 5A, upper panel). However, we did observe a reduction in steady state levels of IRF9 when ORF63 was present (Fig 5A). To confirm that ORF63 affects IRF9 expression, we transduced TRFs with AdORF63/AdTA with decreasing concentrations of Dox to obtain increasing ORF63 expression levels. Western blot analyses revealed that increasing levels of ORF63 inversely correlated with decreasing IRF9 levels (Fig 5B and 5C). Interestingly, reduced IRF9 expression was observed in both unstimulated and IFN-stimulated samples (Fig 5B), suggesting that ORF63 reduces IRF9 regardless of IFN signaling. To confirm that ORF63 does not affect STAT2 levels we transduced TRFs with AdORF63/AdTA in the presence of decreasing amounts of Dox. We observed a reduction in STAT2 levels in cells expressing high levels of ORF63 (0.1 and 0 ng/ul Dox) (Fig 5D). However, STAT1 and GAPDH expression were also affected in these samples, but when we normalized STAT1 and STAT2 expression to GAPDH expression the reduction in STAT1 or STAT2 levels was not significant when ORF63 was expressed (Fig 5E). To determine whether ORF63 affects the transcription of IRF9 we studied IRF9 mRNA levels in TRFs transduced with AdORF63/AdTA in the presence of decreasing amounts of Dox. However, we did not observe a reduction of IRF9 mRNA levels upon increasing transcription of ORF63 suggesting that ORF63 does not affect IRF9 transcription (Fig 5F).

To analyze if ORF63 promotes IRF9 degradation via the proteasome we incubated ORF63-expressing cells with increasing concentrations of the proteasome inhibitor MG132 for 16 hours prior to lysing the cells. Treating ORF63-expressing cells (+) with increasing concentrations of MG132 reversed IRF9 degradation, but we also observed a slight increase in IRF9 levels in control cells (-) (Fig 6A). To determine if the rescue of IRF9 expression in ORF63-expressing cells was due to reduced turnover of residual IRF9 or due to actively blocking ORF63-mediated IRF9 degradation, we averaged the ratio of IRF9 and GAPDH expression in MG132-treated (10 μM) control or ORF63-expressing cells in four independent experiments (Fig 6B). While MG132-treatment increased IRF9 expression in control cells, the difference was not statistically significant. In contrast, MG132-treatment of ORF63-expressing cells resulted in a fivefold increase in IRF9 expression (Fig 5B, p = 0.007). From these data we conclude that ORF63 promotes IRF9 degradation in a proteasome-dependent manner. In addition to IRF9, related IRF proteins play an important role in the regulation of the expression of IFN and ISGs [47–49]. To determine whether SVV ORF63 affects the expression of IRFs other than IRF9, we monitored the steady state expression of IRF1 and IRF3 in TRFs transduced with AdORF63/AdTA in the presence of decreasing amounts of Dox. Increasing levels of ORF63 reduced IRF9 expression levels, but not that of IRF1 or IRF3 (Fig 6C). While we cannot formally rule out that SVV ORF63 might affect other IRFs it seems likely that SVV ORF63 specifically induces the degradation of IRF9 and thus preventing IFN-mediated ISG induction.

IRF9 degradation by SVV ORF63 is responsible for inhibition of ISG expression
To determine whether the reduction of IRF9 was sufficient to prevent ISG-induction, we attempted to recapitulate the ORF63-effect by reducing IRF9 levels using small hairpin RNA (shRNA). Telomerized human fibroblasts (THF) stably expressing ISRE-luciferase (THF-ISRE) were transduced with lentivectors expressing IRF9-specific shRNA. Western blots of IRF9 showed that IRF9 expression levels were reduced in cells expressing shRNA-1 and completely absent in cells expressing shRNA-2 and -3 (Fig 7A). To compare steady state levels of IRF9 upon translational inhibition by shRNA to that of post-translational degradation by ORF63, we transduced THF-ISRE cells with AdORF63/AdTA in the presence of decreasing amounts of Dox. In these human cells, optimal ORF63 levels were observed at 0.1 ng/ml Dox or in the absence of Dox without reduction of GAPDH levels whereas only partial induction of ORF63 was observed at 1 ng Dox. A reduction of IRF9 levels consistent with ORF63 expression was observed. Reduced steady state levels of IRF9 were comparable to the partial reduction of IRF9 observed in shRNA-1 expressing cells while IRF9 was completely absent from shRNA-2 and 3 expressing cells (Fig 7B). However, when the THF-ISRE cells expressing the three shRNAs were incubated with uIFN for 4 or 8 hours, ISG54 mRNA expression was only partially reduced in the presence of shRNA-1, whereas shRNA-2 and shRNA-3 largely prevented ISG induction (Fig 7C). Analysis of IFN-induced ISG54-expression by western blotting confirmed these results (Fig 7D). These data demonstrate that removal of IRF9 is sufficient to inhibit ISG-expression and they are consistent with ORF63 affecting IRF9 protein turnover rather than IRF9 transcription or translation.

To further determine whether IRF9 is the primary JAK/STAT-associated target of ORF63, we took advantage of the fact that HEK 293T cells do not respond efficiently to type I IFN unless IRF9 is overexpressed [50]. Co-transfection of a plasmid encoding an ISRE-luciferase reporter with increasing amounts of rhesus IRF9-expressing plasmid resulted in an IRF9-dependent increase of luciferase expression upon treatment with IFN at 24 hours post transfection (Fig 8A; black lined graph). Interestingly, optimal luciferase stimulation was observed with 25–100 ng of the IRF9 plasmid whereas higher IRF9 concentrations resulted in decreased luciferase activity. This decrease might be due to the fact that higher IRF9 levels activate ISRE activity even in the absence of IFN-treatment (Fig 8B), and a prolonged stimulation might induce negative regulators of IFN signaling [51]. Co-transfection of 1 μg ORF63-expressing plasmid resulted in the inhibition of IRF9-induced luciferase expression both in the presence or absence of IFN (Fig 8A and 8B; gray lined graphs) suggesting that ORF63 inhibition cannot be overcome by increasing IRF9 levels. However, ORF63 needs to be in excess of IRF9 for complete inhibition since complete inhibition of IRF9-dependent ISG-induction was only observed when at least 250 ng of ORF63 plasmid was co-transfected with 50ng IRF9-expressing plasmid (Fig 8C). Together with the finding that ORF63 inhibited IRF9-dependent ISRE-transcription even in the absence of IFN-stimulation (Fig 8B) these data further support the conclusion that degradation of IRF9 is the major mechanism by which ORF63 inhibits JAK/STAT signaling.

Inhibition of JAK-STAT signaling by a conditionally ORF63/ORF70-expressing mutant virus
To study the role of ORF63 in the context of virus-induced inhibition of JAK-STAT signaling we constructed a conditionally ORF63/ORF70-expressing mutant using a recombinant bacterial artificial chromosome containing the complete SVV genome (SVV BAC) [39]. Recently, using the SVV BAC an ORF63/ORF70 SVV mutant was constructed by introducing stop codons in both genes. These mutations severely affected replication of the virus in vitro consistent with ORF63 being essential for viral growth [29]. To generate a mutant virus in which expression levels of ORF63 could be conditionally regulated, we fused the destabilizing domain (DD) of dihydrofolate reductase (DHFR) to the C-termini of ORF63 and ORF70 using two-step red-mediated mutagenesis (Fig 9A) [40]. The addition of DD-DHFR to any protein results in rapid proteasomal degradation of the fusion protein unless DHFR is stabilized with trimethoprim (TMP) [52]. We were able to recover DD-DHFR-tagged SVV in the presence of TMP and growth curves confirmed that viral growth was reduced by about 50% during the first 96 hours of infection compared to that of unmodified BAC-derived SVV when grown in 10 μM TMP (Fig 9B). A more severe reduction in viral growth was observed beyond that time point. To study the effect of TMP-removal on ORF63/70-DHFR SVV replication, we infected Vero cells in the presence of 10 μM TMP until viral plaques were detected (Fig 9C, left panel). Removal of TMP and passaging ORF63/70-DHFR SVV-infected Vero cells resulted in very few plaques (Fig 9C, middle panel). However, virus in these cultures could be rescued when 10 μM of TMP was added back to the culture (Fig 9C, right panel). These observations confirmed that the virus expressing DD-DHFR-tagged ORF63/ORF70 was able to replicate in tissue culture in the presence of TMP and viral growth can be regulated by removal of TMP. To determine whether DHFR-fusion to ORF63/ORF70 affected viral inhibition of the JAK-STAT pathway, we infected TRFs with wild type or ORF63/70-DHFR SVV in the presence of 10 μM TMP and studied expression of STAT2 and IRF9 as well as phosphorylation of STAT2 after 20 minutes of stimulation with IFN (Fig 9D). We observed a reduction in STAT2 and IRF9 expression levels as well as a complete inhibition of STAT2 phosphorylation in both wild type - and ORF63/70-DHFR-infected cells, indicating that the evasion of JAK-STAT signaling was not affected by the presence of the DHFR domains (Fig 9D). Since the removal of TMP from the culture media triggers degradation of ORF63-DHFR and ORF70-DHFR, we infected TRF with ORF63/70-DHFR SVV and cultured the cells in varying concentrations of TMP. We observed a dose-dependent decrease in ORF63 expression and ORF63 was no longer detectable when the virus was grown in 0.02 μM or less TMP (Fig 9E). This reduction in ORF63 levels led to increased levels of IRF9, indicating that expression of these proteins was inversely correlated (Fig 9E). However, the expression of ORF31 (glycoprotein B) was also affected by reducing TMP concentrations, similar to ORF63 expression (Fig 9E, western blot and graph). This result revealed that ORF63/70 is required for ORF31 expression and most likely for the expression of other SVV genes as well [24]. Restoration of IFN-sensitivity could therefore not be unequivocally assigned to the absence of ORF63 since SVV ORF31 was absent as well.

The JAK-STAT evasion strategies employed by SVV are conserved for VZV
In the experiments described above, we established that SVV inhibits JAK-STAT signaling by interfering with IFN-induced phosphorylation of STAT2 and by modulating the degradation of STAT2 and IRF9. Previous reports have shown that VZV interferes with IFN-mediated signaling [12, 17], however, the evasion mechanisms involved in this inhibition are largely unknown. To determine if VZV inhibits JAK-STAT signaling similar to SVV, we infected human fibroblasts, MRC-5 cells, with VZV.eGFP for 48 hours and activated JAK-STAT signaling by incubating the cells with uIFN for 20 minutes prior to harvesting the cells. Similar to SVV, expression levels of IRF9 and STAT2 were reduced in VZV-infected cells (Fig 10A, upper panel). Moreover, complete inhibition of IFN-induced STAT2 phosphorylation was observed, indicating that VZV employs mechanisms of JAK-STAT evasion that are comparable to those of SVV (Fig 10A, upper panel). However, unlike SVV (Fig 5A), IFN-induced STAT1 phosphorylation is blocked by VZV (Fig 10A, lower panel). To determine whether VZV ORF63 prevents ISG-induction, we co-transfected HEK 293T cells with the ISRE-luciferase reporter plasmid and plasmids encoding VZV ORF63 or, as a control, FLAG-tagged SVV ORF32. The cells were stimulated with uIFN for 6 hours at 42 hours post transfection, after which ISRE-luciferase expression was measured. In contrast to untransfected controls or SVV ORF32, expression of VZV ORF63 caused a reduction in IFN-induced luciferase expression (Fig 10B). Expression of the viral proteins was confirmed by western blot (Fig 10C). To assess if VZV ORF63 plays a role in the reduction of IRF9 expression observed in VZV-infected cells (Fig 10A), we expressed the viral protein using the tetracycline-inducible adenovirus system described in Fig 4. MRC5 cells were co-transduced with VZV AdORF63 and AdTA, incubated with the indicated concentrations of Dox and after 48 hours steady state levels of IRF9 were analyzed using western blot. We observed a prominent decrease in IRF9 expression levels in cells that expressed VZV ORF63 (Fig 10D, left panel). To determine if the reduction in STAT2 expression levels and phosphorylation observed in VZV-infected cells was due to VZV ORF63, we stimulated the transduced MRC5 with uIFN for 20 minutes to activate the pathway and studied STAT2 by western blot. STAT2 steady state levels and phosphorylation were unaffected by the presence of VZV ORF63 (Fig 10D, left panel). The same experiment was performed in TRFs and we observed a VZV ORF63-induced reduction of IRF9, but not of STAT2, in those cells as well (Fig 10D, right panel). Taken together these data show that SVV and VZV employ similar mechanisms to interfere with JAK-STAT signaling and that the ORF63 proteins of both viruses contribute to this inhibition by mediating the degradation of IRF9. VZV ORF63 induced IRF9 degradation in both MRC5 and TRFs (Fig 10D), indicating that the pathway’s inhibitory target is conserved between human and rhesus cells.

Discussion
The data presented here show that both SVV and VZV inhibit IFN-mediated ISG induction and reduce the expression of IRF9. In addition, we observed a marginal decrease in STAT2 levels and a complete inhibition of IFN-dependent phosphorylation of STAT2 in both SVV - and VZV-infected cells. Since IRF9 is essential for JAK-STAT signaling, the degradation of IRF9 by ORF63 is thus part of a multi-pronged inhibition of IFN-mediated antiviral gene induction. These observations are consistent with previous reports showing that VZV prevents induction of Mx-1 in brain fibroblasts and reduces STAT2 levels in infected brain fibroblasts [53]. We further demonstrate that SVV ORF63 promotes the degradation of IRF9 in a proteasome-dependent manner, but does not affect STAT2 protein levels or its phosphorylation. SVV ORF63 and VZV ORF63 share 52% amino acid identity and there is evidence that these proteins are expressed in latently infected ganglia [5, 21, 22, 28, 45, 54–56]. We observed that inhibition of IFN-signaling by both proteins correlated with degradation of IRF9 whereas neither protein affected STAT2 expression or phosphorylation. Together with the previous report by Ambagala et al. demonstrating IFN-sensitivity of ORF63-deficient VZV [17] our data thus strongly suggest that ORF63 plays a central role in IFN-signaling inhibition by both SVV and VZV. In addition, our observations reveal that both viruses likely encode additional ORFs responsible for the inhibition of STAT2 phosphorylation and the reduction of STAT2 expression.
The reduction in IRF9 levels observed in SVV/VZV-infected cells and ORF63-expressing cells did not result from reduced transcription (Fig 5F and S2A Fig). Rather, ORF63-mediated the proteasomal degradation of IRF9 independently of IFN-signaling. Although this is the first description of IFN-evasion by IRF9 targeting for a herpesvirus, IRF9 (p48) targeting has been demonstrated for several other viruses. The non-structural protein (NSP) 1 of the simian rotavirus strain SA11-4F was shown to induce proteasomal degradation of all IRF proteins that contain an IRF association domain (IAD), which include IRF3, IRF5, IRF7 and IRF9 [57]. In contrast, our data suggest that ORF63 targets IRF9, but not IRF3 which in VZV is degraded by ORF61 [15]. Adenovirus E1A protein blocks IFN-induced protein expression by reducing IRF9 expression levels, yet the mechanism of this immune evasion is unknown [46, 58]. Incidentally, the presence of E1A in HEK293 cells [59, 60] could be responsible for the requirement of exogenous IRF9 for IFN-dependent ISG-induction. In our co-transfection experiments ORF63 thus seemed to be able to eliminate IRF9 once the E1A-dependent IRF9-inhibition was breached. Additionally, the reovirus type 1 Lang μ2 protein causes nuclear accumulation of IRF9, independent of IFN-stimulation, which results in severely impaired JAK-STAT signaling [61]. Finally, the human papillomavirus (HPV) 16 E7 oncoprotein was shown to interact with IRF9 thereby preventing ISGF3 formation [62]. We did not observe an interaction between IRF9 and ORF63 in immunoprecipitations studies performed in SVV-infected and ORF63-expressing TRFs. Therefore, the exact mechanism by which ORF63 elicits the degradation of IRF9 still needs to be elucidated.
IRF9 is a key player in the JAK-STAT signaling pathway activated by type I IFN: unphosphorylated STAT2 is complexed with IRF9 and the pair continuously shuttles between the cytoplasm and the nucleus, which is driven by the nuclear localization signal of IRF9 and the nuclear export signal (NES) of STAT2 [63]. Upon activation of JAK1 and TYK2, phosphorylated STAT1 and STAT2 dimerize, which results in the loss of the NES of STAT2 [63]. The requirement of IRF9 for anti-viral immune responses was demonstrated by Kimura et al. using IRF9 knock out murine cells. Replication of herpes simplex virus type 1 and vesicular stomatitis virus (a rhabdovirus) was greatly enhanced in IFNα-treated cultures of infected IRF9-/- cells, while the cytokine limited viral replication in wild type cells [64]. In addition, Maiwald et al. created a mathematical model based on experimental data that shows that IRF9 determines the peak time and intensity of type I IFN-induced responses [65]. Our data using IRF9-specific shRNA are consistent with these studies and demonstrate the requirement of IRF9 for efficient ISG induction in human cells. Whereas shRNA-expressing cells seemed to express lower levels of IRF9 than ORF63-expressing cells it needs to be considered that ORF63 needs protein expression to act on IRF9 whereas shRNAs prevent protein expression itself. Thus, we concluded that reduced IRF9 levels are responsible for inhibition of IFN-induced ISG expression in both shRNA and ORF63-expressing cells. We therefore propose that that ORF63 blocks the JAK-STAT pathway by reducing IRF9 levels. This conclusion is further supported by the ability of ORF63 to counteract IRF9-mediated ISG-induction in HEK 293T cells both in the presence and absence of IFN (Fig 8).
Since IRF9-degradation was also observed in VZV-infected and VZV ORF63-transduced cells (Fig 10) it seems highly likely that the restoration of IRF9-levels was responsible for the previously described hyper-sensitivity of ORF63-deficient VZV [17]. The VZV deletion mutant used in this study contained a truncation of ORF63 in which only the first 24 amino acids of ORF63 was expressed. This mutant did not replicate in the presence of IFNα, but was able to replicate, albeit at reduced levels, in some cell types such as the osteosarcoma cell line U2OS [17]. To determine whether IRF9-depletion would restore the ability of this VZV ORF63 deletion virus to replicate in fibroblasts we infected THF-ISRE and THF-ISRE shRNA-3 cells with the VZV-deletion mutant (kindly provided by Jeff Cohen). However, we did no observe an increased growth of this virus upon IRF9-depletion. This indicates that additional functions of ORF63, either directly affecting host pathways or indirectly via other viral proteins, contribute to the reduced growth of the deletion virus.
Introducing stop codons in ORF63 and ORF70 severely impaired the growth of SVV in IFN-deficient Vero cells [29]. Similarly, when we fused a DHFR domain to the C-terminus of both ORF63 and ORF70 to create an inducible knock out for both proteins, removal of TMP resulted not only in the degradation of ORF63 and ORF70 but also prevented expression of other SVV genes that depend on ORF63 function. Because of the requirement of ORF63 for viral replication we were unable to directly address the biological significance of IRF9-degradation for evasion of JAK-STAT signaling by these varicelloviruses. Cohen et al. found that cells infected with the VZV ORF63 deletion virus were highly susceptible to IFN treatment [17], suggesting that ORF63-induced degradation of IRF9 plays a prominent role in evasion of this pathway. However, since ORF63 is required for viral early gene expression and viral replication [23–25], deletion of protein could also affect expression of the as yet unknown inhibitor of STAT2-phorphorylation as well. Thus, ORF63 likely impacts IFN-resistance both directly, by reducing IRF9 expression, and indirectly by regulating the expression of other IFN-inhibitory genes. Our data are thus consistent with a multipronged inhibition of JAK/STAT signaling by SVV and VZV to ensure efficient evasion of this innate immune pathway. It is not uncommon for a virus to target a signaling pathway at multiple levels. For example, VZV codes for at least three independent strategies devoted to inhibiting IRF3-driven expression of IFNs, which including ORF61 [15], ORF47 [14] and IE62 [13]. In addition, it is conceivable that IRF9-inhibition by the immediate early gene ORF63 precedes STAT2 inhibition due to sequential expression of the respective inhibitory proteins during viral infection. The relative contribution of each inhibitory pathway during viral infection thus still needs to be elucidated.
We observed that both SVV and VZV reduce levels and phosphorylation of STAT2, whereas STAT1 levels were not affected. In contrast, reduced phosphorylation of STAT1 was only observed in VZV-infected cells. The diminished STAT2 levels did not result from reduced transcription (S2B Fig). Previous reports demonstrated diminished levels of STAT1 in VZV-infected human fibroblasts treated with IFNγ [66] and reduced STAT1 phosphorylation in VZV-infected skin xenografts in the SCIDhu model [12]. A recent report confirmed VZV-mediated downregulation of STAT2, but downregulation of STAT1 was inconsistent between the experiments and inhibition of STAT1 phosphorylation was not observed [53]. Since STAT1 is shared between type I and type II IFN signal transduction pathways, it represents an attractive target for viral innate immune evasion. However, observations concerning STAT1 expression and phosphorylation in VZV-infected cells are inconsistent (our data, [12, 53, 66]), possibly due to the use of type II IFN and different cell lines. Therefore, we cannot rule out that SVV targets the phosphorylation of STAT1 in cell types other than fibroblasts. Since STAT1 phosphorylation was not reduced in SVV-infected TRFs it is unlikely that SVV interferes upstream of STAT1/STAT2 since the binding of IFN to its receptor triggers the activation of the tyrosine kinases JAK1 and TYK2 which in turn phosphorylate STAT1 and STAT2 resulting in the formation of the ISGF3 complex [67, 68]. If SVV would target either JAK1 or TYK2 one would expect a reduction in STAT1 phosphorylation. For VZV however, this possibility cannot be ruled out since STAT1 phosphorylation is inhibited. Although SVV and VZV-infected cells displayed lower expression levels of STAT2 the remaining STAT2 was not phosphorylated. Therefore, SVV and VZV most likely affect STAT2 phosphorylation directly.
Primary VZV infection starts with respiratory mucosal inoculation [69]. Recently, type III IFNs (IFNλ1–3) have been implicated in playing an important role in limiting (herpes)viral replication in mucosal tissues [70–72]. These cytokines bind to the IL28Rα/IL10Rβ receptor complex [73, 74], which is predominantly expressed on epithelial cells [75]. Engagement of the receptor results in the activation of the JAK-STAT pathway and induction ISG expression [74]. The Kaposi’s sarcoma-associated herpesvirus protein vIRF2 was shown to inhibit both IFNα and IFNλ-mediated ISG expression, by reducing the levels of STAT1 and IRF9 [76, 77]. Similarly, the inhibition of JAK-STAT signaling by VZV is likely to block IFNλ-mediated signaling in epithelial cells, thereby immediately limiting the host anti-viral responses to allow further spreading.
In addition to inhibiting IFN-dependent signal transduction, VZV inhibits activation of the IFN-gene itself and IFN-independent ISG-induction. The VZV proteins IE62, ORF47, and ORF61 all target IRF3-mediated induction of IFNβ and ISG genes, such as ISG15, ISG54 and IS56 [14, 15, 22]. VZV ORF61 also blocks the TNFα-mediated activation of NFκB by inhibiting IκBα [16]. Taken together with our observations, it appears that multiple VZV and SVV ORFs are devoted to interfering at sequential steps along the induction of IFN and anti-viral ISG.
Despite efficiently counteracting IFN activation and IFN-dependent signaling, several reports have indicated the importance of type I IFNs in limiting VZV replication and spread in vivo. Children that were treated for leukemia and were suffering from varicella showed significantly reduced dissemination of the virus in response to the administration of intra-muscular IFNα [78]. In addition, experiments in severe combined immunodeficiency mice engrafted with VZV-infected human skin (SCIDhu mice) showed that preventing IFN signaling with antibodies specific for the interferon receptor resulted in larger cutaneous lesions compared to mice that were untreated [12]. These in vivo responses are most likely explained by the fact that the establishment of an anti-viral state prior to infection is more difficult to overcome than IFN-responses in infected cells. IFNα-treatment of melanoma cells prior to VZV infection led to a reduction in plaque formation [17]. Thus, IFN likely prevents viral spread during the acute phase of infection by inducing an antiviral state in target cells. However, further sensitizing VZV to IFN by therapeutically blocking viral IFN-evasion mechanisms could improve the ability of IFN to prevent spread and/or reactivation of VZV in vivo. This concept could be experimentally tested in non-human primates using the SVV model. Since ORF63 expression have been demonstrated in latently-infected ganglia of SVV-infected rhesus macaques [5], it is also conceivable that IFN evasion is essential for maintaining viral latency. Dissecting the role of ORF63 in limiting IFN signaling could thus lead to a better understanding of the role of IFN in viral latency and reactivation.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. Gilden D, Cohrs RJ, Mahalingam R, Nagel MA. Varicella zoster virus vasculopathies: diverse clinical manifestations, laboratory features, pathogenesis, and treatment. Lancet Neurol. 2009; 8 : 731–740. doi: 10.1016/S1474-4422(09)70134-6 19608099
2. Matsunaga Y, Yamanishi K, Takahashi M. Experimental infection and immune response of guinea pigs with varicella-zoster virus. Infect Immun. 1982; 37 : 407–412. 6288560
3. Wroblewska Z, Devlin M, Reilly K, van Trieste H, Wellish M, Gilden DH. The production of varicella Zoster virus antiserum in laboratory animals. Brief report. Arch Virol. 1982; 74 : 233–238. 6299237
4. Gray WL. Simian varicella: a model for human varicella-zoster virus infections. Rev Med Virol. 2004; 14 : 363–381. 15386593
5. Messaoudi I, Barron A, Wellish M, Engelmann F, Legasse A, Planer S, et al. Simian varicella virus infection of rhesus macaques recapitulates essential features of varicella zoster virus infection in humans. PLoS Pathog. 2009; 5: e1000657. doi: 10.1371/journal.ppat.1000657 19911054
6. Wang BX, Fish EN. The yin and yang of viruses and interferons. Trends Immunol. 2012; 33 : 190–197. doi: 10.1016/j.it.2012.01.004 22321608
7. Levy DE, Garcia-Sastre A. The virus battles: IFN induction of the antiviral state and mechanisms of viral evasion. Cytokine Growth Factor Rev. 2001; 12 : 143–156. 11325598
8. Wilkins C, Gale M Jr. Recognition of viruses by cytoplasmic sensors. Curr Opin Immunol. 2010; 22 : 41–47. doi: 10.1016/j.coi.2009.12.003 20061127
9. Seya T, Kasamatsu J, Azuma M, Shime H, Matsumoto M. Natural killer cell activation secondary to innate pattern sensing. J Innate Immun. 2011; 3 : 264–273. doi: 10.1159/000326891 21454965
10. Le Bon A, Tough DF. Links between innate and adaptive immunity via type I interferon. Curr Opin Immunol. 2002; 14 : 432–436. 12088676
11. Tovey MG, Lallemand C, Thyphronitis G. Adjuvant activity of type I interferons. Biol Chem. 2008; 389 : 541–545. 18953720
12. Ku CC, Zerboni L, Ito H, Graham BS, Wallace M, Arvin AM. Varicella-zoster virus transfer to skin by T Cells and modulation of viral replication by epidermal cell interferon-alpha. J Exp Med. 2004; 200 : 917–925. 15452178
13. Sen N, Sommer M, Che X, White K, Ruyechan WT, Arvin AM. Varicella-zoster virus immediate-early protein 62 blocks interferon regulatory factor 3 (IRF3) phosphorylation at key serine residues: a novel mechanism of IRF3 inhibition among herpesviruses. J Virol. 2010; 84 : 9240–9253. doi: 10.1128/JVI.01147-10 20631144
14. Vandevenne P, Lebrun M, El Mjiyad N, Ote I, Di Valentin E, Habraken Y, et al. The varicella-zoster virus ORF47 kinase interferes with host innate immune response by inhibiting the activation of IRF3. PLoS One. 2011; 6: e16870. doi: 10.1371/journal.pone.0016870 21347389
15. Zhu H, Zheng C, Xing J, Wang S, Li S, Lin R, et al. Varicella-zoster virus immediate-early protein ORF61 abrogates the IRF3-mediated innate immune response through degradation of activated IRF3. J Virol. 2011; 85 : 11079–11089. doi: 10.1128/JVI.05098-11 21835786
16. Sloan E, Henriquez R, Kinchington PR, Slobedman B, Abendroth A. Varicella-zoster virus inhibition of the NF-kappaB pathway during infection of human dendritic cells: role for open reading frame 61 as a modulator of NF-kappaB activity. J Virol. 2012; 86 : 1193–1202. doi: 10.1128/JVI.06400-11 22090112
17. Ambagala AP, Cohen JI. Varicella-Zoster virus IE63, a major viral latency protein, is required to inhibit the alpha interferon-induced antiviral response. J Virol. 2007; 81 : 7844–7851. 17507475
18. Shuai K, Liu B. Regulation of JAK-STAT signalling in the immune system. Nat Rev Immunol. 2003; 3 : 900–911. 14668806
19. Davison AJ, Scott JE. The complete DNA sequence of varicella-zoster virus. J Gen Virol. 1986; 67 (Pt 9): 1759–1816. 3018124
20. Mueller NH, Walters MS, Marcus RA, Graf LL, Prenni J, Gilden D, et al. Identification of phosphorylated residues on varicella-zoster virus immediate-early protein ORF63. J Gen Virol. 2010; 91 : 1133–1137. doi: 10.1099/vir.0.019067-0 20089801
21. Mahalingam R, Wellish M, Cohrs R, Debrus S, Piette J, Rentier B, et al. Expression of protein encoded by varicella-zoster virus open reading frame 63 in latently infected human ganglionic neurons. Proc Natl Acad Sci U S A. 1996; 93 : 2122–2124. 8700895
22. Zerboni L, Sobel RA, Ramachandran V, Rajamani J, Ruyechan W, Abendroth A, et al. Expression of varicella-zoster virus immediate-early regulatory protein IE63 in neurons of latently infected human sensory ganglia. J Virol. 2010; 84 : 3421–3430. doi: 10.1128/JVI.02416-09 20106930
23. Jackers P, Defechereux P, Baudoux L, Lambert C, Massaer M, Merville-Louis MP, et al. Characterization of regulatory functions of the varicella-zoster virus gene 63-encoded protein. J Virol. 1992; 66 : 3899–3903. 1316489
24. Kost RG, Kupinsky H, Straus SE. Varicella-zoster virus gene 63: transcript mapping and regulatory activity. Virology. 1995; 209 : 218–224. 7747473
25. Bontems S, Di Valentin E, Baudoux L, Rentier B, Sadzot-Delvaux C, Piette J. Phosphorylation of varicella-zoster virus IE63 protein by casein kinases influences its cellular localization and gene regulation activity. J Biol Chem. 2002; 277 : 21050–21060. 11912195
26. Desloges N, Rahaus M, Wolff MH. The varicella-zoster virus-mediated delayed host shutoff: open reading frame 17 has no major function, whereas immediate-early 63 protein represses heterologous gene expression. Microbes Infect. 2005; 7 : 1519–1529. 16039898
27. Hood C, Cunningham AL, Slobedman B, Arvin AM, Sommer MH, Kinchington PR, et al. Varicella-zoster virus ORF63 inhibits apoptosis of primary human neurons. J Virol. 2006; 80 : 1025–1031. 16379003
28. Gray WL, Starnes B, White MW, Mahalingam R. The DNA sequence of the simian varicella virus genome. Virology. 2001; 284 : 123–130. 11352673
29. Brazeau E, Wellish M, Kaufer BB, Tischer BK, Gray W, Zhou F, et al. Simian varicella virus open reading frame 63/70 expression is required for efficient virus replication in culture. J Neurovirol. 2011; 17 : 274–280. doi: 10.1007/s13365-011-0025-6 21479719
30. Robinson BA, Estep RD, Messaoudi I, Rogers KS, Wong SW. Viral interferon regulatory factors decrease the induction of type I and type II interferon during rhesus macaque rhadinovirus infection. J Virol. 2012; 86 : 2197–2211. doi: 10.1128/JVI.05047-11 22156526
31. Gierasch WW, Zimmerman DL, Ward SL, Vanheyningen TK, Romine JD, Leib DA. Construction and characterization of bacterial artificial chromosomes containing HSV-1 strains 17 and KOS. J Virol Methods. 2006; 135 : 197–206. 16647145
32. DeFilippis VR, Sali T, Alvarado D, White L, Bresnahan W, Fruh KJ. Activation of the interferon response by human cytomegalovirus occurs via cytoplasmic double-stranded DNA but not glycoprotein B. J Virol. 2010; 84 : 8913–8925. doi: 10.1128/JVI.00169-10 20573816
33. Mahalingam R, Wellish M, White T, Soike K, Cohrs R, Kleinschmidt-DeMasters BK, et al. Infectious simian varicella virus expressing the green fluorescent protein. J Neurovirol. 1998; 4 : 438–444. 9718136
34. Eisfeld AJ, Yee MB, Erazo A, Abendroth A, Kinchington PR. Downregulation of class I major histocompatibility complex surface expression by varicella-zoster virus involves open reading frame 66 protein kinase-dependent and -independent mechanisms. J Virol. 2007; 81 : 9034–9049. 17567702
35. Lenac Rovis T, Bailer SM, Pothineni VR, Ouwendijk WJ, Simic H, Babic M, et al. Comprehensive analysis of varicella-zoster virus proteins using a new monoclonal antibody collection. J Virol. 2013; 87 : 6943–6954. doi: 10.1128/JVI.00407-13 23596286
36. Hardy S, Kitamura M, Harris-Stansil T, Dai Y, Phipps ML. Construction of adenovirus vectors through Cre-lox recombination. J Virol. 1997; 71 : 1842–1849. 9032314
37. Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci U S A. 1992; 89 : 5547–5551. 1319065
38. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25 : 402–408. 11846609
39. Gray WL, Zhou F, Noffke J, Tischer BK. Cloning the simian varicella virus genome in E. coli as an infectious bacterial artificial chromosome. Arch Virol. 2011; 156 : 739–746. doi: 10.1007/s00705-010-0889-4 21487663
40. Tischer BK, von Einem J, Kaufer B, Osterrieder N. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques. 2006; 40 : 191–197. 16526409
41. Gray WL, Mahalingam R. A cosmid-based system for inserting mutations and foreign genes into the simian varicella virus genome. J Virol Methods. 2005; 130 : 89–94. 16043236
42. Reich NC, Liu L. Tracking STAT nuclear traffic. Nat Rev Immunol. 2006; 6 : 602–612. 16868551
43. Erazo A, Kinchington PR. Varicella-zoster virus open reading frame 66 protein kinase and its relationship to alphaherpesvirus US3 kinases. Curr Top Microbiol Immunol. 2010; 342 : 79–98. doi: 10.1007/82_2009_7 20186610
44. Kinchington PR, Fite K, Seman A, Turse SE. Virion association of IE62, the varicella-zoster virus (VZV) major transcriptional regulatory protein, requires expression of the VZV open reading frame 66 protein kinase. J Virol. 2001; 75 : 9106–9113. 11533174
45. Mahalingam R, Gilden DH, Wellish M, Pugazhenthi S. Transactivation of the simian varicella virus (SVV) open reading frame (ORF) 21 promoter by SVV ORF 62 is upregulated in neuronal cells but downregulated in non-neuronal cells by SVV ORF 63 protein. Virology. 2006; 345 : 244–250. 16242745
46. Leonard GT, Sen GC. Effects of adenovirus E1A protein on interferon-signaling. Virology. 1996; 224 : 25–33. 8862396
47. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014; 14 : 36–49. doi: 10.1038/nri3581 24362405
48. Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, et al. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity. 2000; 13 : 539–548. 11070172
49. Yanai H, Chen HM, Inuzuka T, Kondo S, Mak TW, Takaoka A, et al. Role of IFN regulatory factor 5 transcription factor in antiviral immunity and tumor suppression. Proc Natl Acad Sci U S A. 2007; 104 : 3402–3407. 17360658
50. Reid SP, Leung LW, Hartman AL, Martinez O, Shaw ML, Carbonnelle C, et al. Ebola virus VP24 binds karyopherin alpha1 and blocks STAT1 nuclear accumulation. J Virol. 2006; 80 : 5156–5167. 16698996
51. Porritt RA, Hertzog PJ. Dynamic control of type I IFN signalling by an integrated network of negative regulators. Trends Immunol. 2015; 36 : 150–160. doi: 10.1016/j.it.2015.02.002 25725583
52. Iwamoto M, Bjorklund T, Lundberg C, Kirik D, Wandless TJ. A general chemical method to regulate protein stability in the mammalian central nervous system. Chem Biol. 2010; 17 : 981–988. doi: 10.1016/j.chembiol.2010.07.009 20851347
53. Nagel MA, James SF, Traktinskiy I, Wyborny A, Choe A, Rempel A, et al. Inhibition of pSTAT1 Nuclear Translocation and Antiviral Protein Expression in Human Brain Vascular Adventitial Fibroblasts Infected with Varicella Zoster Virus. J Virol. 2014.
54. Grinfeld E, Kennedy PG. Translation of varicella-zoster virus genes during human ganglionic latency. Virus Genes. 2004; 29 : 317–319. 15550771
55. Kennedy PG, Grinfeld E, Bell JE. Varicella-zoster virus gene expression in latently infected and explanted human ganglia. J Virol. 2000; 74 : 11893–11898. 11090189
56. Lungu O, Panagiotidis CA, Annunziato PW, Gershon AA, Silverstein SJ. Aberrant intracellular localization of Varicella-Zoster virus regulatory proteins during latency. Proc Natl Acad Sci U S A. 1998; 95 : 7080–7085. 9618542
57. Arnold MM, Barro M, Patton JT. Rotavirus NSP1 mediates degradation of interferon regulatory factors through targeting of the dimerization domain. J Virol. 2013; 87 : 9813–9821. doi: 10.1128/JVI.01146-13 23824805
58. Leonard GT, Sen GC. Restoration of interferon responses of adenovirus E1A-expressing HT1080 cell lines by overexpression of p48 protein. J Virol. 1997; 71 : 5095–5101. 9188575
59. Graham FL, Smiley J, Russell WC, Nairn R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J Gen Virol. 1977; 36 : 59–74. 886304
60. Haeussler DJ, Evangelista AM, Burgoyne JR, Cohen RA, Bachschmid MM, Pimental DR. Checkpoints in adenoviral production: cross-contamination and E1A. PLoS One. 2011; 6: e23160. doi: 10.1371/journal.pone.0023160 21826235
61. Zurney J, Kobayashi T, Holm GH, Dermody TS, Sherry B. Reovirus mu2 protein inhibits interferon signaling through a novel mechanism involving nuclear accumulation of interferon regulatory factor 9. J Virol. 2009; 83 : 2178–2187. doi: 10.1128/JVI.01787-08 19109390
62. Barnard P, McMillan NA. The human papillomavirus E7 oncoprotein abrogates signaling mediated by interferon-alpha. Virology. 1999; 259 : 305–313. 10388655
63. Banninger G, Reich NC. STAT2 nuclear trafficking. J Biol Chem. 2004; 279 : 39199–39206. 15175343
64. Kimura T, Kadokawa Y, Harada H, Matsumoto M, Sato M, Kashiwazaki Y, et al. Essential and non-redundant roles of p48 (ISGF3 gamma) and IRF-1 in both type I and type II interferon responses, as revealed by gene targeting studies. Genes Cells. 1996; 1 : 115–124. 9078371
65. Maiwald T, Schneider A, Busch H, Sahle S, Gretz N, Weiss TS, et al. Combining theoretical analysis and experimental data generation reveals IRF9 as a crucial factor for accelerating interferon alpha-induced early antiviral signalling. FEBS J. 2010; 277 : 4741–4754. doi: 10.1111/j.1742-4658.2010.07880.x 20964804
66. Abendroth A, Slobedman B, Lee E, Mellins E, Wallace M, Arvin AM. Modulation of major histocompatibility class II protein expression by varicella-zoster virus. J Virol. 2000; 74 : 1900–1907. 10644363
67. Gauzzi MC, Velazquez L, McKendry R, Mogensen KE, Fellous M, Pellegrini S. Interferon-alpha-dependent activation of Tyk2 requires phosphorylation of positive regulatory tyrosines by another kinase. J Biol Chem. 1996; 271 : 20494–20500. 8702790
68. Muller M, Briscoe J, Laxton C, Guschin D, Ziemiecki A, Silvennoinen O, et al. The protein tyrosine kinase JAK1 complements defects in interferon-alpha/beta and -gamma signal transduction. Nature. 1993; 366 : 129–135. 8232552
69. Zerboni L, Sen N, Oliver SL, Arvin AM. Molecular mechanisms of varicella zoster virus pathogenesis. Nat Rev Microbiol. 2014; 12 : 197–210. doi: 10.1038/nrmicro3215 24509782
70. Mordstein M, Neugebauer E, Ditt V, Jessen B, Rieger T, Falcone V, et al. Lambda interferon renders epithelial cells of the respiratory and gastrointestinal tracts resistant to viral infections. J Virol. 2010; 84 : 5670–5677. doi: 10.1128/JVI.00272-10 20335250
71. Pott J, Mahlakoiv T, Mordstein M, Duerr CU, Michiels T, Stockinger S, et al. IFN-lambda determines the intestinal epithelial antiviral host defense. Proc Natl Acad Sci U S A. 2011; 108 : 7944–7949. doi: 10.1073/pnas.1100552108 21518880
72. Ank N, West H, Bartholdy C, Eriksson K, Thomsen AR, Paludan SR. Lambda interferon (IFN-lambda), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo. J Virol. 2006; 80 : 4501–4509. 16611910
73. Sheppard P, Kindsvogel W, Xu W, Henderson K, Schlutsmeyer S, Whitmore TE, et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol. 2003; 4 : 63–68. 12469119
74. Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, et al. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol. 2003; 4 : 69–77. 12483210
75. Sommereyns C, Paul S, Staeheli P, Michiels T. IFN-lambda (IFN-lambda) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. 2008; 4: e1000017. doi: 10.1371/journal.ppat.1000017 18369468
76. Fuld S, Cunningham C, Klucher K, Davison AJ, Blackbourn DJ. Inhibition of interferon signaling by the Kaposi's sarcoma-associated herpesvirus full-length viral interferon regulatory factor 2 protein. J Virol. 2006; 80 : 3092–3097. 16501120
77. Mutocheluh M, Hindle L, Areste C, Chanas SA, Butler LM, Lowry K, et al. Kaposi's sarcoma-associated herpesvirus viral interferon regulatory factor-2 inhibits type 1 interferon signalling by targeting interferon-stimulated gene factor-3. J Gen Virol. 2011; 92 : 2394–2398. doi: 10.1099/vir.0.034322-0 21697347
78. Arvin AM, Kushner JH, Feldman S, Baehner RL, Hammond D, Merigan TC. Human leukocyte interferon for the treatment of varicella in children with cancer. N Engl J Med. 1982; 306 : 761–765. 6174864
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 5
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Human Cytomegalovirus miR-UL112-3p Targets TLR2 and Modulates the TLR2/IRAK1/NFκB Signaling Pathway
- Paradoxical Immune Responses in Non-HIV Cryptococcal Meningitis
- Expression in the Fat Body Is Required in the Defense Against Parasitic Wasps in
- Survives with a Minimal Peptidoglycan Synthesis Machine but Sacrifices Virulence and Antibiotic Resistance
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy