
Viral Activation of MK2-hsp27-p115RhoGEF-RhoA Signaling Axis Causes Cytoskeletal Rearrangements, P-body Disruption and ARE-mRNA Stabilization
We have only scratched the surface in understanding how viruses control host gene expression. Several viruses disrupt important sites of post-transcriptional control of gene expression known as processing bodies (PBs), but underlying regulatory mechanisms and biological relevance remain poorly understood in most cases. Our study shows that the Kaposin B (KapB) protein of Kaposi's sarcoma (KS)-associated herpesvirus, known to block the degradation of a class of labile host mRNAs, does so by constitutively activating a signaling axis involving MK2, hsp27, p115RhoGEF and RhoA, thereby dispersing PBs. Thus, PB disruption may support the secretion of host pro-inflammatory cytokines and angiogenic factors that underlies KS tumor formation. Furthermore, by activating RhoA, KapB also causes cytoskeletal rearrangements, accelerated cell migration and angiogenesis in an endothelial cell model. Our findings position KapB as a key contributor to viral reprogramming of endothelial cells.
Published in the journal:
. PLoS Pathog 11(1): e32767. doi:10.1371/journal.ppat.1004597
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004597
Summary
We have only scratched the surface in understanding how viruses control host gene expression. Several viruses disrupt important sites of post-transcriptional control of gene expression known as processing bodies (PBs), but underlying regulatory mechanisms and biological relevance remain poorly understood in most cases. Our study shows that the Kaposin B (KapB) protein of Kaposi's sarcoma (KS)-associated herpesvirus, known to block the degradation of a class of labile host mRNAs, does so by constitutively activating a signaling axis involving MK2, hsp27, p115RhoGEF and RhoA, thereby dispersing PBs. Thus, PB disruption may support the secretion of host pro-inflammatory cytokines and angiogenic factors that underlies KS tumor formation. Furthermore, by activating RhoA, KapB also causes cytoskeletal rearrangements, accelerated cell migration and angiogenesis in an endothelial cell model. Our findings position KapB as a key contributor to viral reprogramming of endothelial cells.
Introduction
Kaposi's sarcoma-associated herpesvirus (KSHV), a.k.a. human herpesvirus-8 (HHV-8) is the infectious cause of Kaposi's sarcoma (KS), the most common malignancy of untreated AIDS patients, and two rare lymphoproliferative disorders, multicentric Castleman's disease (MCD) and primary effusion lymphoma (PEL) [1]–[3]. Like all herpesviruses, KSHV establishes persistent, life-long infection of its human host. The primary proliferative elements of KS lesions are latently infected endothelial cells (ECs) with an abnormal spindle-shaped morphology, commonly known as ‘spindle cells’. In latency, the viral episome persists in a reversible latent state and viral gene expression is limited to 6 consensus protein products (LANA, v-cyclin, v-FLIP, Kaposins A, B, and C) and 12 pre-miRNAs that are processed into at least 17 mature miRNAs (reviewed in [4], [5], [6]). Spindle cells display actin cytoskeleton rearrangements, enhanced cell motility and an aberrant angiogenic phenotype (recently reviewed in [7], [8]); all of these features can be recapitulated during in vitro infection of primary ECs [8]–[11]. Several KSHV latent gene products have been shown to contribute to these dramatic alterations in EC physiology (reviewed in [8]), but our understanding of their relative contribution to tumor-initiating events remains incomplete.
During KSHV infection, a complex translational program involving translation initiation at non-canonical CUG codons and decoding sets of GC-rich repeats results in the generation of multiple kaposin protein products, including Kaposin B (KapB). We have previously shown that KapB regulates the expression of pathogenetically important pro-inflammatory cytokines and angiogenic factors at the post-transcriptional level [12]. This is achieved by direct binding and activation of mitogen-activated protein kinase (MAPK)-associated protein kinase-2 (MK2), a nodal kinase that regulates the turnover of mRNAs bearing AU-rich instability elements (AREs) within their 3′UTRs [13], [14]. AREs are commonly found in labile mRNAs encoding tightly regulated, potent effector molecules, including many cytokines, angiogenic factors and proto-oncogenes [15], [16]. MK2 phosphorylates a variety of target proteins, including several ARE-binding proteins (ARE-BPs) that govern ARE-mRNA stability, with the net effect of causing ARE-mRNA stabilization. For example, the ARE-BP tristetraprolin (TTP) normally promotes ARE-mRNA turnover by facilitating interactions between bound mRNAs and the cytoplasmic mRNA degrading enzymes associated within exosomes and processing bodies (p-bodies, PBs) (reviewed in [17]). Phosphorylation of TTP by MK2 creates a binding site for 14-3-3 scaffolding proteins, thereby preventing association of ARE-mRNAs with the decay machinery. Accordingly, MK2 activation during latent KSHV infection or in response to ectopic expression of KapB coincided with dramatic stabilization of ARE-mRNAs and increased production of a number of canonical products of ARE-mRNAs, including IL6 and CSF2 [12]. Importantly, KapB also stabilized the ARE-mRNA encoding PROX1 [18], a master regulator of lymphatic reprogramming of vascular endothelial cells, thereby providing a molecular mechanism for KSHV-mediated cell fate reprogramming [1]. Altogether, these findings suggest that KapB makes key contributions to the reprogramming of ECs in KS lesions.
PBs are small ribonucleoprotein (RNP) granules that contain the requisite enzymes to mediate rapid mRNA deadenylation, decapping and exonucleolytic degradation in a 5′ to 3′ direction [19]–[23]. PBs also contain RNA induced silencing complexes (RISC), translational repressors (rck/p54) and many RNA-binding proteins such as the ARE-BP, TTP. PBs are constitutively present in most cells, but they are also dynamic structures; PB number and size increase in response to a variety of environmental stresses, when 5′ to 3′-exonucleolytic decay is blocked, or when translation is inhibited [24]. PB formation involves aggregation of RNA binding proteins, utilizing mRNA itself as an organizing scaffold; as such, PBs disperse after treatment of cells with RNase [25]. Not exclusively sites of ARE-RNA decay, PBs have been shown to have important roles in nonsense-mediated decay, RNA interference, and they can also harbor translationally-silenced mRNAs that have the potential to escape PBs and resume translation [26], [27]. PBs have intimate links to both the microtubule and the actin cytoskeleton (reviewed in [28]). Stationary PBs associate with actin bundles while mobile PBs connect to the microtubule network [29]. They are linked to microtubules via the microtubule-associated protein nesprin-1 and travel along microtubules using the retrograde motor protein dynein [30], [31]. More recently, it was shown that PB accretion and ARE-mRNA turnover was modulated by the cytoskeletal regulator, RhoA GTPase (RhoA) [32]. Relatively little is known about how RhoA and other upstream signaling proteins govern PB assembly and function.
The Rho family of small GTPases (Rho, Rac and Cdc42) are molecular switches that cycle between an inactive GDP-bound configuration and an active GTP-bound form with the aid of guanine nucleotide exchange factors (GEFs) (reviewed in [33], [34]). RhoA regulates actin cytoskeleton dynamics to facilitate normal cell attachment, the formation of actin stress fibers, cell migration and angiogenesis (reviewed in [35]–[37]). Inactive cytosolic RhoA translocates to membranes upon activation by G-protein coupled receptors, that link the G-protein Gα13 to RhoA activation via numerous GEFs including p115RhoGEF [34], [38]–[40]. There, in its active conformation, RhoA can bind numerous downstream effectors. The most extensively studied RhoA effector is the Rho-associated kinase ROCK, a serine/threonine kinase with two isoforms, ROCK1 and ROCK2, bearing 64% overall sequence identity, and many overlapping activities [41]. Upon binding to RhoA, ROCK1/2 promote actomyosin contractility and stress fiber formation by phosphorylating target proteins including focal adhesion kinase (FAK), LIM kinase 1 (LIMK1), myosin light chain (MLC) and myosin phosphatase target (MYPT-1) [42]–[45]. The effects of MK2 are not limited to regulation of ARE-mRNA turnover; numerous studies have pinpointed a role for MK2 in the actin cytoskeletal remodeling required to promote EC migration and angiogenesis [46]–[51] [52], [53]. Active MK2 phosphorylates the small heat shock protein (hsp) 27 and suppresses its actin filament capping activity, releasing the molecule from the barbed end of the actin fiber to permit actin fiber growth [53]–[55]. MK2 also phosphorylates the serine/threonine kinase, LIM kinase 1 (LIMK1) [56], which subsequently phosphorylates and inactivates the actin-severing protein cofilin [45]. Thus, the available data indicates that both MK2 and RhoA support EC cytoskeletal rearrangements, migration and angiogenesis. However, to date, little is known about how these two pathways are functionally integrated.
By studying KSHV latency, we have elucidated the functional integration of the MK2 and RhoA signaling pathways in ECs. We show that Kaposin B activates a signaling axis involving MK2, hsp27, p115RhoGEF and RhoA. The consequences of activation of this pathway in ECs include the formation of actin stress fibers, increased cell migration and angiogenesis, and dispersal of PBs. Because PBs are important sites of ARE-mRNA decay, KapB-mediated PB dispersal supports its role in potent stabilization of ARE-mRNAs. Taken together, these observations position KapB as a key contributor to viral reprogramming of ECs, capable of eliciting many of the phenotypes characteristic of KS tumor cells, and strongly contributing to the post-transcriptional control of EC gene expression and secretion.
Results
KapB activates RhoA GTPase to stimulate actin stress fiber formation, endothelial cell migration and tubule formation
Latent KSHV-infected ECs display marked alterations in cytoskeletal morphology. The latent vFLIP protein has been shown to modulate actin and cause spindling of ECs in an NF-kB-dependent manner [9], but the impact of the remaining latent gene products on the cytoskeleton remains largely unexplored. We previously reported that the latent KapB protein binds and activates MK2 kinase, known to be a major regulator of actin remodeling [12]. For this reason, we investigated the ability of KapB to modulate the actin cytoskeleton in human umbilical vein endothelial cells (HUVECs). Cells ectopically expressing KapB displayed thick parallel actin stress fibers (Fig. 1B). Actin stress fibers have been observed in cells following activation of the small GTPase RhoA [57], [58]. They have also been observed in cells with increased activation of the p38/MK2 signaling pathway or by expression of the constitutively active form of the kinase, MK2 (MK2-EE) or a phosphomimicking form of hsp27 (hsp27-DDD) [59] [60], [61]. Consistent with this, we observed that expression of either MK2-EE or hsp27DDD in HUVECs caused the formation of actin stress fibers (Figs. 1C, 1D). Activation of the p38/MK2 pathway by KapB also causes the increased secretion of pro-inflammatory cytokines that can then act in an autocrine or paracrine fashion to potentiate p38 pathway activation [12]. To address whether the appearance of actin stress fibers in response to KapB expression was mediated largely by KapB-mediated secretion of inflammatory molecules (ie. paracrine effects), we treated HUVECs with conditioned media from KapB-expressing cells. No stress fibers were observed in control cells treated with media from KapB-expressing cells (S1 Fig.), indicating that KapB-mediated rearrangement of the actin cytoskeleton was a cell autonomous effect.

Selective chemical inhibitors were used to investigate the mechanism of KapB-mediated actin rearrangements. Treatment of KapB-expressing HUVECs with a selective MK2 inhibitor [62] prevented formation of actin stress fibers, whereas p38 MAPK inhibition had no effect (Fig. 2A). This data is consistent with the notion that KapB binds and activates MK2 downstream of p38 MAPK. Treatment of cells with a selective inhibitor of the Rho kinases ROCK1 and ROCK2 (hereafter referred to as ROCK), which are RhoA substrates known to play a role in actin stress fiber formation, also inhibited KapB-mediated stress fibers (Fig. 2A). Taken together, these data suggest that KapB-mediated actin rearrangements depend upon activity of both MK2/hsp27 and RhoA/ROCK signaling axes. Because previous studies suggested a functional link between MK2/hsp27 and RhoA activation [63], we measured RhoA activity in KapB-expressing cells using a pull-down assay that isolates only the active (GTP-bound) form of RhoA [64]. KapB expression activated RhoA, both in the absence (Fig. 2B, lane 4) and presence (Fig. 2B, lane 2) of the canonical RhoA activator, LPA [65]. Interestingly, we also observed increased pull-down of the active form of Rho from cells transfected with MK2-EE and hsp27-DDD (Fig. 2B, lanes 5 and 6). Thus, RhoA was activated in cells where MK2 activity was mimicked or stimulated by direct KapB-MK2 binding. This is consistent with previous reports of a non-canonical MK2/hsp27/p115RhoGEF/RhoA signaling pathway in arachadonic acid-treated epithelial cells (63) (Fig. 3).
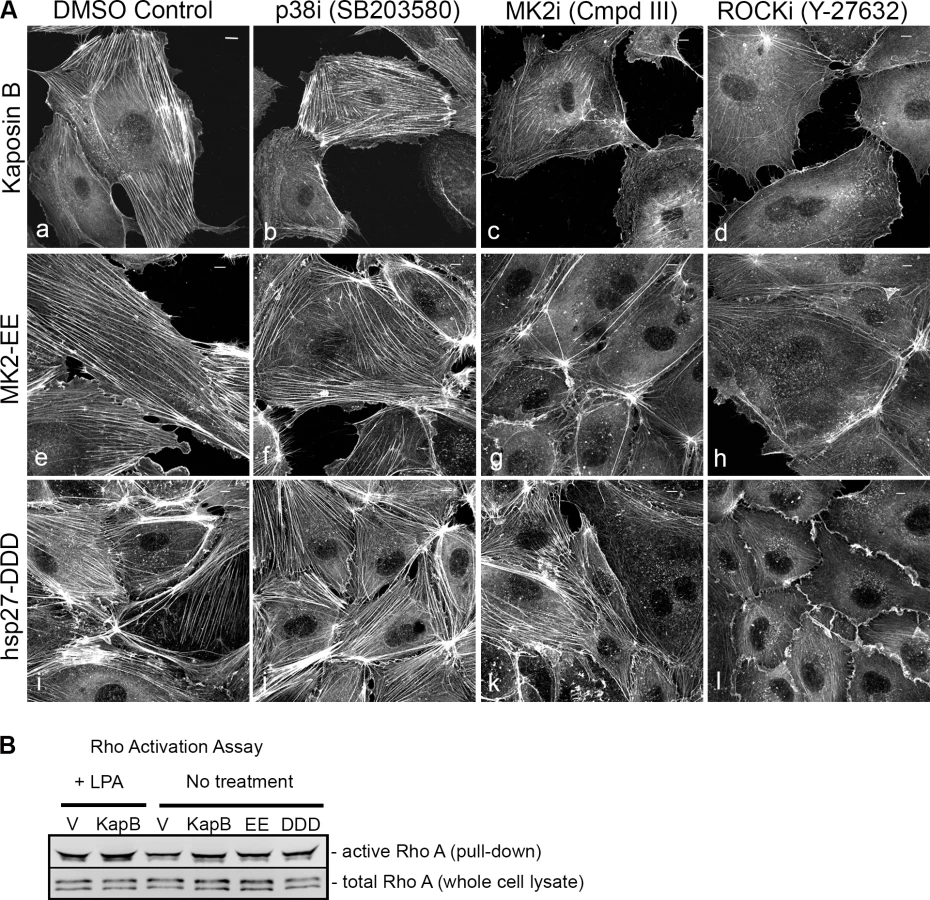

In addition to spurring actin stress fiber formation, activation of Rho family GTPases and the p38/MK2/hsp27 MAPK pathway has previously been linked to increased cell migration, and in the case of ECs, increased angiogenesis [33], [49]–[51], [56], [60]. We observed that ectopic KapB expression in HUVECs promoted cell migration in a wound-healing assay; KapB-expressing cells displayed 59% wound closure compared to 25% wound closure by control cells over a 6-hour period (S2A Fig.). KapB also promoted migration of HUVECs across a gelatin-coated semi-permeable membrane (S2B Fig.). Interestingly, KapB-mediated enhancement of cell migration was detectable only in the absence of the potent endothelial angiogenic molecule, vascular endothelial growth factor (VEGF), though VEGF treatment has no appreciable effect on KapB expression level (S3 Fig.). The p38/MK2/hsp27 pathway plays a clearly defined role mediating the migration of ECs in response to VEGF [60]; however, our results suggest that the activation of this pathway by KapB also mediates EC migration when VEGF levels are low. This supports the notion that KapB targets nodal kinases commonly stimulated during EC migration.
Endothelial cells form tubules on matrigel in an in vitro angiogenesis assay that mimics the formation of blood vessels [56], [66]. We examined the effect of KapB expression on tubule formation compared to control HUVECs expressing either empty vector or the constitutively active from of MK2 (MK2-EE). Both KapB - and MK2-EE-expressing HUVECs formed tubules in matrigel (S4 Fig.). This is consistent with the well-described role of the p38/MK2 pathway in promoting EC tubule formation [50], [56]. In both cases, tubule network formation was reduced after treatment with the ROCK inhibitor Y-27632 (S4B Fig.). Thus, by activating MK2 and RhoA, KapB deregulates several processes in primary ECs contributing to a migratory and angiogenic phenotype.
KapB causes p-body dispersion in a RhoA-dependent manner
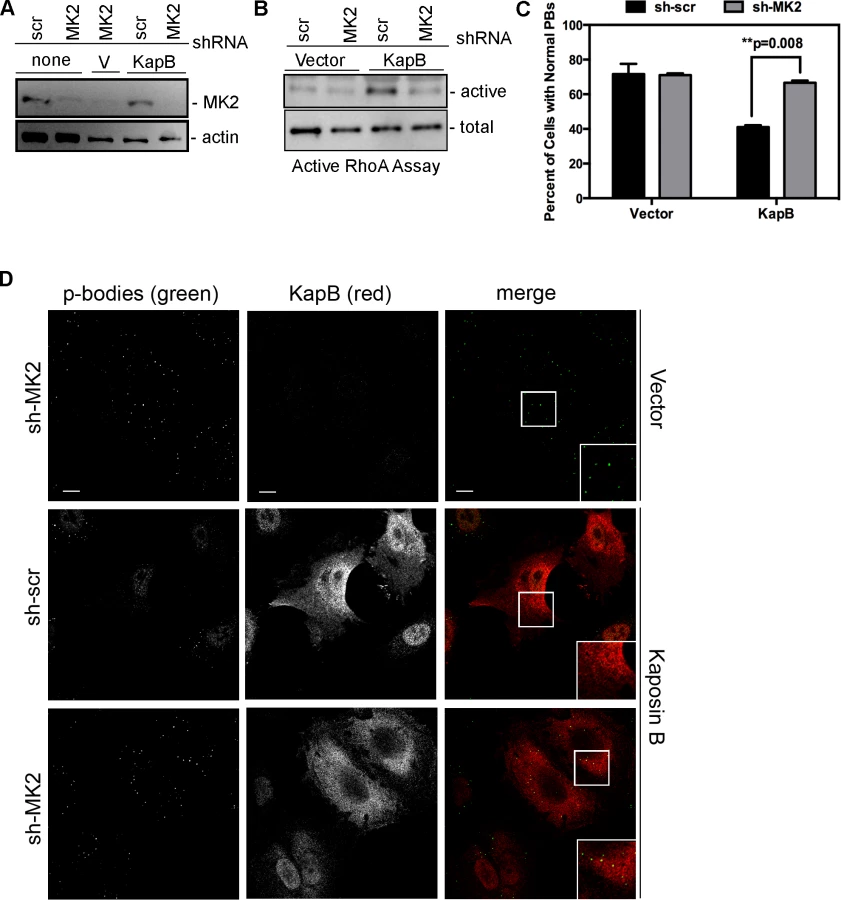
KapB stabilizes labile host cell ARE-mRNAs. Interestingly, a known site of ARE-mRNA translational repression and degradation is the processing body (PB), which has intimate links to both the actin and the microtubule cytoskeleton (reviewed in [28]); stationary PBs associate with actin bundles while mobile PBs connect to the microtubule network [29]. More recently, it was shown that RhoA GTPase activity modulates PB formation [32]. Because KapB activates RhoA we reasoned that PB disruption might contribute to KapB-mediated ARE-mRNA stabilization. PBs were visualized by immunofluorescent staining for two PB resident proteins, hedls or DDX6 [67]. Indeed, KapB-expressing HUVECs displayed a decrease in the number of cells containing PBs of expected dimensions (approximately 0.3 µm in diameter, [68]) (Fig. 4A–B). This effect was quantified by counting the number of KapB-expressing cells that contained one or more PBs of normal size compared to control (empty vector) transduced cells. We observed that in control cell populations, 64% of cells contained normal PBs, compared to 30% in KapB-expressing cells. HUVECs were also stained for the actin cytoskeleton, and KapB-expressing cells displayed thick parallel actin stress fibers (Fig. 4B), consistent with our previous observations that KapB induces actin polymerization and RhoA activation (Figs. 1–2).

It is not known how RhoA activation modulates PBs. RhoA GTPase can be activated by the microtubule-bound guanine exchange factor GEF-H1; microtubule disruption causes release of GEF-H1 and concomitant RhoA activation [69]. To test whether KapB-mediated activation of RhoA and disruption of PB accretion were related to a disruption of the microtubular network, we stained KapB-expressing HUVECs for α-tubulin. We did not observe altered tubulin staining intensity nor did we observe any striking differences in the appearance of the microtubule network in cells expressing KapB compared to controls (Fig. 4C), indicating that KapB-mediated suppression of PBs is independent of changes to microtubule cytoskeleton, and suggesting that RhoA activation is unlikely a result of the release of GEF-H1 from microtubules.
When Takahashi et al. [32] observed that the overexpression of RhoA mediated an alteration to PB dynamics, they concluded that RhoA induced an increase in the number of PBs while causing a marked reduction in average PB size. Using retroviral transduction, we expressed a constitutively active (CA) version or dominant negative (DN) version of RhoA fused to GFP (Rho-CA-eGFP or Rho-DN-eGFP) in primary ECs [37], [70]. We observed both a marked loss of total PBs and a reduction in remaining PB size in HUVECs expressing Rho-CA-eGFP, whereas PBs in cells expressing Rho-DN-eGFP were similar to eGFP control (Fig. 5A). This is consistent with our previous observations that chemical activators of RhoA, including LPA and nocodazole, disrupted PB accretion [67]. Cells expressing CA-RhoA-eGFP also display marked actin stress fiber formation that is lacking in control cells or cells expressing the Rho-dominant negative (DN) construct [37], [70] (Fig. 5A). Irreversible inactivation of RhoA by C3 transferase, which ADP-ribosylates the Asn41 residue, disrupts RhoA-mediated re-organization of actin filaments [38], [71]. To determine whether RhoA activity is required for KapB-mediated actin polymerization and disruption of PBs, we treated KapB-expressing HUVECs with C3. In the absence of C3, the number of cells containing normal PBs was reduced two-fold by KapB expression; when treated with C3, this number was restored to control levels (Fig. 5B, D). C3 also abrogated the ability of KapB to promote actin stress fibers. We also co-expressed KapB with RhoA-DN by sequential transduction of HUVECs. Like C3 treatment, Rho-DN expression also prevented the ability of KapB to disperse PBs (Fig. 5E). However, when KapB-expressing HUVECs were treated with ROCK inhibitor there was no effect on KapB-mediated PB disruption (Fig. 5D). By contrast, ROCK inhibition prevented KapB-mediated stress fiber formation (Fig. 2). This observation uncouples the effect that RhoA activation has on PBs from canonical RhoA effects on actin and migration - processes that clearly require ROCK. Taken together, these experiments show that KapB-mediated PB disruption is dependent on RhoA, but not ROCK, consistent with previous findings [32] (Fig. 3).

KapB activation of MK2 is required for RhoA activity and PB disruption
We hypothesized that KapB expression activates a little-known signaling axis that links upstream activation of the p38/MK2 MAPK pathway to RhoA activity (Fig. 3) (63). To examine the role of MK2 in KapB-mediated RhoA activation, we generated retroviruses expressing short hairpin (sh)-RNAs directed against MK2. Efficient silencing of endogenous MK2 expression (Fig. 6A) prevented RhoA activation in KapB-expressing HUVECs (Fig. 6B). Furthermore, MK2 silencing prevented efficient KapB-mediated PB dispersion (Figs. 6C–D). These data confirm a role for MK2 in the mechanism of KapB-mediated RhoA activation and PB dispersion.
KapB-mediated RhoA activation contributes to AU-rich mRNA stabilization
McCormick and Ganem [12] previously demonstrated that KapB expression caused a dramatic stabilization of AU-rich element (ARE)-mRNAs. To examine the role of RhoA in ARE-mRNA turnover, we utilized a reporter assay developed in our lab and described in detail in [72]. Briefly, HeLa Tet-Off cells were co-transfected with an empty plasmid vector or KapB expression vector, along with a doxycycline (dox)-responsive reporter plasmid encoding firefly luciferase linked to a canonical ARE derived from the labile CSF2 transcript. After 24 hours, transcription was arrested by the addition of dox, and 24 hours later, lysates were harvested for luciferase assays. Co-transfected dox-responsive Renilla luciferase reporter lacking an ARE served as a normalization control. KapB expression caused a striking increase in normalized luciferase activity, as did expression of MK2-EE, hsp27-DDD and RhoA-CA (Fig. 7A). These results are consistent with previous reports of control of ARE-mRNA turnover by RhoA [32] and p38/MK2 signaling pathways [13], [14], [73]. To further elucidate the role of RhoA in ARE-mRNA turnover, dominant negative RhoA (RhoA-DN) was introduced into this system. When KapB, MK2-EE and hsp27-DDD were co-expressed with RhoA-DN, the normalized luciferase activity was markedly reduced, indicating reduced stability of the firefly luciferase transcript (Fig. 7B). Phosphorylation of MK2 substrate hsp27 in cells expressing KapB or MK2-EE was confirmed by immunoblotting (Fig. 7C). These data suggest that KapB requires RhoA activation in order to achieve maximal ARE-RNA stabilization; furthermore, they confirm that MK2 activation precedes RhoA activation (Fig. 3). Thus, KapB causes ARE-mRNA stabilization via the direct binding and activation of MK2 which in turn causes RhoA activation.

Constitutively active MK2 and hsp27 mediate PB dispersal by activating RhoA
Previous work demonstrated that p38/MK2-mediated RhoA activation depends on the phosphorylation of hsp27 on serines 15, 78 and 82, and the formation of a complex comprising phosphorylated hsp27 (p-hsp27), RhoA and the guanine exchange factor (GEF), p115RhoGEF [63]. Therefore, we hypothesized that KapB-mediated RhoA activation may require p-hsp27 and p115RhoGEF. To test this, we undertook a series of experiments that examined the importance of hsp27, p115RhoGEF and RhoA in modulating PB and actin dynamics in response to upstream activators. Expression of MK2-EE and hsp27-DDD in HUVECs promoted the disruption of PBs (Fig. 8A–C). Consistent with our hypothesis, we found that inhibition of RhoA using C3 transferase or co-expression of RhoA-DN restored PB levels to that of controls in MK2-EE-expressing HUVECs (Fig. 8A, D, E). However, when RhoA was inhibited (by either C3 or RhoA-DN) in hsp27-DDD-expressing HUVECs, PB levels were not restored (Fig. 8B–E). Treatment of MK2-EE - and hsp27-DDD-expressing HUVECs with the ROCK inhibitor had no effect on PB disruption (Fig. 8D), as previously observed for KapB (Fig. 5D).

We reasoned that KapB and MK2 activate RhoA by mediating complex formation between p115RhoGEF, RhoA and p-hsp27 (Fig. 3) (63). To investigate the role of p-hsp27, we utilized a dominant negative form of hsp27 (hsp27-AAA) in which the three canonical phosphorylation sites (serines 15, 78, 82) had been mutated to alanines [74]. Hsp27-AAA was co-expressed with KapB, MK2-EE and hsp27-DDD in HUVECs. In all cases, expression of the hsp27-DN prevented PB disruption, restoring PBs to normal levels (Fig. 9A–C). These data support an important role for hsp27 phosphorylation in KapB-mediated activation of RhoA and resulting PB dispersion.

Knockdown of the Rho guanine exchange factor (GEF) p115 prevents KapB-mediated p-body dispersion
Rho GTPase activity is regulated by numerous GEFs, which are in turn are regulated by upstream signals including G-protein activation (in the case of G-protein coupled receptors), phosphorylation (such as by receptor tyrosine kinases) or, as described by Garcia et al. [63], complex formation with phosphorylated hsp27. To confirm a role for the p115RhoGEF in our model of KapB-induced RhoA activation and PB disruption (Fig. 3), we generated lentiviruses expressing short hairpin (sh)-RNAs directed against p115RhoGEF and another RhoA GEF, namely GEF-H1. H1 is bound to microtubules and is released from upon their disruption (e.g. with nocodozole) to mediate activation of RhoA. When HUVECs were transduced with these lentiviral vectors, they expressed eGFP and displayed reduced expression of target genes (S5 Fig.). HUVECs expressing three different shRNA constructs targeting p115RhoGEF were unable to mediate PB disruption in cells expressing KapB, MK2-EE or hsp27-DDD (Fig. 10, S6 Fig.). By contrast, silencing of microtubule-bound GEF-H1 had no appreciable effect on PBs (Fig.10, S6 Fig.). This is consistent with our observation that microtubules are not disrupted in response to KapB expression (Fig. 4). Together, these data indicate that p115RhoGEF is essential for KapB-mediated RhoA activation and PB disruption.

PBs are disrupted during latent KSHV infection
HUVECs were infected with KSHV and establishment of latency was confirmed by LANA immunostaining (Fig. 11A). KapB expression during latent infection of HUVECs was confirmed by immunoblotting (S3 Fig.). LANA-positive cells displayed a marked reduction in the number of cells with normal-sized PBs at 24, 48 and 72 hours post-infection (Fig. 11 A, B). To implicate kaposin gene products in PB disruption, we transduced cells with shRNAs targeting the kaposin transcript (shKAP1, shKAP2). These shRNAs would be expected to silence expression of kaposin gene products (translated from spliced, cytoplasmic kaposin mRNA), but have no effect on Drosha-dependent processing of kaposin transcript-derived miRNAs in the nucleus. KSHV infection of cells bearing kaposin shRNAs revealed either a partial or full restoration in PB levels to that observed in uninfected cells (shKAP1 and shKAP2, respectively, Fig. 11C, D). These data indicate that at least one of the kaposin proteins is required for KSHV to alter PB dynamics during latent infection. Moreover, in parallel shRNA knockdown experiments we demonstrated that p115RhoGEF is essential for PB disruption in latently infected ECs, whereas GEF-H1 was dispensable (Fig. 12). Taken together, these findings indicate that during KSHV latency a product of the kaposin locus, likely KapB, activates the MK2-hsp27-p115RhoGEF-RhoA signaling pathway, thereby disrupting PBs.


Discussion
Latent KSHV infection of primary ECs in vitro causes dramatic changes in cellular physiology that largely reflect observations of KS tumor cells. Infected cells display marked alterations in signal transduction and gene expression, extended life span, and enhanced motility and angiogenic properties. Despite intensive efforts, the precise contributions of individual viral gene products to alterations in EC physiology remain incompletely understood. The data presented in the current study position KapB as a key contributor to viral reprogramming of ECs; ectopic expression of KapB leads to actin stress fiber formation and altered cell morphology, increased motility and an angiogenic phenotype; all of which are characteristic of the KS tumor cells (see model, Fig. 13). Moreover, KapB was sufficient to disrupt PBs, sites of mRNA translational repression and decay. These diverse phenotypes are linked to a signaling axis, comprising MK2, hsp27, p115RhoGEF and RhoA (Fig. 3), which likely evolved to respond to acute, transient stress and promote cell survival. By encoding the KapB protein that directly binds to the nodal kinase MK2, KSHV achieves constitutive activation of this pathway.

Several viruses have been shown to modulate PBs during infection, but the functional relevance of these observations are not yet clear (reviewed in [75]). Many viral RNAs associate with PB resident proteins [76] [77], and there have been reports of viral RNA recruitment to PBs [78]. Thus, PB dispersion might be an attractive mechanism for viral evasion of translational repression and RNA decay. Furthermore, some viruses have been shown to co-opt key PB resident proteins to support viral replication. For example, the flaviviruses West Nile virus and hepatitis C virus recruit DDX6 and Lsm1 to viral replication centers [79], [80] [81], and in so doing cause PB dispersion. By contrast, little is known about the impact of PBs on herpesvirus infection. Human cytomegalovirus has recently been shown to increase PBs in infected cells using a mechanism that requires active synthesis of cellular mRNA [82]. However, disruption of PB accretion by selective knock-down of several PB component proteins had no effect on virus replication in primary fibroblasts, making is unclear how enhanced PB formation benefits viral fitness.
We previously reported PB disruption during lytic KSHV infection, and demonstrated that the lytic viral gene vGPCR was sufficient to disrupt PBs in an ectopic expression model [67]. We now show that PB dispersal is also a cell autonomous feature of KSHV latency, and elucidate the molecular mechanism for KapB-mediated PB disruption. A common feature of these studies was strong correlation between PB dispersal and stabilization of endogenous [67] and exogenous ([67] and Fig. 7) ARE-containing RNAs, and resulting increases in protein production of the encoded products. Thus, multiple KSHV gene products converge on the regulation of ARE-mRNA turnover, providing an attractive mechanism for the overproduction of pro-inflammatory cytokines and angiogenic factors characteristic of KS lesions.
Our study reveals a previously unappreciated mechanism for the control of PB formation, involving MK2-mediated activation of RhoA. Our data fits well with a proposed model by Garcia et al. who observed that arachadonic acid-mediated activation of RhoA depended on prior activation of p38/MK2 and phosphorylation of the MK2 substrate hsp27 [63]. Hsp27 phosphorylation is an important ‘switch’ that allows local recruitment of p115RhoGEF and RhoA activation. In our experiments, we investigated the role of each of these signaling molecules in KapB-mediated PB dispersion. RhoA inhibition, either using the irreversible C3 transferase or expression of a dominant negative construct, blocked PB dispersion (Figs. 5, 8), as did the expression of a dominant negative version of hsp27 (Fig. 9). p115RhoGEF silencing using three different shRNA constructs also restored PB numbers to control levels in cells ectopically expressing KapB, MK2-EE, or hsp27-DDD or in latently KSHV-infected HUVECs (Figs. 10, 12, and S6 Fig.).
Ours is the first study to link MK2 to regulation of PBs, and confirm a previously identified role for RhoA [32]. However, the precise mechanism of PB dispersion downstream of RhoA activation remains to elucidated. Active RhoA signals through numerous effectors, the most extensively studied of which are the Rho-associated kinases (ROCKs), ROCK1 and ROCK2; binding of active RhoA relieves ROCK autoinhibition. PB disruption by KapB is insensitive to treatment with a ROCK inhibitor, suggesting that this process requires RhoA but not its downstream effector ROCK kinases (Figs. 5, 8). Overexpression of ROCK and another RhoA effector, the formin-family member mDia, likewise did not alter PBs [32]. PBs associate and traffic along microtubules [29], [83] and mDia activates microtubule polymerization and cause microtubule bundling [84], [85]. In isolation, these observations would make mDia an attractive candidate effector for PB dispersion. However, because the microtubule cytoskeleton is unchanged in KapB-expressing cells (Fig. 4), this suggests that KapB does not activate mDia. Moreover, shRNA knockdown of the microtubule-bound GEF-H1 had no effect on PB dispersion during latent KSHV infection or in cells ectopically expressing KapB (Figs. 10, 12), also suggesting that microtubules and mDia do not play a role in the mechanism of KapB-mediated PB dispersion. Future elucidation of the mechanism of MK2/hsp27/p115RhoGEF/RhoA-dependent PB dispersal will be challenging, but may be accelerated by careful inspection of PB resident proteins that might either be recruited by active RhoA, or phosphorylated by MK2.
Latent KSHV infection reprograms gene expression in ECs at transcriptional and post-transcriptional levels, but relative contributions of viral gene products to reprogramming remains incompletely understood. Our studies position Kaposin B as a chief post-transcriptional regulator of gene expression, binding and activating the nodal kinase MK2, thereby stabilizing and enhancing the translation of a variety of ARE-mRNAs encoding pathogenetically important pro-inflammatory cytokines and angiogenic factors. The effects of KapB on EC physiology are striking; formation of actin stress fibers, accelerated cell migration, and a strong angiogenic phenotype. Furthermore, KapB-mediated ARE-mRNA stabilization coincided with dispersal of PBs, a major site of ARE-mRNA decay in mammalian cells. By studying KapB, we gained new insight into the fundamental regulation of these processes by a recently identified signaling axis involving MK2, hsp27, p115RhoGEF and RhoA. Stress fiber formation, cell migration and angiogenesis were dependent on the activity of the RhoA substrate kinase ROCK, whereas PB dispersion occurred in a ROCK-independent manner. Taken together, these observations suggest that KapB is a key contributor to viral reprogramming of ECs, capable of eliciting many of the phenotypes characteristic of KS tumor cells, and strongly contributing to the post-transcriptional control of EC gene expression and secretion.
Materials and Methods
Reagents
Doxycycline (dox), blasticidin, puromycin, valproic acid, human AB serum, polyethyleneimine (PEI) and polybrene were purchased from Sigma-Aldrich Canada. C3 transferase (Rho inhibitor I) was from Cytoskeleton, Inc. MK2 inhibitor-III was purchased from Calbiochem. ROCK inhibitor Y-27632 was from Sigma.
To irreversibly inhibit RhoA-GTPase, HUVECs were treated with 1 µg/ml C3 transferase for 6 h in SF medium. Serum-free conditions are important to minimize baseline RhoA activity in the context of C3 exposure. However, since RhoA inhibition by C3 is irreversible after treatment, C3 and serum-free control cells were incubated for 1 h in normal medium to restore baseline PB levels.
Cells
HeLa Tet-Off (Clontech), Phoenix-Amphotropic (a kind gift from G. Nolan, Stanford), and HEK293T cells (ATCC) were maintained at 37°C in a 5% CO2 atmosphere in Dulbecco's modified Eagle's medium containing 100 U of penicillin and streptomycin per ml and 10% heat-inactivated fetal bovine serum. Primary human umbilical vein endothelial cells (HUVECs) were purchased from Lonza. Cultures were expanded in EGM-2 medium (Lonza) on tissue culture plates coated with 0.1% (wt/vol) gelatin (in phosphate-buffered saline [PBS]) and used between passages 5 and 7 for experiments. The BCBL-1 primary effusion lymphoma (PEL) cell line was cultured in RPMI medium containing 10% heat-inactivated fetal bovine serum and 55 µM ß-mercaptoethanol.
Plasmids
pcDNA3-MK2EE was a kind gift from Paul Anderson (Harvard University), and its creation is described in [86]. Briefly, to create pcDNA3-Flag-MK2-EE the cDNA encoding constitutively active murine MK2 was amplified with primers G42/G43 from pcDNA3mycMK2T205E/T317E [13]. The amplicons were digested with BamHI and XhoI, and inserted into the BamHI and XhoI sites of pcDNA3-Flag-BAK. An expression vector encoding the phosphomimicking version of small heat shock protein 27 (hsp27) in which serine residues at positions 15, 78 and 82 have been substituted with aspartic acid (pcDNA3.1-HAhsp27DDD) was generously obtained from Matthias Gaestel and its creation is described [54]. To generate the pcDNA3.1 HA-hsp27-AAA clone, two sequential reactions of Phusion site directed mutagenesis was performed on pcDNA3.1-HA-hsp27-DDD according to the instructions of the manufacturer (NEB) and using the following primers: Reaction 1. D15A forward 5′ - GGCCCCGCCTGGGACCCC-3′, D15A reverse 5′ - CCGCAGGAGCGAGAAGGGG-3′; Reaction 2. D78AD82A forward 5′ - ACTCGCCAGCGGGGTCTCG-3′, D78AD82A reverse 5′ - TGCCGGGCGAGCGCGCGG-3′. The expression vector for KapB (pCR3.1-kapB) and the ARE-RNA reporter plasmids (pTRE2-Rluc, pTRE2-Fluc-ARE, pTRE2-BBB, pTRE2-BBB-ARE, pTRE2-d1EGFP, and pTRE2-d1EGFP-ARE) have been previously described [12], [67], [72]. pCB6-eGFP-RhoA-CA and pCB6-eGFP-RhoA-DN plasmids were a generous gift from Dr. Roy Duncan, Dalhousie University.
Retroviral expression plasmids
To create the pBMN-GFP-IP plasmid, the pEGFP-N1 plasmid (Clontech) was digested with NotI, subjected to a standard fill-in reaction with Klenow DNA polymerase (NEB), and further digested with BglII, releasing the eGFP ORF. To prepare the recipient pBMN-IP vector (G. Nolan lab, Stanford U.), XhoI digestion was performed, followed by a Klenow fill-in reaction, and a BamHI digest. These fragments were ligated to create pBMN-GFP-IP, which permits the expression of GFP and the puromycin resistance gene from a single bicistronic mRNA. pBMN-kapB-IP was generated by BamHI/EcoRI digestion of pCR3.1-kapB, releasing the 636 bp KapB ORF (derived from a pulmonary KS isolate). This fragment was subsequently ligated into the BamHI/EcoRI digested pBMN-IP vector. To create pBMN-MK2EE-IP, pcDNA3-Flag-MK2EE was amplified with primers G42/G43 from pcDNA3mycMK2T205ET317E [13]. The amplicons were digested with BamHI and XhoI, and inserted into BamHI/XhoI digested pBMN-IP vector. To create pBMN-hsp27DDD-IP, HA-hsp27DDD was excised from pcDNA3.1-HAhsp27DDD by sequential XbaI digest, Klenow fill-in to create a blunt end, and EcoRI digest. Following this, the insert was ligated into pBMN-IP, which had been prepared by sequential XhoI digest, Klenow fill-in to create a blunt end, and EcoRI digest. To generate pBMN-IP or pBMN-IB vectors containing RhoA-CA-eGFP and RhoA-DN-eGFP, the ORFs from pCB6-eGFP-RhoA-CA and pCB6-eGFP-RhoA-DN were amplified using Phusion High Fidelity Polymerase (NEB) chain reaction and the following primers: eGFP-N forward (5′-in database-3′) and reverse 5′-ATGCGAATTCTTATTATTACAAGACAAGGCACCCAGATT-3′. The resulting products were digested with BamHI and EcoRI and ligated into the pBMN-IP or pBMN-IB vector backbone. To generate HA-tagged versions of these clones, the ORFs of pCB6-eGFP-RhoA-CA and pCB6-eGFP-RhoA-DN were amplified using Phusion High Fidelity Polymerase (NEB) chain reaction and the following primers: forward 5′ - GCATGGATCCACCATGGAGTACCCATACGATGTTCCAGATTACGCTCCCAGAGCTGCCATCCGGAAGAAAC-3′ and reverse 5′-ATGCGAATTCTTATTATTACAAGACAAGGCACCCAGATT-3′. The resulting PCR products were digested with BamHI and EcoRI and ligated into the pBMN-IP or pBMN-IB vector backbone. To create pBMN-IB-HA-Hsp27-AAA, the ORF of pCDNA3.1-HA-HSP27-AAA was amplified using Phusion High Fidelity Polymerase (NEB) chain reaction and the following primers: forward 5′ - GGTGGAATTCATGGCTTACC-3′ and reverse 5′-ATGCCTCGAGTTATTATTACTTGGCGGCAGTCTCAT-3′. The resulting PCR product was digested with EcoRI and XhoI and ligated into the pBMN-IB vector backbone.
shRNA retroviral plasmids
Retroviral shRNA expression vectors were created via PCR amplification of template oligonucleotides, and cloning into XhoI/EcoRI restriction sites in pSMP (Open Biosystems). Briefly, the following 97-mer template oligonucleotides were synthesized, sh-scrambled: TGCTGTTGACAGTGAGCGAGCACAAGCTGGAGTACAACTATAGTGAAGCCACAGATGTATAGTTGTACTCCAGCTTGTGCCTGCCTACTGCCTCGGA, shMK2: TGCTGTTGACAGTGAGCGCGCCTGAGAATCTCTTATACACTAGTGAAGCCACAGATGTAGTGTATAAGAGATTCTCAGGCTTGCCTACTGCCTCGGA. These sequences were PCR amplified with Xho pSMP forward primer (5′-CAGAAGGCTCGAGAAGGTATATTGCTGTTGACAGTGAGCG-3′) and Eco pSMP reverse primer (5′-CTAAAGTAGCCCCTTGAATTCCGAGGCAGTAGGCA-3′) and Pfu Ultra High Fidelity DNA polymerase (Stratagene). 110-bp amplicons were then digested with XhoI/EcoRI and ligated into XhoI/EcoRI digested pSMP retroviral vector.
Lentiviral plasmids
pMD.2G (envelope) and pSPAX2 (packaging) plasmids were purchased from Addgene. All pGIPZ shRNA constructs used to knock down expression of the RhoA-specific guanine exchange factors (GEFs) listed below were purchased from Open Biosystems (oligo ID in parentheses): p115-3 (V3LHS_317458), p115-4 (V3LHS_317456), p115-9 (V2LHS_37090), H1-1 (V3LHS_317146), H1-2 (V3LHS_317143), and H1-7 (V2LHS_36680). pIPZ was created by PCR amplification of the enhanced CMV promoter of pGIPZ using a forward XbaI primer and a reverse NotI primer. After digestion of both the PCR product and pGIPZ vector with XbaI and NotI, the product was ligated into the vector backbone to create pIPZ. This new vector lacks the ORF for turbo eGFP and places the CMV promoter proximal to the IRES. To create pIPZ shRNA constructs used to knock down expression of the kaposin gene, we used previously generated shRNAs against kaposin in the pSM2 vector. Two different shRNA sequences that target the kaposin ORF were used and are named according to the position of the starting nucleotide. The 22-mer sequences for KapB 692 and KapB 746 shRNAs are 5′-TGTCCCGGATGTGTTACTAAAT-3′ and 5′-ACTCGTTTGTCTGTTGGCGATT-3′, respectively. In order to transfer these from pSM2 to pIPZ, the pSM2 vectors were digested with MluI and XhoI and inserted into pIPZ.
Retrovirus and lentivirus preparation and infections
Retrovirus stocks were produced in 15-cm cell culture dishes by PEI-mediated transfection using 54 µl of PEI and 18 µg of the gene/shRNA of interest, contained in either the pBMN-IP, pBMN-IB or pSMP vector backbone, into the Phoenix amphotropic packing cell line (a kind gift from G. Nolan, Stanford). The transfection medium was replaced after 6 hours. Virus-containing supernatants were harvested 48 h after transfection. These supernatants were spinoculated onto target HUVEC cell monolayers for 2 h at 2,000 rpm in the presence of 5 µg/ml Polybrene (Sigma), and after 24 h, 1 µg/ml puromycin or 10 µg/ml blasticidin was added to select for transductants. Lentivirus stocks were produced in 15-cm cell culture dishes by PEI-mediated co-transfection of three plasmids: 10 µg of the shRNA of interest (in pGIPZ or pIPZ), 3 µg pMD.2G (envelope), 6 µg pSPAX2 (packaging) and 54 µl of PEI into HEK293T cells. Virus-containing supernatants were collected after 48 hours, diluted 1∶2, and added to target HUVEC monolayers for 4-5 hours at 37°C in the presence of 5 µg/ml polybrene (Sigma). After 24 h, 1 µg/ml puromycin was added to select for transductants.
KSHV preparation and infections
Wild-type KSHV virus was produced from lytic reactivation of the BCBL-1 PEL cell line. Briefly, KSHV was induced to lytically reactivate from BCBL-1 cells at a cell concentration of 2×105/ml using 0.3 mM valproic acid. After 7 days of induction, the suspension culture was pre-cleared by centrifugation for 10 minutes at 800×g before being filtered using 0.45 µm Millipore filters. The virus-containing filtrate was then centrifuged for 2 hours at 25,000×g and the supernatant discarded. The virus pellet was resuspended in 1/100 of the original culture volume of DMEM containing 10% FBS and stored at −80°C. Infectious viral titer was determined by immunofluorescent staining with anti-LANA (see method below). For the experiments reported in this paper, a 1∶25 dilution of stock virus was spinoculated onto HUVECs for 45 minutes at 2000 x g and 30°C without the addition of polybrene. The viral inoculum was left on the cells for an additional hour at 37°C before being replaced with normal EGM-2 media according to the methods of [87] [88]. Latently infected cells were fixed at the indicated times post infection.
Luciferase assay
The luciferase reporter assay for identification of modulators of ARE-mediated mRNA decay is described in detail in [72]. Briefly, 105 HeLa Tet-Off cells were cotransfected with 100 ng of a reporter plasmid master mix (pTRE2-Fluc-ARE and pTRE-2-Rluc, at a ratio of 9∶1); 900 ng of an expression vector or an empty vector control; and 3 µl of Fugene HD (Roche) according to the instructions of the manufacturer. Twenty-four hours after transfection, Dox was added (1 µg/ml) to stop de novo transcription from the pTRE reporter plasmids. Twenty-four hours after the addition of Dox, transfected cells were lysed in 200 µl of 1× passive lysis buffer, and samples were processed using the dual-luciferase assay kit (Promega) according to the instructions of the manufacturer. Firefly and Renilla luminescence was determined using the GloMax 20/20 luminometer (Promega). Firefly luminescent signal, expressed in relative light units (RLUs), was normalized to that of Renilla luciferase, to eliminate off-target effects of our expression plasmids or the transfection procedure. Results are expressed as normalized luciferase activity.
Protein preparation and western blotting
Six-well plates of cells were washed once with phosphate-buffered saline and lysed directly in 1× sodium dodecyl sulfate (SDS) sample buffer. Equivalent amounts of protein (10 or 25 µg) were subjected to SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes (Amersham). Membranes were blocked in Tris-buffered saline–Tween-20 (TBST) containing 5% bovine serum albumin unless otherwise indicated and probed overnight at 4°C using anti-KapB (a generous gift from D. Ganem used at 1∶5000 in 5% milk), anti-phospho(ser82)Hsp27 (1∶1000), anti-p115 (1∶1000), anti-H1 (1∶1000), anti-GAPDH (1∶1000, Abcam), anti-RhoA (1∶667), anti-MK2 (1∶1000) or anti-β-actin (1∶2500) antibody. Horseradish peroxidase-conjugated goat anti-rabbit and anti-mouse immunoglobulin secondary antibodies were used at a 1∶2000 dilution. All antibodies were purchased from Cell Signaling Technologies unless otherwise indicated. Secondary antibody was detected using ECL Plus detection reagents (Amersham Biosciences) according to the manufacturer's instructions. The chemiluminescent signal was detected using the Kodak Image Station 4000 mm PRO with no excitation or emission filter.
Rho activation assay
6-well cluster dishes of HeLa-Tet Off cells were subjected to PEI-mediated transfection with 100 ng of a green fluorescent reporter protein, 1.0 µg of expression plasmids for KapB (pcr3.1 KapB), constitutively active MK2 (pcDNA3 FlagMK2-EE), phosphomimicking heat shock protein 27 (pcDNA3.1 HA-hsp27-DDD) or empty vector pcDNA3.1 and 3.3 µl of PEI per well. The transfection medium was replaced after 6 hours. 48 hours post transfection, 105 transfected cells were seeded at sub-confluent density in a 10cm tissue culture dish. 24 hours later, cells were treated with starvation medium containing 0.1% FBS for 24 hours. The next day, cells were starved in medium without FBS for an additional 3–4 hours before being treated or not treated with LPA for 3 minutes. Alternately, confluent 6-well plates of either KapB - or vector control-expressing HUVECs were starved in low-serum EBM-2 medium for 24 hours and in serum-free medium for 4 hours before use. Cells were then immediately washed in ice-cold PBS, and lysed in 500 ul of 1 x Lysis buffer containing aprotinin, pepstatin, leupeptin, and PMSF on ice following the instructions of the Active Rho Detection Kit (Cell Signaling Technologies). Fresh lysates were clarified by centrifugation at 21,000 x g for 5 minutes at 4°C, kept cold at all times, and used immediately for active rho pull downs as recommended by the manufacturer. After binding for one hour, the unbound protein lysate was reserved and used for assay of total protein. For immunoblot analysis 10–20 µg of each total protein lysate and proportional amounts of Rho pull-down reaction (1∶10 ratio of lysate:pull-down) were subjected to 12% - SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes (Amersham). Membranes were blocked Tris-buffered saline–Tween-20 (TBST) containing 5% bovine serum albumin and probed overnight at 4°C using anti-RhoA primary antibody (1∶667, CST) and horseradish peroxidase-conjugated goat anti-rabbit secondary antibody (1∶2000) and developed and imaged according to the above protocol.
Immunofluorescent staining and microscopy
After transduction and selection, HUVECs were seeded on coverslips for microscopy. Twenty-four hours later, cells were either not treated or treated with the appropriate inhibitors or activators as described above and in the figure legends. After treatment, cells were fixed at room temperature in 4% paraformaldehyde (in PBS) for 10 min and then permeabilized with 0.1% Triton X-100 for 10 min. For staining microtubules, fixation was performed at 37°C in pre-warmed 4% paraformaldehyde (in D-PBS) at 37°C before permeabilization as indicated above. Cells were subsequently washed 3 times with PBS and then blocked in 1% human AB serum in PBS for 1 h at room temperature. To stain PB resident proteins, fixed cells were incubated with mouse anti-Hedls antibody (1∶1000; Santa Cruz) or rabbit anti-DDX6 antibody (C terminus, 1∶1000; Bethyl Laboratories) in 1% human AB serum overnight at 4°C. When appropriate and according to figure legends, cells were also incubated overnight at 4°C with rabbit anti-HA (1∶1600, CST), mouse anti-FLAG (1∶1600, Sigma), rabbit anti-LANA (1∶1000, a generous gift from D. Ganem), mouse anti-tubulin (1∶200, Santa Cruz) or rabbit anti-KapB antibody (1∶1000, a generous gift from D. Ganem) for 30 min at room temperature. Primary antibodies were removed by three 5-minute washes of PBS. Goat anti-rabbit Alexa 555, chicken anti-mouse Alexa 488, chicken anti-mouse Alexa 647 or goat anti-rabbit Alexa 647 secondary antibodies (Molecular Probes) were added for 1 h at room temperature in the dark. After being washed as described above, indicated cells were incubated with for one hour at room temperature with 1∶100 phalloidin in PBS (conjugated to either Alexa 555 or 647; Molecular Probes). Finally, cells were washed 3 more times and mounted on microscope slides with ProLong Gold antifade mounting medium (Invitrogen) and visualized using a Zeiss LSM 510 META laser-scanning confocal microscope and the 40× or 63x objective.
Quantification of PBs
HUVECs transduced with vector were examined at 400x or 630x magnification, and the number of cells per field of view that contained normal-sized (approximately 300 nm in diameter; see [67]) PBs were counted. After counting between 100 and 200 cells (usually 4 fields of view), the percentage of cells containing normal PBs was determined. The effect of viral infection or ectopic expression on PB accretion was determined by counting only those cells that were positive by immune fluorescence for infection/gene expression. Results are displayed as the average fold reduction in cells with PBs compared to that of the untreated vector control (±SE).
Wound healing assay
HUVEC cell monolayers were grown on gelatin-coated coverslips that had been etched with a reference marker, transduced with either the KapB retrovirus or an empty vector control and selected with puromycin as described above. After incubating cells in medium devoid of serum or growth factors for 1 hour, cell monolayers were wounded with a p200 pipette tip (by scraping off cells near to the reference marker), washed, and incubated in either complete media or media supplemented with VEGF (10 ng/ml). The ability of cells to repair the wound was monitored over time. Images at the time of wounding (t = 0) or at six hours (t = 6 hours) were captured using an Olympus CKX41 Inverted microscope, and the surface area (SA) of the initial and the remaining wound was determined using Image J. The percent of wound closure after the six hours was calculated using the equation SA(t = 0) –SA(t = 6)/SA(t = 0) ×100. Each experiment was performed in duplicate and the results presented are one representative experiment of three.
Cell migration assays
Cell migration was assayed using a modified Boyden chamber assay [61]. HUVECs, transduced to express either KapB or an empty vector control, were harvested with trypsin, counted, centrifuged and resuspended in supplement-free EBM-2 medium containing 0.1% FBS (0.1%-EBM-2). 7.5×104 cells were added to each 8.0 um pore size gelatinized polycarbonate membrane (Corning) separating the two chambers of a 6.5 mm transwell. After one hour of adhesion, either 0.1%-EBM-2 alone or media containing VEGF (1 or 10 ng/ml) was added to the lower chamber. After 4 hours, non-migratory cells remaining on the upper side of the membrane were removed by cotton swabbing and the cells on the underside of the membrane were fixed with 4% paraformaldehyde before staining with 0.2% crystal violet for 1–2 hours. The filters were dried, removed from the chambers, and visualized by microscopic examination. The number of migrated cells on the lower face of the filter was counted in five random fields at 400x magnification. Assays were done in duplicate and were repeated in three independent experiments. The data represents the average of two technical duplicates and three independent experiments +/ − SE.
Tubule formation assays
Wells of a 48-well plate were coated with 200 µl Matrigel (BD Biosciences). HUVECs, transduced to express KapB, or constitutively active MK2 (MK2-EE) or an empty vector control, were harvested with trypsin, counted, centrifuged and resuspended in basal EBM-2 medium. 5×104 cells were added to the top of each matrigel-containing well in basal media. Cells were incubated at 37°C and observed every hour. Over time, HUVECs progress from individual cells to connected tubules by first forming sprouted cells and then connections between small groups of cells. As tubulogenesis progresses, cells form connected tubes, enclosed polygons and complex meshwork (layered tubes of cells) as described in [66]. At 5 hours, extensive tubules, often with the presence of polygons and complex mesh, had formed and were visualized using an Olympus CKX41 inverted microscope. Representative images were captured using Image Pro Plus. To quantify the angiogenic potential of KapB, an angiogenic score was calculated by adapting the methods of [66]. For each condition, 5 random fields of view at 200x magnification were observed and the number of enclosed polygons was counted. Further, the presence of a complex meshwork was given the following score: 1 = no complex mesh, 2 = presence of complex mesh, and 3 = complex mesh of cell thickness >4 cells. The angiogenic score was then calculated as the product of the number of enclosed polygons and the complex mesh score. Assays were done in duplicate and were repeated in three independent experiments. The data represents the average of two technical duplicates and three independent experiments +/ − SE.
Statistical analysis
Graphing and statistical analyses were performed using GraphPad Prism software. All data are presented as mean +/ − SEM of three independent experiments unless otherwise indicated. Paired parametric t-test was used for comparison between two groups. One-way repeated measures ANOVA was used for comparison between multiple groups where the mean of each group was compared to the mean of the control group. P values are displayed when p<0.05.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. BoshoffC, SchulzTF, KennedyMM, GrahamAK, FisherC, et al. (1995) Kaposi's sarcoma-associated herpesvirus infects endothelial and spindle cells. Nat Med 1 : 1274–1278.
2. CesarmanE, ChangY, MoorePS, SaidJW, KnowlesDM (1995) Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med 332 : 1186–1191 doi:10.1056/NEJM199505043321802
3. SoulierJ, GrolletL, OksenhendlerE, CacoubP, Cazals-HatemD, et al. (1995) Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 86 : 1276–1280.
4. SpeckSH, GanemD (2010) Viral Latency and Its Regulation: Lessons from the Gamma-Herpesviruses. Cell Host and Microbe 8 : 100–115 doi:10.1016/j.chom.2010.06.014
5. AriasC, WeisburdB, Stern-GinossarN, MercierA, BellareP, et al. (2014) KSHV 2.0: A Comprehensive Annotation of the Kaposi's Sarcoma-Associated Herpesvirus Genome Using Next - Generation Sequencing Reveals Novel Genomic and Functional Features. PLoS Pathog 10 : 1–23.
6. UmbachJL, CullenBR (2010) In-depth analysis of Kaposi's sarcoma-associated herpesvirus microRNA expression provides insights into the mammalian microRNA-processing machinery. Journal of Virology 84 : 695–703 doi:10.1128/JVI.02013-09
7. GanemD (2010) KSHV and the pathogenesis of Kaposi sarcoma: listening to human biology and medicine. J Clin Invest 120 : 939–949 doi:10.1172/JCI40567
8. OjalaPM, SchulzTF (2014) Manipulation of endothelial cells by KSHV: Implications for angiogenesis and aberrant vascular differentiation. Seminars in Cancer Biology 26C: 69–77 doi:10.1016/j.semcancer.2014.01.008
9. GrossmannC, PodgrabinskaS, SkobeM, GanemD (2006) Activation of NF - κB by the Latent vFLIP Gene of Kaposi's Sarcoma-Associated Herpesvirus Is Required for the Spindle Shape of Virus-Infected Endothelial Cells and Contributes to Their Proinflammatory Phenotype. Journal of Virology 80 : 7179–7185 doi:10.1128/JVI.01603-05
10. NaranattPP, AkulaSM, ZienCA, KrishnanHH, ChandranB (2003) Kaposi's Sarcoma-Associated Herpesvirus Induces the Phosphatidylinositol 3-Kinase-PKC - -MEK-ERK Signaling Pathway in Target Cells Early during Infection: Implications for Infectivity. Journal of Virology 77 : 1524–1539 doi:10.1128/JVI.77.2.1524-1539.2003
11. CiufoDM, CannonJS, PooleLJ, WuFY, MurrayP, et al. (2001) Spindle cell conversion by Kaposi's sarcoma-associated herpesvirus: formation of colonies and plaques with mixed lytic and latent gene expression in infected primary dermal microvascular endothelial cell cultures. Journal of Virology 75 : 5614–5626 doi:10.1128/JVI.75.12.5614-5626.2001
12. McCormickC (2005) The Kaposin B Protein of KSHV Activates the p38/MK2 Pathway and Stabilizes Cytokine mRNAs. Science 307 : 739–741 doi:10.1126/science.1105779
13. WinzenR, KrachtM, RitterB, WilhelmA, ChenCY, et al. (1999) The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. The EMBO Journal 18 : 4969–4980 doi:10.1093/emboj/18.18.4969
14. NeiningerA, KontoyiannisD, KotlyarovA, WinzenR, EckertR, et al. (2002) MK2 targets AU-rich elements and regulates biosynthesis of tumor necrosis factor and interleukin-6 independently at different post-transcriptional levels. Journal of Biological Chemistry 277 : 3065–3068 doi:10.1074/jbc.C100685200
15. ChenCY, ShyuAB (1995) AU-rich elements: characterization and importance in mRNA degradation. Trends in Biochemical Sciences 20 : 465–470.
16. BakheetT, WilliamsBRG, KhabarKSA (2006) ARED 3.0: the large and diverse AU-rich transcriptome. Nucleic Acids Research 34: D111–D114 doi:10.1093/nar/gkj052
17. SandujaS, BlancoFF, DixonDA (2010) The roles of TTP and BRF proteins in regulated mRNA decay. WIREs RNA 2 : 42–57 doi:10.1002/wrna.28
18. YooJ, KangJ, LeeHN, AguilarB, KafkaD, et al. (2010) Kaposin-B Enhances the PROX1 mRNA Stability during Lymphatic Reprogramming of Vascular Endothelial Cells by Kaposi's Sarcoma Herpes Virus. PLoS Pathog 6: e1001046 doi:10.1371/journal.ppat.1001046
19. ParkerR, ShethU (2007) P Bodies and the Control of mRNA Translation and Degradation. Molecular Cell 25 : 635–646 doi:10.1016/j.molcel.2007.02.011
20. Fenger-GrønM, FillmanC, NorrildB, Lykke-AndersenJ (2005) Multiple Processing Body Factors and the ARE Binding Protein TTP Activate mRNA Decapping. Molecular Cell 20 : 905–915 doi:10.1016/j.molcel.2005.10.031
21. StoecklinG, MayoT, AndersonP (2006) ARE-mRNA degradation requires the 5′-3′ decay pathway. EMBO Rep 7 : 72–77 doi:10.1038/sj.embor.7400572
22. Lykke-AndersenJ (2005) Recruitment and activation of mRNA decay enzymes by two ARE-mediated decay activation domains in the proteins TTP and BRF-1. Genes & Development 19 : 351–361 doi:10.1101/gad.1282305
23. FranksTM, Lykke-AndersenJ (2007) TTP and BRF proteins nucleate processing body formation to silence mRNAs with AU-rich elements. Genes & Development 21 : 719–735 doi:10.1101/gad.1494707
24. KedershaN, StoecklinG, AyodeleM, YaconoP, Lykke-AndersenJ, et al. (2005) Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. The Journal of Cell Biology 169 : 871–884 doi:10.1083/jcb.200502088
25. EulalioA, Behm-AnsmantI, IzaurraldeE (2007) P bodies: at the crossroads of post-transcriptional pathways. Nat Rev Mol Cell Biol 8 : 9–22 doi:10.1038/nrm2080
26. BrenguesM, TeixeiraD, ParkerR (2005) Movement of eukaryotic mRNAs between polysomes and cytoplasmic processing bodies. Science 310 : 486–489 doi:10.1126/science.1115791
27. BhattacharyyaSN, HabermacherR, MartineU, ClossEI, FilipowiczW (2006) Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell 125 : 1111–1124 doi:10.1016/j.cell.2006.04.031
28. KulkarniM, OzgurS, StoecklinG (2010) On track with P-bodies. Biochem Soc Trans 38 : 242 doi:10.1042/BST0380242
29. AizerA, BrodyY, LerLW, SonenbergN, SingerRH, et al. (2008) The Dynamics of Mammalian P Body Transport, Assembly, and Disassembly In Vivo. Molecular Biology of the Cell 19 : 4154–4166 doi:10.1091/mbc.E08
30. RajgorD, MelladJA, SoongD, RattnerJB, FritzlerMJ, et al. (2014) Mammalian microtubule P-body dynamics are mediated by nesprin-1. The Journal of Cell Biology 205 : 457–475 doi:10.1083/jcb.201306076
31. LoschiM, LeishmanCC, BerardoneN, BoccaccioGL (2009) Dynein and kinesin regulate stress-granule and P-body dynamics. Journal of Cell Science 122 : 3973–3982 doi:10.1242/jcs.051383
32. TakahashiS, SakuraiK, EbiharaA, KajihoH, SaitoK, et al. (2011) RhoA activation participates in rearrangement of processing bodies and release of nucleated AU-rich mRNAs. Nucleic Acids Research 39 : 3446–3457 doi:10.1093/nar/gkq1302
33. SchmitzA (2000) Rho GTPases: Signaling, Migration, and Invasion. Experimental Cell Research 261 : 1–12 doi:10.1006/excr.2000.5049
34. BuchsbaumRJ (2007) Rho activation at a glance. Journal of Cell Science 120 : 1149–1152 doi:10.1242/jcs.03428
35. HallA (1998) Rho GTPases and the actin cytoskeleton. Science 279 : 509–514.
36. HallA (2009) The cytoskeleton and cancer. Cancer Metastasis Rev 28 : 5–14 doi:10.1007/s10555-008-9166-3
37. RidleyAJ (2001) Rho GTPases and cell migration. Journal of Cell Science 114 : 2713–2722.
38. FujiharaH, WalkerLA, GongMC, LemichezE, BoquetP, et al. (1997) Inhibition of RhoA translocation and calcium sensitization by in vivo ADP-ribosylation with the chimeric toxin DC3B. molecular biology of the cell 8 : 2437–2447.
39. SiehlerS (2009) Regulation of RhoGEF proteins by G 12/13-coupled receptors. British Journal of Pharmacology 158 : 41–49 doi:10.1111/j.1476-5381.2009.00121.x
40. DubashAD, WennerbergK, Garcia-MataR, MenoldMM, ArthurWT, et al. (2007) A novel role for Lsc/p115 RhoGEF and LARG in regulating RhoA activity downstream of adhesion to fibronectin. Journal of Cell Science 120 : 3989–3998 doi:10.1242/jcs.003806
41. BryanBA, D'AmorePA (2007) What tangled webs they weave: Rho-GTPase control of angiogenesis. Cell Mol Life Sci 64 : 2053–2065 doi:10.1007/s00018-007-7008-z
42. MongPY, WangQ (2009) Activation of Rho Kinase Isoforms in Lung Endothelial Cells during Inflammation. The Journal of Immunology 182 : 2385–2394 doi:10.4049/jimmunol.0802811
43. SomlyoAP, SomlyoAV (2000) Signal transduction by G-proteins, rho-kinase and protein phosphatase to smooth muscle and non-muscle myosin II. J Physiol (Lond) 522 Pt 2 : 177–185.
44. OhashiK, NagataK, MaekawaM, IshizakiT, NarumiyaS, et al. (2000) Rho-associated Kinase ROCK Activates LIM-kinase 1 by Phosphorylation at Threonine 508 within the Activation Loop*. Journal of Biological Chemistry 275 : 3577–3582.
45. SumiT, MatsumotoK, TakaiY, NakamuraT (1999) Cofilin Phosphorylation and Actin Cytoskeletal Dynamics Regulated by Rho - and Cdc42-activated LIM-kinase 2. The Journal of Cell Biology 147 : 1519–1532.
46. KotlyarovA, GaestelM (2002) Is MK2 (mitogen-activated protein kinase-activated protein kinase 2) the key for understanding post-transcriptional regulation of gene expression? Biochem Soc Trans 30 : 959–963.
47. KotlyarovA, YannoniY, FritzS, LaassK, TelliezJ-B, et al. (2002) Distinct cellular functions of MK2. Molecular and Cellular Biology 22 : 4827–4835.
48. LandryJ, HuotJ (1999) Regulation of actin dynamics by stress-activated protein kinase 2 (SAPK2)-dependent phosphorylation of heat-shock protein of 27 kDa (Hsp27). Biochem Soc Symp 64 : 79–89.
49. RousseauS, HouleF, HuotJ (2000) Integrating the VEGF signals leading to actin-based motility in vascular endothelial cells. Trends in Cardiovascular Medicine 10 : 321–327.
50. LamaliceL, Le BoeufF, HuotJ (2007) Endothelial Cell Migration During Angiogenesis. Circulation Research 100 : 782–794 doi:10.1161/01.RES.0000259593.07661.1e
51. GamellC, SusperreguiAG, BernardO, RosaJL, VenturaF (2011) The p38/MK2/Hsp25 Pathway Is Required for BMP-2-Induced Cell Migration. PLoS ONE 6: e16477 doi:10.1371/journal.pone.0016477
52. XuL, ChenS, BerganRC (2006) MAPKAPK2 and HSP27 are downstream effectors of p38 MAP kinase-mediated matrix metalloproteinase type 2 activation and cell invasion in human prostate cancer. Oncogene 25 : 2987–2998 doi:10.1038/sj.onc.1209337
53. CoteMC, LavoieJR, HouleF, PoirierA, RousseauS, et al. (2010) Regulation of Vascular Endothelial Growth Factor-induced Endothelial Cell Migration by LIM Kinase 1-mediated Phosphorylation of Annexin 1. Journal of Biological Chemistry 285 : 8013–8021 doi:10.1074/jbc.M109.098665
54. RogallaT, EhrnspergerM, PrevilleX, KotlyarovA, LutschG, et al. (1999) Regulation of Hsp27 oligomerization, chaperone function, and protective activity against oxidative stress/tumor necrosis factor alpha by phosphorylation. Journal of Biological Chemistry 274 : 18947–18956.
55. HuangJ, XieL-D, LuoL, ZhengS-L, WangH-J, et al. (2014) Silencing heat shock protein 27 (HSP27) inhibits the proliferation and migration of vascular smooth muscle cells in vitro. Mol Cell Biochem 390 : 115–121 doi:10.1007/s11010-014-1962-1
56. KobayashiM, NishitaM, MishimaT, OhashiK, MizunoK (2006) MAPKAPK-2-mediated LIM-kinase activation is critical for VEGF-induced actin remodeling and cell migration. The EMBO Journal 25 : 713–726 doi:10.1038/sj.emboj.7600973
57. AmanoM (1997) Formation of Actin Stress Fibers and Focal Adhesions Enhanced by Rho-Kinase. Science 275 : 1308–1311 doi:10.1126/science.275.5304.1308
58. MaekawaM (1999) Signaling from Rho to the Actin Cytoskeleton Through Protein Kinases ROCK and LIM-kinase. Science 285 : 895–898 doi:10.1126/science.285.5429.895
59. KayyaliUS (2002) Cytoskeletal Changes in Hypoxic Pulmonary Endothelial Cells Are Dependent on MAPK-activated Protein Kinase MK2. Journal of Biological Chemistry 277 : 42596–42602 doi:10.1074/jbc.M205863200
60. RousseauS, HouleF, KotanidesH, WitteL, WaltenbergerJ, et al. (2000) Vascular endothelial growth factor (VEGF)-driven actin-based motility is mediated by VEGFR2 and requires concerted activation of stress-activated protein kinase 2 (SAPK2/p38) and geldanamycin-sensitive phosphorylation of focal adhesion kinase. Journal of Biological Chemistry 275 : 10661–10672.
61. RousseauS, HouleF, LandryJ, HuotJ (1997) p38 MAP kinase activation by vascular endothelial growth factor mediates actin reorganization and cell migration in human endothelial cells. Oncogene 15 : 2169–2177 doi:10.1038/sj.onc.1201380
62. AndersonDR, MeyersMJ, VernierWF, MahoneyMW, KurumbailRG, et al. (2007) Pyrrolopyridine Inhibitors of Mitogen-Activated Protein Kinase-Activated Protein Kinase 2 (MK-2). J Med Chem 50 : 2647–2654 doi:10.1021/jm0611004
63. GarciaMC, RayDM, LackfordB, RubinoM, OldenK, et al. (2009) Arachidonic Acid Stimulates Cell Adhesion through a Novel p38 MAPK-RhoA Signaling Pathway That Involves Heat Shock Protein 27. Journal of Biological Chemistry 284 : 20936–20945 doi:10.1074/jbc.M109.020271
64. Wittchen ES, Burridge K (2008) Chapter 14 Analysis of Low Molecular Weight GTPase Activity in Endothelial Cell Cultures. Angiogenesis - In Vitro Systems. Methods in Enzymology. Elsevier, Vol. 443. pp. 285–298. doi:10.1016/S0076-6879(08)02014-4.
65. YamadaT, OhokaY, KogoM, InagakiS (2005) Physical and Functional Interactions of the Lysophosphatidic Acid Receptors with PDZ Domain-containing Rho Guanine Nucleotide Exchange Factors (RhoGEFs). Journal of Biological Chemistry 280 : 19358–19363 doi:10.1074/jbc.M414561200
66. ArandaE, OwenGI (2009) A semi-quantitative assay to screen for angiogenic compounds and compounds with angiogenic potential using the EA.hy926 endothelial cell line. Biological Research 42 : 377–389.
67. CorcoranJA, KhaperskyyDA, JohnstonBP, KingCA, CyrDP, et al. (2012) Kaposi's Sarcoma-Associated Herpesvirus G-Protein-Coupled Receptor Prevents AU-Rich-Element-Mediated mRNA Decay. Journal of Virology 86 : 8859–8871 doi:10.1128/JVI.00597-12
68. CougotN, CavalierA, ThomasD, GilletR (2012) The Dual Organization of P-bodies Revealed by Immunoelectron Microscopy and Electron Tomography. Journal of Molecular Biology 420 : 17–28 doi:10.1016/j.jmb.2012.03.027
69. KrendelM, ZenkeFT, BokochGM (2002) Nucleotide exchange factor GEF-H1 mediates cross-talk between microtubules and the actin cytoskeleton. Nat Cell Biol 4 : 294–301 doi:10.1038/ncb773
70. FeigLA (1999) Tools of the trade: use of dominant-inhibitory mutants of Ras-family GTPases. Nat Cell Biol 1: E25–E27 doi:10.1038/10018
71. WildeC, AktoriesK (2001) The Rho-ADP-ribosylating C3 exoenzyme from Clostridiumbotulinum and related C3-like transferases. Toxicology 39 : 1647–1660.
72. CorcoranJA, KhaperskyyDA, McCormickC (2011) Assays for monitoring viral manipulation of host ARE-mRNA turnover. Methods 55 : 172–181 doi:10.1016/j.ymeth.2011.08.005
73. HittiE, IakovlevaT, BrookM, DeppenmeierS, GruberAD, et al. (2006) Mitogen-activated protein kinase-activated protein kinase 2 regulates tumor necrosis factor mRNA stability and translation mainly by altering tristetraprolin expression, stability, and binding to adenine/uridine-rich element. Molecular and Cellular Biology 26 : 2399–2407 doi:10.1128/MCB.26.6.2399-2407.2006
74. KnapinskaAM, GratacosFM, KrauseCD, HernandezK, JensenAG, et al. (2011) Chaperone Hsp27 Modulates AUF1 Proteolysis and AU-Rich Element-Mediated mRNA Degradation. Molecular and Cellular Biology 31 : 1419–1431 doi:10.1128/MCB.00907-10
75. ReinekeLC, LloydRE (2013) Diversion of stress granules and P-bodies during viral infection. Virology 436 : 255–267 doi:10.1016/j.virol.2012.11.017
76. WardAM, BidetK, YinglinA, LerSG, HogueK, et al. (2011) Quantitative mass spectrometry of DENV-2 RNA-interacting proteins reveals that the DEAD-box RNA helicase DDX6 binds the DB1 and DB2 3′ UTR structures. RNA Biology 8 : 1173–1186 doi:10.4161/rna.8.6.17836
77. NathansR, ChuC-Y, SerquinaAK, LuC-C, CaoH, et al. (2009) Cellular microRNA and P bodies modulate host-HIV-1 interactions. Molecular Cell 34 : 696–709 doi:10.1016/j.molcel.2009.06.003
78. PijlmanGP, FunkA, KondratievaN, LeungJ, TorresS, et al. (2008) A highly structured, nuclease-resistant, noncoding RNA produced by flaviviruses is required for pathogenicity. Cell Host and Microbe 4 : 579–591 doi:10.1016/j.chom.2008.10.007
79. ChaharHS, ChenS, ManjunathN (2013) P-body components LSM1, GW182, DDX3, DDX6 and XRN1 are recruited to WNV replication sites and positively regulate viral replication. Virology 436 : 1–7 doi:10.1016/j.virol.2012.09.041
80. PagerCT, SchützS, AbrahamTM, LuoG, SarnowP (2013) Modulation of hepatitis C virus RNA abundance and virus release by dispersion of processing bodies and enrichment of stress granules. Virology 435 : 472–484 doi:10.1016/j.virol.2012.10.027
81. ReedJC, MolterB, GearyCD, McNevinJ, McElrathJ, et al. (2012) HIV-1 Gag co-opts a cellular complex containing DDX6, a helicase that facilitates capsid assembly. The Journal of Cell Biology 198 : 439–456 doi:10.1083/jcb.201111012
82. SetoE, InoueT, NakataniY, YamadaM, IsomuraH (2014) Processing bodies accumulate in human cytomegalovirus-infected cells and do not affect viral replication at high multiplicity of infection. Virology 458–459 : 151–161 doi:10.1016/j.virol.2014.04.022
83. SweetTJ, BoyerB, HuW, BakerKE, CollerJ (2007) Microtubule disruption stimulates P-body formation. RNA 13 : 493–502 doi:10.1261/rna.355807
84. IshizakiT, MorishimaY, OkamotoM, FuruyashikiT, KatoT, et al. (2001) Coordination of microtubules and the actin cytoskeleton by the Rho effector mDia1. Nat Cell Biol 3 : 8–14 doi:10.1038/35050598
85. PalazzoAF, CookTA, AlbertsAS, GundersenGG (2001) mDia mediates Rho-regulated formation and orientation of stable microtubules. Nat Cell Biol 3 : 723–729 doi:10.1038/35087035
86. StoecklinG, StubbsT, KedershaN, WaxS, RigbyWFC, et al. (2004) MK2-induced tristetraprolin: 14-3-3 complexes prevent stress granule association and ARE-mRNA decay. The EMBO Journal 23 : 1313–1324 doi:10.1038/sj.emboj.7600163
87. YooS-M, AhnA-K, SeoT, HongHB, ChungM-A, et al. (2008) Centrifugal enhancement of Kaposi's sarcoma-associated virus infection of human endothelial cells in vitro. Journal of Virological Methods 154 : 160–166 doi:10.1016/j.jviromet.2008.07.026
88. BruloisKF, ChangH, LeeAS-Y, EnsserA, WongL-Y, et al. (2012) Construction and manipulation of a new Kaposi's sarcoma-associated herpesvirus bacterial artificial chromosome clone. Journal of Virology 86 : 9708–9720 doi:10.1128/JVI.01019-12
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 1
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Infections in Humans and Animals: Pathophysiology, Detection, and Treatment
- : Trypanosomatids Adapted to Plant Environments
- Environmental Drivers of the Spatiotemporal Dynamics of Respiratory Syncytial Virus in the United States
- Dengue Virus RNA Structure Specialization Facilitates Host Adaptation
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy