
TLR3 Signaling in Macrophages Is Indispensable for the Protective Immunity of Invariant Natural Killer T Cells against Enterovirus 71 Infection
Enterovirus 71 (EV71) is a major causative pathogen of hand, foot and mouth disease. EV71 infection occurs mainly in children but rarely in adults. The factors that determine the susceptibility of children to EV71 infection remain elusive. Here, we found that the paucity of invariant natural killer T (iNKT) cells in new-born mice was associated with their susceptibility to EV71 infection. Furthermore, iNKT cells played a critical role in protecting older young mice from EV71 infection before their adaptive immune systems were fully developed. Mechanistically, TLR3 signaling in macrophages, but not in dendritic cells, was essentially required for iNKT cell activation during EV71 infection. Both interleukin (IL)-12 production and endogenous lipid antigens presented by macrophages were required for full iNKT cell activation. iNKT cells tended to prevent the dissemination of EV71 into central nervous system. Taken together, our findings provide a new insight into the susceptibility of children to EV71 infection, and imply that the manipulation of iNKT cells might represent a potential therapeutic strategy for HFMD and other viral infectious diseases in children.
Published in the journal:
. PLoS Pathog 11(1): e32767. doi:10.1371/journal.ppat.1004613
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004613
Summary
Enterovirus 71 (EV71) is a major causative pathogen of hand, foot and mouth disease. EV71 infection occurs mainly in children but rarely in adults. The factors that determine the susceptibility of children to EV71 infection remain elusive. Here, we found that the paucity of invariant natural killer T (iNKT) cells in new-born mice was associated with their susceptibility to EV71 infection. Furthermore, iNKT cells played a critical role in protecting older young mice from EV71 infection before their adaptive immune systems were fully developed. Mechanistically, TLR3 signaling in macrophages, but not in dendritic cells, was essentially required for iNKT cell activation during EV71 infection. Both interleukin (IL)-12 production and endogenous lipid antigens presented by macrophages were required for full iNKT cell activation. iNKT cells tended to prevent the dissemination of EV71 into central nervous system. Taken together, our findings provide a new insight into the susceptibility of children to EV71 infection, and imply that the manipulation of iNKT cells might represent a potential therapeutic strategy for HFMD and other viral infectious diseases in children.
Introduction
EV71 is a single-stranded, positive-sense RNA (+ssRNA) virus and belongs to the picornaviridae family. EV71 infects mainly children aged less than 5 years [1–3]. Patients with EV71 infection develop sores on the hands, feet, and sometimes buttocks and mouth, namely hand, foot and mouth disease (HFMD). Although many other enteroviruses cause HFMD in children, EV71 infection is more frequently associated with severe central-nervous-system complications in HFMD patients and thereby is a major cause of fatalities [1,4]. Thus, EV71 is considered the most virulent pathogen among the HFMD-associated enteroviruses. EV71 was first isolated from a sick child in California in 1969, and EV71 outbreaks subsequently occurred in Europe in the 1970s. Currently, HFMD is a major endemic infectious disease, with over one million cases annually in China and Southeastern Asia [3,5–7]. So far, the factors that determine the age-dependent susceptibility of children to EV71 infection remain largely unknown.
An early study by Khong et al. has shown that 2-week-old and younger immunodeficient AG129 mice, which lack type I and II interferon receptors, are susceptible to EV71 infection [8]. Their finding suggests that both type I and II interferons (IFNs) are crucial in controlling EV71 infection. Both +ssRNA and -ssRNA are produced in the life cycle of EV71. The recognition of these RNA components by TLR3, TLR7, RIG-I and MDA-5 molecules expressed by host cells potentially induces type I IFN production and limits EV71 infection. Surprisingly, the production of type I IFNs is almost absent in EV71-infected cells presumably because of inhibition by 2C and 3C proteases of the virus [9–11]. Consistently, only very low levels of type I IFNs have been detected in EV71-infected mice [8]. Furthermore, a recent study by Shih’s group has shown that mice deficient for interferon alpha receptor were not susceptible to EV71 infection. In contrast, mice deficient for interferon gamma (IFN-γ) receptor were very susceptible to EV71 infection [12]. Altogether, accumulative evidences suggest that IFN-γ, rather than type I IFNs, is likely important in limiting EV71 infection. Interestingly, it has been reported that severe HFMD patients with pulmonary edema have lower numbers of circulating leukocytes, including natural killer (NK) cells and T cells, in comparison to patients with mild disease [13]. Because technically the report has not distinguished NK cells from invariant natural killer T (iNKT) cells, it is entirely possible that iNKT cells, also IFN-γ-producing cells, may also be decreased in severe patients, and could be a link to the development of the disease.
iNKT cells are a distinct subpopulation of T cells that express an invariant αβ T cell receptor (TCR) and share a number of cell surface markers in common with NK cells. iNKT cells recognize glycolipid antigens presented by the invariant MHC class I-like molecule CD1d, which is expressed mainly on dendritic cells (DCs) and macrophages. Following lipid antigen stimulation, iNKT cells express CD40L molecule [14] and rapidly produce high levels of cytokines and chemokines [15,16]. iNKT cells are considered the first line of immune defense and are important mediators that modulate the innate and adaptive immune responses. iNKT cells are implicated in different types of viral infections, helping to control viral load or participate in disease development [17,18]. Therefore, modulating iNKT cell activation has immense clinical potential against viral infectious diseases.
In addition to microbial lipid antigens, iNKT cells recognize self-lipid antigens and are involved in the immune response to infection by microbes, which do not express glycolipids [19–25]. Paget et al. demonstrated that stimulation with TLR4, TLR7, TLR8 or TLR9 ligands renders DCs capable of promoting iNKT cell activation in a self-antigen-dependent manner. In terms of TLR9 ligand (CpG)-stimulation, iNKT cell activation fully requires type I interferon or IL-12 in dependence of the types of antigen-presenting cells in the context [24,26]. These results suggest that DNA viruses that contain CpG may trigger iNKT cell activation through the CpG-receptor TLR9. Evidently TLR9 signaling is required for iNKT cell activation in the infection by murine cytomegalovirus (MCMV), a DNA virus [27,28]. The iNKT cell activation does not require CD1d signaling but does require IL-12 production by DCs [27]. The CD1d independence and IL-12 dependence of iNKT cell activation in MCMV infection is unexpected and paradoxical to Paget’s initial observation [24] and to a later finding on microbial infections [19]. The discrepancy and the opposite role of iNKT cells [17,18] indicate that the requirement of iNKT cell activation may vary in different viral infections.
In this study, we found that lack of iNKT cells in neonatal mice was associated with their vulnerability to EV71 infection. In addition, iNKT cells played a crucial protective role in EV71-infected young mice aged less than 3 weeks when their adaptive immune systems were not fully developed. The protective immunity mediated by iNKT cells was dependent on TLR3-signaling in macrophages and the production of IL-12 and endogenous lipid ligands by macrophages. Our findings provide a new insight into the susceptibility of children to infectious diseases caused by RNA viruses, and may have implications for potential immune intervention in these diseases.
Results
Immaturity of the immune system is related to the susceptibility of young mice to EV71 infection
To obtain a mouse-adaptive EV71 (EV71M), we inoculated a clinical EV71 isolate (EV71H) into the brains of one-day-old ICR mice 5 times and then passaged the virus in L929 cells. EV71M still replicated as efficiently as the parental isolate in RD cells (Fig. 1A). In contrast to the parental EV71H virus, EV71M also replicated efficiently (increased replication up to 100-fold) in murine L929 cells 48 hours after infection (Fig. 1B). This result suggests that the mouse-adaptive EV71M gained the capability to infect murine cells.

We infected 1-, 3-, 7 - or 14-day-old ICR mice with 2×106 PFU of EV71M. All the one-day-old and 50% of the 3-day-old neonates developed hind limb weakness or paralysis and showed signs of encephalitis manifested by hunched posture, lethargy, and ataxia. None of the mice that developed paralysis survived after 9 days post infection. All 7-day-old mice developed at least one of the above symptoms after 6 days post infection, but their death rates dropped to 15%. All 14-day-old mice survived without any symptoms after the infection (Fig. 1C). In all, the susceptibility of EV71 infection in young mice appeared to be age-dependent.
The age-dependent susceptibility of disease development made us wonder whether the immaturity of the immune system is related to the vulnerability of neonatal mice to EV71 infection. The percentage and numbers of T, NK and NKT cells within the total lymphocyte population in the thymus and spleen of naive mice were therefore determined by flow cytometry on days 3, 7 and 14 after birth. Similar to the previous reports using C57BL/6 mice [29], the percentages of T cells were 2–3-times less in ICR neonates on days 3 and 7 of life compared with 14-day-old mice. Similarly, the percentages of NK and NKT cells were significantly lower in the younger mice. Noticeably, NKT cells were nearly undetectable in the mice before the first week of life (Fig. 1D and S1 Fig.). Thus, the small proportion of iNKT cells tends to be related to the susceptibility of neonates to EV71 infection.
iNKT cells are activated by EV71-infected macrophages
IFN-γ has been suggested to play a critical role in the control of EV71 infection [8,12]. To determine the cellular resources of IFN-γ production after EV71 infection, we cultured splenocytes from C57BL/6 mice with EV71M and analyzed IFN-γ production in the culture supernatants. We observed that the proportion of IFN-γ-producing cells increased 5–20 times upon EV71 infection compared with the controls (Fig. 2A and S2 Fig.). The majority of IFN-γ-producing cells were NK cells, but iNKT cells and NK1.1-negative cells also produced IFN-γ (Fig. 2A). Because NK and iNKT cells both produced IFN-γ after EV71M infection, we wondered which cells were essentially required for the IFN-γ production. RAG1-knockout mice lack NKT cells but have NK cells and other innate components. We therefore cultured RAG1-knockout splenocytes with EV71M and then examined the IFN-γ production in the cultures. We found that the IFN-γ production by the splenocytes of RAG1-knockout mice was less than one quarter of wild-type mice, suggesting that the IFN-γ production of NK cells can not be primarily triggered by EV71 infection (Fig. 2B). These observations suggest that EV71 infection primarily activates iNKT cells, and in return, presumably promotes the activation of NK cells.

To further corroborate the activation of iNKT cells by EV71 infection, we infected adult C57BL/6 mice with EV71M and examined the activation status of iNKT cells in the livers and spleens. Flow cytometry analysis revealed that iNKT cells had upregulated CD25 and CD69 expression and indeed displayed signs of activation in both tissues at 16–24 hours post-infection (Fig. 2C). Similar activation of iNKT cells was observed in the spleens of young mice (S3 Fig.).
iNKT cells can be activated by either DCs or macrophages, which are triggered by TLR ligands or microbial infections [19,24,25,30]. We next sought to determine whether EV71 infection of DCs or macrophages could activate iNKT cells. We cultured purified iNKT or NK cells together with EV71M-infected DCs or macrophages and then examined IFN-γ production in the culture supernatants. As expected, IFN-γ production was negligible in the culture supernatants of DCs, NK or iNKT cells alone. In addition, co-culture of NK cells with EV71M-infected DCs did not significantly upregulate IFN-γ production by NK cells. To our surprise, the IFN-γ production of iNKT cells was also negligible in the co-culture with EV71M-infected DCs (Fig. 2D upper panel). In contrast, the EV71M-infected macrophages dramatically induced IFN-γ production by iNKT cells (Fig. 2D lower panel). The ability of EV71M-infected macrophages to activate iNKT cells appeared not to be linked with higher level of viral replication than DCs, as the viral titers of macrophages were not significantly different from those of DCs after EV71M infection (S4 Fig.). Therefore, these results reveal that macrophages intrinsically possess the capability to specifically activate iNKT cells upon EV71 infection.
TLR3 signaling is indispensable for iNKT cell activation in EV71 infection
TLR-mediated signals and cytokines produced by antigen-presenting cells can activate iNKT cells during microbial infections in an endogenous lipid antigen-dependent manner [19,20,24]. To identify the potential pattern recognition receptor that recognizes EV71 and triggers activation of iNKT cells, we cultured splenocytes from WT mice or mice deficient for TLR3, TLR7 or MyD88 with or without EV71M and measured the production of IFN-γ in the supernatants of the cultures. TLR7 - or MyD88-deficient splenocytes produced a similar level of IFN-γ as WT, TLR3-deficient splenocytes failed to produce IFN-γ in the presence of EV71M (Fig. 3A). Consistently, intracellular staining of IFN-γ revealed that iNKT cells of splenocytes from TLR3-deficient mice failed to upregulate IFN-γ expression upon EV71M challenge (Fig. 3B). Thus, these results indicate that TLR3 may be indispensable for iNKT activation in EV71M infection. To further confirm the role of TLR3 signaling in iNKT cell activation, we cultured iNKT cells purified from WT mice together with EV71M-infected macrophages from TLR3-deficient or WT mice and then examined the IFN-γ expression of iNKT cells. Our results revealed that TLR3-deficient macrophages failed to induce the production of IFN-γ by iNKT cells (Fig. 3C). It has been reported that the activation of iNKT cells by pathogens in vitro does not always reflect the activation requirements in vivo [31]. We therefore examined the expression of CD69 on iNKT cells obtained from EV71M-infected mice and WT mice. We observed that CD69 expression in vivo was significantly reduced on the iNKT cells from EV71-infected, TLR3-deficient mice in comparison with WT mice (Fig. 3D). Altogether, TLR3 expression in macrophages is indispensable for iNKT cell activation during EV71 infection.

Paradoxically, it has been shown that DCs stimulated with polyribo-inosinic-polyribocytidylic acid (pI:C), a TLR3 ligand, are not able to activate iNKT cells [24]. Likely macrophages harbor a specific capacity to activate iNKT cells upon TLR3 triggering. To address this possibility, we stimulated bone marrow-derived dendritic cells (BMDCs) and macrophages with pI:C and then co-cultured them with purified iNKT cells. Consistent with the previous finding [24], BMDCs stimulated with pI:C failed to upregulate IFN-γ production in iNKT cells. In contrast, macrophages stimulated with pI:C significantly upregulated the IFN-γ production of iNKT cells (Fig. 3E). This result demonstrates a cell type-specific role of TLR3 in iNKT cell activation.
To address the potential role of TLR3 in the development and/or control of disease caused by EV71 infection, WT and TLR3-deficient mice aged 7 days were infected with 1×106 PFU of EV71M. At this dose of virus, half of the WT mice were paralyzed and died within 8 days post-infection, while none of the TLR3-deficient mice survived beyond 5 days post-infection (Fig. 3F). This result indicates that the TLR3-triggered immune response is indispensable for controlling EV71-caused disease in young mice.
Endogenous CD1d ligands and IL-12 are both required for full activation of iNKT cells in EV71 infection
Because EV71 per se does not encode an enzyme to synthesize lipid ligands for activating iNKT cells, we next sought to investigate whether endogenous ligands presented by CD1d molecule are required for iNKT cell activation triggered by EV71M-infected macrophages. We found that the addition of blocking antibody against CD1d molecule significantly reduced approximately 50% of the IFN-γ production of iNKT cells co-cultured with EV71M-infected macrophages (Fig. 4A). CD1d deficiency in macrophages also showed dramatically reduced IFN-γ production by iNKT cells (Fig. 4B). N-(n-butyl)-deoxy-galactonojirimycin (NB-DGJ) is an inhibitor of lactase phlorizin hydrolase, which is the first enzyme responsible for the biosynthesis of endogenous lipid antigens [24,32]. We observed that the addition of NB-DGJ also significantly inhibited the IFN-γ response of iNKT cells to EV71 infection in culture (Fig. 4C). Furthermore, we adoptively transferred purified iNKT cells into 7-day-old WT mice and CD1d-/- mice and determined the CD69 expression levels of iNKT cells upon EV71M infection. We observed that CD69 expression levels were significantly upregulated on iNKT cells from WT recipient mice but not on those from CD1d-/- recipient mice (Fig. 4D). Thus, iNKT cell activation in EV71 infection requires both endogenous lipid antigen synthesis and CD1d presentation.
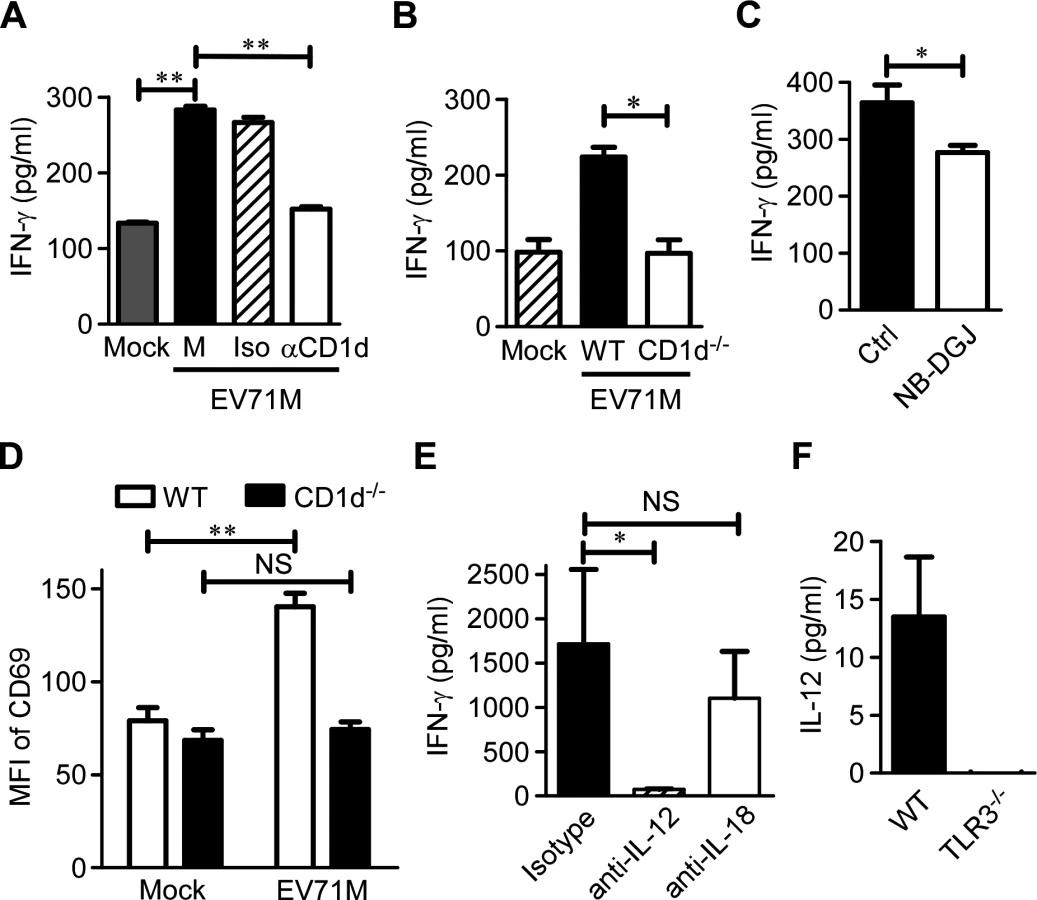
It has been shown that IL-12 and IL-18 are involved in activating iNKT cells after TLR-mediated stimulation of DCs [19–21,23,24,33]. To investigate the contribution of cytokines that are produced by macrophages to iNKT cell activation in response to EV71 infection, we performed experiments with blocking antibodies against IL-12 and IL-18. The presence of anti-IL-12 antibody dramatically inhibited the IFN-γ production of iNKT cells co-cultured with macrophages and EV71M compared to the isotype antibody. The IL-18-blocking antibodies did not significantly affect IFN-γ production (Fig. 4E). Therefore, IL-12 production is required for iNKT cell activation.
We next investigated the effect of TLR3 signaling on IL-12 production by macrophages in response to EV71 infection. IL-12 was measured in the culture supernatant of EV71M-infected WT or TLR3-knockout macrophages. Although significant quantities of IL-12p70 were detected in the culture supernatants of EV71M-infected WT macrophages, it was nearly undetectable in the culture supernatants of EV71M-infected TLR3-knockout macrophages (Fig. 4F). Thus, IL-12 production by EV71-infected macrophages depends on TLR3 signaling.
iNKT cells protect young mice from EV71 infection
We next sought to investigate the protective or pathogenic role of iNKT cells in EV71 infection. We infected 7-day-old, CD1d-deficient or WT mice with EV71M and monitored disease development in the infected mice. At the lower dose of EV71M infection (2×104 PFU), only 10% (1/10) of WT mice exhibited ruffled hair and hunchback, while 40% (6/15) CD1d-deficient mice had symptoms of limb weakness or paresis and higher average clinical scores (Fig. 5A). At the higher dose of EV71M infection (2×105 PFU), all 16 CD1d-deficient mice developed limb paresis after 7 days post-infection, and none survived beyond 14 days. In contrast, none of the WT mice died of EV71M infection, although 30% (3/10) of the mice developed mild symptoms, fluffy hair, hunchback and/or limb weakness (Fig. 5B). Similarly, all the Jα18-knockout mice that lack of type I iNKT cells developed limb paresis and died within 12 days after being infected with the higher dosage of EV71M (Fig. 5C).

We further examined the virus loads in the tissues of EV71M-infected mice. We observed that the virus loads were significantly higher in the spleens and spinal cords of CD1d-deficient mice compared with WT mice on days 2 and 4 post-infection (Fig. 5D, E). Noticeably, the virus loads in the brain of CD1d-deficient mice were not significantly different from that of WT mice on day 2 post-infection but were over 1000-times higher on day 4 (Fig. 5D, E). Altogether, these results indicate iNKT cells protect young mice from EV71 infection by preventing virus spread to the neuronal system.
Discussion
EV71 infection causes illness mainly in infants or young children. Similarly, disease development after EV71 infection only occurred in neonatal or young mice aged less than 2 weeks after adaptation [34,35]. Older human beings and mice appear to resist EV71 infection. Currently, the factors that contribute to age-dependent resistance remain elusive. Here, we found that the age-dependent susceptibility of mice to EV71 infection was reversely associated with iNKT cell development. EV71 infection triggered iNKT cell activation in a TLR3-dependent manner. Interestingly, TLR3-triggering in macrophages, rather than in BMDCs, activated iNKT cells during EV71 infection. Both IL-12 production and endogenous CD1d ligands were required for the activation of iNKT cells triggered by EV71-infected macrophages. iNKT cells appeared to prevent the spreading of EV71 into central nervous system. Furthermore, TLR3 and CD1d-deficient young mice developed more severe disease post EV71 infection. These novel findings not only help to explain the age-dependent susceptibility of host to EV71 infection but also provide initial knowledge about how infections by RNA viruses trigger activation of iNKT cells.
iNKT cells begin to develop in mice approximately 7 days after birth (Fig. 1D and S1 Fig.) [36]. We found that wild-type mice are susceptible to EV71M infection under this age. iNKT-deficient mice under 3 weeks of age are more susceptible to EV71M infection when compared with wild-type mice. It is known that the adaptive immune system begins to mature at this time [37]. Therefore, the availability of iNKT cells in early life is an important factor to control EV71 infection. In other words, iNKT cells play a critical role in protecting mice from the virus infection between the second and third weeks of their life, before the adaptive immune system becomes fully functional.
The mechanism underlying how infections of RNA viruses trigger iNKT cell activation has not been well-elucidated. Nevertheless, it has been reported that DCs stimulated by ligands of TLR7 and TLR8, pattern recognition receptors sensing RNA viruses, are capable of activating iNKT cells. Paradoxically, DCs stimulated by the dsRNA analog pI:C, TLR3 ligand, fail to activate iNKT cells [24]. Thus, one can expect that the ssRNA sensors TLR7 and TLR8 are required for iNKT cell activation, and that TLR3 signaling unlikely contributes to iNKT cell activation during RNA virus infection. In contrast to the assumption, our present study revealed that TLR3, rather than TLR7 and other TLRs, was crucial in iNKT cell activation during EV71 infection. Macrophages but not DCs were indispensable antigen-presenting cells for activation of iNKT cells.
The incapability of TLR3-mediated signaling in DCs to activate iNKT cells highlights that there is an intrinsic difference in TLR3 signaling pathways between macrophages and DCs. This merits further investigation. However, one caveat is that DCs in our and others’ experiments were in vitro-cultured BMDCs. Unlike in vitro BMDCs, the DCs in lymphoid organs and other tissues of humans and mice are heterogeneous in phenotype and functionality [38]. Thus, it is impossible to exclude some subsets of DCs that may activate iNKT cells upon RNA virus infection in vivo. Furthermore, fecal-oral infection is a natural route of transmission of EV71 infection. CD1d also expresses on the intestine epithelial cells and is involved in the induction of oral tolerance and protection from mucosal infections [39]. We also observed that 75% of CD1d-knockout mice upon oral infection of EV71M developed disease and had significantly higher clinical score than WT mice, as all the wild type mice survived without any palpable symptoms after the infection (S5 Fig.). Therefore, it is also possible that intestine epithelial cells also play a role in the mucosal immunity elicited by iNKT cells against EV71 infection. It is also of great interest to investigate the cell type-dependent activation of iNKT cells in EV71 infection as well as other viral infections in future.
Viruses mostly do not encode enzymes responsible for synthesizing glycolipids that bind to CD1d molecule. Our results showed that blocking CD1d-mediated antigen presentation with antibody and biosynthesis of endogenous lipid antigens in EV71-infected macrophages with an inhibitor significantly decreased the activation of iNKT cells. In addition, CD1d-deficient macrophages post EV71 infection had significantly reduced ability to activate iNKT cells compared to WT macrophages. Furthermore, adoptive transfer of purified iNKT cells also slightly but significantly prolonged the survival of EV71M-infected 3-day-old WT mice, but this protective effect did not occur in CD1d-deficient recipients (S6 Fig.). Similarly, adoptive transfer experiment also suggested that activation of iNKT cells in vivo in EV71M infection also requires CD1d molecule (Fig. 4D). These results all suggested that full iNKT cell activation during EV71 infection required CD1d molecule and its presentation of endogenous lipid antigens. This is in contrast to the findings showing that iNKT cell activation triggered by the infection of MCMV is not dependent on CD1d molecule [27,28]. MCMV encodes a CD1d-like protein, m157, that can directly bind to the Ly49H molecule on NK cells and activate the NK cells [40,41]. Ly49H is still expressed on NK1.1-positive cells in Nfil3-deficient mice that have no NK cells but still have iNKT cells [42], suggesting that iNKT cells may potentially express Ly49H. The potential Ly49H-m157 interaction may thus contribute to the CD1d independence of iNKT cell activation. Altogether, the discrepancy in the CD1d requirement in viral infections may support a notion that different viruses may trigger distinct pathways to activate iNKT cells.
Overall, we showed that iNKT cells are crucial antiviral effectors against EV71 infection before the adaptive immune system becomes fully functional in mice. Our result of human peripheral blood mononuclear cells (PBMCs) demonstrated that human iNKT cells can also be activated by EV71 infection (S7 Fig.). Children under the age of 5 years are more frequently susceptible to EV71 infection as well as other RNA virus infections [5,6], including infections by influenza A virus, respiratory syncytial virus, human metapneumovirus, coronaviruses and rhinovirus than adults [43–45]. Thus, our results encourage further investigations on the role of iNKT cells in childhood diseases caused by these viruses and exploring therapeutic strategies against these viral infections by manipulating iNKT cells.
Materials and Methods
Ethics statement on human subjects
Use of human PBMCs from healthy donors was approved by the Institutional Ethics Committee of the Institut Pasteur of Shanghai (Permit Number: IPS-2012001).
Ethics statement on animal subjects
All animal experiments were performed in strict accordance with the regulations in the Guide for the Care and Use of Laboratory Animals issued by the Ministry of Science and Technology of the People's Republic of China. All efforts were made to minimize suffering. The protocol was approved by the Institutional Animal Care and Use Committee of the Institut Pasteur of Shanghai (Permit Number: A2011006).
Mice
C57BL/6 and ICR mice were obtained from Shanghai Laboratory Animal Center (SLAC). TLR3-/- mice were kindly provided by Dr. Richard Flavell and have been backcrossed to C57BL/6 background for more than eight generations. RAG1-/- mice and MyD88-/- mice with the C57BL/6 background were provided by the Model Animal Research Center of Nanjing University. B6.129S1-Tlr7tm1Flv/J (TLR7-/-) mice in C57BL/6 background were purchased from the Jackson Laboratory. Jα18-/- mice in C57BL/6 background were originally obtained from M. Taniguchi (RIKEN Research Center, [46]). Vα14-Jα18 transgenic mice in C57BL/6 background were generated as previously described [47]. All mice were kept under specific pathogen-free (SPF) conditions in the SLAC. Infections were performed in containment isolators under SPF conditions. Animals were infected intraperitoneally (i.p.) with the indicated dose of EV71M in 50 μl RPMI-1640 medium. The clinical scores of the mice after EV71M infection were as follows: 0, healthy; 1, ruffled hair and hunchback; 2, limb weakness; 3, paralysis in 1 limb; 4, paralysis in both limbs; and 5, death.
Reagents and antibodies
pI:C, PMA, ionomycin and bredfeldin A were purchased from Sigma-Aldrich. Anti-CD4, anti-CD3, anti-TCRβ, anti-CD19, anti-CD25, anti-CD69, anti-DX5, anti-CD11b, anti-CD11c, anti-NK1.1, anti-IFN-γ and isotype-matched control antibodies were purchased from eBioscience. Blocking antibodies against CD1d, IL-12 and IL-18 were also purchased from eBioscience.
Cells and viruses
RD cells were cultured in Dulbecco's modified Eagle’s medium (HyClone) supplemented with 10% fetal calf serum (FCS). Mouse fibroblast L929 cells were grown in RPMI-1640 medium (HyClone) with 10% FCS. The clinical strain EV71H was originally isolated from a child who suffered EV71 encephalomyelitis in 2008 [48].
The mouse-adapted virus strain (EV71M) was generated following a previously described method [49]. In brief, 1-day-old ICR mice (n = 8) were intracerebrally inoculated with 1×104 PFU of the parental strain. Brains of the infected mice were collected and pooled 4 days post-infection, and homogenates of the brains were prepared in HBSS at a ratio of 10% weight/volume. The virus in the homogenate was cultured in L929 cells. The stock virus was harvested at the fifth passage and titrated in RD cells. The final virus stock was 4×108 PFU/ml. All experiments with EV71 virus were performed in a biosafety level-2 laboratory at the Institut Pasteur of Shanghai.
RD and L929 cell monolayers in 24-well plates (2–3×105 cells) were infected with the parental strain EV71H or EV71M at a multiplicity of infection (MOI) of 0.1 for 1 hour at 37°C. The cells were washed twice with PBS and then cultured in DMEM containing 2% FCS. Samples were harvested at various indicated time points. At each time point, the supernatant was stored in aliquots at -80°C and later titrated by a plaque assay using RD cell monolayers in 24-well plates.
BMDCs and peritoneal macrophages
BMDCs were generated as described previously [50]. Briefly, bone marrow was harvested from tibias and femurs of different mice. After lysis of red blood cells, bone marrow cells were cultured for 6 days in RPMI-1640 medium supplemented with 10% FCS and supernatant from GM-CSF-expressing J5 cells, which were kindly provided originally by Dr. Herman Eisen. CD11c+ cells were further purified by positive selection using MACS separation columns (Miltenyi Biotec) according to the manufacturer’s instructions. The purified cells were above 96% CD11c positive.
The murine peritoneal macrophages were harvested from 6–8-week-old C57BL/6 mice after the i.p. injection of 1 ml of 4% thioglycollate medium (Sigma-Aldrich) as described previously [51]. Before EV71 infection, the macrophages were cultured overnight in serum-free RPMI-1640 with antibiotics to minimize the influence of FBS.
Purification of iNKT cells
For iNKT cell adoptive transfer experiments, a single-cell suspension was prepared from Vα14-Jα18 transgenic mouse spleens. iNKT cells were prepared using the modified protocol of Pei [52]. Briefly, freshly isolated spleens were mashed and passed through a 40-μm nylon strainer to give a single-cell suspension. Cells from three spleens were pooled, and RBCs were removed by lysis. The splenocytes were stained with CD1d-tetramer or antibodies against NK1.1 and TCRβ and then sorted using a FACS Aria II cell sorter (BD Biosciences) (≥97% pure).
Infection of DCs and macrophages and co-culture with iNKT cells
Macrophages or DCs (2×105 cells/well in a U-bottomed, 96-well plate) were incubated with EV71 (MOI = 10) or TLR agonists for 24 hours, extensively washed with PBS, and cultured for 24 hours with spleen iNKT cells (1×105 /well) in U-bottomed, 96-well plates containing RPMI 1640 medium supplemented with 10% FBS. In some cases, neutralizing or control antibodies were added during the co-culture. The IFN-γ concentrations in the co-culture supernatants were measured by ELISA (eBioscience). To inhibit GSL synthesis, macrophages were treated with 100 μg/ml NB-DGJ for 24 hours prior to the iNKT cell assays. NB-DGJ was also present during the stimulation experiments at 100 μg/ml.
Isolation and cultivation of human PBMCs
Human PBMCs were isolated from whole blood via Ficoll-Hypaque density gradient centrifugation and subsequently washed with RPMI-1640 before further use. For examining iNKT cell activation, EV71-infected or mock-treated PBMCs were resuspended in RPMI-1640 supplemented with 10% FCS and seeded at 5×106 cells/ml in 24-well plates for 16 hours.
Flow cytometry
Cells were washed and blocked in staining buffer (PBS, 0.3% BSA and 0.1% sodium azide) containing anti-CD16/CD32 for 10 minutes at 4°C then stained with fluorophore-conjugated antibodies. After washing twice with staining buffer, data were collected on an LSRII flow cytometer (BD Biosciences). For intracellular staining, 2.5 μg/ml BFA was added during the last 4 hours of stimulation to block the secretion of cytokines. The cells were washed and stained for cell-surface markers. After fixation and permeabilization with Cytofix/Cytoperm Kit (BD pharmingen) according to the manufacturer’s protocol, the cells were stained with FITC-anti-mouse IFN-γ or isotype control and analyzed with an LSR II flow cytometer. The data were analyzed using FlowJo software (Tree Star).
RNA extraction, cDNA synthesis and real-time PCR
Total RNA was isolated from EV71-infected or un-infected cells or tissues with TRIzol reagent (Invitrogen), and cDNAs were synthesized from 1 μg of total RNA with random hexamer primers and Superscript reverse transcriptase (TAKARA Biotechnology Co., Ltd) according to standard procedures. cDNAs were used as templates for PCR amplification using the SYBR Green PCR Master Mix (Takara Biotechnology Co., Ltd) and the ABI 7900HT Fast Real-Time PCR System (Applied Biosystems, Foster City, CA). Primers for EV71 and GAPDH were previously described by Xu et al. and Paget et al., respectively [53] [24].
Cytokine analysis
Samples were harvested and stored at -80°C before analysis. The IFN-γ and IL-12 levels were quantified using ELISA Ready-SET-Go (eBioscience).
Statistical analyses
Statistical analyses for continuous data were performed with Prism5 for Windows software (Prism Graph-Pad Software Inc) using two-tailed Student’s t-tests. P < 0.05 was considered significant. Statistical differences in mouse survival were determined by Gehan-Breslow-Wilcoxon Test analysis. Graphs were produced and statistical analyses were performed using GraphPad Prism.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Solomon T, Lewthwaite P, Perera D, Cardosa MJ, McMinn P, et al. (2010) Virology, epidemiology, pathogenesis, and control of enterovirus 71. Lancet Infect Dis 10 : 778–790. doi: 10.1016/S1473-3099(10)70194-8 20961813
2. Deng C, Yang C, Wan J, Zhu L, Leng Q (2011) Irregular poliovirus vaccination correlates to pulmonary edema of hand, foot, and mouth disease. Clin Vaccine Immunol 18 : 1589–1590. doi: 10.1128/CVI.05132-11 21752953
3. Tan X, Huang X, Zhu S, Chen H, Yu Q, et al. (2011) The persistent circulation of enterovirus 71 in People's Republic of China: causing emerging nationwide epidemics since 2008. PLoS One 6: e25662. doi: 10.1371/journal.pone.0025662 21980521
4. Huang CC, Liu CC, Chang YC, Chen CY, Wang ST, et al. (1999) Neurologic complications in children with enterovirus 71 infection. N Engl J Med 341 : 936–942. 10498488
5. Ho M, Chen ER, Hsu KH, Twu SJ, Chen KT, et al. (1999) An epidemic of enterovirus 71 infection in Taiwan. Taiwan Enterovirus Epidemic Working Group. N Engl J Med 341 : 929–935. 10498487
6. Chan LG, Parashar UD, Lye MS, Ong FG, Zaki SR, et al. (2000) Deaths of children during an outbreak of hand, foot, and mouth disease in sarawak, malaysia: clinical and pathological characteristics of the disease. For the Outbreak Study Group. Clin Infect Dis 31 : 678–683. 11017815
7. Seiff A (2012) Cambodia unravels cause of mystery illness. Lancet 380 : 206. 22826835
8. Khong WX, Yan B, Yeo H, Tan EL, Lee JJ, et al. (2011) A non mouse-adapted Enterovirus 71 (EV71) strain exhibits neurotropism causing neurological manifestations in a novel mouse model of EV71 infection. J Virol.
9. Lei X, Liu X, Ma Y, Sun Z, Yang Y, et al. (2010) The 3C protein of enterovirus 71 inhibits retinoid acid-inducible gene I-mediated interferon regulatory factor 3 activation and type I interferon responses. J Virol 84 : 8051–8061. doi: 10.1128/JVI.02491-09 20519382
10. Lei X, Sun Z, Liu X, Jin Q, He B, et al. (2011) Cleavage of the adaptor protein TRIF by enterovirus 71 3C inhibits antiviral responses mediated by Toll-like receptor 3. J Virol 85 : 8811–8818. doi: 10.1128/JVI.00447-11 21697485
11. Wang B, Xi X, Lei X, Zhang X, Cui S, et al. (2013) Enterovirus 71 protease 2Apro targets MAVS to inhibit anti-viral type I interferon responses. PLoS Pathog 9: e1003231. doi: 10.1371/journal.ppat.1003231 23555247
12. Liao CC, Liou AT, Chang YS, Wu SY, Chang CS, et al. (2014) Immunodeficient Mouse Models with Different Disease Profiles by in vivo Infection with the Same Clinical Isolate of Enterovirus 71. J Virol.
13. Wang SM, Lei HY, Huang KJ, Wu JM, Wang JR, et al. (2003) Pathogenesis of enterovirus 71 brainstem encephalitis in pediatric patients: roles of cytokines and cellular immune activation in patients with pulmonary edema. J Infect Dis 188 : 564–570. 12898444
14. Tomura M, Yu WG, Ahn HJ, Yamashita M, Yang YF, et al. (1999) A novel function of Valpha14+CD4+NKT cells: stimulation of IL-12 production by antigen-presenting cells in the innate immune system. J Immunol 163 : 93–101. 10384104
15. Stetson DB, Mohrs M, Reinhardt RL, Baron JL, Wang ZE, et al. (2003) Constitutive cytokine mRNAs mark natural killer (NK) and NK T cells poised for rapid effector function. J Exp Med 198 : 1069–1076. 14530376
16. Kronenberg M, Gapin L (2002) The unconventional lifestyle of NKT cells. Nat Rev Immunol 2 : 557–568. 12154375
17. Diana J, Lehuen A (2009) NKT cells: friend or foe during viral infections? Eur J Immunol 39 : 3283–3291. doi: 10.1002/eji.200939800 19830742
18. Juno JA, Keynan Y, Fowke KR (2012) Invariant NKT cells: regulation and function during viral infection. PLoS Pathog 8: e1002838. doi: 10.1371/journal.ppat.1002838 22916008
19. Brigl M, Tatituri RV, Watts GF, Bhowruth V, Leadbetter EA, et al. (2011) Innate and cytokine-driven signals, rather than microbial antigens, dominate in natural killer T cell activation during microbial infection. J Exp Med.
20. Brigl M, Bry L, Kent SC, Gumperz JE, Brenner MB (2003) Mechanism of CD1d-restricted natural killer T cell activation during microbial infection. Nat Immunol 4 : 1230–1237. 14578883
21. Mattner J, Debord KL, Ismail N, Goff RD, Cantu C 3rd, et al. (2005) Exogenous and endogenous glycolipid antigens activate NKT cells during microbial infections. Nature 434 : 525–529. 15791258
22. Skold M, Behar SM (2005) The role of group 1 and group 2 CD1-restricted T cells in microbial immunity. Microbes Infect 7 : 544–551. 15777730
23. Salio M, Speak AO, Shepherd D, Polzella P, Illarionov PA, et al. (2007) Modulation of human natural killer T cell ligands on TLR-mediated antigen-presenting cell activation. Proc Natl Acad Sci U S A 104 : 20490–20495. 18077358
24. Paget C, Mallevaey T, Speak AO, Torres D, Fontaine J, et al. (2007) Activation of invariant NKT cells by toll-like receptor 9-stimulated dendritic cells requires type I interferon and charged glycosphingolipids. Immunity 27 : 597–609. 17950005
25. Brennan PJ, Tatituri RV, Brigl M, Kim EY, Tuli A, et al. (2011) Invariant natural killer T cells recognize lipid self antigen induced by microbial danger signals. Nat Immunol 12 : 1202–1211. doi: 10.1038/ni.2143 22037601
26. Paget C, Bialecki E, Fontaine J, Vendeville C, Mallevaey T, et al. (2009) Role of invariant NK T lymphocytes in immune responses to CpG oligodeoxynucleotides. J Immunol 182 : 1846–1853. doi: 10.4049/jimmunol.0802492 19201836
27. Tyznik AJ, Tupin E, Nagarajan NA, Her MJ, Benedict CA, et al. (2008) Cutting edge: the mechanism of invariant NKT cell responses to viral danger signals. J Immunol 181 : 4452–4456. 18802047
28. Wesley JD, Tessmer MS, Chaukos D, Brossay L (2008) NK cell-like behavior of Valpha14i NK T cells during MCMV infection. PLoS Pathog 4: e1000106. doi: 10.1371/journal.ppat.1000106 18636102
29. Berzins SP, Cochrane AD, Pellicci DG, Smyth MJ, Godfrey DI (2005) Limited correlation between human thymus and blood NKT cell content revealed by an ontogeny study of paired tissue samples. Eur J Immunol 35 : 1399–1407. 15816002
30. Barral P, Polzella P, Bruckbauer A, van Rooijen N, Besra GS, et al. (2010) CD169(+) macrophages present lipid antigens to mediate early activation of iNKT cells in lymph nodes. Nat Immunol 11 : 303–312. doi: 10.1038/ni.1853 20228797
31. Holzapfel KL, Tyznik AJ, Kronenberg M, Hogquist KA (2014) Antigen-dependent versus-independent activation of invariant NKT cells during infection. J Immunol 192 : 5490–5498. doi: 10.4049/jimmunol.1400722 24813205
32. Andersson U, Butters TD, Dwek RA, Platt FM (2000) N-butyldeoxygalactonojirimycin: a more selective inhibitor of glycosphingolipid biosynthesis than N-butyldeoxynojirimycin, in vitro and in vivo. Biochem Pharmacol 59 : 821–829. 10718340
33. Nagarajan NA, Kronenberg M (2007) Invariant NKT cells amplify the innate immune response to lipopolysaccharide. J Immunol 178 : 2706–2713. 17312112
34. Wang YF, Chou CT, Lei HY, Liu CC, Wang SM, et al. (2004) A mouse-adapted enterovirus 71 strain causes neurological disease in mice after oral infection. J Virol 78 : 7916–7924. 15254164
35. Miyamura K, Nishimura Y, Abo M, Wakita T, Shimizu H (2011) Adaptive mutations in the genomes of enterovirus 71 strains following infection of mouse cells expressing human P-selectin glycoprotein ligand-1. J Gen Virol 92 : 287–291. doi: 10.1099/vir.0.022418-0 20943886
36. Hammond K, Cain W, van Driel I, Godfrey D (1998) Three day neonatal thymectomy selectively depletes NK1.1+ T cells. Int Immunol 10 : 1491–1499. 9796916
37. Adkins B, Leclerc C, Marshall-Clarke S (2004) Neonatal adaptive immunity comes of age. Nat Rev Immunol 4 : 553–564. 15229474
38. Shortman K, Liu YJ (2002) Mouse and human dendritic cell subtypes. Nat Rev Immunol 2 : 151–161. 11913066
39. Zeissig S, Kaser A, Dougan SK, Nieuwenhuis EE, Blumberg RS (2007) Role of NKT cells in the digestive system. III. Role of NKT cells in intestinal immunity. Am J Physiol Gastrointest Liver Physiol 293: G1101–1105. 17717040
40. Smith HR, Heusel JW, Mehta IK, Kim S, Dorner BG, et al. (2002) Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc Natl Acad Sci U S A 99 : 8826–8831. 12060703
41. Arase H, Mocarski ES, Campbell AE, Hill AB, Lanier LL (2002) Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science 296 : 1323–1326. 11950999
42. Firth MA, Madera S, Beaulieu AM, Gasteiger G, Castillo EF, et al. (2013) Nfil3-independent lineage maintenance and antiviral response of natural killer cells. J Exp Med 210 : 2981–2990. doi: 10.1084/jem.20130417 24277151
43. Fairchok MP, Martin ET, Chambers S, Kuypers J, Behrens M, et al. (2010) Epidemiology of viral respiratory tract infections in a prospective cohort of infants and toddlers attending daycare. J Clin Virol 49 : 16–20. doi: 10.1016/j.jcv.2010.06.013 20650679
44. Venter M, Lassauniere R, Kresfelder TL, Westerberg Y, Visser A (2011) Contribution of common and recently described respiratory viruses to annual hospitalizations in children in South Africa. J Med Virol 83 : 1458–1468. doi: 10.1002/jmv.22120 21678450
45. Nair H, Brooks WA, Katz M, Roca A, Berkley JA, et al. (2011) Global burden of respiratory infections due to seasonal influenza in young children: a systematic review and meta-analysis. Lancet 378 : 1917–1930. doi: 10.1016/S0140-6736(11)61051-9 22078723
46. Cui J, Shin T, Kawano T, Sato H, Kondo E, et al. (1997) Requirement for Valpha14 NKT cells in IL-12-mediated rejection of tumors. Science 278 : 1623–1626. 9374462
47. Griewank K, Borowski C, Rietdijk S, Wang N, Julien A, et al. (2007) Homotypic interactions mediated by Slamf1 and Slamf6 receptors control NKT cell lineage development. Immunity 27 : 751–762. 18031695
48. Yang C, Deng C, Wan J, Zhu L, Leng Q (2011) Neutralizing antibody response in the patients with hand, foot and mouth disease to enterovirus 71 and its clinical implications. Virol J 8 : 306. doi: 10.1186/1743-422X-8-306 21679417
49. Ong KC, Badmanathan M, Devi S, Leong KL, Cardosa MJ, et al. (2008) Pathologic characterization of a murine model of human enterovirus 71 encephalomyelitis. J Neuropathol Exp Neurol 67 : 532–542. doi: 10.1097/NEN.0b013e31817713e7 18520772
50. Inaba K, Inaba M, Romani N, Aya H, Deguchi M, et al. (1992) Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med 176 : 1693–1702. 1460426
51. Zhu QY, Liu Q, Chen JX, Lan K, Ge BX (2010) MicroRNA-101 targets MAPK phosphatase-1 to regulate the activation of MAPKs in macrophages. J Immunol 185 : 7435–7442. doi: 10.4049/jimmunol.1000798 21068409
52. Pei B, Speak AO, Shepherd D, Butters T, Cerundolo V, et al. (2011) Diverse endogenous antigens for mouse NKT cells: self-antigens that are not glycosphingolipids. J Immunol 186 : 1348–1360. doi: 10.4049/jimmunol.1001008 21191069
53. Xu W, Liu CF, Yan L, Li JJ, Wang LJ, et al. (2012) Distribution of enteroviruses in hospitalized children with hand, foot and mouth disease and relationship between pathogens and nervous system complications. Virol J 9 : 8. doi: 10.1186/1743-422X-9-8 22230340
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 1
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Infections in Humans and Animals: Pathophysiology, Detection, and Treatment
- : Trypanosomatids Adapted to Plant Environments
- Environmental Drivers of the Spatiotemporal Dynamics of Respiratory Syncytial Virus in the United States
- Dengue Virus RNA Structure Specialization Facilitates Host Adaptation
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy