
ExoT Induces Atypical Anoikis Apoptosis in Target Host Cells by Transforming Crk Adaptor Protein into a Cytotoxin
We have previously demonstrated that ExoT is both necessary and sufficient to induce potent apoptosis in host epithelial cells in a manner that depends primarily on its ADP-ribosyltransferase (ADPRT) domain activity. However, the molecular basis underlying ExoT/ADPRT-induced apoptosis remains unknown. In this study, we demonstrate that ExoT/ADPRT by targeting the adaptor protein Crk, transforms this innocuous cellular protein into a cytotoxin that induces atypical anoikis by disrupting the focal adhesion sites which in turn interferes with the integrin-mediated pro-survival signaling.
Published in the journal:
. PLoS Pathog 11(5): e32767. doi:10.1371/journal.ppat.1004934
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004934
Summary
We have previously demonstrated that ExoT is both necessary and sufficient to induce potent apoptosis in host epithelial cells in a manner that depends primarily on its ADP-ribosyltransferase (ADPRT) domain activity. However, the molecular basis underlying ExoT/ADPRT-induced apoptosis remains unknown. In this study, we demonstrate that ExoT/ADPRT by targeting the adaptor protein Crk, transforms this innocuous cellular protein into a cytotoxin that induces atypical anoikis by disrupting the focal adhesion sites which in turn interferes with the integrin-mediated pro-survival signaling.
Introduction
Pseudomonas aeruginosa is a Gram-negative opportunistic pathogen that targets immunocompromised individuals and those with injured epithelia, making it one of the leading causes of nosocomial infections and the leading cause of morbidity and mortality in cystic fibrosis patients [1–3]. P. aeruginosa boasts a large arsenal of cell surface-associated and secreted virulence factors [4]. Prominent amongst them is the Type III Secretion System (T3SS) which contributes to the virulence of a large number of Gram-negative pathogens [5,6]. This conduit allows P. aeruginosa to directly translocate a set of peptide virulence factors, termed effector proteins, into the eukaryotic host cell, where they subvert host signal transduction pathways to advance P. aeruginosa infection [7]. To date, four T3SS effectors have been identified in P. aeruginosa: ExoU, ExoT, ExoS, and ExoY. ExoU is a potent phospholipase that induces necrotic cytotoxicity in eukaryotic cells [8,9]. ExoS and ExoT are homologous bifunctional proteins with an N-terminal GTPase activating protein (GAP) domain and a C-terminal ADP-ribosyltransferase (ADPRT) domain [10,11]. The GAP domains of ExoT and ExoS inhibit RhoA, Rac1, and Cdc42, small GTPases [12–15], while the ADPRT domains of ExoS and ExoT modify non-overlapping host targets. The ExoS ADPRT domain targets many host proteins including Ras, Ral, Rab, and Rac, and the ADPRT activity of ExoT modifies CrkI/II (the two isoforms of Crk) adaptor proteins and the glycolytic enzyme PGK1 [16–21]. Finally, ExoY is an adenylate cyclase that functions as an edema factor [22,23].
Unlike exoS, exoU, and exoY which are encoded in subsets of clinical isolates, exoT is present in almost all P. aeruginosa virulent clinical strains studied thus far [24,25], suggesting a more fundamental role for this virulence factor in P. aeruginosa pathogenesis. Indeed, P. aeruginosa strains defective in ExoT exhibit reduced virulence and are impaired in dissemination in mice [11,18,26]. Moreover, Balachandran et al. recently demonstrated an elegant host defense mechanism involving ubiquitin ligase Cbl-b that specifically targets ExoT, but not ExoS or ExoU, for proteasomal degradation [26]. This finding further highlights the importance of ExoT in P. aeruginosa pathogenesis and host responses to this pathogen. We and others have demonstrated that ExoT alters actin cytoskeleton, causes cell rounding, inhibits cell migration, functions as an anti-internalization factor, blocks cell division by targeting cytokinesis at multiple steps, and inhibits wound healing [12,13,18,27]. More recently, we demonstrated that ExoT is both necessary and sufficient to induce apoptosis in HeLa cells in a manner that is primarily dependent on its ADPRT domain activity [28]. However, the mechanism underlying the ExoT-induced apoptosis in epithelial cells remains unknown.
In this report, we demonstrate that ExoT-induced apoptosis is mediated by the Crk adaptor protein. Our data strongly suggest that ExoT/ADPRT activity, by ADP-ribosylating Crk, transforms this innocuous cellular protein into a cytotoxin that causes atypical anoikis by interfering with integrin-mediated survival signaling.
Results
ExoT/ADPRT induces atypical anoikis apoptosis
Most ExoT or ExoT/ADPRT-intoxicated HeLa cells exhibited movement after cell rounding and prior to succumbing to death, as determined by the uptake of propidium iodide (PI) impermeant nuclear stain, which fluoresces red in dead or dying cells [28,29] (Fig 1A, S1 Movie). This type of cell death morphologically resembled an apoptotic programmed cell death known as anoikis, which occurs as a consequence of loss of cell adhesion and/or inappropriate cell/matrix interaction [30]. Depending on the cell line or the environmental cues, anoikis can be initiated and executed by different pathways, including the intrinsic and the extrinsic apoptotic pathways [30]. However, some common features have emerged. The common hallmarks of anoikis include: enhanced and persistent activation of p38β and JNK by phosphorylation, which is required for anoikis cell death; degradation of p130Cas and paxillin focal adhesion proteins; down activation of FAK, and down-regulation of integrin-mediated survival signaling [30–32].

We conducted a time course infection study to evaluate the possibility that ExoT/ADPRT activity induced anoikis in epithelial cells. HeLa cells were infected at a multiplicity of infection (MOI) of 10 with isogenic mutants of the PA103 strain, a clinical isolate which encodes and expresses ExoU and ExoT [24,25], including PA103∆exoU (∆U) which carries an in-frame deletion in the exoU gene but expresses ExoT; PA103∆exoU/exoT(R149K) (∆U/T(G-A+)) which carries an in-frame deletion in the exoU gene but expresses ExoT with a mutant GAP but functional ADPRT domain; or PA103 pscJ::gentR (T3SS mutant, unable to deliver ExoT into host cells) (S1 Table). To be able to focus on ExoT-induced cytotoxicity, we conducted these studies in the exoU-deleted PA103 genetic background (∆U), as we have previously described [27,28]. Moreover, because the T3SS alone induces necrotic cytotoxicity, which is completely abrogated by ExoT [28], we also left out the effectorless T3SS proficient strain (∆U∆T).
In line with our hypothesis, infection with ExoT or ExoT/ADPRT expressing P. aeruginosa strains resulted in substantial and persistent activation of both p38β and JNK by 4hr post-infection (Fig 1B). To ensure that ExoT/ADPRT activity was sufficient to activate p38β and JNK in the absence of other bacterial factors, HeLa cells were transiently transfected with pIRES2 mammalian expression vectors, harboring wild-type ExoT (pExoT), ExoT with functional ADPRT (pExoT(G-A+)), or the inactive form of ExoT (pExoT(G-A-)), all C-terminally fused to GFP, or empty vector (pGFP) (S1 Table). GFP fusion does not alter ExoT’s virulence functions [27,28]. JNK and p38β activation was evaluated by immunofluorescent (IF) microscopy of fixed cells ~24 hr post transfection. Despite reduced expression of ExoT and ExoT(G-A+) (Fig 1G) due to Cbl-b mediated proteasomal degradation of ExoT and ExoT(G-A+) [26], transient transfection with pExoT-GFP and pExoT(G-A+)-GFP resulted in significant increases in p38β and JNK activation as compared to pExoT(G-A-)-GFP or the pGFP empty vector (Fig 1C–1F), indicating that ExoT/ADPRT activity is sufficient to activate p38β and JNK.
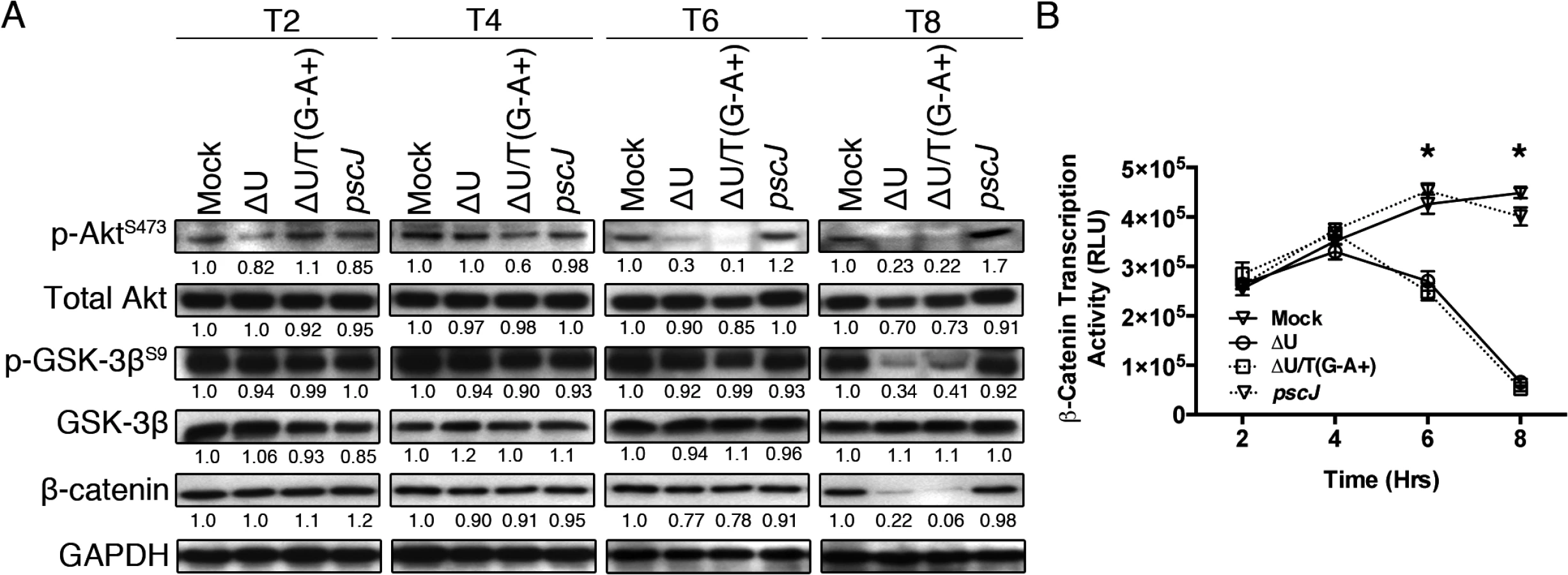
Activation of p38β and JNK leads to disruption in integrin-mediated survival signaling, culminating in anoikis [30,31,33]. We conducted similar infection studies to evaluate the impact of ExoT/ADPRT on integrin-mediated survival signaling using Akt activation and β-catenin activity as readouts. The Akt/β-catenin pathway is an important integrin-mediated survival signaling pathway whose disruption results in anoikis [32,34–36]. Integrin interaction with extracellular matrix (ECM) leads to activation (phosphorylation) of Akt. Phospho-Akt (p-Akt) in turn activates β-catenin by blocking its inhibitor GSK-3β through phosphorylation. (Unphosphorylated GSK-3β is an inhibitor of Akt/β-catenin mediated survival signaling as it targets β-catenin for proteasomal degradation [32,34–36]). Once activated, the β-catenin transcription factor turns on the expression of pro-survival proteins [34]. In line with our hypothesis, infection with ExoT or ExoT/ADPRT-expressing strains (∆U and ∆U/T(G-A+) respectively) reduced Akt activation (p-AktS473) levels by 6hr and p-Akt-mediated GSK-3β phosphorylation (inactivation) by 8hr post-infection (Fig 2A). This resulted in substantial reduction in β-catenin levels (Fig 2A) and β-catenin activity, particularly by 8hr post-infection (Fig 2B, n = 3, p<0.001).
HeLa S3 cells are derived from HeLa cells through planktonic growth and are frequently used as anoikis resistant cells because their survival is adhesion independent [37,38]. We reasoned that if ExoT or ExoT/ADPRT induced anoikis in the target cells, HeLa S3 should be resistant to their cytotoxicity. To test this hypothesis, we infected HeLa and HeLa S3 cells with ExoT-expressing ∆U, ExoT/ADPRT-expressing ∆U/T(R149K), T3SS mutant pscJ, or PBS (Mock). Cytotoxicity was observed by time-lapse videomicroscopy and measured every 15 minutes using PI uptake as a marker for cell death [29,39]. Consistent with our previous report [28], HeLa cells were sensitive to ExoT and ExoT/ADPRT-induced cytotoxicity (Fig 3A and 3C and S2 Movie). Despite similar levels of ExoT intoxication (Fig 3E), HeLa S3 cells displayed resistance to ExoT and ExoT/ADPRT-induced cytotoxicity (Fig 3B and 3D and S3 Movie). Taken together, these data indicated that ExoT and ExoT/ADPRT induced anoikis by interfering with integrin survival signaling.

The dominant negative mutant form of Crk phenocopies ExoT-induced apoptosis
The ADPRT domain of ExoT ADP-ribosylates a conserved arginine residue in the SH2 domain of CrkI and CrkII isoforms of Crk, disrupting Crk SH2 interactions with its cognate substrates [17,40]. Crk has been implicated in cell death, although its role in cytotoxicity remains controversial. Depending on cell type or physiological condition, Crk has been found to be either pro-apoptotic [41–45] or pro-survival [46,47]. We wished to examine the possible role of Crk in the ExoT/ADPRT-induced apoptosis in HeLa cells.
We transfected HeLa cells with a mammalian expression vector harboring wild type CrkI (pCrkI) or the CrkI/R38K mutant form (in which the conserved arginine 38 residue in the SH2 domain was mutated to lysine), fused at their C-termini to GFP (S1 Table), and assessed apoptosis by IF videomicroscopy in the presence of PI, as we described previously [28]. Because of low transfection efficiencies, we were unable to assess the impact of CrkII or CrkII/R38K mutation on survival in HeLa cells. Of note, CrkI/R38K has been shown to act as a DN mutant, interfering with CrkI and CrkII associated cellular activities [48,49]. Despite similar transfection efficiencies (Fig 4D), transfection with CrkI/R38K SH2 DN phenocopied ExoT and ExoT/ADPRT-induced apoptosis as it significantly increased Z-VAD sensitive apoptotic cell death in HeLa cells (Fig 4A–4C and S4 Movie; n = 610; p<0.001). The time to death, defined as the start time of gene expression as determined by GFP expression, to the time of death as determined by PI uptake, in the presence CrkI/R38K SH2 DN mutant was 9.1 ± 1.1 hr which is also nearly identical to the observed time to death in the presence of ExoT (8.5 ± 1.3 hr) or ADPRT (8.6 ± 0.8 hr) ([28] and Fig 4E). Similar to ExoT and ADPRT, transfection with CrkI/R38K SH2 DN also resulted in activation of JNK and p38β in HeLa cells (S1 Fig, n = 122 and n = 124 respectively, one-way ANOVA, p<0.001). Moreover, HeLa S3 cells were also completely resistant to CrkI/R38K SH2 DN-induced apoptosis (S2 Fig). Combined, these data indicated that ExoT-mediated apoptosis is phenocopied by the CrkI/R38K SH2 DN mutant form.

ExoT/ADPRT transforms Crk into a cytotoxin
So far our data indicate that ExoT/ADPRT induces anoikis in a manner that likely involves Crk. The two most likely scenarios are: 1) Crk is required for survival and its modification by ExoT/ADPRT prevents it from performing its survival function; or 2) Crk is not required for survival but when it is ADP-ribosylated by ExoT/ADPRT, it is transformed into a cytotoxin which disrupts survival functions inside the cell. We favored the second scenario because; although, Crk-null (Crk-/-) mice die shortly after birth [50], Crk-/- cells not only survive, they are actually more resistant to apoptosis [42]. These reports strongly suggest that while Crk is required for development, Crk function is not essential for cellular survival per se.
We reasoned that if ExoT or ExoT/ADPRT-induce apoptosis in epithelial cells by transforming Crk, Crk-/- cells should be resistant to ExoT and ExoT/ADPRT-induced cytotoxicity. In contrast, complementing Crk-/- cells with Crk should then restore their sensitivity to ExoT and ExoT/ADPRT-induced apoptosis. To address these possibilities, we complemented Crk-/- mouse embryonic fibroblasts (MEFs) with either CrkI-GFP or GFP alone by transfection. The pCrkI-GFP or pGFP-transfected Crk-/- cells were then infected with ExoT-expressing ∆U, ExoT/ADPRT-expressing ∆U/T(G-A+) or T3SS mutant pscJ and assessed for cytotoxicity by IF timelapse videomicroscopy. Although more cytotoxicity was observed in Crk-/- cells that were infected with P. aeruginosa, regardless of the strain, when compared to HeLa cells, Crk-/- cells complemented with CrkI-GFP were significantly more sensitive to ExoT or ExoT/ADPRT-induced cytotoxicity, as compared to GFP complemented Crk-/- cells (Fig 5B and S5 and S6 Movies. Representative frames are shown in Fig 5A). Additionally, the time to death, (defined as the time of infection to the time cell death manifested by PI staining), in the presence of ∆U or ∆U/T(R149K), was also significantly faster in Crk-/- cells complemented with CrkI-GFP, as compared to the GFP-complemented Crk-/- cells that succumbed to death in response to infection with P. aeruginosa (Fig 5C).

If CrkI SH2 domain modification by ExoT ADP-ribosylation or by mutagenesis (CrkI/R38K) renders CrkI into a cytotoxin, expression of CrkI/R38K SH2 mutant, but not CrkI, should also result in cytotoxicity in Crk-/- cells, phenocopying ExoT’s effect in CrkI-complemented Crk-/- cells. Consistent with this view, transient transfection with the CrkI/R38K SH2 mutant resulted in significantly more cytotoxicity in Crk-/- cells, as compared to CrkI (Fig 6 and S7 Movie). Transfection with CrkI/R38K, W170K, which harbors null mutations in both SH2 and SH3 domains of CrkI [48], did not result in cytotoxicity in Crk-/- (Fig 6), indicating that the SH3 domain of CrkI must be functional for CrkI/R38K mutant to function as a dominant negative (DN) and a cytotoxin. Corroborating this hypothesis, complementing Crk-/- cells with CrkI/R38K, W170K double mutant did not render Crk-/- cells sensitive to ExoT or ExoT/ADPRT-induced cell death (S3 Fig, S8 Movie), indicating that the SH3 domain of CrkI must be functional for the ADP-ribosylated CrkI to act as a cytotoxin. As expected, Crk-/- cells did not express CrkI/II isoforms of Crk (S4 Fig).

ExoT and ExoT/ADPRT, and CrkI/R38K interfere with integrin survival signaling by disrupting focal adhesion sites
We next sought to determine how ADP-ribosylation of CrkI by ExoT/ADPRT domain activity or by mutagenesis (R38K mutation) could potentially transform this cellular protein disruptive to integrin-mediated survival signaling. We hypothesized that CrkI ADP-ribosylation by ExoT/ADPRT could disrupt integrin-survival signaling by destabilizing the focal adhesion (FA) sites. We based this hypothesis on the knowledge that FAK activation and FAK/p130Cas interactions at FA sites stabilize the integrin/extracellular matrix (ECM) interactions and are required for survival signaling [51–53]. During FA assembly, integrin/ECM interaction results in activation (phosphorylation) of focal adhesion kinase (FAK), which in turn phosphorylates p130Cas and paxillin [54,55]. Phosphorylated p130Cas and paxillin then recruit Crk to FA by directly interacting with its SH2 domain [56]. Although ExoT’s impact on FA has not been investigated, Crk interactions with p130Cas and paxillin have been shown to be disrupted by ExoT/ADPRT activity [40]. Therefore, while FAK, p130Cas, and paxillin localization to FA is upstream of Crk, disruption of Crk activity by ExoT or by CrkI/R38K mutation could potentially disrupt FAK, p130Cas, and paxillin subcellular localization, and/or their maintenance, and/or their activation at FA sites. This would in turn interfere with integrin/ECM survival signaling and would lead to anoikis [53,57].
To test this hypothesis, we performed the aforementioned time-course infection studies with ExoT-expressing ∆U, ExoT/ADPRT-expressing ∆U/T(G-A+), and the T3SS mutant pscJ, and assessed the impact of ExoT or ExoT/ADPRT on FA sites by determining the total number of FAK and p-130Cas positive FA puncta per cell and the intensity of FAK and p-130Cas stainings per FA puncta by IF microscopy (described in Methods and S5 Fig). Infection with ∆U or ∆U/T(G-A+) bacteria resulted in substantial reduction in FAK and p130Cas localization to FA sites by 4h post-infection, as manifested by reduced number of FA puncta (Fig 7A–7D) and reduced staining intensities of FAK and p130Cas per FA puncta (S6A and S6B Fig). As expected, transient transfection with expression vectors harboring ExoT (pExoT) or the ExoT/ADPRT domain (pExoT(G-A+)) also resulted in significant reduction in FAK and p130Cas localization to the FA sites (Fig 7E–7H and S6C and S6D Fig), indicating that ExoT/ADPRT activity is sufficient to disrupt FA sites. Interestingly, infection with ∆U or ∆U/T(G-A+) did not significantly affect the total cellular levels of activated (phosphorylated) FAK (p-FAKY397) or activated (phosphorylated) p-130Cas (p-p130CasY165) as measured by Western blotting (Fig 7I). These data indicated that FAK and p130Cas are able to get to the FA sites where they become phosphorylated but they are not maintained at FA sites in the presence of ExoT or ExoT/ADPRT activity. Of note, in HeLa cells transfected with pCrkI-GFP, CrkI co-localized with FAK and p130Cas at FA sites (Fig 8A–8F, pCrkI panels). In contrast, CrkI/R38K did not localize to FA sites and disrupted FAK and p130Cas localization to the FA sites in HeLa cells (Fig 8A–8F, pCrkI/R38K panels), phenocopying the adverse effect of ExoT and ExoT/ADPRT on FA and supporting our hypothesis that ExoT ADP-ribosylation of CrkI renders CrkI disruptive to FA sites.


It is generally believed that Crk is an essential component of FA sites [56,58]. However, our data strongly suggested that although Crk may be recruited to FA, its function may not require for FA assembly or its dynamics. Consistent with this view, we found FAK to localize to FA sites in Crk-/- cells (Fig 8G–8I), indicating that FAK subcellular localization to FA sites and its maintenance in that compartment does not require Crk in this cell line. Interestingly, in Crk-/- cells that were transfected with CrkI-GFP, CrkI co-localized with FAK at FA sites (Fig 8G) indicating that CrkI can be recruited and maintained at FA sites in this cell line as well. Similar to HeLa, CrkI/R38K SH2 DN mutant did not localize to FA sites and disrupted FAK localization to FA sites in Crk-/- cells that were transfected with CrkI/R38K-GFP expression vector (Fig 8G–8I). Of note, CrkI/R38K, W170K (SH2 and SH3 double mutant) did not localize to FA sites and failed to disrupt FAK localization to FA sites in Crk-/- and HeLa cells (Fig 8G–8I), indicating that the SH3 domain must be functional for CrkI/R38K to act as a dominant negative.
We next asked whether CrkI presence in FA sites affected FA structures and/ or their function in in Crk-/- cells. We evaluated the impact of CrkI on FA structure and function in Crk-/- cells by determining how CrkI affected: (i) the number of FA puncta per cell, (ii) the ability of Crk-/- cells to adhere to the surface, as determined by their surface area, and (iii) the ability of Crk-/- cells to migrate, as determined by distance these cells travelled within 24 hr after transfection with CrkI. The data indicated that CrkI presence in FA sites in Crk-/- cells, complemented with CrkI, did not affect their FA structures or functions (S7 Fig). Collectively, these data indicate that while CrkI can be recruited to FA, its function is not essential for FA assembly or survival. However, when modified by ExoT or by mutagenesis, it can disrupt FA sites, which would result in anoikis.
Discussion
We have recently reported that Pseudomonas aeruginosa ExoT is both necessary and sufficient to induce potent apoptosis in target host cells in a manner that primarily depends on its ADPRT domain activity [28] but the mechanism underlying ExoT/ADPRT-induced apoptosis has not been determined. We now report that ExoT/ADPRT induces anoikis by disrupting FA sites. Our data demonstrate that within 4h of exposure to ExoT or ExoT/ADPRT, FA sites are destabilized (Fig 7). Concomitant with FA disruption, intoxication with ExoT or ExoT/ADPRT leads to persistent activation of p38β and JNK (Fig 1), which are known drivers of anoikis [33,59]. This in turn leads to dampened integrin-mediated survival signaling by 6–8 hr post-exposure, as manifested by reduced Akt activation and β-catenin-mediated survival signaling (Fig 2). ExoT/ADPRT-induced anoikis is atypical in that we did not observe degradation of p130Cas or down-activation of FAK (Fig 7I), both of which have been reported to occur during anoikis and be required for cytotoxicity [30–32,53,60]. Instead, ExoT or ExoT/ADPRT interfered with FAK and p130Cas subcellular localization to the FA sites (Fig 7A–7H). Our data suggest that FA sites may serve as cellular survival centers. Consistent with this notion, JNK localization to FA sites and its interaction with FAK and p130Cas in that compartment has been shown to be required for survival [57]. Of note, ExoT and ExoT/ADPRT also disrupted JNK localization at FA sites (S8 Fig and Fig 1C, compare JNK staining at FA sites in the untransfected and pGFP - transfected cells to the pExoT or pExoT(G-A+)-transfected cells which lack JNK at FA sites).
Crk is generally believed to be an essential component of FA [56,58], although it is not clear whether it is CrkI or CrkII or both that function in FA. Moreover, Crk has also been implicated in cellular survival and/or apoptosis but Crk’s role in these processes remain controversial in that it is found to be either pro-apoptotic [41–45] or pro-survival [46,47]. Our data strongly suggest that Crk function may not be essential for FA or for cellular survival per se, but when modified by ExoT or by mutagenesis, Crk can be transformed into a cytotoxin that interferes with survival signaling by disrupting FA (Figs 5, 6 and 8). How does modification of CrkI SH2 domain by ExoT/ADPRT-mediated ADP-ribosylation render CrkI disruptive to integrin-mediated survival signaling? We propose that ADP-ribosylation by ExoT transforms CrkI into a dominant negative mutant that is disruptive to FA structures. Our reasoning is that while these ADP-ribosylated CrkI and CrkI/R38K are unable to interact through the SH2 domain with their cognate substrates such as p130Cas and paxillin [40], they would be able to interact with a number of essential FA components, such as C3G [61,62] through the SH3 domain. This prevents their localization to FA sites, and destabilizes FA structures, thus culminating in anoikis as our data demonstrate (Figs 7 and 8). Consistent with this hypothesis, CrkI/(R38K, W170K) which harbors mutations in both SH2 and SH3 domains failed to disrupt FA sites (Fig 8), or induce apoptosis in Crk-/- cells (Fig 6), or renders Crk-/- cells sensitive to ExoT or ExoT/ADPRT cytotoxicity (S3 Fig, S8 Movie).
Crk has been shown to function in various host defenses against bacterial pathogens, such as inhibiting EPEC-induced actin-based pedestal formation, increasing bacterial clearance through phagocytosis of IgG-opsonized pathogens by Fcγ receptors, and potentially enhancing innate immune activation through pattern recognition receptors (PRR) by sequestering bacterial virulence factors, such as EPEC’s Tir, which interfere with PRR signaling [63–65]. Therefore, it is not surprising that pathogens have evolved mechanisms to target this central host protein in order to advance their own agenda. For example, EPEC induces phosphorylation of the major regulatory tyrosine in CrkII, preventing CrkII from sequestering Tir, thus freeing this virulence factor to promote pedestal formation [63]. Additionally, P. aeruginosa blocks host cell proliferation by inhibiting Crk’s essential function for cytokinesis [27].
In summary, we propose that ExoT by ADP-ribosylating Crk transforms this cellular protein into a cytotoxin, which induces anoikis by disrupting FA structures and interfering with the integrin survival signaling.
Materials and Methods
Cell culture and reagents
HeLa (ATCC), HeLa S3 (ATCC), and Crk-/- cells were cultured in complete DMEM (Life Technologies) containing phenol red supplemented with 10% FCS, 1% penicillin/streptomycin, and 1% L-glutamine at 37°C in the presence of 5% CO2. For transfection experiments, 0.4μg plasmid DNA was used with Effectene (Qiagen) according to the manufactures protocol. Antibodies were from Cell Signaling Technologies (CST) unless otherwise noted: pAKT (#9271); Crk (BD 610036); GAPDH (GenScript A00191); pJNK (#4668); JNK (#9252); p130Cas (BD 610271); p-p130Cas(Y165) (#4015); p38β (#9212); p-p38β (#9215); FAK (BD 610087); pFAK(Y397) (#3283).
Cytotoxicity measurement by time-lapse videomicroscopy was performed as we described previously [27,28]
Briefly, HeLa, HeLa S3, and Crk-/- cells were grown in DMEM without phenol red with (for transfection studies) or without antibiotics (for infection studies) for 24 hr. These cells were then transfected with indicated expression vectors or infected with indicated strains as described [28]. 1h after transfection or at time of infection, cells were given 7μg/ml propidium iodide (Sigma) and then placed on an AxioVert Z1 microscope (Zeiss) fitted with a live-imaging culture box (Pecon) maintaining 37°C in the presence of 5% CO2. Time-lapse videos were taken using AxioVision 4.2.8 software. Video analysis was performed with ImageJ 1.47 software (NIH).
Western blot
Samples were assessed by Western blot as described previously [29,66]. Briefly, cells were lysed following infection with 1% TX-100 containing a protease inhibitor cocktail (Roche Diagnostics), 100mM PMSF, and 100mM Na3VO4. Lysates were mixed with 4X SDS loading buffer and loaded onto 10% SDS-polyacrylamide gels. After resolving, gels were transferred to PVDF membranes, blocked with 5% milk, and probed overnight with primary antibody at 4°C. After washing, blots were probed with HRP-conjugated secondary antibody (Cell Signaling Technologies). Blots were developed with ECL or ECL+ reagent (GE Healthcare). Films were developed with an autoprocessor.
Bacteria strains and plasmids
All bacterial strains and expression vectors and their sources are indicated in S1 Table. These isogenic strains were in PA103 genetic background. For infection studies, bacteria were cultured overnight in Luria-Bertani (LB) broth at 37°C without shaking. Bacteria were added at M.O.I of 10.
Luciferase reporter assay
β-catenin transcriptional activity assay was performed as previously described [67]. HeLa cells were transfected with 400ng TOPFlash plasmid (Upstate) using Effectene (Qiagen). After 24 hr, the cells were washed with PBS and given fresh media either with our without antibiotics. The cells were either mock infected (PBS) or infected with the aforementioned strains which were resuspended in PBS. Luciferase levels were measured according to the manufacturers protocol using the Luciferase Assay System (Promega) using a Moonlight 2010 Luminometer (BD Bioscience) and normalizing to Renilla luciferase’s baseline luminance. Experiments were performed in triplicate for each time point.
Immunofluorescent microscopy
Immunofluorescent microscopy was carried out as previously described [28,66,68]. Briefly, coverslips were treated with poly-l-lysine and 40μg/ml human fibronectin (Millipore) before seeding cells. After 24 hr, cells were either transfected for 17h or infected for 4h. At each end-point, cells were fixed with 10% ice-cold TCA for 10min. Cells were permeablized with 0.2% Triton X-100 (Sigma) for 15min at RT, blocked with 3% FCS for 1H at 37°C before staining overnight with primary antibody. Next, cells were washed 3x with PBS before staining with conjugated secondary antibody, AF594 or AF488 (Life Technologies) for 1hr at 37°C. The coverslip was mounted on DAPI containing VectaMount (Vector Laboratories). Cells were imaged AxioVision 4.2.8 software using an AxioVert Z1 microscope (Zeiss) using a 63X objective.
Focal adhesion analysis
Immunofluorescent microscopy images were processed using ImageJ v1.47 (NIH). Cell outlines were first saved using the selection manager. A background subtraction was applied to the channel containing the focal adhesion marker. Next, a threshold level was applied to that channel followed by particle analysis to identify the number of puncta per cell selection and staining intensity per puncta. Each image set was processed using equal threshold and particle analysis values.
Statistical analysis
Two-tailed Student t-tests, one-way ANOVA with Tukey post-hoc test, and Chi-squared analyses were used to assess significance with p<0.05 considered significant. Analysis was carried out with Prism 6.0 (GraphPad).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Hauser AR, Cobb E, Bodi M, Mariscal D, Valles J, et al. (2002) Type III protein secretion is associated with poor clinical outcomes in patients with ventilator-associated pneumonia caused by Pseudomonas aeruginosa. Crit Care Med 30 : 521–528. 11990909
2. Engel J, Balachandran P (2009) Role of Pseudomonas aeruginosa type III effectors in disease. Curr Opin Microbiol 12 : 61–66. doi: 10.1016/j.mib.2008.12.007 19168385
3. Engel JN (2003) Molecular pathogenesis of acute Pseudomonas aeruginosa infections. In: Hauser A, Rello J, editors. Severe Infections Caused by Pseudomonas aeruginosa. New York City: Kluwer Academic/Plenum Press. pp. 201–230.
4. Salyers AA, Whitt DD, editors (1994) Bacterial pathogenesis: a molecular approach. Washington, D. C.: ASM Press. 260–268 p.
5. Stavrinides J, McCann HC, Guttman DS (2008) Host-pathogen interplay and the evolution of bacterial effectors. Cell Microbiol 10 : 285–292. 18034865
6. Hueck C (1998) Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol and Molec Biol Rev 62 : 379–433. 9618447
7. Hauser AR (2009) The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat Rev Microbiol 7 : 654–665. doi: 10.1038/nrmicro2199 19680249
8. Sato H, Frank DW (2004) ExoU is a potent intracellular phospholipase. Mol Microbiol 53 : 1279–1290. 15387809
9. Sato H, Feix JB, Hillard CJ, Frank DW (2005) Characterization of phospholipase activity of the Pseudomonas aeruginosa type III cytotoxin, ExoU. J Bacteriol 187 : 1192–1195. 15659695
10. Henriksson ML, Sundin C, Jansson AL, Forsberg A, Palmer RH, et al. (2002) Exoenzyme S shows selective ADP-ribosylation and GTPase-activating protein (GAP) activities towards small GTPases in vivo. Biochem J 367 : 617–628. 12132999
11. Shaver CM, Hauser AR (2004) Relative contributions of Pseudomonas aeruginosa ExoU, ExoS, and ExoT to virulence in the lung. Infect Immun 72 : 6969–6977. 15557619
12. Garrity-Ryan L, Kazmierczak B, Kowal R, Commolli J, Hauser A, et al. (2000) The arginine finger domain of ExoT is required for actin cytoskeleton disruption and inhibition of internalization of Pseudomonas aeruginosa by epithelial cells and macrophages. Infect Immun 68 : 7100–7113. 11083836
13. Krall R, Schmidt G, Aktories K, Barbieri JT (2000) Pseudomonas aeruginosa ExoT is a Rho GTPase-activating protein. Infect Immun 68 : 6066–6068. 10992524
14. Wurtele M, Wolf E, Pederson KJ, Buchwald G, Ahmadian MR, et al. (2001) How the Pseudomonas aeruginosa ExoS toxin downregulates Rac. Nat Struct Biol 8 : 23–26. 11135665
15. Braun M, Stuber K, Schlatter Y, Wahli T, Kuhnert P, et al. (2002) Characterization of an ADP-ribosyltransferase toxin (AexT) from Aeromonas salmonicida subsp. salmonicida. J Bacteriol 184 : 1851–1858. 11889090
16. Barbieri JT, Sun J (2004) Pseudomonas aeruginosa ExoS and ExoT. Rev Physiol Biochem Pharmacol 152 : 79–92. 15375697
17. Sun J, Barbieri JT (2003) Pseudomonas aeruginosa ExoT ADP-ribosylates CT10 regulator of kinase (Crk) proteins. J Biol Chem 278 : 32794–32800. 12807879
18. Garrity-Ryan L, Shafikhani S, Balachandran P, Nguyen L, Oza J, et al. (2004) The ADP ribosyltransferase domain of Pseudomonas aeruginosa ExoT contributes to its biological activities. Infection and immunity 72 : 546–558. 14688136
19. Sato H, Frank DW, Hillard CJ, Feix JB, Pankhaniya RR, et al. (2003) The mechanism of action of the Pseudomonas aeruginosa-encoded type III cytotoxin, ExoU. Embo J 22 : 2959–2969. 12805211
20. Barbieri JT (2000) Pseudomonas aeruginosa exoenzyme S, a bifunctional type-III secreted cytotoxin. Int J Med Microbiol 290 : 381–387. 11111915
21. Yahr T, Mende-Mueller LM, Friese MB, Frank DW (1997) Identification of type III secreted products of the Pseudomonas aeruginosa exoenzyme S regulon. J Bacteriol 179 : 7165–7168. 9371466
22. Vance RE, Rietsch A, Mekalanos JJ (2005) Role of the type III secreted exoenzymes S, T, and Y in systemic spread of Pseudomonas aeruginosa PAO1 in vivo. Infect Immun 73 : 1706–1713. 15731071
23. Yahr TL, Vallis AJ, Hancock MK, Barbieri JT, Frank DW (1998) ExoY, an adenylate cyclase secreted by the Pseudomonas aeruginosa type III system. Proc Natl Acad Sci U S A 95 : 13899–13904. 9811898
24. Feltman H, Khan S, Jain M, Peterson L, Hauser A (2001) Type III secretion genotypes of clinical and environmental Pseudmonas aeruginosa isolates. ASM abstracts: D22.
25. Feltman H, Schulert G, Khan S, Jain M, Peterson L, et al. (2001) Prevalence of type III secretion genes in clinical and environmental isolates of Pseudomonas aeruginosa. Microbiology 147 : 2659–2669. 11577145
26. Balachandran P, Dragone L, Garrity-Ryan L, Lemus A, Weiss A, et al. (2007) The ubiquitin ligase Cbl-b limits Pseudomonas aeruginosa exotoxin T-mediated virulence. J Clin Invest 117 : 419–427. 17235393
27. Shafikhani SH, Engel J (2006) Pseudomonas aeruginosa type III-secreted toxin ExoT inhibits host-cell division by targeting cytokinesis at multiple steps. Proceedings of the National Academy of Sciences of the United States of America 103 : 15605–15610. 17030800
28. Shafikhani SH, Morales C, Engel J (2008) The Pseudomonas aeruginosa type III secreted toxin ExoT is necessary and sufficient to induce apoptosis in epithelial cells. Cellular microbiology 10 : 994–1007. 18053004
29. Wood S, Pithadia R, Rehman T, Zhang L, Plichta J, et al. (2013) Chronic alcohol exposure renders epithelial cells vulnerable to bacterial infection. PLoS One 8: e54646. doi: 10.1371/journal.pone.0054646 23358457
30. Chiarugi P, Giannoni E (2008) Anoikis: a necessary death program for anchorage-dependent cells. Biochem Pharmacol 76 : 1352–1364. doi: 10.1016/j.bcp.2008.07.023 18708031
31. Bouchard V, Harnois C, Demers MJ, Thibodeau S, Laquerre V, et al. (2008) B1 integrin/Fak/Src signaling in intestinal epithelial crypt cell survival: integration of complex regulatory mechanisms. Apoptosis 13 : 531–542. doi: 10.1007/s10495-008-0192-y 18322799
32. Vachon PH (2011) Integrin signaling, cell survival, and anoikis: distinctions, differences, and differentiation. Journal of signal transduction 2011 : 738137. doi: 10.1155/2011/738137 21785723
33. Cardone MH, Salvesen GS, Widmann C, Johnson G, Frisch SM (1997) The regulation of anoikis: MEKK-1 activation requires cleavage by caspases. Cell 90 : 315–323. 9244305
34. Orford K, Orford CC, Byers SW (1999) Exogenous expression of beta-catenin regulates contact inhibition, anchorage-independent growth, anoikis, and radiation-induced cell cycle arrest. J Cell Biol 146 : 855–868. 10459019
35. Fang D, Hawke D, Zheng Y, Xia Y, Meisenhelder J, et al. (2007) Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J Biol Chem 282 : 11221–11229. 17287208
36. Frisch SM, Schaller M, Cieply B (2013) Mechanisms that link the oncogenic epithelial-mesenchymal transition to suppression of anoikis. J Cell Sci 126 : 21–29. doi: 10.1242/jcs.120907 23516327
37. Cheng TL, Lai CH, Jiang SJ, Hung JH, Liu SK, et al. (2014) RHBDL2 Is a Critical Membrane Protease for Anoikis Resistance in Human Malignant Epithelial Cells. Scientific World Journal 2014 : 902987. doi: 10.1155/2014/902987 24977233
38. Puck TT, Marcus PI (1955) A Rapid Method for Viable Cell Titration and Clone Production with Hela Cells in Tissue Culture: The Use of X-Irradiated Cells to Supply Conditioning Factors. Proc Natl Acad Sci U S A 41 : 432–437. 16589695
39. Goldufsky J, Wood S, Hajihossainlou B, Rehman T, Majdobeh O, et al. (2015) Pseudomonas aeruginosa exotoxin T induces potent cytotoxicity against a variety of murine and human cancer cell lines. J Med Microbiol 64 : 164–173. doi: 10.1099/jmm.0.000003-0 25627204
40. Deng Q, Sun J, Barbieri JT (2005) Uncoupling Crk signal transduction by Pseudomonas exoenzyme T. J Biol Chem 280 : 35953–35960. 16123042
41. Kar B, Reichman CT, Singh S, O'Connor JP, Birge RB (2007) Proapoptotic function of the nuclear Crk II adaptor protein. Biochemistry 46 : 10828–10840. 17764157
42. Austgen K, Johnson ET, Park TJ, Curran T, Oakes SA (2012) The adaptor protein CRK is a pro-apoptotic transducer of endoplasmic reticulum stress. Nat Cell Biol 14 : 87–92. doi: 10.1038/ncb2395 22179045
43. Smith JJ, Richardson DA, Kopf J, Yoshida M, Hollingsworth RE, et al. (2002) Apoptotic regulation by the Crk adapter protein mediated by interactions with Wee1 and Crm1/exportin. Mol Cell Biol 22 : 1412–1423. 11839808
44. Parrizas M, Blakesley VA, Beitner-Johnson D, Le Roith D (1997) The proto-oncogene Crk-II enhances apoptosis by a Ras-dependent, Raf-1/MAP kinase-independent pathway. Biochem Biophys Res Commun 234 : 616–620. 9175762
45. Evans EK, Lu W, Strum SL, Mayer BJ, Kornbluth S (1997) Crk is required for apoptosis in Xenopus egg extracts. Embo J 16 : 230–241. 9029144
46. Cho SY, Klemke RL (2000) Extracellular-regulated kinase activation and CAS/Crk coupling regulate cell migration and suppress apoptosis during invasion of the extracellular matrix. J Cell Biol 149 : 223–236. 10747099
47. Salameh A, Galvagni F, Bardelli M, Bussolino F, Oliviero S (2005) Direct recruitment of CRK and GRB2 to VEGFR-3 induces proliferation, migration, and survival of endothelial cells through the activation of ERK, AKT, and JNK pathways. Blood 106 : 3423–3431. 16076871
48. Lamorte L, Royal I, Naujokas M, Park M (2002) Crk adapter proteins promote an epithelial-mesenchymal-like transition and are required for HGF-mediated cell spreading and breakdown of epithelial adherens junctions. Mol Biol Cell 13 : 1449–1461. 12006644
49. Rodrigues SP, Fathers KE, Chan G, Zuo D, Halwani F, et al. (2005) CrkI and CrkII function as key signaling integrators for migration and invasion of cancer cells. Molecular cancer research: MCR 3 : 183–194. 15831672
50. Park TJ, Boyd K, Curran T (2006) Cardiovascular and craniofacial defects in Crk-null mice. Mol Cell Biol 26 : 6272–6282. 16880535
51. Kim SH, Turnbull J, Guimond S (2011) Extracellular matrix and cell signalling: the dynamic cooperation of integrin, proteoglycan and growth factor receptor. J Endocrinol 209 : 139–151. doi: 10.1530/JOE-10-0377 21307119
52. Cabodi S, Di Stefano P, Leal Mdel P, Tinnirello A, Bisaro B, et al. (2010) Integrins and signal transduction. Adv Exp Med Biol 674 : 43–54. 20549939
53. Frisch SM, Vuori K, Ruoslahti E, Chan-Hui PY (1996) Control of adhesion-dependent cell survival by focal adhesion kinase. J Cell Biol 134 : 793–799. 8707856
54. Kanchanawong P, Shtengel G, Pasapera AM, Ramko EB, Davidson MW, et al. (2010) Nanoscale architecture of integrin-based cell adhesions. Nature 468 : 580–584. doi: 10.1038/nature09621 21107430
55. Zaidel-Bar R, Milo R, Kam Z, Geiger B (2007) A paxillin tyrosine phosphorylation switch regulates the assembly and form of cell-matrix adhesions. J Cell Sci 120 : 137–148. 17164291
56. Feller SM (2001) Crk family adaptors-signalling complex formation and biological roles. Oncogene 20 : 6348–6371. 11607838
57. Almeida EA, Ilic D, Han Q, Hauck CR, Jin F, et al. (2000) Matrix survival signaling: from fibronectin via focal adhesion kinase to c-Jun NH(2)-terminal kinase. J Cell Biol 149 : 741–754. 10791986
58. Turner CE (2000) Paxillin interactions. J Cell Sci 113 Pt 23 : 4139–4140. 11069756
59. Owens TW, Valentijn AJ, Upton JP, Keeble J, Zhang L, et al. (2009) Apoptosis commitment and activation of mitochondrial Bax during anoikis is regulated by p38MAPK. Cell Death Differ 16 : 1551–1562. doi: 10.1038/cdd.2009.102 19662026
60. Wei L, Yang Y, Zhang X, Yu Q (2004) Cleavage of p130Cas in anoikis. J Cell Biochem 91 : 325–335. 14743392
61. Wozniak MA, Modzelewska K, Kwong L, Keely PJ (2004) Focal adhesion regulation of cell behavior. Biochim Biophys Acta 1692 : 103–119. 15246682
62. Parsons JT (2003) Focal adhesion kinase: the first ten years. J Cell Sci 116 : 1409–1416. 12640026
63. Nieto-Pelegrin E, Meiler E, Martin-Villa JM, Benito-Leon M, Martinez-Quiles N (2014) Crk adaptors negatively regulate actin polymerization in pedestals formed by enteropathogenic Escherichia coli (EPEC) by binding to Tir effector. PLoS Pathog 10: e1004022. doi: 10.1371/journal.ppat.1004022 24675776
64. Lee WL, Cosio G, Ireton K, Grinstein S (2007) Role of CrkII in Fcgamma receptor-mediated phagocytosis. J Biol Chem 282 : 11135–11143. 17308335
65. Martinez-Quiles N, Feuerbacher LA, Benito-Leon M, Hardwidge PR (2014) Contribution of Crk Adaptor Proteins to Host Cell and Bacteria Interactions. Biomed Res Int 2014 : 372901. doi: 10.1155/2014/372901 25506591
66. Wood S, Sivaramakrishnan G, Engel J, Shafikhani SH (2011) Cell migration regulates the kinetics of cytokinesis. Cell Cycle 10 : 648–654. 21293189
67. Kumar A, Zloza A, Moon RT, Watts J, Tenorio AR, et al. (2008) Active beta-catenin signaling is an inhibitory pathway for human immunodeficiency virus replication in peripheral blood mononuclear cells. J Virol 82 : 2813–2820. doi: 10.1128/JVI.02498-07 18199649
68. Shafikhani SH, Mostov K, Engel J (2008) Focal adhesion components are essential for mammalian cell cytokinesis. Cell Cycle 7 : 2868–2876. 18787414
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 5
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Human Cytomegalovirus miR-UL112-3p Targets TLR2 and Modulates the TLR2/IRAK1/NFκB Signaling Pathway
- Paradoxical Immune Responses in Non-HIV Cryptococcal Meningitis
- Expression in the Fat Body Is Required in the Defense Against Parasitic Wasps in
- Survives with a Minimal Peptidoglycan Synthesis Machine but Sacrifices Virulence and Antibiotic Resistance
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy