
The Recent Evolution of a Maternally-Inherited Endosymbiont of Ticks Led to the Emergence of the Q Fever Pathogen,
How virulent infectious diseases emerge from non-pathogenic organisms is a challenging question. Here, we address this evolutionary issue in the case of Q fever. Its causative agent, the intracellular bacterium Coxiella burnetii, is extremely infectious to humans and a variety of animals. However, uncertainty persists regarding its evolutionary origin, including the identity and lifestyle of its ancestors. In this article, we show that C. burnetii arose from a rare evolutionary transformation of a maternally-inherited endosymbiont of ticks into a specialized and virulent pathogen of vertebrates. While arthropod symbionts are typically transmitted maternally and thought not to be infectious to vertebrates, we establish here that one Coxiella symbiont has evolved the necessary adaptations to exploit the vertebrate cell, leading to the emergence of Q fever.
Published in the journal:
. PLoS Pathog 11(5): e32767. doi:10.1371/journal.ppat.1004892
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004892
Summary
How virulent infectious diseases emerge from non-pathogenic organisms is a challenging question. Here, we address this evolutionary issue in the case of Q fever. Its causative agent, the intracellular bacterium Coxiella burnetii, is extremely infectious to humans and a variety of animals. However, uncertainty persists regarding its evolutionary origin, including the identity and lifestyle of its ancestors. In this article, we show that C. burnetii arose from a rare evolutionary transformation of a maternally-inherited endosymbiont of ticks into a specialized and virulent pathogen of vertebrates. While arthropod symbionts are typically transmitted maternally and thought not to be infectious to vertebrates, we establish here that one Coxiella symbiont has evolved the necessary adaptations to exploit the vertebrate cell, leading to the emergence of Q fever.
Introduction
‘Query fever’ (Q fever) is a highly infectious zoonotic disease first identified in 1937 [1,2,3,4,5]. The causative agent, the obligate intracellular bacterium Coxiella burnetii, infects a variety of vertebrate species, including humans. Sporadic cases in humans occur annually worldwide, but occasional outbreaks are also common [1,2,3,4]. For example, in the Netherlands more than 4,000 human cases were reported between 2007 and 2010 [6]. While most human cases are self-limiting with fever and fatigue, acute forms range from mild flu-like symptoms to pneumonia or hepatitis. The disease can also become chronic (mainly endocarditis), and, though rarely fatal, remains highly debilitating even when treated with antibiotics [1,2]. Most human cases are linked to contact with infected livestock, especially goats and sheep, which suffer abortion and reproductive disorders. Infection usually occurs by the inhalation of aerosolized resistant small cell variants that are present in the excretions of infected animals. Other modes of transmission including ingestion of unpasteurized milk or dairy products; human-to-human contact is also possible but considered rare [1,2,4,5]. One of the most virulent reference strains of C. burnetii (strain RSA 493 / Nine Mile I [7]) was isolated from a guinea pig on which field-collected Rocky Mountain wood ticks Dermacentor andersoni had fed, suggesting that transmission through tick bites may also occur [8]. The small cell variants of the bacterium can survive and remain highly infectious for long periods in the environment, leading to the classification of C. burnetii as potential bioterrorism agent [9].
The evolutionary origin of Q fever is unclear since the C. burnetii ancestor and its primary lifestyle remain entirely unknown. Historically, C. burnetii was assigned to the taxonomic order Rickettsiales (Alphaproteobacteria), but it has been recently considered more closely related to the Legionellales order (Gammaproteobacteria) because of its genetic proximity to the Legionnaires' disease agent, Legionella pneumophila [10]. The Legionellales order includes many other intracellular bacteria infecting non-vertebrate species, such as, for instance, Rickettsiella species that are both widespread and biologically diverse in arthropods [11,12,13]. Within the Coxiella genus, the only known relative of C. burnetii which has been formally identified is C. cheraxi, a pathogen of crayfishes [14]. Many past descriptions of Coxiella were likely biased toward the detection of pathogenic strains since most C. burnetii isolates were collected from humans or domestic ruminants during Q fever outbreaks [1,5,15]. However, the advent of 16S rRNA gene sequencing as a universal DNA barcoding marker in bacteria has led to the description of a few novel Coxiella-like organisms in non-vertebrate species (listed in [16]), and particularly in ticks [17,18,19,20,21,22,23,24,25]. All these Coxiella-like organisms are closely related, but genetically distinct to C. burnetii, suggesting that some diversity exists within the Coxiella genus. The highly conserved nature of the 16S rRNA gene sequences has prevented researchers from establishing the exact relationship between C. burnetii and Coxiella-like organisms, and a sister clade relationship is commonly assumed [16,18,21,22,23].
The Coxiella-like organisms differ from C. burnetii in their biological traits and some may behave as subtle symbionts engaged in intricate interactions with ticks. In ticks belonging to Ornithodoros, Amblyomma and Rhipicephalus genera, Coxiella-like organisms were found to massively infect ovaries and to be maternally inherited through the egg cytoplasm [18,20,21,26]. In these tick species, the presence of the bacteria in the Malpighian tubules further suggests a possible role in nutrition by potentially provisioning their hosts with essential nutriments [20,21,26]. Indeed, the elimination of these bacteria with an antibiotic treatment was shown to negatively impact the fitness of the lone star tick A. americanum [27]. Accordingly, when the Coxiella-like bacterium found in A. americanum was recently sequenced [16], no recognizable virulence genes were found, indicating that this bacterium is likely non-pathogenic. In contrast, its genome encodes major vitamin and cofactor biosynthesis pathways, suggesting that it may be a vitamin-provisioning endosymbiont. This interaction exhibits the typical hallmarks of maternally-inherited symbionts with essential roles in arthropod biology [28,29]. Such patterns have been found in other exclusive blood-feeding species like bedbugs [30] and tsetse flies [31], two insect groups which rely on a single food source throughout their developmental cycle and harbor beneficial microbes that provide nutrients absent from their restricted diets. The Coxiella-like organisms of ticks share obvious similarities with these beneficial endosymbionts.
Here, we examine the origin of the Q fever pathogen, C. burnetii, by inferring the evolutionary processes that have shaped diversity within the entire Coxiella genus. To this aim, we first sampled an extensive range of ticks, with 58 tick species examined, and developed a sensitive detection method that reveals a wider Coxiella diversity than recognized in past studies. Second, instead of relying solely on the 16S rRNA gene, a molecular marker that is notoriously inadequate for inferring reliable fine-scale phylogenies [32], we used a novel multilocus typing method, allied to Whole Genome Sequencing (WGS) data, and conducted phylogenetic analyses on a large amount of DNA sequence data. Third, we examined two major ecological features of Coxiella-like organisms, i.e. their ability to be maternally-inherited through the tick egg cytoplasm and to grow in a vertebrate cell environment suitable to C. burnetii. Altogether, this corpus of data has led to the characterization of a large genetic and ecological diversity within the Coxiella genus, far beyond the C. burnetii type species. The Coxiella-like organisms of ticks form an ancient lineage of maternally-inherited tick endosymbionts that do not lie as a sister-clade to C. burnetii but rather form a basal lineage illustrative of the ancestral Coxiella life style.
Results
Ticks commonly harbor Coxiella-like organisms
We performed an extensive screening for the presence of Coxiella in 916 tick specimens from 58 species belonging to the two main tick families, Ixodidae (hard ticks, 36 species) and Argasidae (soft ticks, 22 species) (Fig 1 and Table 1). Except for 37 specimens (6 species) derived from laboratory colonies, all other tick specimens were sampled from natural populations in Europe, Americas, Africa, Oceania and Asia (n = 112 localities). In these populations, ticks were collected either in the host habitat or directly on hosts (Table 1). To detect C. burnetii and its relatives, we developed a detection method based on a nested polymerase chain reaction (PCR) using total tick DNA extracts to amplify a 539–542 base-pair (bp) fragment of the Coxiella rpoB gene (Table A in S1 Text).


Using this procedure, all the tick-borne bacteria we detected belong to the Legionellales order and can be unambiguously assigned either to Coxiella or to its sister genus, Rickettsiella. Whole tick DNA extracts from more than two thirds of the specimens (637 out of 916, 69.6%) and the species (40 out of 58, 70.0%) were found to be positive for Coxiella (Fig 1 and Table 1). Coxiella was found in most tested genera of hard ticks (Rhipicephalus, Ixodes, Amblyomma, Dermacentor, Haemaphysalis) and soft ticks (Ornithodoros, Argas). In almost all infected species, Coxiella was detected in >90% of the examined specimens, indicating high Coxiella prevalence in diverse tick species. For example, infection was apparently fixed in populations of most Rhipicephalus and Ornithodoros species (Table 1). In contrast, Coxiella was frequently absent in Ixodes species and displayed highly variable prevalence in the five infected species (out of 12 screened).
Other Legionellales bacteria of the genus Rickettsiella were found in 52 specimens (5.7%) from six Ornithodoros species and three Ixodes species (Fig 1 and Table 1). In two of the three Rickettsiella-infected Ixodes species, i.e. I. ricinus and I. uriae, Coxiella was also found, but in different individuals and in distinct populations (i.e., no co-infection by Coxiella and Rickettsiella occurred at individual and population levels; Table 1). Adding the Rickettsiella-positive samples, we found that 689 of the 916 examined tick specimens (75.2%) and 44 of 58 screened species (76%) harbored either Coxiella or one of its relatives.
High genetic diversity among tick-borne Coxiella
To characterize Coxiella genetic diversity, we developed a multi-locus typing method based on five conserved bacterial genes including rpoB and four other housekeeping genes: 16S rRNA, 23S rRNA, GroEL and dnaK (Table A and Fig A in S1 Text). Multi-locus sequences were obtained from a subsample of 85 Coxiella - and 12 Rickettsiella-positive tick specimens (one to four specimens per infected species were examined). All five bacterial genes were successfully amplified from 71 Coxiella - and 12 - Rickettsiella positive specimens representing 35 Coxiella - and six Rickettsiella-infected tick species. For five other Coxiella-infected species (i.e., 14 individual ticks), only three to four bacterial genes were successfully amplified. The sequences were easily readable without double peaks, indicating that there was no coinfection of Coxiella/Rickettsiella strains in any specimen.
The overall dataset included 33 to 40 alleles per bacterial gene (Table 2) and 51 new multi-locus genotypes (43 in Coxiella and eight in Rickettsiella). Within the Coxiella genus, all pairs of 16S rRNA gene sequences are at least 93% identical (Table 2) and range in threshold values typically used to delineate other Legionellales genera such as Legionella [33] and Rickettsiella [34]. Each of the infected tick species harbored a specific bacterial genotype or a set of closely related genotypes. None of the Coxiella multi-locus genotypes identified in ticks was identical to those of the 15 C. burnetii reference strains (Table B in S1 Text), although some showed moderate levels of nucleotide identity: pairwise identity between the two groups ranged from 77.8% to 97.7%. For each bacterial gene, the genetic diversity was significantly higher in the Coxiella strains of ticks than in C. burnetii as illustrated by the metrics on their respective genetic diversity (Table 2, paired t test, all P < 0.02).

Coxiella burnetii originated from a tick-borne Coxiella ancestor
We constructed a multi-gene phylogeny of the entire Coxiella genus using a dataset that included the Coxiella and Rickettsiella sequences from ticks, the 15 C. burnetii reference genomes, as well as sequences from Legionella spp. and more distant outgroups that were available in GenBank (Table B in S1 Text). The concatenated sequences included 3009 unambiguously aligned base pairs (bp). Prior recombination tests showed that Coxiella and Rickettsiella strains did not exhibit a strictly clonal structure, but rather experienced significant genetic exchanges. We thus applied a sequence-based network approach that does not force relationships to be tree-like but rather incorporates recombination into the phylogenetic reconstruction. The network results (Fig 2), as well as the results from the Maximum Likelihood (ML) tree-based analysis (Fig B in S1 Text), consistently showed that the Coxiella genus can be split into four main clades (labeled A-to-D hereafter) with each clade clustering the Coxiella genotypes found in five to 15 tick species. The phylogenetic analyses also highlight that all C. burnetii isolates cluster into a unique subclade embedded within the A clade (Fig B and C in S1 Text and Fig 2). Notably, the closest relatives of C. burnetii are the Coxiella strains from soft ticks of the Ornithodoros and Argas genera, suggesting that the common ancestor of C. burnetii originated from a Coxiella hosted by soft ticks.

The partitioning of Coxiella diversity among tick species revealed a complex structure, indicating a role for both co-divergence and horizontal transfer events in the evolution of this bacterial group. Closely related Coxiella-like organisms were frequently found in closely related tick species, a pattern suggestive of co-divergence between Coxiella and ticks (Fig B in S1 Text and Fig 2). For instance, all the Coxiella-like organisms found in the 12 examined Rhipicephalus tick species cluster together within the C clade, whereas all the Coxiella-like organisms in the Ixodes species cluster within the B clade (Fig B in S1 Text and Fig 2). Conversely, some Coxiella-like organisms found in related tick species are only distantly related and do not cluster together (e.g., the Coxiella-like organisms of Ornithodoros soft ticks are scattered among the A, B and C clades), a pattern suggestive of horizontal transfers among tick species.
Further analyses were conducted by examining public repositories of DNA sequencing data generated by the whole genome sequencing (WGS) projects of the cattle tick R. microplus and the deer tick I. scapularis. Using the 1,995,281 bp C. burnetii (str. Nine Mile I RSA 493) genome as a probe, we found clear evidence of Coxiella infections in R. microplus, but not in I. scapularis. A total of 31 contigs (514–2,349 bp, totaling 34,990 bp) from R. microplus sequencing were uniquely attributable to Coxiella. They matched 50 genes of C. burnetii with 68-to-100% nucleotide identity (Fig A and Table C in S1 Text and Fig 3A). Alignment of the 31 Coxiella contigs to other bacterial genomes, including the 15 C. burnetii reference genomes (19,304 unambiguously aligned bp), corroborates the finding of our prior five loci-based analyses: the Coxiella strain identified in R. microplus is evolutionarily related, but distinct, to C. burnetii (Fig 3B).
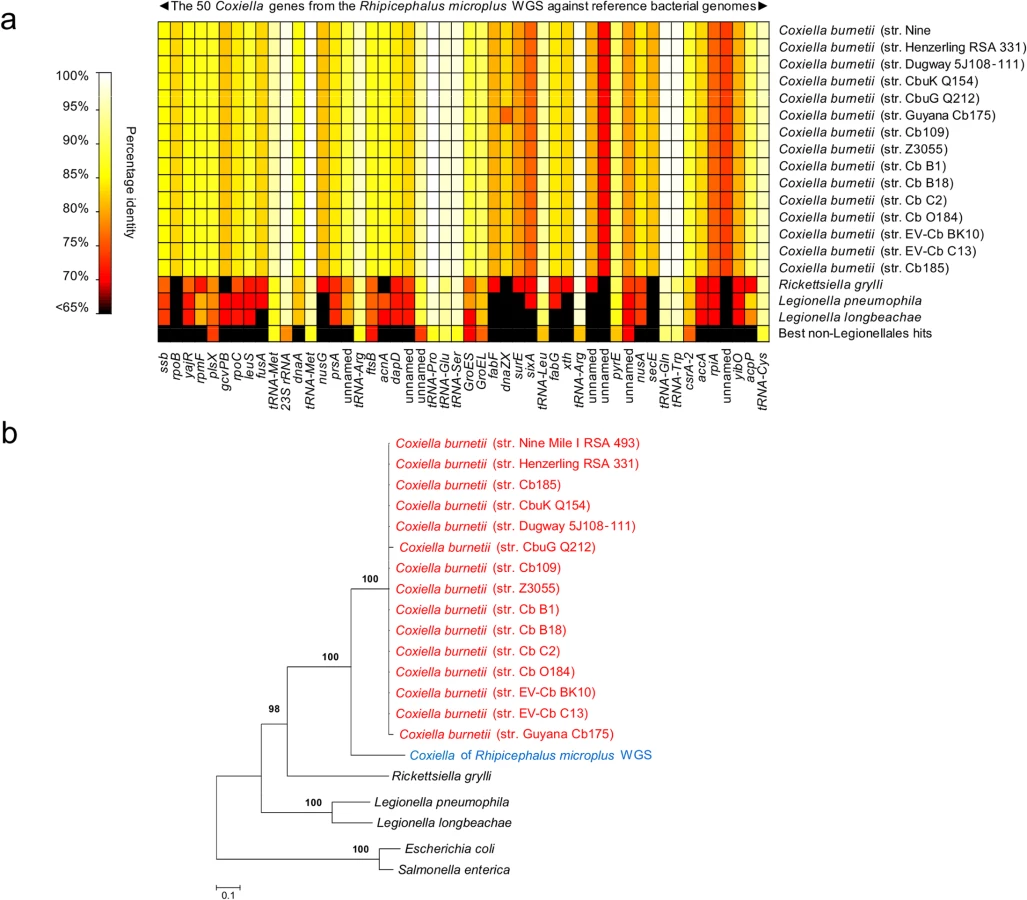
Coxiella-like organisms are maternally inherited in ticks
It should be noted that the R. microplus WGS DNA examined above was extracted from eggs of an inbred strain of ticks (Deutsch strain), first derived from a few field specimens sampled in Texas in 2001, and reared for at least seven generations in the laboratory. The presence of Coxiella DNA in the WGS of R. microplus eggs thus raised the issue of their maternal inheritance in ticks. To address this question, 24 gravid females of four Coxiella-positive tick species were collected either from seabird nests (O. maritimus, n = 8 females), from a dog (R. sanguineus, n = 1) or from laboratory colonies (R. microplus, n = 7; A. americanum, n = 8) in order to test for the presence of Coxiella-infection in the cytoplasm of their progeny (8 to 14 surface-sterilized eggs per female were individually examined; i.e., 244 eggs in total). The occurrence of maternal transmission was detected in all four tick species and in almost all eggs: O. maritimus—79 Coxiella-positive eggs out of 80, R. sanguineus—14 out of 14, R. microplus—68 out of 70, and A. americanum—80 of 80. The mean transmission rate can thus be estimated at 0.988 (95% confidence interval, 0.965–0.994), demonstrating highly efficient maternal transmission of Coxiella in ticks. Maternal inheritance is thus widespread in the Coxiella genera, being found in three different clades (A: Coxiella-like organism of O. maritimus; C: R. sanguineus and R. microplus; D: A. americanum).
Differences in metabolic requirements of Coxiella-like organisms and C. burnetii
We next compared the metabolic requirements of Coxiella-like organisms with those of C. burnetii by assessing their ability to replicate in both an axenic medium ACCM2 (mimicking the environment of the acidified lysosome-like vacuoles of phagocytes typically colonized by C. burnetii; [35]) and directly inside vertebrate host cells. First, ACCM2 was inoculated with Coxiella-like organisms extracted from eggs of either O. maritimus, R. microplus or A. americanum. Inoculated media were incubated for 10 days under standard conditions used to amplify C. burnetii. Although our C. burnetii positive controls readily replicated in the media, the Coxiella obtained from eggs of the three tick species did not grow. We then incubated egg homogenates from ticks of O. maritimus and R. microplus with mammalian cell cultures for seven days. Similar to results under axenic conditions, the incubation of vertebrate cell lines with Coxiella-like organisms failed to produce Coxiella-containing vacuoles, whereas the same cell lines incubated with C. burnetii under the same conditions were readily infected. The apparent inability to amplify tick-borne Coxiella through standardized protocols, well-characterized for C. burnetii, suggests that, despite their phylogenetic proximity, the Coxiella-like bacteria are adapted to radically different environments.
Discussion
Since its original description, C. burnetii infections have been characterized in a wide variety of hosts. While only two species have been formally identified within the Coxiella genus, we show here that a far greater diversity of Coxiella exists in ticks. We detect the presence of Coxiella-like organisms in many more tick species than previously known [17,18,19,20,21,22,23,24,25] and describe a far wider genetic diversity among these bacteria than previously suspected. The incidence of Coxiella, as well as of its sister genus Rickettsiella, in ticks is exceptionally high, with approximately three quarters of tick species infected. Although possible tick-borne transmission of C. burnetii has been reported [1,2,8], none of the 43 new Coxiella genotypes identified here are identical to C. burnetii. We also demonstrate for genetically divergent Coxiella strains (i.e., members of the A, C and D clades) found in four tick species that infection is primarily transmitted maternally via the egg cytoplasm. These results converge to support the hypothesis that these Coxiella-like organisms are specific endosymbionts of ticks. Phylogenetic evidence further shows that one of the Coxiella-like organisms belonging to the A clade and primarily hosted by soft ticks has served as the progenitor of C. burnetii.
Three complementary lines of argument indicate a much longer evolutionary history for Coxiella-tick associations than for vertebrate-Coxiella associations. The first lies in the broad distribution of Coxiella and Rickettsiella bacteria across tick species, genera and families. The second concerns the extensive genetic diversity found in tick-borne Coxiella strains compared to C. burnetii strains, as illustrated by the clear subdivision of this genus into four highly divergent clades (A-D). Finally, the clustering of all C. burnetii strains within one of the clades of tick-borne Coxiella shows that the ancestor of C. burnetii was a tick-associated bacterium which succeeded in infecting vertebrate cells. The remarkably low genetic diversity of C. burnetii, previously noted in other studies [36,37], indicates a unique and recent emergence of this highly infectious vertebrate pathogen. Interestingly, this hypothesis was initially raised a decade ago from observations of the profound differences in genome architecture of C. burnetii relative to other pathogenic intracellular bacteria [38]. It was again emphasized from the genome sequencing of new C. burnetii strains [39]. Our data brings further support to this hypothesis by demonstrating that C. burnetii roots within the Coxiella phylogeny. Comparative genome sequences of C. burnetii [38, 39] and of the Coxiella-like organism from A. americanum [16] also suggest that Coxiella bacteria differ substantially in terms of genome size and gene content. The C. burnetii genome (A clade) has a size of ca 2Mb [38, 39], whereas the genome of the Coxiella-like organism isolated in A. americanum (D clade) is only about a 1/3 of this size (ie. 0.66 Mb) with a large percentage of missing genes [16]. This reduction in genome size may limit the transition to pathogenicity, and suggests that some Coxiella-like organisms may have evolved towards exclusive and irreversibly specialized interactions with their tick hosts. Overall, the diversity of genome sizes emphasizes that members of the different Coxiella clades may have retained a variety of evolutionary strategies to favour their spread and persistence in their hosts.
We identified Coxiella as a major emerging clade of bacterial endosymbionts allied to ticks. Coxiella-like organisms are maternally-transmitted through the egg cytoplasm at high frequency with 98–100% mother-to-offspring transmission, a pattern also reported in previous studies [20,21,26]. This transmission pattern is the rule for a variety of bacterial endosymbionts that live exclusively within arthropod cells [28,29,40]. While some, like Wolbachia, are globally common symbionts estimated to infect ca. 40% of insect species [41,42], others are globally rare, but common and important in particular arthropod groups [28]. This is precisely the case for Coxiella-like organisms; although they have not been found in other arthropod species, they are commonly associated with ticks. This leads to the obvious question of the phenotypic consequences of Coxiella-tick interactions. In some cases, Coxiella-like organisms of ticks likely act as obligate mutualistic symbionts required to support normal tick development, potentially provisioning their hosts with essential nutriments absent in vertebrate blood [16,20,21,26,27]. The ubiquity of Coxiella in some tick groups-such as in the Rhipicephalus genus in which infection is at fixation - corroborates the hypothesis of an obligate endosymbiont. This is not, however, the case for all tick species since some, such as I. ricinus and I. uriae, harbour Coxiella-like organisms at much lower frequencies. In these tick species, Coxiella is more likely to behave as a conditional mutualist-i.e., that confers advantages under certain environmental conditions - or as a reproductive parasite-i.e., that manipulates host reproduction toward the production of daughters (the transmitting sex), as commonly observed in arthropods with a variety of facultative symbionts [28,40]. It should also be noted that other endosymbionts also occur in ticks and may have evolved under complex multispecific interactions [17,24]. For instance, whereas the soft tick O. moubata was not found to be infected by a Coxiella-like organism in the present study, this tick species has been found to be infected by an endosymbiont belonging to the Francisella genus [17]. Endosymbionts other than Coxiella may thus interact with ticks, a pattern suggesting that endosymbiotic systems can be dynamic across tick lineages. These different hypotheses will now require specific testing.
Another question remains concerning the degree of vertebrate infection risk by the Coxiella-like organisms of ticks. Ticks are found worldwide and blood-feed on many different hosts; a combination of traits that may facilitate tick-to-vertebrate transfers of Coxiella. However, the bacteria observed in this study seem confined to ticks and, to our knowledge, none have ever been isolated from a vertebrate or associated with clinical symptoms. This suggests that these tick-associated bacteria currently pose a much lower infection risk to vertebrates than C. burnetii. As discussed above, the genome reduction of the Coxiella-like organism isolated in A. americanum, with the lack of nearly all the genes associated with pathogenicity [16], corroborates this view. Moreover, the inability to grow tick-borne bacteria in vertebrate cells highlights the significant barrier that must be overcome by the bacteria to successively achieve tick-to-vertebrate transmission. This type of transmission may, nonetheless, occasionally occur; an avian Coxiella-like organism was recently reported to induce fatal systematic infections in domestic birds [43,44,45]. A very similar infection pattern was found for another maternally inherited endosymbiont, Arsenophonus, a widespread bacterium in different insect groups [41,46,47,48,49]. In particular, some Arsenophonus strains were detected in the phloem of plants fed on by infected phytophagous insects and were assumed to be opportunistic plant pathogens [50]. In such cases, the plant host may act as an ecological arenas for the global exchange of endosymbionts like Arsenophonus, serving as a possible intermediate host for the horizontal transfer of bacteria among insect species [48]. In the case of Coxiella-like organisms, the extent of exchange between different tick species via the vertebrate host is yet to be established, but could be favoured by tick co-feeding (ticks feeding in close proximity on the host). The genetic similarity between Coxiella-like organisms found in unrelated tick species highlights the capacity to shift tick host species. Future research is now needed to assess the potential of different Coxiella-like organisms to infect vertebrates.
The reasons why C. burnetii is a highly virulent pathogen of vertebrates, but not Coxiella-like organisms (especially those from the A clade) remain unknown. As an intracellular pathogen with airborne transmission, C. burnetii has evolved specific mechanisms to survive in the abiotic environment, as well as to infect and exploit vertebrate cells [15]. Several evolutionary pathways may explain the acquisition of the genetic material necessary for this major lifestyle transition; this includes spontaneous genetic mutations in the genome of a tick-Coxiella ancestor, or the more likely transfer and integration of virulence genes from a co-infecting pathogen. The opportunity of gene transfer among bacteria, irrespective of their pathogenic or symbiotic properties, relies on their frequent co-occurrence within the same tick host [25,51,52]. The Coxiella-like organisms of the A clade may have dynamic genomes as observed in many arthropod symbionts: although they reside in confined intracellular environments, arthropod symbionts commonly experience variable degrees of recombination and gene transfer [53,54,55,56,57]. These gene transfers have served as immediate and powerful mechanisms of rapid adaptation in many endosymbionts, such as Wolbachia [56] and Hamiltonella [55,57]. This mechanism may explain the evolutionary transition from a Coxiella tick-symbiont of the A clade to the vertebrate pathogen C. burnetii. Other genetic connections are also possible; several C. burnetii genes that may contribute to major virulence traits, such as tissue tropism, are similar to eukaryotic genes and may have been acquired through lateral gene transfers from eukaryotes [38,39]. Detailed studies of virulence genes in C. burnetii and their homology with Coxiella-like organisms of the A clade will now be necessary to understand the remarkable emergence of the Q fever agent.
The evolutionary transition observed within the Coxiella genus is one of the rare cases reported to date of an arthropod-inherited symbiont evolving metabolic adaptations leading to the emergence of a vertebrate infectious disease. Another such transition occurred in the Rickettsia genus. The best-known members of this genus are transmitted by blood-feeding arthropods and are pathogenic in the vertebrate host. However, in recent years, many maternally-inherited Rickettsia endosymbionts found exclusively in arthropods have been discovered [58,59]. The examination of the evolutionary history of the Rickettsia genus revealed that this bacterium originated from endosymbionts of invertebrates and only secondarily became vertebrate pathogens [58,59]. Like Coxiella, some Rickettsia species of blood-feeding hosts have underwent a horizontal transmission through a vertebrate host, leading to pathogen emergence. Other bacteria, such as Arsenophonus [47,48] and Sodalis [60] may have had similar life cycle transitions, but the case of Coxiella is unique in that the arthropod host is no longer required to complete its life cycle.
In conclusion, we show that C. burnetii arose from a rare and recent event: the evolutionary transformation of a maternally inherited endosymbiont of ticks into a specialized and virulent pathogen of vertebrates. This raises a series of exciting questions related to both how Coxiella endosymbionts made the major evolutionary transition leading to the emergence of Q fever and their role in the population dynamics of ticks. Identifying the evolutionary processes that transform symbiotic bacteria into emerging pathogens will require further exploration into the biology of the entire Coxiella genus.
Methods
Tick collection
The examined specimens represent the two main tick families, nine genera, 58 species and 112 populations from around the world (Table 1). Field specimens were sampled on various host species belonging to major mammal and bird families or from their habitats. We also used specimens from laboratory colonies reared in captivity for at least three generations for six tick species (derived from field specimens collected in North America, South America, Africa and China). All samples were preserved in 70–90% ethanol at room temperature until use. Before storage, tick eggs collected under laboratory conditions were surface-sterilized with 2.6% sodium hypochlorite and 0.5% SDS for 1 min and washed with sterile water to avoid external bacterial contamination.
Coxiella screening and typing
Tick DNA was individually extracted using the DNeasy Blood & Tissue Kit (QIAGEN) following manufacturer instructions. DNA template quality was systematically verified by PCR amplification of the 18S ribosomal RNA (18S rRNA) or the cytochrome oxydase 1 (C01) arthropod primers (Table A in S1 Text). Tick DNA samples were then tested for Coxiella presence using a nested PCR assay and sequencing of the rpoB gene using Coxiella-specific primers. The use of nested PCR was efficient at decreasing the probability of contamination from unwanted amplification products. Additional PCR assays on the 16S rRNA, 23S rRNA, GroEL and dnaK genes were conducted on a subsample of Coxiella-positive tick DNA to obtain additional DNA sequences for phylogenetic analyses. We used 15 recently published genomes of C. burnetii (mainly isolated from humans and ruminants) and the genome of Rickettsiella grylli from woodlice (listed in Table B in S1 Text) as references to design PCR primers. The efficiency of our typing method was ascertained through positive PCR amplification and clear sequences for the five loci in four cultured reference strains of C. burnetii (Table B in S1 Text). Gene features, primers and PCR conditions are detailed in Table A in S1 Text. All PCR products were visualized through electrophoresis in a 1.5% agarose gel. Positive PCR products were purified and sequenced in both directions (EUROFINS). The chromatograms were manually inspected and cleaned with CHROMAS LITE (http://www.technelysium.com.au/chromas_lite.html) and sequence alignments were done using CLUSTALW [61], both implemented in MEGA [62].
Coxiella sequences were also searched for in the whole genome sequence (WGS) data of R. microplus and I. scapularis (GenBank accession numbers ADMZ02000000 and ABJB000000000, respectively) using the 1,995,281 bp C. burnetii genome (str. Nine Mile I RSA 493, GenBank accession number NC002971) as a probe and the Basic Local Alignment Search Tool (BLAST) with default parameters. Table C in S1 Text reports the number and content of Coxiella contigs that were detected in the R. microplus WGS data.
Molecular and phylogenetic analyses
The GBLOCKS program [63] with default parameters was used to remove poorly aligned positions and to obtain non-ambiguous sequence alignments. Sequences of individual genes that differed by one or more nucleotides were assigned distinct allele numbers using DNASP [64], with the option of excluding sites with alignment gaps and/or missing data. Tick-borne Coxiella strains are defined as each unique combination of alleles. The genetic diversity estimates (Ps, number of polymorphic sites; Ad, allelic diversity; π, nucleotide diversity; D, average number of nucleotide differences between sequences) were computed using DNASP. Other statistical analyses were carried out using the R statistical package. All sequence alignments were checked for putative recombinant regions using the GENECONV [65] and RDP [66] methods available in the RDP3 computer analysis package [67].
Phylogenetic analyses were based on single and concatenated sequences of the five bacterial genes used in the multi-locus typing scheme and on the 50 Coxiella genes found in the R. microplus WGS data. Sequence alignments included Coxiella and Rickettsiella sequences obtained in this study from tick DNA, as well as sequences available in GenBank from reference strains of C. burnetii, Rickettsiella grylli, Legionella pneumophila, L. longbeacheae, and two more distantly related bacteria, Escherichia coli and Salmonella enterica (Table B in S1 Text). The evolutionary models fitting the sequence data most closely were determined using the Akaike information criterion with the program MEGA. For each data set examined, the best-fit approximation was the general time reversible model with gamma distribution and invariant sites (GTR+G+I). Network-based phylogenetic analyses were done using SplitsTree, implementing the evolutionary model under the agglomerating NeighborNet algorithm [68]. Tree-based phylogenetic analyses were done using maximum-likelihood (ML) analyses. A ML heuristic search using a starting tree obtained by neighbor-joining was conducted in MEGA. Clade robustness was assessed by bootstrap analysis using 1,000 replicates.
Culture assays
We first assessed the ability of tick-borne Coxiella to replicate in an axenic medium as follows. Tick eggs were surface-sterilized as described above and homogenized by hand in sterile water. Eggs homogenates were used to inoculate 2ml of the axenic medium ACCM2 [35] and incubated three days in a humidified atmosphere of 5% CO2 and 2.5% O2 at 37°C. 50μl of each culture were then diluted in 2ml of fresh ACCM2 and further incubated under the same conditions for 10 days to assess bacterial growth. We then assessed the ability of tick-borne Coxiella to replicate inside vertebrate host cells as follows. Surface-sterilized tick eggs were homogenized by hand in 1 ml of 10% Fœtal Bovine Sérum (FBS) supplemented MEM medium (GIBCO). The homogenate (0.5 ml) was diluted in 25 ml of 10% SVF-MEM and centrifuged at 4000 rpm (2000g) at 4°C for 30 min. Ten ml of the supernatant was mixed with 10 ml of 10% FSB-MEM and again centrifuged at 2000g at 4°C for 30 min. Ten ml of the supernatant was harvested and filtered through a sterile 0.45 μm pore size filter (MILLIPORE). Two flasks containing confluent Sheep Fœtal Thymus cells (SFT) cells were inoculated with 5 ml of the obtained filtrate and incubated at 35°C and allowed to grow for 12 weeks. Cell culture flasks were observed daily for the presence of contamination or growth signs such as vacuoles containing Coxiella, during the first week then once a week. As a positive control, a homogenate of C. burnetii was used following the same protocol.
Accession codes
Nucleotide sequences of PCR-amplified fragments of tick-borne Coxiella and Rickettsiella genes have been deposited in the GenBank nucleotide database under accession codes KP994768-KP994862 (16S rRNA), KP994678-KP994767 (23S rRNA), KP985445-KP985537 (GroEL), KP985265-KP985357 (rpoB) and KP985358-KP985444 (dnaK).
Supporting Information
Zdroje
1. Madariaga MG, Rezai K, Trenholme GM, Weinstein RA. Q fever: a biological weapon in your backyard. Lancet Infect Dis. 2003;3 : 709–721. 14592601
2. Raoult D, Marrie T, Mege J. Natural history and pathophysiology of Q fever. Lancet Infect Dis. 2005;5 : 219–226. 15792739
3. Vanderburg S, Rubach MP, Halliday JE, Cleaveland S, Reddy EA, Crump JA. Epidemiology of Coxiella burnetii infection in Africa: a OneHealth systematic review. PLoS Negl Trop Dis. 2014;8: e2787. doi: 10.1371/journal.pntd.0002787 24722554
4. Angelakis E, Raoult D. Q Fever. Vet Microbiol. 2010;140 : 297–309. doi: 10.1016/j.vetmic.2009.07.016 19875249
5. Bewley KR. Animal models of Q fever (Coxiella burnetii). Comp Med. 2013;63 : 469–476. 24326221
6. van der Hoek W, Dijkstra F, Schimmer B, Schneeberger PM, Vellema P, Wijkmans C, et al. Q fever in the Netherlands: an update on the epidemiology and control measures. Euro Surveill 2010;15.
7. Russell-Lodrigue KE, Andoh M, Poels MWJ, Shive HR, Weeks BR, Zhang GQ, et al. Coxiella burnetii isolates cause genogroup-specific virulence in mouse and guinea pig models of acute Q fever. Infect Immun. 2009;77 : 5640–5650. doi: 10.1128/IAI.00851-09 19786560
8. McDade JE. Historical aspects of Q fever. In: Marie TJ, editor. Q Fever Volume 1: The Disease. Boca Raton: CRC Press; 1990. pp. 5–21.
9. Oyston PC, Davies C. Q fever: the neglected biothreat agent. J Med Microbiol. 2011;60 : 9–21. doi: 10.1099/jmm.0.024778-0 21030501
10. Weisburg WG, Dobson ME, Samuel JE, Dasch GA, Mallavia LP, Baca O, et al. Phylogenetic diversity of the Rickettsiae. J Bacteriol. 1989;171 : 4202–4206. 2753854
11. Leclerque A, Kleespies RG. Type IV secretion system components as phylogenetic markers of entomopathogenic bacteria of the genus Rickettsiella. FEMS Microbiol Ecol. 2008;279 : 167–173. doi: 10.1111/j.1574-6968.2007.01025.x 18179586
12. Bouchon D, Cordaux R, Grève P. Rickettsiella, intracellular pathogens of arthropods. In: Zchori-Fein E, Bourtzis K, editors. Manipulative Tenants. Boca Raton: CRC Press; 2012. pp. 127–148.
13. Tsuchida T, Koga R, Horikawa M, Tsunoda T, Maoka T, Matsumoto S, et al. Symbiotic bacterium modifies aphid body color. Science 2010;330 : 1102–1104. doi: 10.1126/science.1195463 21097935
14. Tan CK, Owens L. Infectivity, transmission and 16S rRNA sequencing of a rickettsia, Coxiella cheraxi sp. nov., from the freshwater crayfish Cherax quadricarinatus. Dis Aquat Organ. 2000;41 : 115–122. 10918979
15. van Schaik EJ, Chen C, Mertens K, Weber MM, Samuel JE. Molecular pathogenesis of the obligate intracellular bacterium Coxiella burnetii. Nat Rev Microbiol. 2013;11 : 561–573. doi: 10.1038/nrmicro3049 23797173
16. Smith TA, Driscoll T, Gillespie JJ, Raghavan R. A Coxiella-like Endosymbiont is a potential vitamin source for the Lone Star Tick. Genome Biol Evol. 2015;7 : 831–838. doi: 10.1093/gbe/evv016 25618142
17. Noda H, Munderloh UG, Kurtti TJ. Endosymbionts of ticks and their relationship to Wolbachia spp. and tick-borne pathogens of humans and animals. Appl Environ Microbiol. 1997;63 : 3926–3932. 9327557
18. Almeida AP, Marcili A, Leite RC, Nieri-Bastos FA, Domingues LN, Ricardo Martins J, et al. Coxiella symbiont in the tick Ornithodoros rostratus (Acari: Argasidae). Ticks Tick Borne Dis. 2012;3 : 203–206. doi: 10.1016/j.ttbdis.2012.02.003 22480930
19. Duron O, Jourdain E, McCoy KD. Diversity and global distribution of the Coxiella intracellular bacterium in seabird ticks. Ticks Tick Borne Dis. 2014;5 : 557–563. doi: 10.1016/j.ttbdis.2014.04.003 24915875
20. Klyachko O, Stein BD, Grindle N, Clay K, Fuqua C. Localization and visualization of a Coxiella-type symbiont within the lone star tick, Amblyomma americanum. Appl Environ Microbiol. 2007;73 : 6584–6594. 17720830
21. Machado-Ferreira E, Dietrich G, Hojgaard A, Levin M, Piesman J, Zeidner NS, et al. Coxiella symbionts in the Cayenne tick Amblyomma cajennense. Microb Ecol. 2011;62 : 134–142. doi: 10.1007/s00248-011-9868-x 21611689
22. Lalzar I, Harrus S, Mumcuoglu KY, Gottlieb Y. Composition and seasonal variation of Rhipicephalus turanicus and Rhipicephalus sanguineus bacterial Communities. Appl Environ Microbiol. 2012;78 : 4110–4116. doi: 10.1128/AEM.00323-12 22467507
23. Jasinskas A, Zhong J, Barbour AG. Highly prevalent Coxiella sp. bacterium in the tick vector Amblyomma americanum. Appl Environ Microbiol. 2007;73 : 334–336. 17085709
24. Clay K, Klyachko O, Grindle N, Civitello D, Oleske D, Fuqua C. Microbial communities and interactions in the lone star tick, Amblyomma americanum. Mol Ecol. 2008;17 : 4371–4381. 19378409
25. Wilkinson DA, Dietrich M, Lebarbenchon C, Jaeger A, Le Rouzic C, Lagadec E, et al. Massive infection of seabird ticks with bacterial species related to Coxiella burnetii. Appl Environ Microbiol. 2014;80 : 3327–3333. doi: 10.1128/AEM.00477-14 24657860
26. Lalzar I, Friedmann Y, Gottlieb Y. Tissue tropism and vertical transmission of Coxiella in Rhipicephalus sanguineus and Rhipicephalus turanicus ticks. Environ Microbiol; 2014;16 : 3657–3668. doi: 10.1111/1462-2920.12455 24650112
27. Zhong J, Jasinskas A, Barbour AG. Antibiotic treatment of the tick vector Amblyomma americanum reduced reproductive fitness. PLoS One. 2007;2: e405. 17476327
28. Moran NA, McCutcheon JP, Nakabachi A. Genomics and evolution of heritable bacterial symbionts. Annu Rev Genet. 2008;42 : 165–190. doi: 10.1146/annurev.genet.41.110306.130119 18983256
29. Wernegreen JJ. Endosymbiosis. Curr Biol. 2012;22: R555–561. doi: 10.1016/j.cub.2012.06.010 22835786
30. Hosokawa T, Koga R, Kikuchi Y, Meng XY, Fukatsu T. Wolbachia as a bacteriocyte-associated nutritional mutualist. Proc Natl Acad Sci USA. 2010;107 : 769–774. doi: 10.1073/pnas.0911476107 20080750
31. Akman L, Yamashita A, Watanabe H, Oshima K, Shiba T, Hattori M, et al. Genome sequence of the endocellular obligate symbiont of tsetse flies, Wigglesworthia glossinidia. Nat Genet. 2002;32 : 402–407. 12219091
32. Husnik F, Chrudimsky T, Hypsa V. Multiple origins of endosymbiosis within the Enterobacteriaceae (gamma-Proteobacteria): convergence of complex phylogenetic approaches. BMC Biol. 2011;9 : 87. doi: 10.1186/1741-7007-9-87 22201529
33. Gomez-Valero L, Rusniok C, Buchrieser C. Legionella pneumophila: population genetics, phylogeny and genomics. Infect Genet Evol. 2009;9 : 727–739. doi: 10.1016/j.meegid.2009.05.004 19450709
34. Leclerque A, Kleespies RG. A Rickettsiella bacterium from the hard tick, Ixodes woodi: molecular taxonomy combining multilocus sequence typing (MLST) with significance testing. PLoS One. 2012;7: e38062. doi: 10.1371/journal.pone.0038062 22675436
35. Omsland A, Beare PA, Hill J, Cockrell DC, Howe D, Virtaneva K, et al. Isolation from animal tissue and genetic transformation of Coxiella burnetii are facilitated by an improved axenic growth medium. Appl Environ Microbiol. 2011;77 : 3720–3725. doi: 10.1128/AEM.02826-10 21478315
36. Sekeyová Z, Roux V, Raoult D. Intraspecies diversity of Coxiella burnetii as revealed by com1 and mucZ sequence comparison. FEMS Microbiol Lett. 1999;180 : 61–67. 10547445
37. Pearson T, Hornstra HM, Sahl JW, Schaack S, Schupp JM, Beckstrom-Sternberg SM, et al. When outgroups fail; phylogenomics of rooting the emerging pathogen, Coxiella burnetii. Syst Biol. 2013;62 : 752–762. doi: 10.1093/sysbio/syt038 23736103
38. Seshadri R, Paulsen IT, Eisen JA, Read TD, Nelson KE, Nelson WC, et al. Complete genome sequence of the Q-fever pathogen Coxiella burnetii. Proc Natl Acad Sci USA. 2003;100 : 5455–5460. 12704232
39. Beare PA, Unsworth N, Andoh M, Voth DE, Omsland A, Gilk SD, et al. Comparative genomics reveal extensive transposon-mediated genomic plasticity and diversity among potential effector proteins within the genus Coxiella. Infect Immun. 2009;77 : 642–656. doi: 10.1128/IAI.01141-08 19047403
40. Engelstadter J, Hurst GDD. The ecology and evolution of microbes that manipulate host reproduction. Annu Rev Ecol Evol Syst. 2009;40 : 127–149.
41. Duron O, Bouchon D, Boutin S, Bellamy L, Zhou L, Engelstadter J, et al. The diversity of reproductive parasites among arthropods: Wolbachia do not walk alone. BMC Biol. 2008;6 : 27. doi: 10.1186/1741-7007-6-27 18577218
42. Zug R, Hammerstein P. Still a host of hosts for Wolbachia: analysis of recent data suggests that 40% of terrestrial arthropod species are infected. PLoS One. 2012;7: e38544. doi: 10.1371/journal.pone.0038544 22685581
43. Shivaprasad HL, Cadenas MB, Diab SS, Nordhausen R, Bradway D, Crespo R, et al. Coxiella-like infection in psittacines and a toucan. Avian Dis. 2008;52 : 426–432. 18939630
44. Woc-Colburn AM, Garner MM, Bradway D, West G, D'Agostino J, Trupkiewicz J, et al. Fatal coxiellosis in Swainson's Blue Mountain Rainbow Lorikeets (Trichoglossus haematodus moluccanus). Vet Pathol. 2008;45 : 247–254. doi: 10.1354/vp.45-2-247 18424842
45. Vapniarsky N, Barr BC, Murphy B. Systemic Coxiella-like infection with myocarditis and hepatitis in an eclectus parrot (Eclectus roratus). Vet Pathol. 2012;49 : 717–722. doi: 10.1177/0300985811409251 21712515
46. Duron O, Wilkes TE, Hurst GDD. Interspecific transmission of a male-killing bacterium on an ecological timescale. Ecol Lett. 2010;13 : 1139–1148. doi: 10.1111/j.1461-0248.2010.01502.x 20545734
47. Duron O, Schneppat UE, Berthomieu A, Goodman SE, Droz B, Paupy C, et al. Origin, acquisition and diversification of heritable bacterial endosymbionts in louse flies and bat flies. Mol Ecol. 2014;23 : 2105–2017. doi: 10.1111/mec.12704 24612422
48. Jousselin E, Coeur d'Acier A, Vanlerberghe-Masutti F, Duron O. Evolution and diversity of Arsenophonus endosymbionts in aphids. Mol Ecol. 2013;22 : 260–270. doi: 10.1111/mec.12092 23106652
49. Novakova E, Hypsa V, Moran NA. Arsenophonus, an emerging clade of intracellular symbionts with a broad host distribution. BMC Microbiol. 2009;9 : 143. doi: 10.1186/1471-2180-9-143 19619300
50. Bressan A. Emergence and evolution of Arsenophonus bacteria as insect-vectored plant pathogens. Infect Genet Evol. 2014;22 : 81–90. doi: 10.1016/j.meegid.2014.01.004 24444593
51. Benson MJ, Gawronski JD, Eveleigh DE, Benson DR. Intracellular symbionts and other bacteria associated with deer ticks (Ixodes scapularis) from Nantucket and Wellfleet, Cape Cod, Massachusetts. Appl Environ Microbiol. 2004;70 : 616–620. 14711698
52. van Overbeek L, Gassner F, van der Plas CL, Kastelein P, Nunes-da Rocha U, Takken W. Diversity of Ixodes ricinus tick-associated bacterial communities from different forests. FEMS Microbiol Ecol. 2008;66 : 72–84. doi: 10.1111/j.1574-6941.2008.00468.x 18355299
53. Duron O. Lateral transfers of insertion sequences between Wolbachia, Cardinium and Rickettsia bacterial endosymbionts. Heredity 2013;111 : 330–337. doi: 10.1038/hdy.2013.56 23759724
54. Baldo L, Werren JH. Revisiting Wolbachia supergroup typing based on WSP: spurious lineages and discordance with MLST. Curr Microbiol. 2007;55 : 81–87. 17551786
55. Degnan PH, Moran NA. Evolutionary genetics of a defensive facultative symbiont of insects: exchange of toxin-encoding bacteriophage. Mol Ecol. 2008;17 : 916–929. doi: 10.1111/j.1365-294X.2007.03616.x 18179430
56. Nikoh N, Hosokawa T, Moriyama M, Oshima K, Hattori M, Fukatsu T. Evolutionary origin of insect-Wolbachia nutritional mutualism. Proc Natl Acad Sci USA. 2014;111 : 10257–10262. doi: 10.1073/pnas.1409284111 24982177
57. Duron O. Arsenophonus insect symbionts are commonly infected with APSE, a bacteriophage involved in protective symbiosis. FEMS Microbiol Ecol. 2014;90 : 184–194. doi: 10.1111/1574-6941.12381 25041857
58. Perlman SJ, Hunter MS, Zchori-Fein E. The emerging diversity of Rickettsia. Proc R Soc Lond B Biol Sci. 2006;273 : 2097–106.
59. Weinert LA, Werren JH, Aebi A, Stone GN, Jiggins FM. Evolution and diversity of Rickettsia bacteria. BMC Biol. 2009;7 : 6. doi: 10.1186/1741-7007-7-6 19187530
60. Clayton AL, Oakeson KF, Gutin M, Pontes A, Dunn DM, von Niederhausern AC, et al. A novel human-infection-derived bacterium provides insights into the evolutionary origins of mutualistic insect–bacterial symbioses. PLoS Genet. 2012;8: e1002990. doi: 10.1371/journal.pgen.1002990 23166503
61. Thompson JD, Gibson TJ, Higgins DG. Multiple sequence alignment using ClustalW and ClustalX. Curr Protoc Bioinformatics 2002; 2 : 3. doi: 10.1002/0471250953.bi0203s00 18792934
62. Kumar S, Tamura K, Nei M. MEGA3: Integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Brief Bioinform. 2004;5 : 150–163. 15260895
63. Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17 : 540–552. 10742046
64. Librado P, Rozas J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009;25 : 1451–1452. doi: 10.1093/bioinformatics/btp187 19346325
65. Sawyer SA. GENECONV: A computer package for the statistical detection of gene conversion. http://www.math.wustl.edu/~sawy.
66. Martin D, Rybicki E. RDP: detection of recombination amongst aligned sequences. Bioinformatics. 2000;16 : 562–563. 10980155
67. Martin D, Lemey P, Lott M, Moulton V, Posada D, Lefeuvre P. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics. 2010;26 : 2462–2463. doi: 10.1093/bioinformatics/btq467 20798170
68. Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006;23 : 254–267. 16221896
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 5
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Human Cytomegalovirus miR-UL112-3p Targets TLR2 and Modulates the TLR2/IRAK1/NFκB Signaling Pathway
- Paradoxical Immune Responses in Non-HIV Cryptococcal Meningitis
- Expression in the Fat Body Is Required in the Defense Against Parasitic Wasps in
- Survives with a Minimal Peptidoglycan Synthesis Machine but Sacrifices Virulence and Antibiotic Resistance
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy