
Widespread Recombination, Reassortment, and Transmission of Unbalanced Compound Viral Genotypes in Natural Arenavirus Infections
The facility with which viruses evolve underlies many of the problems they cause. Virus evolution is the reason we need a new flu vaccine each year. It’s how HIV and other viruses develop drug resistance. And it enables viruses to occasionally jump from animals to humans and cause new diseases. It is therefore important to understand how and under what circumstances viruses evolve. The arenaviruses are a group of viruses that infect mammals and snakes. Mammalian arenaviruses normally infect rodents but they can also infect humans, and, when they do severe and sometimes fatal disease can result. In this study, we studied genetic diversity in arenaviruses infecting captive snakes. We discovered an astonishing amount of diversity. Most snakes are infected by more than one virus strain, and these strains are merging and shuffling their genes (they are undergoing recombination and reassortment). The extent to which this is happening is exceptional, and has likely been caused by the importation and mixing in captivity of infected snakes from the wild. This provides an excellent opportunity to study the processes of virus evolution and may be an example of human activity altering its course.
Published in the journal:
. PLoS Pathog 11(5): e32767. doi:10.1371/journal.ppat.1004900
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004900
Summary
The facility with which viruses evolve underlies many of the problems they cause. Virus evolution is the reason we need a new flu vaccine each year. It’s how HIV and other viruses develop drug resistance. And it enables viruses to occasionally jump from animals to humans and cause new diseases. It is therefore important to understand how and under what circumstances viruses evolve. The arenaviruses are a group of viruses that infect mammals and snakes. Mammalian arenaviruses normally infect rodents but they can also infect humans, and, when they do severe and sometimes fatal disease can result. In this study, we studied genetic diversity in arenaviruses infecting captive snakes. We discovered an astonishing amount of diversity. Most snakes are infected by more than one virus strain, and these strains are merging and shuffling their genes (they are undergoing recombination and reassortment). The extent to which this is happening is exceptional, and has likely been caused by the importation and mixing in captivity of infected snakes from the wild. This provides an excellent opportunity to study the processes of virus evolution and may be an example of human activity altering its course.
Introduction
Several mechanisms generate viral genetic diversity [1–3]. All work by producing populations of viral genomes, a small fraction of which may exhibit an adaptive advantage in a new host species, different tissue, or in the face of drug or immune pressure. Replication of RNA viral genomes by error-prone polymerases results in relatively high mutation frequencies, and RNA viruses replicate as collections of closely related variant genotypes [2,4–7]. Viral genomes can shed content or acquire new loci from their hosts by horizontal gene transfer. In coinfected cells, recombination between different viral strains or species can produce chimeric progeny [8–11]. And in cells coinfected by segmented viruses, reassortment generates virions containing shuffled mixtures of segments from the parental genotypes [11,12].
Understanding basic mechanisms of viral adaptation is essential in order to better combat, prevent, and predict viral diseases. For example, the ability of pandemic influenza virus strains to efficiently replicate in and be transmitted between humans frequently results from de novo mutation and reassortment [13]. The continuous emergence of drug-resistant genotypes is a major hindrance to the effective treatment of human immunodeficiency virus-1 and other pathogens [14,15]. And, recombination between individually attenuated strains present in the oral poliovirus vaccine results in neuropathic progeny strains, and complicates eradication efforts [16].
Viruses in the family Arenaviridae have bi-segmented single-stranded RNA genomes with a characteristic organization and gene repertoire [17–21]. The larger genome segment (L) is about 7 kb in length and encodes the viral RNA-dependent RNA polymerase (L) and a small zinc-binding RING domain protein (Z). The smaller segment (S) is about half as long and encodes the glycoprotein precursor (GPC) and nucleoprotein (NP). On each segment, the two viral genes are in opposite coding orientations and are separated by an intergenic region (IGR) that is predicted to form stable hairpin structures.
Two major lineages of arenaviruses have been described: those that primarily infect rodents and those that infect snakes. The rodent arenaviruses (proposed genus Mammarenavirus) typically establish chronic mild infections in their natural hosts but can be transmitted to humans and other mammals [22]. Severe disease such as Lassa hemorrhagic fever can result from these zoonotic infections. The snake arenaviruses (proposed genus Reptarenavirus) were first identified in US cases of inclusion body disease, a progressive and sometimes fatal disease best described in members of the Boidae and Pythonidae families (boas and pythons) [23,24]. The identification and study of snake arenaviruses in captive snakes in Europe corroborated and extended this finding [25–27]. One major difference between the snake and mammalian arenaviruses is the provenance of their GPC genes, with the snake virus gene being more closely related to the glycoprotein gene of filoviruses and some avian retroviruses [23,28].
Several mechanisms of arenavirus evolution have been described [17,29–31]. Like all RNA viruses, arenavirus genome replication is relatively error prone, and arenaviruses replicate as collections of related variants in vivo [32]. Recombination is thought to have given rise to the ancestral S segment of a clade of the New World rodent arenaviruses [33–35]. Recombination and reassortment between co-infecting arenaviruses has been observed in the laboratory [36–38]. And, it has been suggested that arenaviruses detected in snakes in Europe might have undergone recombination [39,40]. However, arenavirus recombination and reassortment have not been confirmed in natural infections involving extant species.
In this study we document and investigate viral genetic complexity of an unanticipated extent and form in naturally infected captive snakes. We determined the complete or near complete sequences of 210 viral genome segments using metagenomic sequencing. Sequencing results were corroborated and extended by discriminating quantitative reverse transcriptase PCR (qRT-PCR) and by tissue culture isolation experiments. We detected widespread recombination and reassortment. We also observed an unbalanced accumulation of multiple distinct viral genotypes in individual infections. These findings provide an opportunity to study basic mechanisms of virus evolution and fitness through the identification of genetic determinants underpinning their action.
Results
Sample collection and whole genome sequencing
In order to further characterize the genetic diversity of the snake arenaviruses, we gathered 123 frozen case and control tissue samples from around the U.S.A. that were collected between 1997 and 2014 (S1 Table). We screened these samples for snake arenavirus RNA using qRT-PCR with degenerate primers targeting the glycoprotein gene. A total of 56 samples tested positive by qRT-PCR for viral RNA. Clinical data of varying detail was available for samples. Histopathology was available for 58 of the 123 samples and detection of viral RNA was well correlated with histopathology-based IBD diagnosis (Table 1). However, many infected snakes displayed no overt clinical signs (S1 Table). Thus, detection of viral RNA was correlated with detection of inclusions in tissue sections, but not with obvious clinical measures in a straightforward fashion, consistent with previous reports [23,25,26,41].

We performed metagenomic sequencing to determine complete viral genome sequences. Samples from 98 animals, including the PCR-positive samples, were sequenced. Samples from 48 of the arenavirus-positive snakes were sequenced to a depth sufficient for assembly of complete or near complete viral genome segment sequences. In many cases, the assemblies included predicted terminal sequences (S1 Fig, [23,42]). In total, just over 1 million reads contributed to the assembly of 210 genome segment sequences (148 L and 62 S sequences) totaling 1.24 Mb.
Genome segment assemblies were validated by re-mapping paired-end reads [43]. Assemblies were well supported, with 99-fold median coverage (S1 Fig). Coverage levels were generally lowest in the intergenic regions (IGR), likely as a result of the predicted hairpin-forming sequences in these regions (S1 Fig). In cases where coverage levels in IGRs fell below 2 reads, PCR was used to confirm assembly continuity.
Genome segments with less than 80% global pairwise nucleotide identity were classified into distinct genotypes (Figs 1 and 2 and S2 Fig) [44]. A total of 11 S and 23 L genotypes were delineated by this criterion, and were designated S1-S11 and L1-L23. Within genotypes, L segment sequences shared a mean value of 96% pairwise nucleotide identity, and between genotypes, sequences shared 65% identity (S2A Fig). For S segments, these values were 96% and 64% (S2B Fig). Multiple alignments of the four coding sequences were used to create Bayesian phylogenies to visualize the inferred evolutionary relationship of these genotypes (Figs 1–3 and S3 Fig).



In 21 of 48 infected snakes, viral genotypes consisted of a single S and a single L segment genotype (Fig 4 and S3 Table, snakes #1–21). For example, snakes #1–3, the annulated tree boas from the California Academy of Sciences described in an earlier report, harbored S genotype 1 and L genotype 1. This S1/L1 genotype corresponds to the virus we referred to as “CAS virus” (CASV; [23]). In snakes #4–6, segments of genotype S2 and L2 were detected, a genotype corresponding to “Golden Gate virus” [23].
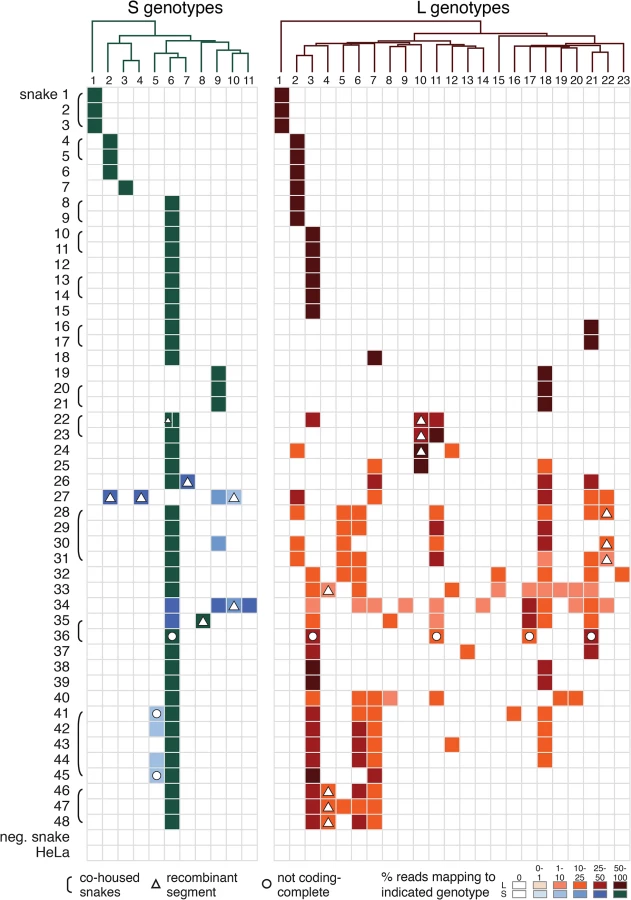
Numerous reassortant genotypes were evident among the singly infected snakes (Fig 4). For example, the L2 segments present in snakes #7 and 8 were nearly identical at 98.6% pairwise identity, but their S segments shared only 73.5% pairwise identity (genotypes S3 and S6), indicating a more distant common ancestor. Similarly, the S6 segments in snakes #15 and 16 were 97.8% identical but the L3 and L21 segments in these snakes were only 62.2% identical. It was not possible in most cases to determine which S/L pairs were ancestral, nor when precisely reassortment had occurred, but it was clear that reassortment had produced these permuted pairings.
However, in the majority of animals (27 of 48 snakes), we observed viral genotypes that were substantially more complex than single S/L pairs. These snakes harbored multiple S and L genome segment sequences (Fig 4; snakes #22–48). The sets of viral genotypes ranged from that which would be expected for a straightforward co-infection by 2 virus strains in snake #45, to more complex combinations such as that in snake #33, which contained the sequences of 1 S and 10 distinct L segments. The animal with the most viral genotypes was snake #34, which contained 15 genotypes (4 S and 11 L) that could combine to produce virions with 44 unique S/L pairs (assuming coinfection of individual cells). The distinct L and S segment sequences within individual snakes shared on average only 65% and 70% pairwise nucleotide identity respectively, a level of variation not consistent with sequencing error or intrahost diversification. Snakes that were housed together often shared similar combinations of genotypes, and in these cases the sequences of these segments were closely related (Figs 1 and 2 and Fig 4).
The accumulation of viral genotypes within individual animals was not balanced. In all cases there were more L than S segment genotypes (Fig 4 and S4 Fig). On average, there were more than twice as many L segment genotypes as S genotypes per animal (mean values of 4.7 L and 2.4 S genotypes per multiply infected animal; S4 Fig). In fact, 18 of 27 animals with multiple L sequences harbored only a single S genotype.
The S6 genotype was dominant in individual animals and at a population level (Fig 1 and Fig 4). This genotype was first detected in a snake from Collierville, TN, and a partial S6 sequence was reported previously [23]. S segments of this genotype were detected in 37 of 48 snakes (77%). These sequences shared 96% average global pairwise nucleotide identity. Remarkably, the S6 segment genotype was found in combination with 21 of the 23 L segment genotypes described in this study.
In addition to reassortant genotypes, we also identified 6 recombinant S segments and 8 recombinant L segments (Figs 1 and 2, Figs 5 and 6, S1 Fig, and S2 Table). We used the RDP4 software to detect and statistically evaluate support for recombination events (Table 2; [45]). Recombination events were well supported by RDP4 analysis and by read coverage levels over recombination junctions (S1 Fig and Table 2). We also confirmed segment continuity by PCR amplification across junctions.


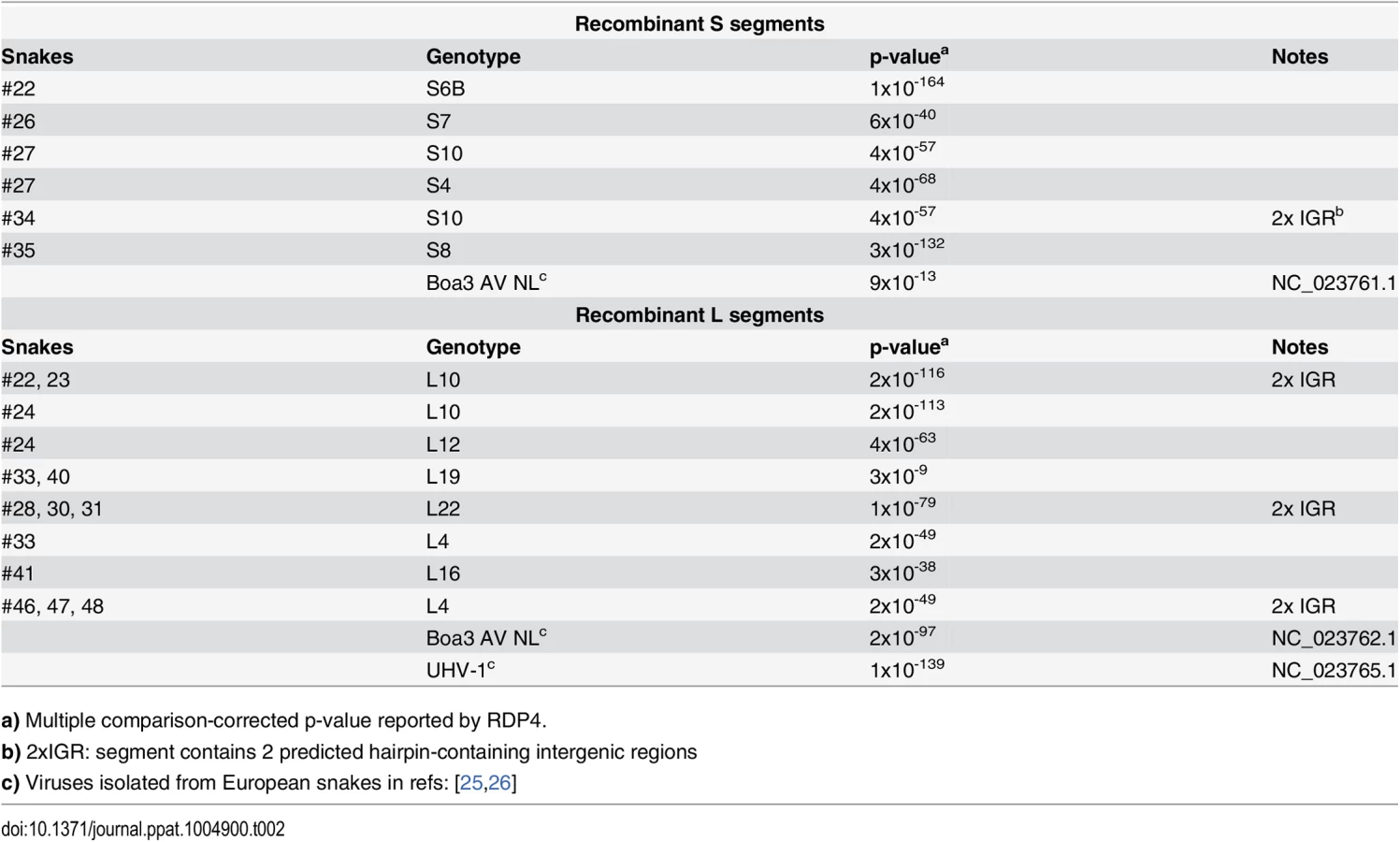
While this analysis provided clear evidence of recombination, it was not always possible to determine which genotypes were parental and which were recombinant. However, it appeared that many of the recombinant segments coexisted in snakes with one of their parental genotypes. For example, in snake #35, 2 S segment sequences were evident. One of these was a canonical S6 genotype. The other segment, designated S8, shared 97% average pairwise nucleotide identity with the S6 segment’s GPC gene, but only 66% identity in the NP gene (Fig 5A). The S8 NP appeared to derive from a segment similar to the S11 NP found in snake #34. Similar patterns were observed for S segments in snakes #22, 26, 27, 34, and 35, and for L segments in snakes #22, 23, 28, 30, 31, 33, 46, 47, and 48 (Figs 1 and 2 and Fig 5B). These may be situations where the parental and progeny genotypes persistently replicated together following a recombination event. Alternatively, the genotypes could have been acquired in independent transmission events.
Some recombination events resulted in unusual genome organizations. For example, the L10 genotype found in snakes #22 and #23 consisted of a full Z coding sequence and a partial L coding sequence from one parental segment concatenated to a partial Z and full L from a second segment (Fig 6). This segment was predicted to contain 2 intergenic hairpin-containing regions (2xIGR) separating the 2 L/Z pairs. Similar double-IGR segments and partial CDS were observed in recombinant S segments as well (Fig 6). An offset template switching event during genome replication may have generated these 2xIGR segments (S5 Fig; [10]).
We used several independent approaches to corroborate the original metagenomic sequencing. First, we completely re-sequenced 41 samples, using independent library preparations, and derived essentially identical results. Second, we developed a panel of PCR primer pairs that discriminated between distinct viral genotypes, and performed qRT-PCR on a subset of samples and genotypes. In all cases, qPCR-based genotyping mirrored sequencing results (S6 Fig). Third, we used tissue culture isolation as another means of determining viral genotype and to confirm that sequences corresponded to infectious virus (see below).
Transmission of compound genotypes
The introduction of an already infected snake (#35) into the proximity of an uninfected snake (#36) in a private collection enabled us to monitor viral transmission (Fig 7). A 2011 blood sample from snake #36 tested negative for snake arenavirus RNA by qRT-PCR and deep sequencing. Snake #36 was then not exposed to other snakes until September 2012, when its owners obtained a second snake, #35. Snake #35 arrived with stomatitis and was anorexic. Nevertheless, after a 4-week quarantine, snakes #35 and #36 were placed in the same enclosure. Snake #35 continued to refuse to feed and died two weeks later. We obtained the body of snake #35 and a blood sample from snake #36 taken in November 2012, and an additional blood sample from snake #36 from January 2013.

We determined that multiple genotypes were transmitted from snake #35 to snake #36 during their cohabitation (Fig 7). Snake #35’s viral genotype consisted of 2 S and 6 L genotypes (S6,8 / L3,8,11,17,18,21; Fig 7B). The November 2012 snake #36 blood sample was arenavirus positive, but the only genotypes detected by sequencing were S6 and L3. The January 2013 snake #36 sample was still positive, but now L genotypes 11, 17, and 21 were also detectable in the blood. Analysis of the viral sequences recovered from the two snakes revealed that they were closely related (98.5–100% identity). This data supports the transmissibility of compound unbalanced snake arenavirus genotypes in the context of cohabitation.
Tissue culture isolation of unbalanced populations of viruses
We performed tissue culture isolation and passaging experiments to investigate whether compound viral genotypes were competent to initiate productive infections. We applied homogenates from samples to cultures of boa constrictor-derived JK cells and monitored levels of virus RNA by qRT-PCR using genotype-discriminating primers. In all cases, we detected replication of all of the viral genotypes identified by our metagenomic sequencing (Fig 8). For example, snake #38 contained viral sequences of genotype S6/L3,18. When a liver homogenate from this snake was applied to a JK culture, replication of all 3 of these segments was detected (Fig 8A). Similarly, when a homogenate from snake #47 was used as inoculum, replication of all 6 expected viral genotypes was observed (S6 and L3,4,5,6,7; Fig 8B). The distinct L segments exhibited approximately equal replication efficiencies in these experiments, and the populations could be passaged to uninfected cell cultures. Thus, sequences of multiple viral genotypes corresponded to replication competent virus, and multiple viral genome segments replicated as stable ensembles in culture.

We also investigated whether an L segment with an unusual 2x IGR organization was competent for replication. Snake #47 L4 contains a partial Z region and 2 predicted IGR hairpins (Fig 6). To track this segment during infection, we performed qRT-PCR using 2 primer pairs: one pair that targeted the L gene of this segment and one pair that spanned the recombination junction (Fig 8C). Throughout the experiment, near equivalent amounts of template were detected using these 2 primer pairs, suggesting that most copies of this segment maintained their unusual structure.
We performed endpoint dilution experiments to determine the genotypes of individual virus particles. We prepared dilution series from liver homogenates from snakes #37 and #47 and inoculated JK cells in 96 well plates. After 7–10 days, we transferred supernatants to new plates and stained cells with anti-NP Ab to determine wells positive for the presence of virus. Positive wells were then genotyped using discriminating qRT-PCR. In most cases, RNA from a single S and a single L genotype were detected in individual wells infected with the most dilute inocula (Fig 9). In 3 of 14 (21%) wells at these highest dilutions, more than one L genotype was detected. This could be the result of stochastic co-infection, clumped virus particles, or virus particles packaging more than one L segment. These results were consistent with the model that most virus particles packaged a single L segment, although we could not exclude the possibility that a minority of particles may package additional segments.

Analysis of intrahost variation of individual genotypes
Intrahost variation for individual genotypes was also evident. For most genome segments, multiple sites with minor allele variants were detected (S7 Fig). The frequency of variants in most of these cases was less than 10 variant sites per kb (i.e., ≤1%; S7 Fig). In several cases, a higher frequency of variant sites was observed, for example the S6 segment of snake #41, which averaged 17 variant sites per kb (1.7% variants sites). This could have resulted from a greater degree of intrahost variation and divergence or from infection by viruses with closely related genotypes whose sequences were too similar to separate using short read assembly.
Discussion
In this study, we surveyed the genetic diversity of arenaviruses infecting captive snakes in the USA. We used metagenomic sequencing and de novo assembly to determine genome sequences of viruses infecting 48 snakes. We found that most snakes were multiply infected by unbalanced ensembles of S and L segment genotypes. In total, we assembled 148 L and 62 S segment sequences that grouped into 23 L and 11 S genotypes. This expands the known diversity within this group of viruses by several fold. The high level of multiple infection has apparently given rise to numerous recombinant and reassortant genotypes, altogether compromising hundreds of unique viral combinations. We also discovered recombinant genotypes with non-canonical genome organizations, including those harboring apparently superfluous content. Metagenomic sequencing results were corroborated by PCR-based approaches, and extended by tissue culture isolation experiments. These findings highlight the utility of performing unbiased whole genome sequencing to determine pathogen genotypes. Indeed, our initial PCR-based screening correctly identified infected animals, but completely failed to uncover the genetic complexity present in the infections.
Although natural infection by multiple arenaviruses has not been previously documented, this phenomenon has been reported for other viruses. For example, infection involving up to 3 influenza viruses has been documented in humans and wild birds [46,47]. And, up to 7 or 9 distinct genotypes of torque teno virus or papillomavirus have been identified in individual human samples [48–50]. In plants, a virus isolate from citrus trees persistently infected by citrus tristeza virus was found to include several genotypes [51]. Shared characteristics of host-pathogen interaction may enable such highly multiple infections. These include persistent, sub-clinical viral replication, the absence of barriers to superinfection, the lack of an immune response capable of clearing infection, and a high prevalence of infection.
Although we detected many instances of snake tissues containing multiple viral genotypes, our results do not prove that individual cells in these animals were multiply infected. However, the detection of recombinant and reassortant genotypes suggests that at least in some cases cells are multiply infected.
Although multiple infection per se is not unprecedented, several aspects of these findings are. One is the apparent disconnect between the dynamics of the two viral genome segments, both in individual animals and at a population level. In individual animals, the accumulation of S and L segments was unbalanced: in all multiply infected animals, there were more L than S segments. In the most extreme case (snake #33), a single S genotype was paired with an ensemble of 10 L genotypes. It is possible that within animals the S and L segments inhabit different fitness landscapes. Another, not mutually exclusive, possibility is that differential replication kinetics or packaging efficiencies of the two segments may underlie the observed imbalance. Additional experiments in vitro and in animals will clarify this issue.
The population level dominance of the S6 genotype was also unexpected and is worthy of additional investigation. Genotype S6 segments were present in 37/48 infections (77%) and in 29 of these, no other S segment was detected. One possible explanation is that the S6 genotype replicates more efficiently within animals, or is more efficiently packaged and transmitted than other competing S segments. Alternatively, the high frequency of this genotype may be a stochastic effect, or may be proportional to viral genotypes in natural virus populations, from which these viruses in captive snakes presumably originate. Alternatively, it is possible that the 23 L genotypes observed here were originally paired with 23 S genotypes in free-ranging hosts. If this were the case, then 12 S genotypes are unaccounted for. Testing of wild-caught snakes could reveal the “missing” S genotypes and original S-L pairings and would reveal whether the S6 genotype has indeed risen to dominance in the context of captive animals.
Whether a similar degree of multiple infection is possible in mammalian arenaviruses is an open question, and one that may be relevant to the possible emergence of new mammalian arenavirus strains with pathogenic potential. It may be that there are larger species barriers for mammalian arenaviruses than there are for snake arenaviruses. Another possibility is that an ecological situation analogous to captive snake breeding has never been created for rodents. Alternatively, characteristics of the mammalian arenavirus host-pathogen interaction may prevent multiple infection. Indeed, superinfection exclusion has been documented in mammalian arenavirus tissue culture experiments [52–57]. And, cross-protection between mammalian arenaviruses has been documented in vivo [58–60]. Assuming that superinfection accounts for at least some of the genotype accumulation observed here, then no such mechanisms are operating in these snakes. Laboratory experiments with mammalian arenaviruses and other segmented viruses could test the generality of this phenomenon.
The discovery of “2xIGR” genome segment configurations was also unanticipated. It would be reasonable to predict that these segments would exhibit decreased fitness or be unstable during replication, given that they carry superfluous content. However, two lines of evidence suggested that these 2xIGR segments are capable of transmission and are stable over multiple rounds of replication. First, several of these segments were detected in co-housed snakes (L10 in snakes #22 and 23; L22 in #28, 30, and 31; L4 in #46–48). Presumably, each of these segments was initially generated via recombination in a single infected snake and then transmitted. Second, in tissue culture these segments replicated stably and could be passaged and isolated (Figs 8C and 9B). More extensive passage experiments in animals and culture will reveal whether maintenance of the 2xIGR configuration is disfavored over the long term.
These novel segment configurations also raise the possibility for the creation of payload-containing arenavirus genome segments. For example, the L10 segment found in snakes #22 and #23 contain 2 intergenic regions and 559 bases of extraneous incomplete coding sequence. If a suitable reverse genetic system were developed, this regions could be replaced with an internal ribosomal entry site and the 516 base NanoLuc luciferase gene or other payloads [61]. Such a tagged virus could be used for example in in vivo pathogenesis studies as an alternative to tri-segmented recombinant arenaviruses [62].
The nomenclature and taxonomy of the snake arenaviruses will likely have to be reconsidered in light of these findings [22]. We propose a nomenclature like that used for influenza A virus (IAV) subtypes, where new S and L genotypes are simply enumerated [63]. We would also propose following the taxonomic scheme for IAV subtypes, which belong to a single species, Influenza A virus. In this case, snake arenavirus genotypes could be grouped into one or possibly more species.
Recombinant genotypes were not limited to those described in this study. Discordance between GPC - and NP-based phylogenies including sequences from viruses detected in snakes in Europe suggested possible S-segment recombination [39,40]. The increased phylogenetic resolution enabled by this study confirmed the recombinant nature of the S and L segments of Boa3 AV NL and the L segment of UHV-1 [25,26] (Table 2, Figs 1 and 2).
It is possible that snake importation and husbandry practices have inadvertently created an ecological context that has enabled this phenomenon. Boa constrictors with different colorations (“color morphs”) are highly valued by collectors and breeders. Such colorations arise in nature as local adaptations and wild-caught snakes are commonly imported for breeding purposes. An estimated 98,500 boa constrictors per year were imported into the USA alone between 2005–2010 [64]. Mammalian arenavirus species have co-evolved with their distinct, geographically isolated rodent hosts, and it may be that snake arenavirus strains have co-evolved similarly in the wild. It is plausible that apparently healthy snakes persistently infected by various arenavirus species have been imported and intermingled in high-density breeding operations. This possible anthropogenic disruption of pathogen ecology is reminiscent of the influenza virus diversity generated in live animal markets [65]. Sampling of viral diversity in free-ranging snake populations are needed to clarify the impact of human activities on the evolution of these viruses and to further assess the disease potential of the resulting recombinant and reassortant genotypes.
In the absence of barriers to superinfection, an incalculable number of novel viral genotype configurations, made even more numerous by frequent intra-segment recombination and an error-prone polymerase, could rapidly evolve and accumulate within individual animals and in breeding facilities. In theory, this situation could be exacerbated by the introduction of mammalian arenavirus-infected rodents as feedstock [66], although it is unknown whether recombinant or reassortant mammalian/reptile arenaviruses are possible or viable. Regardless, further investigation of high-density reptile breeding and feed rodent facilities should be considered.
Materials and Methods
Sample collection
Samples were collected between 1997 and 2014 from California, Washington, Arkansas, Tennessee, Louisiana, Georgia, and Florida. Veterinarians in private practice or at university teaching hospitals collected samples. Samples were submitted to the University of Florida or the University of California San Francisco for further processing and storage. Blood samples were collected by cardiac or tail vein puncture and frozen until further processing. Tissue samples were collected during necropsy and frozen until further analysis or placed in formalin for histopathology. For histopathology, samples were preserved in 10% formalin, paraffin embedded, sectioned, and stained with hematoxylin and eosin. Board-certified pathologists blinded to infection status of samples examined H&E stained sections.
qRT-PCR
RNA extracted from tissues (500 ng) or tissue culture supernatant was denatured for 5 min at 65°C then cooled on ice and added to 10 μl RT reactions containing 100 pmoles random hexamer oligonucleotide (MDS-286), 1× reaction buffer, 5 mM dithiothreitol, 1.25 mM (each) deoxynucleoside triphosphates (dNTPs), and 100 U SuperScript III (Life Technologies). Reaction mixtures were incubated for 5 min at 25°C then for 60 min at 42°C and then for 15 min at 70°C. cDNA was diluted 1 : 10 in 10mM Tris pH8, 0.1 mM EDTA. qPCR reactions contained 5 μl diluted cDNA, 0.5 μM each primer, 10 mM Tris pH 8.8, 50 mM KCl, 1.5 mM MgCl2, 0.2 mM each dNTP, 5% glycerol, 0.08% NP-40, 0.05% Tween-20, 0.15 μl Taq DNA polymerase, and 1x Sybr Green (Life Technologies) in each 15 μl reaction. Primer sequences are listed in S2 Table. Degenerate primers targeting the glycoprotein gene (MDS-400 and -402) were used to screen for infection. qRT-PCR to screen for individual S and L genotypes was performed using panels of primers that were designed to discriminate between the different sequences. Primer pair efficiencies were determined using template dilution series [67,68].
Library preparation and sequencing
RNA was extracted from tissues as previously described [23]. Sequencing libraries were prepared as previously described [69]. Paired-end 2x135 bp sequencing was performed on an Illumina HiSeq 2500 in the Center for Advanced Technology at UCSF., producing an average of 2.0x106 read pairs per sample.
Sequence analysis
A stepwise pipeline was used to process sequencing data. First, data was demultiplexed. Then, 5 bases were trimmed of the 5′ end of reads and 1 base off the 3′ end. Next, low quality read pairs with any 10 base window with an average quality score below 30 were discarded. Then, reads sharing >98% global nucleotide identity (likely PCR duplicates) were collapsed using the cd-hit-est software version 4.6 [44]. Adapter sequences were trimmed from the ends of reads. Then, host-derived sequences were filtered as previously described [23]. Viral genome sequences were assembled from the remaining reads
An iterative strategy was used to assemble genome segment sequences. First, from each dataset, post-filtering reads were aligned using Bowtie2 to a database composed of all already-described snake arenavirus genome segments sequences [43]. Alignment parameters were set stringently (minimum alignment score of 150 in local mode alignment) so that only reads closely matching already described sequences aligned. Alignments were converted into BAM format using SAMTools software and inspected in Geneious software [70,71]. Sites differing from reference sequences were corrected to generate new draft genome sequences. Remaining virus-derived reads (determined by BLASTx, as described below) that didn’t align to an existing virus sequence were used to seed assemblies using the PRICE targeted de novo assembler [72]. PRICE contigs were added to the set of genome segment sequences and the process was reiterated until all reads were accounted for as described below.
Once a complete set of genome segment sequences was assembled, we used Bowtie2 to remap all reads from each dataset to the set of genome segment sequences derived from that dataset. These alignments were manually inspected and were used to generate coverage metrics. For each aligned base, coverage was only counted if the preceding and succeeding 3 bases also aligned. This “continuous coverage” metric is more conservative than a simple coverage metric and was implemented to identify possible incorrect assemblies. BAM files from these alignments, as well as FASTQ files of raw sequencing data for all snakes, have been deposited in the NCBI Short Read Archive (SRA; accession SRP057522). Genome segment sequences have been deposited in GenBank with accessions KP071471-KP071680.
We employed the following analysis strategy to confirm that we were accounting for the full viral genetic complexity in our datasets, i.e. that we were not overlooking any viral genome segments. We used the BLASTx tool to align translated reads from each dataset to a database containing all available snake arenavirus protein sequences, including the ones from this study. Because BLASTx alignments are based on protein sequence similarity, it is possible to use them to detect sequences with relatively distant homology. Thus, the number of reads with BLASTx alignments to arenavirus protein sequences (with E-value ≤ 10–8) determined a minimum expected number of virus sequences in each dataset. Then, we used the Bowtie2 aligner to stringently map reads from each dataset to the coding sequences of the assemblies generated from that dataset as described above. This allowed us to confirm that the assemblies accounted for all of the arenavirus-derived sequences in each dataset. To calculate the fractional abundance of individual genotypes, we divided the number of read pairs mapping to that genotype by the total number of arenavirus-mapping reads from that dataset.
Phylogenetic analyses
We performed phylogenetic analyses to infer evolutionary relationships between viral genotypes. We created multiple sequence alignments of the coding sequences for each of the 4 viral gene coding sequences, using MAFFT version v7.017 with default parameters [73]. These alignments were trimmed using the Gblocks software version 0.91b using default parameters except allowing up to half gaps in columns (parameters:-t = d-b5 = h [74]). We used these trimmed alignments and the JModelTest software v2.1.6 to identify a best-fit nucleotide substitution model (GTR; [75,76]). We ran this software with parameters:-s 11-f-i-g 4-AIC-BIC-AICc-DT-p-a-w. We used MrBayes 3.2.2 to create Bayesian phylogenies from these alignments, using commands lset nst = 6 rates = propinv and mcmc ngen = 2000000 [77]. These phylogenies were visualized using FigTree software (http://tree.bio.ed.ac.uk/software/figtree/).
To create phylogenies including representative snake and mammalian arenavirus sequences, we first downloaded all sequences from the NCBI nucleotide database w/ query: “txid11617[Organism:exp]”, i.e. all sequences annotated as being of arenavirus origin. We removed sequences that were not complete or not coding-complete. We extracted NP and L CDS from these sequences. We used cd-hit-est to create a set of representative sequences, with sequences sharing >80% pairwise nucleotide identity collapsed (-c 0.8) [44]. We then created and trimmed multiple alignments and phylogenies as described above.
Detection of recombinant genotypes
Global multiple sequence alignments of all S and all L segments were created using MAFFT software version 7.017 with default parameters [73]. Full genome sequences for described European snake arenavirus isolates were included in these alignments (University of Helsinki virus (UHV-1) and Boa arenavirus NL; NCBI accessions NC_023766.1, NC_023765.1, NC_023761.1, and NC_023762.1). Alignments were analyzed with the RDP4 recombination detection program version 4.39 using default parameters except to specify linear molecule topology [45]. We required that recombination events be detected by at least 2 of the methods implemented in the software. Putative recombinant segments were validated by examination of phylogenetic discordance and pairwise sequence alignments.
RNA secondary structure analysis
Genome segments sequences were divided into 140 nt sliding windows (the approximate size of intergenic regions) offset by 5 nt. CentroidFold v0.0.9 was used to calculate the minimum free energy of folding for each window using parameters-g 4-e CONTRAfold [78].
Analysis of variant sites
Variant sites were called using SAMTools version 0.1.19, using command mpileup—I. We required that variant sites be supported by at least 4 reads in the context of a minimum coverage level of 20 total reads. The number of variant sites per genome segment was calculated and normalized to the length of each segment.
Tissue culture
RIC For inoculation experiments, frozen tissue samples were thawed on ice and homogenized in MEM + 25mM HEPES (SF-MEM) using a Dounce homogenizer. Homogenates were clarified by centrifugation at 10,000g for 2 minutes then passed through a 0.45 μm filter. Filtrates were diluted 1 : 10 in SF-MEM then added to cultures of near confluent JK cells. Culture medium was replaced periodically and supernatants were stored at -80°C until further analysis.
Monitoring of viral replication in culture
We used qRT-PCR to measure viral RNA levels in culture supernatant. We extracted RNA from 180 μl supernatant using the Zymo viral RNA kit (Zymo Research). 5 μl RNA (25% of eluate) was used as template in RT reactions as above. Resulting cDNA was diluted and used in qPCR reactions as above, with primers listed in S2 Table. Primer pair efficiencies were calculated as above and used to determine quantities of viral RNA relative to the amount of S segment RNA present in the first time point sample.
End-point dilution experiments
JK cells were grown as described above and were plated at a density of 5000 cells per well in 96-well plates. One day later diluted virus stocks were added to cells. Cells were incubated for 7–10 days and then supernatants were transferred to new 96 well plates. Then wells were stained with anti-GGV-NP antibody, which cross-reacts with the NPs of the viruses used in these experiments. Staining and washing was performed as previously described [23]. Stained plates were scanned on an Odyssey Licor instrument to identify infected wells. Supernatant from NP-positive wells were transferred to 24-well plate wells plated the day prior with 75,000 JK cells per well. One day later cell culture supernatant was replaced. After an additional 3 days of incubation, culture supernatant was collected and clarified by centrifugation at 10,000g for 1 minute. RNA was isolated from these supernatants using the ZR Viral RNA kit according to the manufacturer’s protocol (Zymo Research). RNA was used as template in qRT-PCR as described above to measure levels of viral RNA of various genotypes.
Ethics statement
This study did not include experiments involving live animals. In some cases, samples (typically blood) were collected from live animals by attending veterinarians. In other cases, tissues were collected during necropsy. All samples were taken and used with owners consent. Some samples were collected in the context of other, related studies: The acquisition of tissue samples at the University of Florida was authorized under University of Florida Institutional Animal Care and Use Committee Protocol A116. The acquisition of samples at the University of California Davis was authorized under IACUC protocol 17450.
Supporting Information
Zdroje
1. Holmes EC. Virus Evolution. In: D M Knipe, Howley P M, editors. Fields Virology. 6th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2013. pp. 286–313.
2. Holland J, Spindler K, Horodyski F, Grabau E, Nichol S, VandePol S. Rapid evolution of RNA genomes. Science. 1982;215 : 1577–1585. 7041255
3. Domingo E, Escarmís C, Sevilla N, Moya A, Elena SF, Quer J, et al. Basic concepts in RNA virus evolution. FASEB J. 1996;10 : 859–864. 8666162
4. Eigen M. Self organization of matter and the evolution of biological macromolecules. Naturwissenschaften. 1971;58 : 465–523. 4942363
5. Domingo E, Sheldon J, Perales C. Viral quasispecies evolution. Microbiol Mol Biol Rev MMBR. 2012;76 : 159–216. doi: 10.1128/MMBR.05023-11 22688811
6. Lauring AS, Andino R. Quasispecies theory and the behavior of RNA viruses. PLoS Pathog. 2010;6: e1001005. doi: 10.1371/journal.ppat.1001005 20661479
7. Holland JJ, De La Torre JC, Steinhauer DA. RNA virus populations as quasispecies. Curr Top Microbiol Immunol. 1992;176 : 1–20. 1600748
8. Hershey AD, Rotman R. Linkage Among Genes Controlling Inhibition of Lysis in a Bacterial Virus. Proc Natl Acad Sci U S A. 1948;34 : 89–96. 16578282
9. Luria SE, Dulbecco R. Genetic Recombinations Leading to Production of Active Bacteriophage from Ultraviolet Inactivated Bacteriophage Particles. Genetics. 1949;34 : 93–125. 17247312
10. Kirkegaard K, Baltimore D. The mechanism of RNA recombination in poliovirus. Cell. 1986;47 : 433–443. 3021340
11. Domingo E. Virus Evolution. In: Knipe D M, Howley P M, editors. Fields Virology. 5th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2007. pp. 389–421.
12. Fields BN, Joklik WK. Isolation and preliminary genetic and biochemical characterization of temperature-sensitive mutants of reovirus. Virology. 1969;37 : 335–342. 5777554
13. Webster RG, Bean WJ, Gorman OT, Chambers TM, Kawaoka Y. Evolution and ecology of influenza A viruses. Microbiol Rev. 1992;56 : 152–179. 1579108
14. Clavel F, Hance AJ. HIV drug resistance. N Engl J Med. 2004;350 : 1023–1035. 14999114
15. Chen R, Quinones-Mateu ME, Mansky LM. Drug resistance, virus fitness and HIV-1 mutagenesis. Curr Pharm Des. 2004;10 : 4065–4070. 15579088
16. Kew OM, Sutter RW, de Gourville EM, Dowdle WR, Pallansch MA. Vaccine-derived polioviruses and the endgame strategy for global polio eradication. Annu Rev Microbiol. 2005;59 : 587–635. 16153180
17. Buchmeier MJ, de la Torre JC, Peters CJ. Arenaviridae: the viruses and their replication. In: Knipe D M, Howley P M, editors. Fields Virology. 5th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2007. pp. 1791–1828.
18. Salvato MS, Clegg JCS, Buchmeier MJ, Charrel RN, Gonzalez JP, Lukashevich IS, et al. Arenaviridae. Virus taxonomy: classification and nomenclature of viruses: Ninth Report of the International Committee on Taxonomy of Viruses. San Diego: Elsevier; pp. 715–723.
19. Emonet SF, de la Torre JC, Domingo E, Sevilla N. Arenavirus genetic diversity and its biological implications. Infect Genet Evol J Mol Epidemiol Evol Genet Infect Dis. 2009;9 : 417–429.
20. Gonzalez JP, Emonet S, de Lamballerie X, Charrel R. Arenaviruses. Curr Top Microbiol Immunol. 2007;315 : 253–288. 17848068
21. Charrel RN, Coutard B, Baronti C, Canard B, Nougairede A, Frangeul A, et al. Arenaviruses and hantaviruses: from epidemiology and genomics to antivirals. Antiviral Res. 2011;90 : 102–114. doi: 10.1016/j.antiviral.2011.02.009 21356244
22. Radoshitzky S, Bào Y, Buchmeier M, Charrel R, Clawson A, Clegg C, et al. Past, Present, and Future of Arenavirus Taxonomy. Arch Virol. 2015; In Press.
23. Stenglein MD, Sanders C, Kistler AL, Ruby JG, Franco JY, Reavill DR, et al. Identification, characterization, and in vitro culture of highly divergent arenaviruses from boa constrictors and annulated tree boas: candidate etiological agents for snake inclusion body disease. mBio. 2012;3: e00180–00112. doi: 10.1128/mBio.00180-12 22893382
24. Chang L-W, Jacobson ER. Inclusion Body Disease, A Worldwide Infectious Disease of Boid Snakes: A Review. J Exot Pet Med. 2010;19 : 216–225.
25. Bodewes R, Kik MJL, Raj VS, Schapendonk CME, Haagmans BL, Smits SL, et al. Detection of novel divergent arenaviruses in boid snakes with inclusion body disease in The Netherlands. J Gen Virol. 2013;94 : 1206–1210. doi: 10.1099/vir.0.051995-0 23468423
26. Hetzel U, Sironen T, Laurinmäki P, Liljeroos L, Patjas A, Henttonen H, et al. Isolation, identification, and characterization of novel arenaviruses, the etiological agents of boid inclusion body disease. J Virol. 2013;87 : 10918–10935. doi: 10.1128/JVI.01123-13 23926354
27. Hepojoki J, Kipar A, Korzyukov Y, Bell-Sakyi L, Vapalahti O, Hetzel U. Replication of Boid Inclusion Body Disease-Associated Arenaviruses Is Temperature Sensitive in both Boid and Mammalian Cells. J Virol. 2015;89 : 1119–1128. doi: 10.1128/JVI.03119-14 25378485
28. Koellhoffer JF, Dai Z, Malashkevich VN, Stenglein MD, Liu Y, Toro R, et al. Structural Characterization of the Glycoprotein GP2 Core Domain from the CAS Virus, a Novel Arenavirus-Like Species. J Mol Biol. 2013; 426 : 1452–1468 doi: 10.1016/j.jmb.2013.12.009 24333483
29. Charrel RN, Lemasson J-J, Garbutt M, Khelifa R, Micco PD, Feldmann H, et al. New insights into the evolutionary relationships between arenaviruses provided by comparative analysis of small and large segment sequences. Virology. 2003;317 : 191–196. 14698659
30. Zapata JC, Salvato MS. Arenavirus variations due to host-specific adaptation. Viruses. 2013;5 : 241–278. doi: 10.3390/v5010241 23344562
31. Emonet S, Lemasson J-J, Gonzalez J-P, de Lamballerie X, Charrel RN. Phylogeny and evolution of old world arenaviruses. Virology. 2006;350 : 251–257. 16494913
32. Sevilla N, de la Torre JC. Arenavirus diversity and evolution: quasispecies in vivo. Curr Top Microbiol Immunol. 2006;299 : 315–335. 16568904
33. Archer AM, Rico-Hesse R. High Genetic Divergence and Recombination in Arenaviruses from the Americas. Virology. 2002;304 : 274–281. 12504568
34. Charrel RN, de Lamballerie X, Emonet S. Phylogeny of the genus Arenavirus. Curr Opin Microbiol. 2008;11 : 362–368. doi: 10.1016/j.mib.2008.06.001 18602020
35. Charrel RN, Feldmann H, Fulhorst CF, Khelifa R, de Chesse R, de Lamballerie X. Phylogeny of New World arenaviruses based on the complete coding sequences of the small genomic segment identified an evolutionary lineage produced by intrasegmental recombination. Biochem Biophys Res Commun. 2002;296 : 1118–1124. 12207889
36. Riviere Y, Oldstone MB. Genetic reassortants of lymphocytic choriomeningitis virus: unexpected disease and mechanism of pathogenesis. J Virol. 1986;59 : 363–368. 2426464
37. Lukashevich IS. Generation of reassortants between African arenaviruses. Virology. 1992;188 : 600–605. 1585636
38. Vezza AC, Bishop DHL. Recombination Between Temperature-Sensitive Mutants of the Arenavirus Pichinde. J Virol. 1977;24 : 712–715. 916035
39. Bodewes R, Raj VS, Kik MJL, Schapendonk CM, Haagmans BL, Smits SL, et al. Updated phylogenetic analysis of arenaviruses detected in boid snakes. J Virol. 2014;88 : 1399–1400. doi: 10.1128/JVI.02753-13 24379418
40. Hetzel U, Sironen T, Laurinmäki P, Liljeroos L, Patjas A, Henttonen H, et al. Reply to “Updated phylogenetic analysis of arenaviruses detected in boid snakes.” J Virol. 2014;88 : 1401. doi: 10.1128/JVI.03044-13 24379419
41. Chang L-W, Fu A, Wozniak E, Chow M, Duke DG, Green L, et al. Immunohistochemical detection of a unique protein within cells of snakes having inclusion body disease, a world-wide disease seen in members of the families Boidae and Pythonidae. PloS One. 2013;8: e82916. doi: 10.1371/journal.pone.0082916 24340066
42. Ladner JT, Beitzel B, Chain PSG, Davenport MG, Donaldson E, Frieman M, et al. Standards for Sequencing Viral Genomes in the Era of High-Throughput Sequencing. mBio. 2014;5: e01360–14. doi: 10.1128/mBio.01360-14 24939889
43. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9 : 357–359. doi: 10.1038/nmeth.1923 22388286
44. Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinforma Oxf Engl. 2006;22 : 1658–1659.
45. Martin DP, Lemey P, Lott M, Moulton V, Posada D, Lefeuvre P. RDP3: a flexible and fast computer program for analyzing recombination. Bioinforma Oxf Engl. 2010;26 : 2462–2463.
46. Sharp GB, Kawaoka Y, Jones DJ, Bean WJ, Pryor SP, Hinshaw V, et al. Coinfection of wild ducks by influenza A viruses: distribution patterns and biological significance. J Virol. 1997;71 : 6128–6135. 9223507
47. Ghedin E, Fitch A, Boyne A, Griesemer S, DePasse J, Bera J, et al. Mixed infection and the genesis of influenza virus diversity. J Virol. 2009;83 : 8832–8841. doi: 10.1128/JVI.00773-09 19553313
48. Takayama S, Yamazaki S, Matsuo S, Sugii S. Multiple Infection ofTT Virus(TTV) with Different Genotypes in Japanese Hemophiliacs. Biochem Biophys Res Commun. 1999;256 : 208–211. 10066448
49. Niel C, Saback FL, Lampe E. Coinfection with multiple TT virus strains belonging to different genotypes is a common event in healthy Brazilian adults. J Clin Microbiol. 2000;38 : 1926–1930. 10790123
50. Schmitt M, Dondog B, Waterboer T, Pawlita M, Tommasino M, Gheit T. Abundance of Multiple High-Risk Human Papillomavirus (HPV) Infections Found in Cervical Cells Analyzed by Use of an Ultrasensitive HPV Genotyping Assay. J Clin Microbiol. 2010;48 : 143–149. doi: 10.1128/JCM.00991-09 19864475
51. Weng Z, Barthelson R, Gowda S, Hilf ME, Dawson WO, Galbraith DW, et al. Persistent Infection and Promiscuous Recombination of Multiple Genotypes of an RNA Virus within a Single Host Generate Extensive Diversity. PLoS ONE. 2007;2: e917. 17878952
52. Lehmann-Grube F. A carrier state of lymphocytic choriomeningitis virus in L cell cultures. Nature. 1967;213 : 770–773. 4961982
53. Nguyen-hong-Diet null, Libíková H. Viral superinfection in cells carrying an arenavirus and/or a togavirus. Acta Virol. 1978;22 : 477–484. 35946
54. Damonte EB, Mersich SE, Coto CE. Response of cells persistently infected with arenaviruses to superinfection with homotypic and heterotypic viruses. Virology. 1983;129 : 474–478. 6312683
55. Bruns M, Zeller W, Lehmann-Grube F. Studies on the mechanism of lymphocytic choriomeningitis virus homologous interference. Med Microbiol Immunol (Berl). 1986;175 : 101–104. 3014286
56. Ellenberg P, Edreira M, Scolaro L. Resistance to superinfection of Vero cells persistently infected with Junin virus. Arch Virol. 2004;149 : 507–522. 14991440
57. Welsh RM, Pfau CJ. Determinants of lymphocytic choriomeningitis interference. J Gen Virol. 1972;14 : 177–187. 4622135
58. Tauraso N, Shelokov A. PROTECTION AGAINST JUNIN VIRUS BY IMMUNIZATION WITH LIVE TACARIBE VIRUS. Proc Soc Exp Biol Med Soc Exp Biol Med N Y N. 1965;119 : 608–611. 14328956
59. Weissenbacher MC, Coto CE, Calello MA. Cross-protection between Tacaribe complex viruses. Presence of neutralizing antibodies against Junin virus (Argentine hemorrhagic fever) in guinea pigs infected with Tacaribe virus. Intervirology. 1975;6 : 42–49. 178627
60. Weissenbacher MC, Coto CE, Calello MA, Rondinone SN, Damonte EB, Frigerio MJ. Cross-protection in nonhuman primates against Argentine hemorrhagic fever. Infect Immun. 1982;35 : 425–430. 6276301
61. Hall MP, Unch J, Binkowski BF, Valley MP, Butler BL, Wood MG, et al. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem Biol. 2012;7 : 1848–1857. doi: 10.1021/cb3002478 22894855
62. Emonet SF, Garidou L, McGavern DB, de la Torre JC. Generation of recombinant lymphocytic choriomeningitis viruses with trisegmented genomes stably expressing two additional genes of interest. Proc Natl Acad Sci U S A. 2009;106 : 3473–3478. doi: 10.1073/pnas.0900088106 19208813
63. A revision of the system of nomenclature for influenza viruses: a WHO Memorandum. Bull World Health Organ. 1980;58 : 585–591. 6969132
64. Collis A, Fenili R. The Modern U.S. Reptile Industry [Internet]. Georgetown Economic Services, LLC; 2011. http://www.whitehouse.gov/sites/default/files/omb/assets/oira_1018/1018_04182011-3.pdf
65. Webster RG. Wet markets—a continuing source of severe acute respiratory syndrome and influenza? The Lancet. 2004;363 : 234–236. 14738798
66. Knust B, Ströher U, Edison L, Albariño CG, Lovejoy J, Armeanu E, et al. Lymphocytic Choriomeningitis Virus in Employees and Mice at Multipremises Feeder-Rodent Operation, United States, 2012. Emerg Infect Dis. 2014;20 : 240–247. doi: 10.3201/eid2002.130860 24447605
67. Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29: e45. 11328886
68. Peirson SN, Butler JN, Foster RG. Experimental validation of novel and conventional approaches to quantitative real-time PCR data analysis. Nucleic Acids Res. 2003;31: e73. 12853650
69. Stenglein MD, Jacobson ER, Wozniak EJ, Wellehan JFX, Kincaid A, Gordon M, et al. Ball Python Nidovirus: a Candidate Etiologic Agent for Severe Respiratory Disease in Python regius. mBio. 2014;5: e01484–14. doi: 10.1128/mBio.01484-14 25205093
70. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinforma Oxf Engl. 2009;25 : 2078–2079.
71. Drummond AJ, Ashton B, Buxton S, Cheung M, Cooper A, Heled J, et al. Geneious v5.1 [Internet]. 2010. http://www.geneious.com
72. Ruby JG, Bellare P, DeRisi JL. PRICE: Software for the Targeted Assembly of Components of (Meta)Genomic Sequence Data. G3 GenesGenomesGenetics. 2013; g3.113.005967.
73. Katoh K, Standley DM. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol Biol Evol. 2013;30 : 772–780. doi: 10.1093/molbev/mst010 23329690
74. Talavera G, Castresana J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol. 2007;56 : 564–577. 17654362
75. Darriba D, Taboada GL, Doallo R, Posada D. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods. 2012;9 : 772–772. doi: 10.1038/nmeth.2109 22847109
76. Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52 : 696–704. 14530136
77. Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinforma Oxf Engl. 2003;19 : 1572–1574.
78. Hamada M, Kiryu H, Sato K, Mituyama T, Asai K. Prediction of RNA secondary structure using generalized centroid estimators. Bioinformatics. 2009;25 : 465–473. doi: 10.1093/bioinformatics/btn601 19095700
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 5
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Human Cytomegalovirus miR-UL112-3p Targets TLR2 and Modulates the TLR2/IRAK1/NFκB Signaling Pathway
- Paradoxical Immune Responses in Non-HIV Cryptococcal Meningitis
- Expression in the Fat Body Is Required in the Defense Against Parasitic Wasps in
- Survives with a Minimal Peptidoglycan Synthesis Machine but Sacrifices Virulence and Antibiotic Resistance
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy