
A Single Protein S-acyl Transferase Acts through Diverse Substrates to Determine Cryptococcal Morphology, Stress Tolerance, and Pathogenic Outcome
Cryptococcus neoformans is a ubiquitous environmental yeast that kills over 625,000 people annually, mainly in developing countries. Healthy humans frequently inhale infectious particles without noticeable symptoms. However, in immunocompromised people, the initial lung infection can spread to other sites, particularly to the central nervous system where it causes lethal brain infection. The infected host responds by deploying immune cells to engulf and kill the yeast, but C. neoformans can survive this engulfment and even multiply within the host cells. To understand the interactions between the invading microbe and host cells we screened 1,201 fungal mutants to identify fungal factors that influence these processes. One mutant, lacking an enzyme that modifies proteins with the lipid palmitate, showed an increase in engulfment by the host along with dramatic defects in morphology, stress resistance, and virulence. We went on to identify the proteins this enzyme modifies and explain how its absence leads to altered cell wall synthesis, signal transduction, and membrane trafficking; these changes explain the behavior of the mutant. We also found that the mutant could not cause disease in an animal model. Our work shows that protein palmitoylation is critical for cryptococcal pathogenesis and presents a potential avenue for antifungal therapy.
Published in the journal:
. PLoS Pathog 11(5): e32767. doi:10.1371/journal.ppat.1004908
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004908
Summary
Cryptococcus neoformans is a ubiquitous environmental yeast that kills over 625,000 people annually, mainly in developing countries. Healthy humans frequently inhale infectious particles without noticeable symptoms. However, in immunocompromised people, the initial lung infection can spread to other sites, particularly to the central nervous system where it causes lethal brain infection. The infected host responds by deploying immune cells to engulf and kill the yeast, but C. neoformans can survive this engulfment and even multiply within the host cells. To understand the interactions between the invading microbe and host cells we screened 1,201 fungal mutants to identify fungal factors that influence these processes. One mutant, lacking an enzyme that modifies proteins with the lipid palmitate, showed an increase in engulfment by the host along with dramatic defects in morphology, stress resistance, and virulence. We went on to identify the proteins this enzyme modifies and explain how its absence leads to altered cell wall synthesis, signal transduction, and membrane trafficking; these changes explain the behavior of the mutant. We also found that the mutant could not cause disease in an animal model. Our work shows that protein palmitoylation is critical for cryptococcal pathogenesis and presents a potential avenue for antifungal therapy.
Introduction
Cryptococcus neoformans is a fungal pathogen that causes over 625,000 deaths per year, mainly in severely immunocompromised individuals. Cryptococcosis is contracted by inhalation of infectious particles from the environment [1], which leads to a primary pulmonary infection. In healthy people this infection is typically cleared, but in immunocompromised hosts the organism can proliferate and disseminate, with a tropism for the central nervous system where it causes lethal meningoencephalitis. As a result, this pathogen is a major threat to AIDS patients and to the rapidly growing population of individuals with other immunosuppressive conditions [2–5]. Host phagocytes, mainly macrophages, are critical for initial control of this facultative intracellular pathogen [6]. However, as the flip side to their positive role as the first line of host defense, these cells may also serve as sites for replication and latency, or potentially as vehicles for yeast dissemination [1]. In line with these activities, several studies have demonstrated a correlation between poor patient outcomes and the capacity of clinical strains to be phagocytosed and/or to proliferate intracellularly [7, 8]. Understanding the opposing roles of macrophages in cryptococcal infection and their interactions with C. neoformans is key to our ability to influence such events in favor of the host. Despite the importance of these interactions to cryptococcal pathogenesis, the critical features of the host and fungus that govern them have not been determined.
We developed an image-based high-throughput screening (HTS) assay to probe fungal-host cell interactions [9] and evaluated a C. neoformans partial deletion collection [10] for altered engulfment by a human macrophage-like cell line. One ‘hit’ lacked a gene that encodes a protein S-acyltransferase (PAT), incriminating protein palmitoylation as a key pathway in cryptococcal pathogenesis. Protein palmitoylation, the reversible addition of palmitate to cysteine, can regulate the stability, localization, and function of target proteins [11]. The enzymes mediating this modification were first identified in the model yeast S. cerevisiae [12, 13] and are now recognized as important effectors in eukaryotic cells [11]. Although protein palmitoylation has been shown to influence infectivity in viruses [14], bacteria [15], and parasites [16–18], its role in fungal pathogenesis has not been explored. The importance of this lipid modification in fungal pathogens is supported by studies of Ras1 localization in Aspergillus fumigatus, C. neoformans, and Candida albicans [19–21], but no other proteins have been shown to be functionally palmitoylated in these organisms. Finally, no PAT has been characterized in a pathogenic fungus. Our studies demonstrate that a single PAT is a major determinant of cryptococcal pathogenesis and, by defining the relevant palmitoylome, we identify the cellular mechanisms by which defects of this fatty acid modification dramatically alter fungal morphology, host interactions, and virulence in vivo.
Results
Identification of fungal genes that influence interactions with host macrophages
C. neoformans engulfment by host cells and subsequent intracellular proliferation has been implicated in dissemination, virulence, and ultimately in patient outcome [8, 22, 23]. However, the full complement of fungal genes that participate in these processes has not been defined, and how individual gene products modulate interactions with host phagocytes is not known. To address cryptococcal interactions with host cells, we used an automated high content imaging method [9] to quantify the interactions between a human monocytic cell line (THP-1) and mutant fungi from a deletion collection made in the highly pathogenic reference strain H99 [10]. Of the 1,201 mutants we screened, 56 (4.7%) showed significant alterations in phagocytic index (Fig 1A). These mutants (S1 Table) were roughly equally distributed between strains with decreased and increased engulfment (30 and 26, respectively); the ten most extreme in each category are shown in Table 1. An example data set from one plate of the mutant collection (Fig 1B) shows strikingly increased phagocytosis of three mutants, two of which, pka1 and rim101, are known to have altered cell surface structures that would explain this phenotype [24, 25]. Additionally, pbx1, the top hit of the high uptake mutants (Table 1), has defects in cell wall structure and capsule assembly that cause increased engulfment by macrophages [26]. These observations validated our strategy for probing the interactions between C. neoformans and macrophages and encouraged us to further pursue novel hits from our screen.


A putative S-acyltransferase regulates C. neoformans uptake and adherence
Another strain (2A12) that consistently demonstrated an elevated phagocytic index (Fig 1B and Table 2) lacks the uncharacterized gene CNAG_03981. This gene is highly homologous to S. cerevisiae PFA4, which encodes a palmitoyl acyltransferase (PAT), and was accordingly given the same name (following guidelines in [27]). PATs are DHHC zinc finger domain-containing enzymes that mediate the reversible addition of palmitate to proteins, thereby regulating their membrane association and biological function [11]. Eukaryotic cells often express multiple DHHC domain proteins, which have similar enzymatic activity but modify variably overlapping groups of substrates [28]. These enzymes play key roles in protein fatty-acylation and membrane targeting [11], but have never been studied in C. neoformans or any other fungal pathogen. There are seven putative PATs encoded in the H99 genome; four of these were deleted in the collection that we screened but only pfa4Δ differed significantly from wild-type cells (Table 2). This suggested that Pfa4 acylates at least one protein that both influences host cell interactions and is not modified by other PATs. Given the limited knowledge of protein palmitoylation in C. neoformans biology and pathogenesis, we chose this mutant for mechanistic study.

We first generated independent pfa4 deletions in C. neoformans reference strain H99 (used for the deletion collection) and its more genetically tractable derivative KN99 [29]. Like 2A12, both mutants showed consistent increases in adherence to and engulfment by macrophages compared to wild-type cells (Fig 1C), with the greater uptake readily visible by confocal microscopy (Fig 2A and S1 and S2 Videos). These phenotypes, which were independent of the method used to label the cells (S1A and S1B Fig), were all reversed by complementation of the mutant with the wild-type gene at the endogenous locus.

The extremely high numbers of internalized mutant cells (Figs 1C and 2A) could potentially alter intracellular trafficking of C. neoformans, which is usually delivered to lysosomes after endocytosis [30]. To test this we used confocal microscopy to assess the progression of pfa4Δ and wild-type cells through various intracellular compartments after their exposure to host phagocytes (S2A Fig). The distribution of wild-type and mutant fungi between the cell surface (adherent cells), early endosomes (marked with EEA1), and lysosomes (marked with LAMP-1) was similar at late time points. The only significant differences were observed soon (15 min) after assay initiation, when a greater fraction of wild-type cells remained surface-accessible (adherent) while more mutant cells had already been phagocytosed (although not yet associated with EEA1). Overall, although the mutant is more efficiently internalized, both strains reach EAA1 and LAMP-1 compartments with similar dynamics. It has recently been suggested that C. neoformans-containing lysosomes do not completely acidify [31]. To test whether acidification differed between lysosomes containing pfa4Δ and wild-type yeast, we performed a phagocytosis assay in the presence of Lysotracker Red, a dye that becomes trapped and fluorescent in acidified organelles. We found that both strains were similarly distributed between unstained phagosomes and lysosomes (positive for Lysotracker; S2B and S2C Fig)
C. neoformans lacking PFA4 exhibits morphological defects and surface changes
In addition to an increased number of internalized pfa4Δ cells, our confocal studies revealed an unusual and dramatic change in their morphology (Fig 2A and S1 and S2 Videos). While wild-type cells are spherical, the mutant cells appeared to have collapsed in on themselves, manifested as membrane staining in either crescent shapes or double rings depending on cell orientation. This aberrant morphology occurred whether the fungi were inside macrophages (Fig 2A) or grown independently (Fig 2B), indicating that the alteration is intrinsic to the mutant rather than induced by the host cells. We tested other dyes to rule out the possibility that the shape change was due to the Lucifer Yellow (LY) stain used in our phagocytosis studies; in all cases we observed a similar phenotype (Fig 2B). Next, to eliminate the possibility that any compound that binds cell wall structures induces cell collapse, we imaged actively growing, unstained wild-type and pfa4Δ mutant cells by brightfield and differential interference contrast (DIC) light microscopy. Under these unstained, actively growing conditions we could easily detect the same aberrant shapes seen in pfa4Δ cells stained with various dyes (S3 Fig), indicating that they represent an intrinsic feature of this mutant. Finally, to get a detailed view of this morphological defect we examined the cells by scanning electron microscopy. Consistent with our light microscopy results, wild-type cells were globular and smooth while pfa4Δ cells were dramatically deformed (Fig 2C). Surprisingly, this has little effect on their ability to replicate at 30°C, where their growth rate is close to that of wild-type cells.
The pfa4Δ mutant showed altered initial interactions with host cells and aberrant morphology. One model that explains both observations is that the mutant has fundamental defects in cell wall structure that alter both surface molecule exposure and cell wall integrity. To probe cell wall organization, we used dye and lectin binding with flow cytometry to assess the accessibility of various cell wall components (Fig 3). We found that chitin accessibility, probed with calcofluor white (CFW), was not significantly altered in pfa4Δ, unlike the decreased signal in a chitin synthase mutant (chs3Δ) included as a control (Fig 3B). In contrast, probes of chitosan (Eosin Y; EoY) and mannans (Concanavalin A lectin; ConA) showed that these glycans were much more accessible in the pfa4Δ mutant (Fig 3B), supporting aberrant wall structure; this was also reflected in an altered staining pattern for ConA (S4 Fig). Similarly, LY and pontamine (Pont), also dyes that bind cell wall (although their specific targets are not defined), showed clear changes in binding the mutant compared to controls (Fig 3B). The abnormal exposure of chitosan and mannans at the surface of pfa4Δ cells could explain their greater recognition by macrophages (see Discussion).

We reasoned that the altered arrangement of cell wall components in the pfa4Δ mutant would threaten overall cell integrity. We tested this hypothesis by plating serial dilutions of pfa4Δ in the presence of various stressors. Compared to wild-type and the complemented mutant, pfa4Δ was sensitive to plasma membrane damaging agents (SDS and H2O2), osmotic stress (KCl and NaCl), cell wall binding dyes (CFW, CR, and LY), and elevated temperature (37°C) (Figs 4A and S5). Only temperature sensitivity could be rescued by sorbitol (Fig 4A), suggesting that the cell integrity defects and temperature sensitivity are caused by perturbation of different pathways. This experiment also indicates that Pfa4 is not absolutely required for growth at high temperatures; in support of this conclusion, the pfa4Δ cells continued to grow slowly at 37°C for over a day even in the absence of sorbitol (S5A Fig). The mutant was also hypersensitive to treatment with cell wall lysing enzymes (S5C Fig), an assay which probes cell wall stability as well as cellular response to cell wall damage [32]. In all cases genomic or plasmid complementation of pfa4Δ restored wild-type phenotypes.

The pleiotropic effects of PFA4 deletion suggested the dysfunction of one or more protein substrates of palmitoylation, which are not lipidated and therefore mislocalized, misfolded and/or degraded. To test whether the enzymatic activity of Pfa4 was indeed responsible for these phenotypes, we mutated its catalytic DHHC sequence to DHAS (S5D Fig); mutation of this cysteine abolishes PAT activity in other systems [12, 13, 16]. When both forms of the protein were expressed in pfa4Δ, only the wild-type rescued the mutant’s sensitivity to cell wall stress (S5E Fig), showing that the observed defects are due to a lack of PAT enzymatic activity.
The inability of pfa4Δ to withstand cell wall stress could reflect defects in cell wall structure, inability to respond to and repair a damaged wall, or both. To investigate cell wall structure we used transmission electron microscopy (TEM). The walls of wild-type strains and of pfa4Δ expressing wild-type PFA4 were fairly uniform in thickness, and showed the expected multilayered organization [33]: an electron-dense inner layer surrounded by a more electron-lucent layer and then an outer rim of capsule (Fig 4B; the capsule layer is thin because the strains were grown in rich medium). In these cells the inner layer was always ≥50% of the total wall width (example shown in Fig 4C, top image). In contrast, the cell walls of the mutant (with or without the catalytically-dead Pfa4AS) were generally thinner, primarily due to a reduction in the inner layer (Fig 4B and 4C, middle image). In ~80% of these cells the inner layer was <50% of the total wall width or was completely absent (Fig 4C, graph); in many of them the existing outer layer was also disorganized (Fig 4C, bottom image).
We next tested whether pfa4Δ cells have defects in cell wall stress signaling that render them unable to respond to environmental changes, by growing serial dilutions of wild-type, mutant, and mutants expressing either PFA4 or the catalytically-dead pfa4AS on media containing caffeine (S5E Fig). Caffeine stimulates the cAMP/PKA pathway, activating PKA1/2 and thereby mimicking cell wall stress. This chemical activation of the cell integrity pathway can be lethal if there is a preexisting defect in the pathway [34]. pfa4Δ could not grow under these conditions, consistent with a signaling defect in response to cell wall stress. Taken together, these results indicate that pfa4Δ cells have both altered cell wall structure and defective transduction of signals from the cell integrity pathway that would normally compensate for such changes. This results in a disordered wall with altered exposure of cell wall components, which in turn likely facilitates recognition by host cells (see Discussion).
A distinguishing feature and major virulence factor of C. neoformans is its polysaccharide capsule, which associates with the cell wall via α-glucan [33, 35]. We observed that pfa4Δ cells were clumpy in culture, a characteristic often seen in hypocapsular cryptococci that suggested these cells might have a capsule defect. Interestingly, this was not observed: the capsules of pfa4Δ cells were morphologically similar to those of wild-type under inducing conditions, although they were slightly smaller overall (Fig 4D).
Pfa4 is required for in vitro and in vivo virulence
C. neoformans survives and proliferates within macrophage phagolysosomes [31, 36, 37]. We assessed the behavior of pfa4Δ cells in this challenging environment and found that host phagocytes rapidly killed them (Fig 5A). In contrast, wild-type and complemented mutant cells showed robust growth in this context (Fig 5A), and even caused host cell numbers to decrease slightly (they were unperturbed by the pfa4Δ mutant).

We further tested the virulence of pfa4Δ in a mouse model of cryptococcosis, monitoring disease progression by weight loss. Infection with wild-type or the complemented mutant killed 50% of the mice in 16 days, with all animals steadily losing weight by about two weeks and succumbing to infection by day 18 (Fig 5B). In contrast, mice infected with pfa4Δ showed a modest (3–5%) and transient (days 8–14) weight loss early in infection, but recovered and grew normally until the study was terminated at day 45; no CFU were recovered from lung or brain at that time. This dramatic effect of a single PAT on fungal pathogenesis is unprecedented.
Identification of Pfa4-specific substrates
Despite the importance of palmitoylation to fundamental processes of cell biology [11], the palmitoylome of C. neoformans, like that of other fungal pathogens, has never been defined, with only one protein (Ras1) shown to be functionally palmitoylated [19]. The dramatic effects of PFA4 deletion, which our active-site mutation studies showed are due to lack of enzymatic activity, indicate that this lipid modification is crucial for cryptococcal cell integrity and virulence. To mechanistically explain these observations, we used fatty acid chemical reporter labeling and bioorthogonal chemistry proteomics to determine the specific set of proteins modified by Pfa4 [38–40]. In this method, cells are metabolically labeled with alk-16, a palmitic acid analog with an alkyne group, which is incorporated into proteins in place of the normal fatty acid (Fig 6A). Proteins modified with alk-16 can then be labeled with azide-functionalized reagents via ‘click chemistry’ for fluorescence detection or proteomic analysis (Fig 6A; [39–41]). We grew wild-type and pfa4Δ cells with alk-16, and then performed labeling reactions with azido-rhodamine for in-gel fluorescence detection (Fig 6B; [38]). The total protein profile of the mutant was similar to that of wild-type, although there were fewer species at high molecular weights; alk-16 labeling did not alter this pattern. The alk-16-labeled proteins of both strains also showed similar profiles, but with a slight decrease in the overall levels of modified proteins in the mutant (Fig 6B).
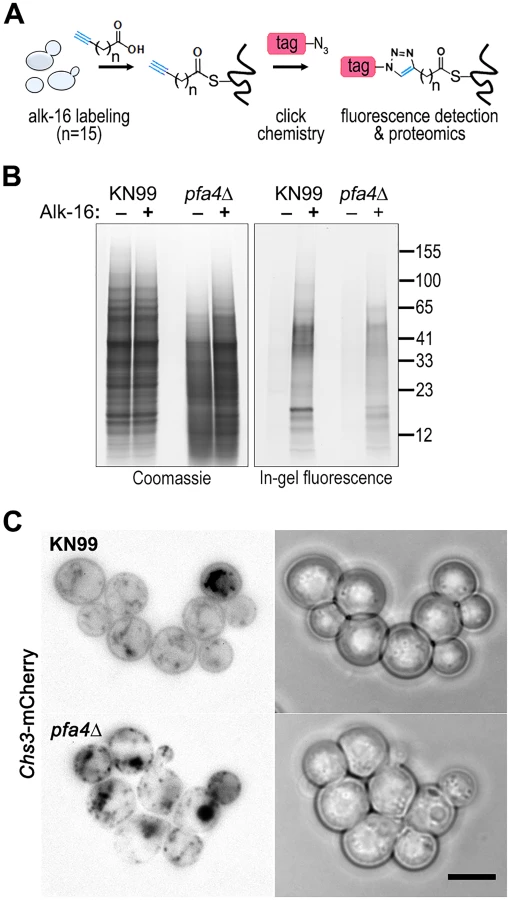
We next reacted alk-16-labeled proteins with azido-biotin, purified the biotinylated proteins with streptavidin beads, and evaluated Pfa4-specific alk-16-labeled proteins by comparative proteomics. Of the 427 proteins identified in two independent experiments with at least 2 unique peptides, 72 showed ≥5-fold enrichment in the wild-type compared to the pfa4Δ mutant in both studies (S1 File). High-confidence Pfa4 substrates included proteins that act in a variety of cell wall processes, including cell wall synthesis, membrane trafficking, signal transduction, and transport (Table 3). At the top of our list was chitin synthase 3 (Chs3), which has been characterized as a Pfa4 substrate in S. cerevisiae [42, 43]. Interestingly, a second chitin synthase (Chs1) is also a Pfa4 substrate.

A number of substrates from our Pfa4-dataset have homologs that are known to be palmitoylated in S. cerevisiae, although not necessarily by Pfa4; these include Sso1 and Sso2, Vac8, Gpa2, Yck1 and Yck2, and Env7 [28]. Some have homologs known to be palmitoylated in other systems, such as Rho11 [44] and Vac8 [16]. Notably, C. neoformans Ras1 was labeled by alk-16 independent of Pfa4 (S1 File). This is consistent with previous studies in budding and fission yeast demonstrating that Ras1 is also a substrate of the Erf2/4 PAT complex, which is intact in our mutant [28, 41, 45]. Together, our results indicate that Pfa4 does not significantly alter global levels of fatty-acylation in C. neoformans, but palmitoylates specific proteins central to stress resistance and consequently to virulence, despite the presence of six other probable PAT genes in the cryptococcal genome.
Chs3 is critical for normal wall synthesis and maintenance [32, 46]. The discovery that it is a major substrate of Pfa4 is consistent with the multiple cell wall-related defects we observed in the pfa4Δ mutant, and explains how Pfa4 influences cell morphology, integrity, and consequently virulence. To establish a direct link between Pfa4-mediated palmitoylation and Chs3 function, we generated strains expressing Chs3-mCherry from the endogenous locus in both wild-type and pfa4Δ backgrounds. Palmitoylated Chs3 localized to internal compartments and to the plasma membrane, seen as a homogeneous rim outlining the cells (Fig 6C, top panel). In contrast, in the pfa4Δ cells, Chs3 is restricted to internal membranes, with occasional staining of vacuoles, suggestive of degradation (Fig 6C, bottom panel). This mislocalization of Chs3 is consistent with lack of palmitoylation of this protein and the cell wall-related defects observed in pfa4Δ cells. Interestingly, pfa4Δ and chs3Δ cells do not have completely congruent phenotypes. For example, chs3Δ does not exhibit the increased phagocytosis that first brought pfa4Δ to our attention (Fig 7A), although both strains show increased sensitivity to some cell wall stressors (Fig 7B) and poor retention of melanin at the cell wall (Fig 7C–7E, and [32]). Differences between the two mutants are likely due to the redundancy of both chitin synthases and palmitoyltransferases in C. neoformans, as well as the reduced palmitoylation of other Pfa4 substrates in the pfa4Δ mutant (see Discussion).

Discussion
Phagocytes play multiple roles in cryptococcal pathogenesis, destroying fungi under some circumstances but also potentially harboring them and enabling them to survive, proliferate, and disseminate [1, 36]. Some outcomes of cryptococcal interactions with macrophages, including fungal engulfment and intracellular proliferation, correlate highly with patient outcome [7, 8]. These observations make host-pathogen interactions a compelling area of study, and raise the question of whether they might present feasible targets for antifungal therapy. Pursuing this question, however, requires mechanistic understanding of these events from the vantage point of both host and pathogen.
As a first step in such investigations, we used a high-content imaging-based assay to screen 1,201 C. neoformans mutants (corresponding to ~17% of the genome). We found 56 mutants that showed significantly altered uptake by host cells, including 29 lacking genes of unknown function that have not previously been investigated. Many of the mutants showing increased engulfment had been reported to be defective in host-pathogen interactions in other models; this validated our screen and provided strong support for uncharacterized hits. The genes deleted in several of the high-uptake mutants encode proteins involved in synthesis or remodeling of the cell wall and/or capsule, surface structures that interact most directly with host cells. Others encode signaling molecules or transcription factors involved in the response to environmental changes, such as would be encountered during infection. Intriguingly, most of the hits with reduced engulfment, more than half of which encode proteins with no known homologs in S. cerevisiae, have never been investigated. Future studies defining their biological roles should increase our understanding of C. neoformans’ interactions with host cells. Notably, the level of engulfment has no simple relationship to overall virulence in animal models, perhaps illustrating the complex role of phagocytosis in cryptococcal infection [36, 47]. For example, two hypervirulent mutants [10] showed opposite uptake results, with one (9A12) very poorly internalized while the other (2G9; lacking RIM101) was avidly engulfed.
One mutant that demonstrated increased uptake by phagocytes lacks PFA4, which encodes a protein containing the well-characterized DHHC domain characteristic of PAT enzymes. PATs catalyze the post-translational addition of palmitate to proteins, a reversible modification that can influence the localization, stability, and/or function of their substrates. The C. neoformans H99 genome contains seven genes encoding DHHC-domain proteins, and functional redundancy is common in this family of enzymes. It was therefore surprising that deletion of PFA4 had such a dramatic effect on C. neoformans morphology, stress sensitivity, and virulence. This suggested that Pfa4 modifies specific substrates that are critical in cryptococcal biology. For this reason, and because of the recent attention to PATs as potential antimicrobial drug targets [48, 49], we investigated the mechanism(s) by which lack of Pfa4 causes these phenotypes.
We postulated that Pfa4 was the primary or sole PAT modifying important cryptococcal proteins required for cell integrity and virulence. Our proteomic analysis supported this hypothesis, identifying 72 proteins as preferentially palmitoylated by Pfa4 (Table 3 and S1 File). As in S. cerevisiae [42], Chs3 is a key Pfa4 target. This protein is one of eight cryptococcal chitin synthases and is responsible for synthesizing the majority of cellular chitin during vegetative growth [32, 46]. If Chs3 does not properly localize and act in pfa4Δ cells as a result of lacking palmitoylation, one would expect to see cell walls with reduced chitin and consequently impaired function. This is exactly what we observe: Chs3-mCherry in the mutant is mostly restricted to internal membranes and is depleted from the plasma membrane compared to in WT cells (Fig 6C); as a consequence, the inner layer of the cell wall, which corresponds to the layer containing chitin [33], is markedly reduced. Chs1, another class IV chitin synthase, is also preferentially modified by Pfa4 and may contribute to these cell wall defects.
Beyond altered chitin synthase activity, cell wall production is likely further compromised in pfa4Δ cells secondary to defects in intracellular traffic. Pfa4 substrates that we identified include several proteins involved in protein secretion that are known to be palmitoylated in S. cerevisiae (Table 3) or other organisms. Since multiple proteins involved in cell wall biogenesis are membrane proteins that travel to their site of action in secretory vesicles, dysfunction of SNARES or other proteins involved in this transport could alter cell surface composition via partial blockade or mislocalization of vesicle cargo.
Aberrant cell wall synthesis probably causes the dramatically altered morphology of pfa4Δ cells (Figs 2 and S3). Such changes were previously only seen in dying cryptococci that had been exposed to harsh conditions, such as digestion with lysosomal extracts in vitro or extended growth in infected animals [50, 51]. In contrast, pfa4Δ shows wall collapse even during normal growth in culture in the absence of any stains or exogenous compounds. Mutant cells are also hypersensitive to salt and sorbitol, suggesting defects in regulating turgor pressure. Regulatory disturbance is further suggested by the sensitivity of pfa4Δ to caffeine, which activates the cell integrity pathway. These phenotypes are consistent with our identification of several proteins that function in signal transduction as Pfa4 substrates (Table 3). These include Rho11, a GTPase that acts in cell integrity signaling via the MAP kinase pathway [52], and an uncharacterized protein similar to Rho GTPase activating protein (GAP) that may be the Rho11 GAP. Mislocalization of these proteins would likely impair the cellular response to cell wall damage. We also identified the α subunit of the large G-protein Gpa1 as a Pfa4 substrate; this protein is upstream of cryptococcal cAMP signaling and is involved in pheromone and mating responses [53], which could explain the mating defects when pfa4Δ strains are crossed to each other (S6 Fig). Perturbation of multiple signaling pathways due to lack of Pfa4 severely limits the mutant cells’ ability to respond appropriately to changing environmental conditions, exacerbating the effects of defective wall synthesis and undermining mutant survival in the host.
We considered the possibility that the increased uptake of pfa4Δ cells by host phagocytes reflected inviability of the yeast. However, we ruled out this possibility by demonstrating viability of the mutant under conditions of our uptake assays (S5 Fig). Furthermore, killing fungi by treatment with heat, ethanol, or azide did not alter uptake of wild-type (S1C Fig) or mutant cells.
The combination of impaired cell wall synthesis and inability to appropriately respond to this condition results in weak and disorganized walls. This may impair other key virulence attributes of C. neoformans, such as the polysaccharide capsule, which associates with the cell wall. A perturbed wall, even in cells where the capsule is only slightly reduced in radius (as with pfa4Δ), may alter the capsule so that it cannot maintain its normal antiphagocytic role and exposes underlying wall components. This, combined with the changed wall arrangement, could explain our observation of abnormally high surface accessibility of specific cell wall components (Fig 3). These included cell wall mannoproteins [54, 55], which can interact with host phagocyte mannose receptors, and chitosan, which also interacts with macrophage receptors and induces a robust inflammatory response [56, 57]. Greater accessibility of these glycans could in turn explain the increased phagocytosis of pfa4Δ cells by macrophages. Once engulfed, these less robust cells, with defects in cell wall, signal transduction, and virulence factor expression, fare poorly (Fig 5A). Potentially reducing the pathogenicity even of cryptococci that remain outside of host phagocytes, we found important membrane transporters that are not correctly palmitoylated in the mutant. These include a putative carbohydrate transporter, a phosphate transporter, and NIC1 and SIT1, which transport nickel and siderophore-iron complexes, respectively [58, 59]. Because these metals are limiting during infection, incorrect processing or targeting of their transporters could influence pathogenesis. Furthermore, melanin, an important virulence factor in this pathogen, is poorly retained at the cell wall (Fig 7C–7E), a phenotype also seen in chs3Δ cells [32] and associated with reduced virulence. Ultimately, all of these factors combine to result in avirulence of the pfa4Δ mutant (Fig 8).

In contrast to our findings, the initial survey of cryptococcal deletion mutants [10] categorized the strain deleted for PFA4 (2A12) as normal in virulence. This may reflect the practical strategy used in that large-scale study, where pools of mutants were assayed, or the timing of those virulence studies. We did observe that animals infected with pfa4Δ initially show mild symptoms of disease, suggesting the initiation of a pathogenic process that might be interpreted as normal infectivity in short-term studies (as in ref. [10]), but that they subsequently clear the infection and recover completely. Consistent with our observations, 2A12 does show reduced virulence in recent studies of this deletion library using both an IV mouse model and invertebrate models of infection (performed at room temperature) [60]. The latter also supports our conclusion above that Pfa4’s contribution to virulence is temperature independent.
As well as demonstrating the key role of Pfa4-dependent palmitoylation in C. neoformans, our work provides valuable data sets to the community from both our screen and our palmitoylome analysis. It also bolsters a model that explains why multiple PATs have been retained during evolution despite the widespread redundancy of these enzymes: key PATs like Pfa4 may modify specific substrates that perform critical functions, in addition to sharing substrates with other PATs. This concept is supported by the recent identification of specific PATs that regulate central pathogenic processes in Toxoplasma and Plasmodium [16–18].
Our determination of the Pfa4-palmitoylome offers new insights into the role of an important regulatory lipid modification in the biology of C. neoformans. Pfa4 in C. neoformans is notable in modifying proteins that exhibit diverse modes of membrane association, including those that are otherwise predicted to be soluble or to have one or many membrane-spanning domains (Table 3). In contrast, its S. cerevisiae homolog appears to be restricted to modifying polytopic membrane proteins [28, 42]. Several cryptococcal Pfa4 substrates are also fungal-specific (e.g., the chitin synthases, vacuolar protein 8, the nickel and siderophore transporters, and the product of CNAG_02114). Finally, cryptococcal Pfa4 is unique among PATs studied to date in that it is essential for virulence in an animal model. The closest human homolog of Pfa4, DHHC6, is considerably distant in sequence homology, with the similarity restricted to the catalytic DHHC domain. These findings encourage efforts towards development of specific PAT inhibitors as novel avenues for therapeutics.
Materials and Methods
Strains, growth conditions, and reagents
Strains used were C. neoformans serotype A strain H99α, its derivative KN99α, and deletions in these backgrounds (see below). The cryptococcal partial deletion collection in H99α [10] was purchased from the Fungal Genetics Stock Center (University of Missouri, Kansas City, MO) and H99α chs3Δ was a generous gift from Jennifer Lodge (Washington University). Fungal strains were maintained at -80°C and grown at 30°C on yeast peptone dextrose (YPD) with antibiotics as appropriate (100 μg/mL of nourseothricin (clonNAT, WERNER BioAgents, Germany) or 100 μg/mL G418 (Geneticin, Life Technologies, USA)).
The human monocytic cell line THP-1 (ATCC TIB-202) was grown in THP-1 complete media (RPMI-1640 with L-glutamine supplemented with 1 mM sodium pyruvate, 0.05% 2-mercaptoethanol, 10% FBS, and 100 units/mL Penicillin - 100 μg/mL Streptomycin solution as indicated) and differentiated with phorbol 12-myristate 12-acetate (PMA, from Sigma, St. Louis, MO) as described in [9]. THP-1 cultures were split every 3–4 days (inoculum of 105 cells/mL) and new batches were thawed every month.
All tissue culture plasticware and media were from BD Falcon and Sigma, fungal media components from Difco, PCR primers from Sigma, biolistic transformation reagents and materials from Bio-Rad, DH5α cells from Life Technologies, and restriction enzymes from New England Biolabs. Reagents for electron microscopy were from Ted Pella (Redding, CA) and Polysciences (Warrington, PA); antibodies for immunofluorescence were from Abcam (ab2900, anti-EEA1 rabbit polyclonal) or the Developmental Studies Hybridoma Bank (clone H4A3, University of Iowa); and antibodies for immunoblotting were from Sigma (clone HA-7 anti-HA mouse monoclonal and anti-FLAG rabbit polyclonal). Reagents for bioorthogonal labeling and click chemistry were from Sigma, except for azido-rhodamine, which was prepared as previously described [38].
Library screening
To screen fungal mutants, THP-1 cells were seeded in 96-well plates (3.33 ×105 cells/mL, 100 μL), incubated for 48 hr (37°C, 5% CO2) in THP-1 complete media, washed three times with 150 μL RPMI-1640, and cultured for one day in serum-free media with antibiotics. In parallel a 96-pin replicator (Nalge Nunc International, Rochester, NY) was used to inoculate strains from the C. neoformans deletion collection into a Nunc Edge—96 well microplate containing 150 μL YPD per well. The microplates were incubated at 30°C overnight on a mini-orbital shaker (BELLCO Biotechnology, Vineland, NJ), followed by transfer of a 35 μL aliquot from each well into a new 96-well flat-bottom microplate (Costar 3904). The transferred cells were washed once with PBS (pH 7.5), once in Mcllvaine’s buffer (pH 6.0), and then resuspended in 100 μL of the same buffer containing 100 μg/mL Lucifer Yellow dye (Sigma L0144). After a 30 min incubation at RT with gentle agitation the cells were collected, washed once with PBS, and opsonized (30 min, 37°C) in 100 μL 40% human serum with gentle agitation. Serum was obtained from healthy donors with informed consent under a protocol approved by the Washington University in St. Louis Institutional Review Board. Finally, the cells were washed three times with PBS, resuspended in 150 μL RPMI-1640, and 35 μL from each well was diluted into 1 mL of pre-warmed RPMI-1640 in a deep-well 96-well plate (Nunc). To initiate the assay, the medium from each well containing THP-1 was aspirated and replaced by 100 μL of the cryptococcal suspension. After a 1 hr incubation (37°C, 5% CO2) the plates were washed vigorously four times with 150 μL PBS using a microplate washer (ELx405TM Select CW, Biotek, Winooski, VT). The samples were then immediately fixed in 150 μL 4% formaldehyde (20 min, 4°C), washed twice with PBS, and permeabilized for 20 min at RT with 0.1% saponin in PBS (150 μL). Samples were next washed twice with PBS, stained (15 min, RT, in the dark) with 2 μg/mL DAPI and 0.25 μg/mL HCS CellMask Deep Red (Life Technologies) in PBS, washed twice more with PBS, and 100 μL of 10 mM NaN3 in PBS was added to each well. Plates were either imaged immediately (on an IN Cell 1000 analyzer, GE, Piscataway, NJ) or stored at 4°C for later analysis. GE INCell Investigator Developer Software was used to identify host cell and fungal borders and calculate the overlap as described in [9]. Fungal cells that overlapped >50% with host cell bodies were considered internalized, ≤50% were considered adherent, and fungal cells with no overlap were not counted. In parallel with the screening assay, an aliquot of each fungal cell suspension was pipetted into empty 96-well plates for enumeration to allow normalization of results to fungal cell number (macrophage uptake of C. neoformans is linear in the range of fungal concentrations used in these assays [9]). The results were analyzed plate-wise (to reveal any systematic errors in different plates) before normalization and calculation of values relative to wild-type.
Fungal genome manipulation
We used the split marker method [61] to delete PFA4 (CNAG_03981) in H99α and KN99α after amplifying NAT resistance split marker fragments from genomic DNA of strain 2A12 (pfa4Δ) from the Madhani deletion collection. For chromosomal complementation, we used a split marker approach to replace the NAT cassette of the mutant with wild-type genomic PFA4 sequences in tandem with a G418 resistance cassette. For endogenous tagging of the CHS3 gene (CNAG_05581) with mCherry, the last 1,674 bp of CHS3 were amplified with a BamHI site replacing the STOP codon and ligated to a BamHI/AvrII-cut fragment composed of mCherry followed by an HA epitope, a STOP codon, and 445 bp of the TRP1 terminator. The ligated fragment was cloned in front of a NAT resistance cassette and 616 bp of the CHS3 terminator (sequences immediately following the STOP codon) were subsequently cloned after the NAT cassette. The resulting plasmid was digested with BglII/MluI to release the 5’ fragment of the split marker and with XmaI/EcoRV to release the 3’ fragment of the split marker. Transformation was by biolistics (Bio-Rad PDS-1000/He) as described in [62].
For plasmid construction, a fragment encompassing the PFA4 coding locus and 226 bp of 3’ sequence was amplified so as to incorporate sequence that encodes 1.5X HA epitope tags in place of the first 2 codons. Fusion PCR was used to ligate this fragment to a second amplicon consisting of 900 bp of 5’ sequence (including the starting ATG) and sequence encoding 1.5X HA epitope tags, so as to reconstitute sequence encoding an N-terminal 3X HA-tagged Pfa4 sequence. This product (~3.5 kb) was cloned into ApaI/KpnI-digested pIBB103 [63] for expression and also used as template for mutagenesis of the DHHC motif into DHAS using overlapping primers containing the codon change. Plasmid transformation was as described in [63].
Fluorescence microscopy and flow cytometry
Cells were grown overnight at 30°C in YPD (with appropriate antibiotics if needed to maintain plasmids), diluted as for phenotyping, washed in PBS, and resuspended at 107/mL for staining as follows (all manipulations at RT): For LY and EoY (Sigma), cells were washed once in McIlvaines buffer, pH 6.0; resuspended in the same; and incubated for ~15 min with 250 μg/mL of the dye. For CFW (Fluorescent Brightener 28, Sigma), UV2B (Polysciences, Inc.) and Pont (Pontamine fast scarlet 4B, Bayer Corp., Robinson, PA), cells were stained in PBS with 100 μg/mL of CFW or UV2B or a 1 : 10,000 dilution of Pont (w/v). For ConA-FITC (Sigma), cells were stained with 30 μg/mL in Hepes-buffered saline, pH 7.0, containing 10 mM each MgCl2 and CaCl2.
For fluorescence microscopy, stained cells were washed twice, resuspended in the same volume of the corresponding buffer, mixed vigorously, spotted onto glass slides, covered, and imaged immediately on a wide field Zeiss Axioskop 2 MOT Plus with appropriate filters (DAPI for CFW and UV2B; FITC for LY, EoY, and ConA-FITC; and Texas Red for Pont). For the Chs3-mCherry strains, overnight cultures grown in YPD were washed twice with PBS, resuspended in 3 mL of PBS, and 6 μl were spotted on polylysine-coated glass slides and imaged immediately. For flow cytometry cells were washed three times, fixed in 3.7% formaldehyde/PBS (10 min; RT) or resuspended in PBS with 10 mM NaN3 and analyzed on an LSRII flow cytometer (Becton Dickinson, Franklin Lakes, NJ) for analysis using FlowJo software (Tree Star Inc., Ashland, OR).
Electron microscopy (EM)
For transmission EM, overnight cultures grown in YPD were diluted 10-fold, grown to OD600 = 0.2 (~107/mL), and washed twice in PBS. The cell pellet was resuspended in 1 mL of primary fixation mix (2.5% paraformaldehyde/2% glutaraldehyde in 100 mM cacodylate buffer, pH 7.2), incubated for 1 hr at room temperature (RT), washed in the same buffer, and post-fixed in 1% osmium tetroxide (Polysciences, Inc.) for 1 hr at RT. Samples were then rinsed in the same buffer, followed by dehydration in a graded series of ethanol and propylene oxide prior to embedding in Eponate 12 resin (Ted Pella, Inc.). Sections of 90 nm were cut with a Leica Ultracut UCT ultramicrotome (Leica Microsystems, Inc., Bannockburn, IL), stained with uranyl acetate and lead citrate, and viewed on a JEOL 1200EX transmission electron microscope (JEOL USA Inc., Peabody, MA) equipped with an AMT 8 megapixel digital camera (Advanced Microscopy Techniques, Woburn, MA).
For scanning EM, cultures were grown and fixed as above but in sodium phosphate buffer, then washed and 8.8 x 106 cells (4 x 106 cells/cm2) were added to wells of a 6-well plate containing a polylysine-coated plastic coverslip. After incubation at 4°C for 1–2 hr the coverslips were washed twice with DPBS, re-fixed in 2% paraformaldehyde, 2.5% glutaraldehyde in 0.1 M Sorensen’s sodium phosphate buffer (potassium-free, pH 7.4), and then sequentially rinsed in buffer and NanoPure Ultra-filtered deionized water. They were next post-fixed in 1% osmium tetroxide (aqueous) for 1 hr, rinsed with NanoPure Ultra-filtered deionized water, dehydrated in ethanol (30%, 50%, 70%, 80%, 90%, 3X 95%, and 3X absolute ethanol), critical point dried (Tousimis Samdri-780, Rockville, MD) via liquid carbon dioxide, mounted on aluminum stubs with double-sided adhesive carbon tabs, and sputter coated (Tousimis Samsputter-2a) with gold-palladium. Images were acquired using a Hitachi S2600 (Hitachi-hitec, Tokyo, Japan) instrument.
Phenotyping
Strains to be tested were grown overnight in YPD, diluted to ~2 x 106/ml, and grown for two doublings. The cultures were then serially diluted (10-fold) and spotted (5 μL) onto buffered (pH 6.8 with 100 mM KPO4 buffer) synthetic dextrose medium with 1 mg/mL calcofluor white or onto YPD with 1.2 M NaCl; 1.2 M KCl; 0.01 and 0.03% SDS; 1, 3, and 5 mM H2O2; 0.1, 0.25, 0.5, 0.75, and 1 mg/mL caffeine; 1 mg/mL Congo red (stock prepared in 70% ethanol); or 25 μg/mL Lucifer Yellow. The same plates were also prepared containing various concentrations of sorbitol (0.5, 1, or 1.5 M). Plates were incubated at 30°C and 37°C for 3–4 days. Sensitivity to lysing enzymes was tested as in [64].
Capsule induction
Cells were grown overnight in YPD, washed twice with DMEM, and 1 mL aliquots (106 cells) were pipetted into 24-well tissue culture plates (3 wells per strain) and incubated (37°C; 5% CO2) for 24 hr. The suspension was washed twice with deionized water (dH2O), resuspended in 24 μL dH2O, mixed with ~8 μL of India ink and visualized on a Zeiss Axioskop 2 MOT Plus microscope. At least 150 cells from each strain (50 per well) were analyzed with ImageJ (NIH) for capsule thickness ((outer capsule diameter minus cell wall diameter)/2).
Melanin assays
For solid media assays, the cells were grown overnight in YPD medium at 30°C, diluted the next morning in 5 mL of YPD, grown to an OD600 of 0.2, washed twice in PBS, and adjusted to 107 cells/mL in PBS. 10-fold serial dilutions were made and 5 μL of each dilution spotted on L-DOPA (1 mM) plates. The plates were incubated at 25°C, 30°C, and 37°C for 3–4 days in the dark.
For assays in liquid medium, cells of each strain were grown similarly overnight, diluted in 25 mL of YPD, and allowed to grow for 2–3 generations. At that point, the cells were washed in PBS, resuspended in 2 mL glucose-free asparagine and salts media, and the cell density was quantified. The strains were adjusted to 5 x 107 cells/ml and incubated at 25°C for 18–24 hr in asparagine medium containing 1 mM L-DOPA. The cultures were spun down at 1000xg for 10 min, and photographed. To quantify the melanin in the media, the OD405 was measured for 100 μL aliquots of the supernatant fractions.
Virulence assays
To assess fungal survival in macrophages, THP-1 cells grown in 12-well plates (250,000 cells per well), were washed with assay medium (RPMI + 1% FBS). In parallel, overnight fungal cultures (OD600 = 0.2–0.4; 1–2 x 107/mL) were washed twice with PBS, resuspended (108 cells/mL) in 40% human serum for opsonization (37°C; 30 min; with rotation), rewashed, resuspended in assay medium, and added to the THP-1 cells at an MOI of 0.1 or 1.0 as indicated. Plates were incubated for 1 hr, rinsed twice with 1 mL Dulbecco’s PBS (DPBS), and incubation continued for 0 hours (to measure initial association) or for the time indicated after addition of RPMI + 1%FBS. At the desired assay time points the medium was aspirated, wells were washed once with 1 mL DPBS, 1 mL of lysis buffer (0.05% SDS, 1 mM EDTA) was added, and the plate was shaken on a plate mixer for ~3 min. The resulting lysate was collected, vortexed vigorously, diluted, and spotted onto YPD media for determination of colony forming units (CFU).
To test virulence in mice, strains were cultured overnight in YPD, collected, washed in PBS, diluted to 106 cells/mL in PBS, and briefly sonicated (to disperse clumps seen in pfa4Δ). Sonication did not adversely affect mutant viability. Aliquots (50 μL) of the suspension were used to intranasally inoculate groups of ten mice (4–6 week-old female A/Jcr mice; National Cancer Institute) and dilutions of the suspension were plated immediately after infection to confirm inocula. Animals were monitored closely and sacrificed if they lost >20% relative to peak weight or at the end of the experiment (45 days). Homogenates of lungs and brains from 3 of the surviving mice infected with pfa4Δ were plated to determine organ burden.
Alk-16 labeling and click chemistry
Whole cell lysates (50 μg) were diluted with SDS buffer (4% SDS, 150 mM NaCl, 50 mM triethanolamine pH 7.4, Roche EDTA-free protease inhibitor cocktail) to 44.5 μL and then reacted with 5.5 μL freshly prepared click chemistry reaction cocktail containing azido-rhodamine (1 μL, 10 mM stock solution in DMSO), tris(2-carboxyethyl)phosphine hydrochloride (TCEP) (1 μL, 50 mM freshly prepared stock solution in deionized water), tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA) (2.5 μL, 10 mM stock solution in DMSO/t-butanol) and CuSO4•5H2O (1 μL, 50 mM freshly prepared stock solution in deionized water)] for 1 h at room temperature. The click reactions were terminated by the addition of ice-cold methanol (1 mL). The mixtures were placed at −20°C overnight and then centrifuged at 18,000×g for 20 min at 4°C to precipitate proteins. The supernatants from the samples were discarded. The protein pellets were washed with methanol twice, allowed to air-dry for 10 min, resuspended in 35 μL of SDS lysis buffer, and diluted with 12.5 μL 4× reducing SDS-loading buffer (40% glycerol, 200 mM Tris-HCl pH 6.8, 8% SDS, 0.4% bromophenol blue) and 2.5 μL 2-mercaptoethanol. The resulting samples were heated for 5 min at 95°C and resolved on 4–20% SDS-PAGE gels (Bio-Rad). For in-gel fluorescence scanning, the gels were destained in 40% methanol, 10% acetic acid for at least 1 h, and then scanned on a GE Healthcare Typhoon 9400 variable mode imager with excitation and emission at 532 nm and 580 nm, respectively. After scanning, gels were also stained with Coomassie Brilliant Blue (Bio-Rad).
Affinity enrichment of palmitoylated proteins and mass spectrometry
For affinity purification of alk-16-modified proteins, 2 mg of cell lysates labeled with alk-16 were subjected to Cu(I)-catalyzed click reaction as described above, except that azido-biotin was substituted for azido-rhodamine. Methanol-precipitated and washed protein pellets were resuspended in 200 μL of 4% SDS buffer (50 mM TEA, 150 mM NaCl, pH 7.4). Equal amounts of protein for each sample were diluted 1/4 by volume with 50 mM TEA buffer (150 mM NaCl, pH 7.4). 60 μl prewashed streptavidin agarose beads (Invitrogen) were added to each sample and the protein and bead mixtures were incubated for 1 h at room temperature on a nutating mixer. The beads were then washed once with PBS and 0.2% (w/v) SDS, three times with PBS and twice with 250 mM ammonium bicarbonate (ABC). Beads were resuspended in 500 μl 8 M urea, reduced with 10 mM DTT for 30 min, and then alkylated with 50 mM iodoacetamide in the dark for another 30 min. Finally, the beads were washed with 25 mM ammonium bicarbonate three times and digested with 0.5 μg of trypsin at 37°C overnight. The supernatant of each sample was collected, dried, and solubilized in 5% acetonitrile/1% formic acid for LC-MS analysis.
LC-MS analysis was performed with a Dionex 3000 nano-HPLC coupled to an Orbitrap XL mass spectrometer (ThermoFisher). Peptide samples were pressure-loaded onto a home-made C18 reverse-phase column (75 μm diameter, 15 cm length). A 180-minute gradient increasing from 95% buffer A (HPLC grade water with 0.1% formic acid) and 5% buffer B (HPLC grade acetonitrile with 0.1% formic acid) to 75% buffer B in 133 minutes was used at 200 nL/min. The Orbitrap XL was operated in top-8-CID-mode with MS spectra measured at a resolution of 60,000@m/z 400. One full MS scan (300–2000 MW) was followed by three data-dependent scans of the nth most intense ions with dynamic exclusion enabled. Acquired tandem MS spectra were extracted using ProteomeDiscoverer v.1.4.0.288 (Thermo, Bremen, Germany) and queried against the Uniprot complete Cryptococcus neoformans var. grubii H99 proteome (UP000010091) database concatenated with common known contaminants using MASCOT v.2.3.02 (Matrixscience, London, UK). Peptides fulfilling a Percolator calculated 1% false discovery rate (FDR) threshold were reported. The abundance of an identified protein was calculated based on the average area of its three most abundant peptides. For a protein to be considered a Pfa4-specific substrate, it had to be at least five-fold more abundant in the wild-type sample compared to the pfa4Δ sample as measured by protein abundance in both of the two independent experiments and be identified with at least two unique peptides.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Sabiiti W, May RC. Mechanisms of infection by the human fungal pathogen Cryptococcus neoformans. Future Microbiol. 2012;7(11):1297–313. Epub 2012/10/19. doi: 10.2217/fmb.12.102 23075448
2. Pyrgos V, Seitz AE, Steiner CA, Prevots DR, Williamson PR. Epidemiology of cryptococcal meningitis in the US: 1997–2009. PLoS One. 2013;8(2):e56269. Epub 2013/03/05. doi: 10.1371/journal.pone.0056269 23457543
3. Pappas PG. Cryptococcal infections in non-HIV-infected patients. Trans Am Clin Climatol Assoc. 2013;124 : 61–79. Epub 2013/07/23. PMCID: 3715903. 23874010
4. Bratton EW, El Husseini N, Chastain CA, Lee MS, Poole C, Sturmer T, et al. Comparison and temporal trends of three groups with cryptococcosis: HIV-infected, solid organ transplant, and HIV-negative/non-transplant. PLoS One. 2012;7(8):e43582. Epub 2012/09/01. doi: 10.1371/journal.pone.0043582 22937064
5. Mirza SA, Phelan M, Rimland D, Graviss E, Hamill R, Brandt ME, et al. The changing epidemiology of cryptococcosis: an update from population-based active surveillance in 2 large metropolitan areas, 1992–2000. Clin Infect Dis. 2003;36(6):789–94. Epub 2003/03/11. doi: 10.1086/368091 12627365
6. Wozniak KL, Hardison S, Olszewski M, Wormley FL Jr. Induction of protective immunity against cryptococcosis. Mycopathologia. 2012;173(5–6):387–94. Epub 2011/12/07. doi: 10.1007/s11046-011-9505-8 22354778
7. Alanio A, Desnos-Ollivier M, Dromer F. Dynamics of Cryptococcus neoformans-macrophage interactions reveal that fungal background influences outcome during cryptococcal meningoencephalitis in humans. MBio. 2011;2(4). Epub 2011/08/11. doi: 10.1128/mBio.00158-11
8. Sabiiti W, Robertson E, Beale MA, Johnston SA, Brouwer AE, Loyse A, et al. Efficient phagocytosis and laccase activity affect the outcome of HIV-associated cryptococcosis. J Clin Invest. 2014;124(5):2000–8. Epub 2014/04/20. doi: 10.1172/JCI72950 24743149
9. Srikanta D, Yang M, Williams M, Doering TL. A sensitive high-throughput assay for evaluating host-pathogen interactions in Cryptococcus neoformans infection. PLoS One. 2011;6(7):e22773. Epub 2011/08/11. doi: 10.1371/journal.pone.0022773 21829509
10. Liu OW, Chun CD, Chow ED, Chen C, Madhani HD, Noble SM. Systematic genetic analysis of virulence in the human fungal pathogen Cryptococcus neoformans. Cell. 2008;135(1):174–88. Epub 2008/10/16. doi: 10.1016/j.cell.2008.07.046 18854164
11. Linder ME, Jennings BC. Mechanism and function of DHHC S-acyltransferases. Biochem Soc Trans. 2013;41(1):29–34. Epub 2013/01/30. doi: 10.1042/BST20120328 23356254
12. Lobo S, Greentree WK, Linder ME, Deschenes RJ. Identification of a Ras palmitoyltransferase in Saccharomyces cerevisiae. J Biol Chem. 2002;277(43):41268–73. Epub 2002/08/24. doi: 10.1074/jbc.M206573200 12193598
13. Roth AF, Feng Y, Chen L, Davis NG. The yeast DHHC cysteine-rich domain protein Akr1p is a palmitoyl transferase. J Cell Biol. 2002;159(1):23–8. Epub 2002/10/09. doi: 10.1083/jcb.200206120 12370247
14. Veit M, Serebryakova MV, Kordyukova LV. Palmitoylation of influenza virus proteins. Biochem Soc Trans. 2013;41(1):50–5. Epub 2013/01/30. doi: 10.1042/BST20120210 23356257
15. Hicks SW, Charron G, Hang HC, Galan JE. Subcellular targeting of Salmonella virulence proteins by host-mediated S-palmitoylation. Cell Host Microbe. 2011;10(1):9–20. Epub 2011/07/20. doi: 10.1016/j.chom.2011.06.003 21767808
16. Beck JR, Fung C, Straub KW, Coppens I, Vashisht AA, Wohlschlegel JA, et al. A Toxoplasma palmitoyl acyl transferase and the palmitoylated armadillo repeat protein TgARO govern apical rhoptry tethering and reveal a critical role for the rhoptries in host cell invasion but not egress. PLoS Pathog. 2013;9(2):e1003162. Epub 2013/02/15. doi: 10.1371/journal.ppat.1003162 23408890
17. Frenal K, Tay CL, Mueller C, Bushell ES, Jia Y, Graindorge A, et al. Global analysis of apicomplexan protein S-acyl transferases reveals an enzyme essential for invasion. Traffic. 2013;14(8):895–911. Epub 2013/05/04. doi: 10.1111/tra.12081 23638681
18. Jones ML, Collins MO, Goulding D, Choudhary JS, Rayner JC. Analysis of protein palmitoylation reveals a pervasive role in Plasmodium development and pathogenesis. Cell Host Microbe. 2012;12(2):246–58. Epub 2012/08/21. doi: 10.1016/j.chom.2012.06.005 22901544
19. Nichols CB, Ferreyra J, Ballou ER, Alspaugh JA. Subcellular localization directs signaling specificity of the Cryptococcus neoformans Ras1 protein. Eukaryot Cell. 2009;8(2):181–9. Epub 2008/12/23. doi: 10.1128/EC.00351-08 19098128
20. Fortwendel JR, Juvvadi PR, Rogg LE, Asfaw YG, Burns KA, Randell SH, et al. Plasma membrane localization is required for RasA-mediated polarized morphogenesis and virulence of Aspergillus fumigatus. Eukaryot Cell. 2012;11(8):966–77. Epub 2012/05/09. doi: 10.1128/EC.00091-12 22562470
21. Piispanen AE, Bonnefoi O, Carden S, Deveau A, Bassilana M, Hogan DA. Roles of Ras1 membrane localization during Candida albicans hyphal growth and farnesol response. Eukaryot Cell. 2011;10(11):1473–84. Epub 2011/09/13. doi: 10.1128/EC.05153-11 21908593
22. McQuiston TJ, Williamson PR. Paradoxical roles of alveolar macrophages in the host response to Cryptococcus neoformans. J Infect Chemother. 2012;18(1):1–9. Epub 2011/11/03. doi: 10.1007/s10156-011-0306-2 22045161
23. Coelho C, Bocca AL, Casadevall A. The intracellular life of Cryptococcus neoformans. Annu Rev Pathol. 2014;9 : 219–38. Epub 2013/09/21. doi: 10.1146/annurev-pathol-012513-104653 24050625
24. O'Meara TR, Holmer SM, Selvig K, Dietrich F, Alspaugh JA. Cryptococcus neoformans Rim101 is associated with cell wall remodeling and evasion of the host immune responses. MBio. 2013;4(1). Epub 2013/01/17. doi: 10.1128/mBio.00522-12
25. Choi J, Vogl AW, Kronstad JW. Regulated expression of cyclic AMP-dependent protein kinase A reveals an influence on cell size and the secretion of virulence factors in Cryptococcus neoformans. Mol Microbiol. 2012;85(4):700–15. Epub 2012/06/22. doi: 10.1111/j.1365-2958.2012.08134.x 22717009
26. Kumar P, Heiss C, Santiago-Tirado FH, Black I, Azadi P, Doering TL. Pbx proteins in Cryptococcus neoformans cell wall remodeling and capsule assembly. Eukaryot Cell. 2014;13(5):560–71. Epub 2014/03/04. doi: 10.1128/EC.00290-13 24585882
27. Inglis DO, Skrzypek MS, Liaw E, Moktali V, Sherlock G, Stajich JE. Literature-based gene curation and proposed genetic nomenclature for Cryptococcus. Eukaryot Cell. 2014;13(7):878–83. Epub 2014/05/13. doi: 10.1128/EC.00083-14 24813190
28. Roth AF, Wan J, Bailey AO, Sun B, Kuchar JA, Green WN, et al. Global analysis of protein palmitoylation in yeast. Cell. 2006;125(5):1003–13. Epub 2006/06/06. doi: 10.1016/j.cell.2006.03.042 16751107
29. Nielsen K, Cox GM, Wang P, Toffaletti DL, Perfect JR, Heitman J. Sexual cycle of Cryptococcus neoformans var. grubii and virulence of congenic a and alpha isolates. Infect Immun. 2003;71(9):4831–41. Epub 2003/08/23. PMCID: 187335. 12933823
30. Wozniak KL, Levitz SM. Cryptococcus neoformans enters the endolysosomal pathway of dendritic cells and is killed by lysosomal components. Infect Immun. 2008;76(10):4764–71. Epub 2008/08/06. doi: 10.1128/IAI.00660-08 18678670
31. Smith LM, Dixon EF, May RC. The fungal pathogen Cryptococcus neoformans manipulates macrophage phagosome maturation. Cell Microbiol. 2014. Epub 2014/11/15. doi: 10.1111/cmi.12394
32. Banks IR, Specht CA, Donlin MJ, Gerik KJ, Levitz SM, Lodge JK. A chitin synthase and its regulator protein are critical for chitosan production and growth of the fungal pathogen Cryptococcus neoformans. Eukaryot Cell. 2005;4(11):1902–12. Epub 2005/11/10. doi: 10.1128/EC.4.11.1902-1912.2005 16278457
33. Reese AJ, Yoneda A, Breger JA, Beauvais A, Liu H, Griffith CL, et al. Loss of cell wall alpha(1–3) glucan affects Cryptococcus neoformans from ultrastructure to virulence. Mol Microbiol. 2007;63(5):1385–98. Epub 2007/01/25. doi: 10.1111/j.1365-2958.2006.05551.x 17244196
34. Kuranda K, Leberre V, Sokol S, Palamarczyk G, Francois J. Investigating the caffeine effects in the yeast Saccharomyces cerevisiae brings new insights into the connection between TOR, PKC and Ras/cAMP signalling pathways. Mol Microbiol. 2006;61(5):1147–66. Epub 2006/08/24. doi: 10.1111/j.1365-2958.2006.05300.x 16925551
35. Reese AJ, Doering TL. Cell wall alpha-1,3-glucan is required to anchor the Cryptococcus neoformans capsule. Mol Microbiol. 2003;50(4):1401–9. Epub 2003/11/19. 14622425
36. Johnston SA, May RC. Cryptococcus interactions with macrophages: evasion and manipulation of the phagosome by a fungal pathogen. Cell Microbiol. 2013;15(3):403–11. Epub 2012/11/07. doi: 10.1111/cmi.12067 23127124
37. Levitz SM, Nong SH, Seetoo KF, Harrison TS, Speizer RA, Simons ER. Cryptococcus neoformans resides in an acidic phagolysosome of human macrophages. Infect Immun. 1999;67(2):885–90. Epub 1999/01/23. PMCID: 96400. 9916104
38. Charron G, Zhang MM, Yount JS, Wilson J, Raghavan AS, Shamir E, et al. Robust fluorescent detection of protein fatty-acylation with chemical reporters. J Am Chem Soc. 2009;131(13):4967–75. Epub 2009/03/14. doi: 10.1021/ja810122f 19281244
39. Yount JS, Moltedo B, Yang YY, Charron G, Moran TM, Lopez CB, et al. Palmitoylome profiling reveals S-palmitoylation-dependent antiviral activity of IFITM3. Nat Chem Biol. 2010;6(8):610–4. Epub 2010/07/06. doi: 10.1038/nchembio.405 20601941
40. Yount JS, Zhang MM, Hang HC. Visualization and Identification of Fatty Acylated Proteins Using Chemical Reporters. Curr Protoc Chem Biol. 2011;3(2):65–79. Epub 2011/05/01. doi: 10.1002/9780470559277.ch100225 23061028
41. Zhang MM, Wu PY, Kelly FD, Nurse P, Hang HC. Quantitative control of protein S-palmitoylation regulates meiotic entry in fission yeast. PLoS Biol. 2013;11(7):e1001597. Epub 2013/07/12. doi: 10.1371/journal.pbio.1001597 23843742
42. Lam KK, Davey M, Sun B, Roth AF, Davis NG, Conibear E. Palmitoylation by the DHHC protein Pfa4 regulates the ER exit of Chs3. J Cell Biol. 2006;174(1):19–25. Epub 2006/07/05. doi: 10.1083/jcb.200602049 16818716
43. Gonzalez Montoro A, Chumpen Ramirez S, Quiroga R, Valdez Taubas J. Specificity of transmembrane protein palmitoylation in yeast. PLoS One. 2011;6(2):e16969. Epub 2011/03/09. doi: 10.1371/journal.pone.0016969 21383992
44. Sanchez-Mir L, Franco A, Martin-Garcia R, Madrid M, Vicente-Soler J, Soto T, et al. Rho2 palmitoylation is required for plasma membrane localization and proper signaling to the fission yeast cell integrity mitogen - activated protein kinase pathway. Mol Cell Biol. 2014;34(14):2745–59. Epub 2014/05/14. doi: 10.1128/MCB.01515-13 24820419
45. Young E, Zheng ZY, Wilkins AD, Jeong HT, Li M, Lichtarge O, et al. Regulation of Ras localization and cell transformation by evolutionarily conserved palmitoyltransferases. Mol Cell Biol. 2014;34(3):374–85. Epub 2013/11/20. doi: 10.1128/MCB.01248-13 24248599
46. Baker LG, Specht CA, Donlin MJ, Lodge JK. Chitosan, the deacetylated form of chitin, is necessary for cell wall integrity in Cryptococcus neoformans. Eukaryot Cell. 2007;6(5):855–67. Epub 2007/04/03. doi: 10.1128/EC.00399-06 17400891
47. Del Poeta M. Role of phagocytosis in the virulence of Cryptococcus neoformans. Eukaryot Cell. 2004;3(5):1067–75. Epub 2004/10/08. 10.1128/EC.3.5.1067-1075.2004. 15470235
48. Blanc M, Blaskovic S, van der Goot FG. Palmitoylation, pathogens and their host. Biochem Soc Trans. 2013;41(1):84–8. Epub 2013/01/30. doi: 10.1042/BST20120337 23356263
49. Goldston AM, Sharma AI, Paul KS, Engman DM. Acylation in trypanosomatids: an essential process and potential drug target. Trends Parasitol. 2014;30(7):350–60. Epub 2014/06/24. doi: 10.1016/j.pt.2014.05.003 24954795
50. Hole CR, Bui H, Wormley FL Jr., Wozniak KL. Mechanisms of dendritic cell lysosomal killing of Cryptococcus. Sci Rep. 2012;2 : 739. Epub 2012/10/18. doi: 10.1038/srep00739 23074646
51. Okagaki LH, Strain AK, Nielsen JN, Charlier C, Baltes NJ, Chretien F, et al. Cryptococcal cell morphology affects host cell interactions and pathogenicity. PLoS Pathog. 2010;6(6):e1000953. Epub 2010/06/30. doi: 10.1371/journal.ppat.1000953 20585559
52. Lam WC, Gerik KJ, Lodge JK. Role of Cryptococcus neoformans Rho1 GTPases in the PKC1 signaling pathway in response to thermal stress. Eukaryot Cell. 2013;12(1):118–31. Epub 2012/11/20. doi: 10.1128/EC.05305-11 23159519
53. Alspaugh JA, Perfect JR, Heitman J. Cryptococcus neoformans mating and virulence are regulated by the G-protein alpha subunit GPA1 and cAMP. Genes Dev. 1997;11(23):3206–17. Epub 1998/02/12. PMCID: 316752. 9389652
54. Levitz SM, Specht CA. The molecular basis for the immunogenicity of Cryptococcus neoformans mannoproteins. FEMS Yeast Res. 2006;6(4):513–24. Epub 2006/05/16. doi: 10.1111/j.1567-1364.2006.00071.x 16696647
55. Teixeira PA, Penha LL, Mendonca-Previato L, Previato JO. Mannoprotein MP84 mediates the adhesion of Cryptococcus neoformans to epithelial lung cells. Front Cell Infect Microbiol. 2014;4 : 106. Epub 2014/09/06. doi: 10.3389/fcimb.2014.00106 25191644
56. Bueter CL, Lee CK, Rathinam VA, Healy GJ, Taron CH, Specht CA, et al. Chitosan but not chitin activates the inflammasome by a mechanism dependent upon phagocytosis. J Biol Chem. 2011;286(41):35447–55. Epub 2011/08/25. doi: 10.1074/jbc.M111.274936 21862582
57. Bueter CL, Lee CK, Wang JP, Ostroff GR, Specht CA, Levitz SM. Spectrum and mechanisms of inflammasome activation by chitosan. J Immunol. 2014;192(12):5943–51. Epub 2014/05/16. doi: 10.4049/jimmunol.1301695 24829412
58. Singh A, Panting RJ, Varma A, Saijo T, Waldron KJ, Jong A, et al. Factors required for activation of urease as a virulence determinant in Cryptococcus neoformans. MBio. 2013;4(3):e00220–13. Epub 2013/05/09. doi: 10.1128/mBio.00220-13 23653445
59. Tangen KL, Jung WH, Sham AP, Lian T, Kronstad JW. The iron - and cAMP-regulated gene SIT1 influences ferrioxamine B utilization, melanization and cell wall structure in Cryptococcus neoformans. Microbiology. 2007;153(Pt 1):29–41. Epub 2006/12/23. doi: 10.1099/mic.0.2006/000927-0
60. Desalermos A, Tan X, Rajamuthiah R, Arvanitis M, Wang Y, Li D, et al. A Multi-Host Approach for the Systematic Analysis of Virulence Factors in Cryptococcus neoformans. J Infect Dis. 2014. Epub 2014/08/13. doi: 10.1093/infdis/jiu441
61. Fu J, Hettler E, Wickes BL. Split marker transformation increases homologous integration frequency in Cryptococcus neoformans. Fungal Genet Biol. 2006;43(3):200–12. Epub 2006/02/25. doi: 10.1016/j.fgb.2005.09.007 16497523
62. Toffaletti DL, Rude TH, Johnston SA, Durack DT, Perfect JR. Gene transfer in Cryptococcus neoformans by use of biolistic delivery of DNA. J Bacteriol. 1993;175(5):1405–11. Epub 1993/03/01. PMCID: 193227. 8444802
63. Skowyra ML, Doering TL. RNA interference in Cryptococcus neoformans. Methods Mol Biol. 2012;845 : 165–86. Epub 2012/02/14. doi: 10.1007/978-1-61779-539-8_11 22328374
64. Gerik KJ, Donlin MJ, Soto CE, Banks AM, Banks IR, Maligie MA, et al. Cell wall integrity is dependent on the PKC1 signal transduction pathway in Cryptococcus neoformans. Mol Microbiol. 2005;58(2):393–408. Epub 2005/10/01. doi: 10.1111/j.1365-2958.2005.04843.x 16194228
65. Feldmesser M, Rivera J, Kress Y, Kozel TR, Casadevall A. Antibody interactions with the capsule of Cryptococcus neoformans. Infect Immun. 2000;68(6):3642–50. Epub 2000/05/19. PMCID: 97654. 10816523
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 5
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Human Cytomegalovirus miR-UL112-3p Targets TLR2 and Modulates the TLR2/IRAK1/NFκB Signaling Pathway
- Paradoxical Immune Responses in Non-HIV Cryptococcal Meningitis
- Expression in the Fat Body Is Required in the Defense Against Parasitic Wasps in
- Survives with a Minimal Peptidoglycan Synthesis Machine but Sacrifices Virulence and Antibiotic Resistance
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy