
Promiscuous RNA Binding Ensures Effective Encapsidation of APOBEC3 Proteins by HIV-1
APOBEC3 proteins are cell-encoded restriction factors that counteract infections, particularly by retroviruses such as HIV-1, and retrotransposons. When packaged into HIV-1 particles, APOBEC3G and APOBEC3F both inhibit reverse transcription and induce destructive hypermutation in viral DNA. The mechanism of APOBEC3 virion packaging awaits elucidation, though a dependency on RNA binding has been established. Here, we employed a cross-linking and next generation sequencing approach to determine which RNAs are bound to A3G and A3F in HIV-1 infected cells. We show that both proteins bind to multiple different RNAs, including viral RNA as well as cellular coding and non-coding RNAs, with relatively little evidence of selectivity. We then developed a complementation assay to address the diversity of RNAs that can act as substrates for A3G/F virion packaging. Consistent with the RNA binding profiles, many RNAs can promote packaging provided that those RNAs are, themselves, packaged. These observations suggest that APOBEC3 packaging lacks selectivity and is driven simply by the non-specific RNA binding capabilities of these proteins. We speculate that this model accounts for the broad range of retro-elements that are susceptible to repression by individual APOBEC3 proteins, and also that such substrates cannot escape APOBEC3-mediated inhibition through sequence variation.
Published in the journal:
. PLoS Pathog 11(1): e32767. doi:10.1371/journal.ppat.1004609
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004609
Summary
APOBEC3 proteins are cell-encoded restriction factors that counteract infections, particularly by retroviruses such as HIV-1, and retrotransposons. When packaged into HIV-1 particles, APOBEC3G and APOBEC3F both inhibit reverse transcription and induce destructive hypermutation in viral DNA. The mechanism of APOBEC3 virion packaging awaits elucidation, though a dependency on RNA binding has been established. Here, we employed a cross-linking and next generation sequencing approach to determine which RNAs are bound to A3G and A3F in HIV-1 infected cells. We show that both proteins bind to multiple different RNAs, including viral RNA as well as cellular coding and non-coding RNAs, with relatively little evidence of selectivity. We then developed a complementation assay to address the diversity of RNAs that can act as substrates for A3G/F virion packaging. Consistent with the RNA binding profiles, many RNAs can promote packaging provided that those RNAs are, themselves, packaged. These observations suggest that APOBEC3 packaging lacks selectivity and is driven simply by the non-specific RNA binding capabilities of these proteins. We speculate that this model accounts for the broad range of retro-elements that are susceptible to repression by individual APOBEC3 proteins, and also that such substrates cannot escape APOBEC3-mediated inhibition through sequence variation.
Introduction
The apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like 3 (APOBEC3, or A3) proteins have been identified as potent antiviral effector proteins [1,2]. There are seven family members in humans, each of which contains one (A3A, A3C and A3H) or two (A3B, A3D, A3F and A3G) characteristic zinc-coordination domains, one of which is catalytically active [3]. These proteins have been identified as inhibitors of retroviruses such as human immunodeficiency virus type-1 (HIV-1) [4], simian immunodeficiency viruses, murine leukaemia virus [5–7] and mouse mammary tumour virus [8], as well as viruses from other families such as hepatitis B virus [9], adeno-associated virus [10] and also endogenous retroelements [11]. Viruses have developed assorted strategies to evade A3-mediated inhibition, the most prominent of which is the expression of the dedicated regulatory protein, Vif, by most lentiviruses. Specifically, HIV-1 Vif counteracts APOBEC3 proteins by inducing their proteasomal degradation through the direct recruitment of CBF-β and a cellular E3 ubiquitin ligase comprising CUL5, ELOB/C, and RBX2 [12–15]. When Vif is absent or defective, APOBEC3 proteins are packaged into progeny virions and transferred to target cells during new infections, where they inhibit reverse transcription and hypermutate nascent cDNAs through excessive cytidine-to-uridine editing [5,7,16–19]. Thus, the encapsidation of APOBEC3 proteins into viral particles is essential for their antiviral activity, and a complete description of APOBEC3 protein function will require a full understanding of the packaging mechanism.
APOBEC3 proteins are RNA binding proteins [20–22]. A3G associates in an RNA-dependent mechanism with multiple ribonucleoprotein (RNP) complexes and accumulates in cytoplasmic RNA-rich microdomains such as P-bodies, stress granules and Staufen-containing granules [23–26]. Localisation to these regions does not appear to be important for antiviral function [27,28], and it has been suggested that sequestration in RNPs may be important for downregulation of APOBEC3 protein activity within cells. These findings further raise the question of how APOBEC3 proteins are packaged into assembling virions. One elegant study has demonstrated that it is newly synthesised protein that is encapsidated, presumably by averting entrapment into cytoplasmic RNPs [29].
The packaging of A3G into HIV-1 particles requires the nucleocapsid (NC) region of the viral Gag protein [30–33]. It has been established that the A3G interaction with NC is RNA-dependent, leading to the consensus view that its packaging is reliant upon its capacity to bind RNA [30–33]. However, although several groups have sought to define specific RNAs that are responsible for A3G packaging, a clear consensus has not yet emerged. In particular, Kahn et al. suggested that viral genomic RNA (gRNA) is required for A3G packaging [34], Wang et al. have concluded that 7SL RNA is the responsible RNA [35], and Svarovskaia et al. proposed that both viral and cellular RNAs play a role [32].
A3G interacts with diverse RNAs such as the 7SL RNA (the RNA component of the signal recognition particle, SRP), Alu RNAs, human Y RNAs and several mRNAs [24,36], many of which are also packaged into retroviruses (reviewed in [37]). In our study, we used an unbiased strategy to identify the RNAs that interact with A3G or A3F in HIV-1 infected cells and in budded virions, and then undertook specific experiments to investigate the involvement of such RNAs in A3G and A3F packaging. To define the interacting RNAs, we employed a cross-linking and immunoprecipitation technique (iCLIP) followed by next generation sequencing. This method has successfully defined RNAs that interact with proteins such as neuro-oncological ventral antigen (NOVA), hnRNP C and Argonautes [38–40].
Here, we demonstrate that A3G and A3F interact with a broad range of RNA molecules, including HIV-1 gRNA, cellular mRNAs and a number of small non-coding RNAs. A series of cell-based assays revealed that no single/unique RNA mediates the encapsidation of A3G or A3F, suggesting that multiple, diverse RNAs can recruit APOBEC3 proteins into viral particles, provided that they are themselves packaged. We therefore propose that A3G and A3F exploit their relatively non-specific RNA binding capabilities to patrol the cytoplasm for nascent retroviral RNA, thereby ensuring effective capture by assembling viruses and resultant antiviral function.
Results
iCLIP reveals that A3G and A3F bind to diverse RNA targets
Packaging of A3G into HIV-1 particles requires the nucleocapsid (NC) region of p55Gag, and it has been established that this interaction is RNA-dependent [30–33]. This led to the consensus view that A3G packaging depends on RNA binding. In order to identify the specific RNAs to which A3G binds for efficient packaging, we first applied an unbiased, deep sequencing method to catalogue the RNAs that are bound to A3G and A3F in living cells productively infected with HIV-1. We also extended this study to determine A3G and A3F target RNAs in cell-free HIV-1 virions.
To generate libraries of RNAs bound to A3G or A3F, we first generated CEM-SS human T-cell lines that stably expressed GFP (negative control), GFP-A3G, GFP-A3F, T7-GFP (negative control), T7-A3G or T7-A3F (S1 Fig.). By using two different immunoprecipitation tags for A3G and A3F, we could identify if either the GFP or T7-epitope tag biased the resulting library. Importantly, our study is distinguished from all others (to the best of our knowledge) by inclusion of GFP-only controls: this is an important addition as it allows the determination of RNA binding enrichment relative to background. These cultures were challenged with vif-deficient HIV-1 such that more than 90% of the cells were infected, as judged by intracellular p24Gag staining (S2 Fig.). 48 h after infection, the supernatants were collected and used to assess A3G and A3F antiviral efficacy. Regardless of the tag, both A3G and A3F were antiviral and inhibited single-cycle virus infectivity by 100 - and 30-fold, respectively (S3 Fig.).
Virus-producing cells and viruses produced from these cells were used to generate iCLIP libraries. Fig. 1A shows RNA cross-linked to the proteins of interest after T7 or GFP directed immunoprecipitation, RNAse digestion (used to shorten the length of bound RNAs for later deep sequencing) and ligation of a linker. Of note, A3G and A3F each cross-link vastly more RNA than GFP, consistent with the fact that these proteins are established RNA binding proteins. The RNAs that migrate at higher molecular weights than the protein of interest were extracted. A primer that anneals to the linker was then used to generate cDNA. The cDNAs were circularised, a further primer annealed over the linker to create a region of double stranded DNA and digested with BamHI. This procedure generated DNAs where the unknown sequence was flanked by linkers, allowing PCR amplification of the library (Fig. 1B) and next generation sequencing.

The sequencing provided a total of 20 million reads of 50 nucleotides each. The libraries obtained for each protein contained between 0.1 and 5 million sequences. We aligned the reads to the human genome and to the HIV-1 genome using bowtie [41]. Only reads that aligned uniquely in the genomes with a maximum of two mismatched nucleotides were considered for analysis. Furthermore, the reads containing the same random barcode and truncated at the same nucleotide were considered PCR artefacts, and only one of such reads was considered. These unique reads represented between 80% and 95% of the total reads for each replicate experiment, demonstrating the high level of sequencing library complexity obtained in the experiments. Human mRNAs were split into regions, specifically the 5’ - and 3’-untranslated regions (UTRs), introns and open reading frames (ORFs), and each region type was analysed independently. The number of reads aligning to a specific sequence or class of sequences was divided by the total number of aligned reads obtained from the library and then compared with the GFP control to provide measurements of enrichment. We performed two independent experiments for each cell line. By comparing these replicates, we observed that the data sets were highly correlated (r>0.98). The data obtained from the differently tagged (GFP versus T7) proteins also exhibited high correlation (r>0.95). Therefore, we present here the averaged data obtained for the four libraries with standard deviations. As summarised in Fig. 1C, A3G and A3F binding to mRNAs was enriched compared to the GFP control. While the binding of both A3G and A3F to the 3’-UTRs was higher (2-fold), only A3G was enriched in the ORF or 5’-UTR regions (2 - and 3-fold, respectively); Fig. 1C and S1 Table).
We next looked in detail at virion-associated RNA: both the gRNA and host RNAs such as tRNAs, U RNAs, 7SL, ribosomal RNAs and Y RNAs [37]. Because these cellular RNAs are transcribed from repeat elements in the human genome, the reads were aligned using the repeat masker software [42] to a library containing consensus sequences of the elements, allowing misalignment of up to 3 nucleotides per read. As before, hits in each repeat consensus were divided by the total number of reads and compared to the GFP negative control. Fig. 1D and S2 Table show that A3G and A3F bound 2-fold more to the viral gRNA compared with GFP. Also, both A3G and A3F bound to Y RNAs (17 - and 7-fold, respectively) and to U RNAs (2-fold), as compared to GFP. Interestingly, A3G bound to 7SL (14-fold) while A3F did not.
As depicted in Fig. 1E, we observed that in infected cells, 20% of the RNA that is bound to A3G or A3F is of viral origin. However, in viral particles this RNA constitutes the majority of the library (80% of reads aligned to HIV-1 gRNA). This implies that, in vif-deficient HIV-1, A3G/F may be packaged mainly through interactions with HIV-1 gRNA, or that APOBEC3 proteins bind to encapsidated cellular RNAs and then transfer to gRNA once inside the viral particles.
In light of the diversity of RNA substrates bound by A3G or A3F, we undertook a series of studies designed to determine which RNAs can mediate the encapsidation of A3G or A3F into budding HIV-1 particles.
HIV-1 genomic RNA is dispensable for A3G and A3F packaging
One obvious RNA species that could potentially mediate the packaging of A3G into particles is the viral gRNA. Indeed, at least one previous report has considered this RNA to be essential for A3G packaging [34]. Since A3G and A3F are clearly able to bind to this RNA in infected cells, we performed packaging assays to test this hypothesis. First, we used lentiviral vectors with or without gRNA (Fig. 2A). Succinctly, 293T cells stably expressing HA-tagged A3G or A3F were co-transfected with the HIV-1-based packaging plasmid (p8.91) [43], the VSV glycoprotein envelope expression vector, and either the pHR’SIN-cPPT-SEW lentiviral vector plasmid (denoted lt vector) [44] that expresses gRNA with an intact packaging signal (Ψ) or a mock plasmid. Immunoblot analysis of particles harvested 48 h post transfection and isolated through a sucrose cushion shows that A3G and A3F were packaged into both viral vectors with almost identical efficiency, indicating that viral gRNA is not required for effective A3G or A3F packaging.

These data were next confirmed using an alternative experimental system where the gRNA also serves as the mRNA for Gag (Fig. 2B). 293T cells stably expressing HA-tagged A3G or A3F were transfected with expression vectors encoding: (i) Gag-Pol, (ii) Gag, (iii) Gag in which NC had been replaced by a leucine zipper domain (Zwt) to allow for Gag multimerisation and VLP production (Gag ΔNC) [45], or (iv) a Gag-Pol vector in which the previously defined gRNA packaging determinant between SL2 and SL3 [46] had been deleted (ΔΨ); (S4 Fig.). The protease region of Pol was rendered inactive in the Gag-Pol constructs to facilitate the detection and comparison of Gag across the different samples. Analysis of viral-like particles (VLPs) confirmed that NC is necessary for efficient A3G and A3F packaging, but showed that the Ψ element is dispensable (Fig. 2B, lanes 3 and 7) [30–33]. Since Gag alone was able to package A3G or A3F efficiently, we conclude that Pol is also not required for the packaging of A3G and A3F (Fig. 2B, compare lanes 4 with 1 and 8 with 5).
We also determined the amount of gRNA packaged into wild type Gag, Gag ΔNC and Gag-Pol ΔΨ VLPs by quantitative real time PCR (qRT-PCR) analysis (Fig. 2C). As anticipated, the Gag ΔNC, and ΔΨ VLPs each contained <5% of the level of gRNA compared to wild type Gag VLPs.
7SL is not necessary for A3G or A3F packaging
We next examined a second RNA, the non-coding 7SL RNA of the SRP, for its importance in A3G/F packaging. This RNA is incorporated into retroviral particles [47–49]. In particular, our iCLIP analyses (Fig. 1D) confirmed earlier work showing that A3G, unlike A3F, binds to 7SL RNA [50]. Of note, one group has previously reported that 7SL is the RNA required for A3G packaging [35], whereas a second concluded the opposite [51].
Over-expression of SRP19, a protein component of the SRP, reduces the amount of 7SL RNA packaging into HIV-1 particles [35], presumably by binding to free cellular 7SL RNA and precluding its interaction with Gag and resultant incorporation into assembling virions. Accordingly, we transfected 293T cells stably expressing A3G or A3F with a plasmid encoding the vif-deficient NL4-3 provirus together with an expression vector for SRP19 or a control vector. Immunoblot analysis 48 h post transfection showed that both A3G and A3F were still packaged into virions, irrespective of the reduction in virion-associated 7SL RNA (Fig. 3A). Interestingly, reducing the amount of packaged 7SL did not influence the infectivity of the viruses or the antiviral activity of the packaged A3G or A3F (S5 Fig.). The data were then confirmed using Gag VLPs, with Gag ΔNC serving as a negative control (Fig. 3B). Quantitative RT-PCR analysis of RNA extracted from these VLPs confirmed that SRP19 overexpression reduced virion-associated 7SL RNA levels by more than 90% (Fig. 3C). Our data therefore rule out a unique requirement for 7SL RNA for efficient A3G or A3F incorporation into virions, suggesting that their packaging may be mediated by other RNAs.
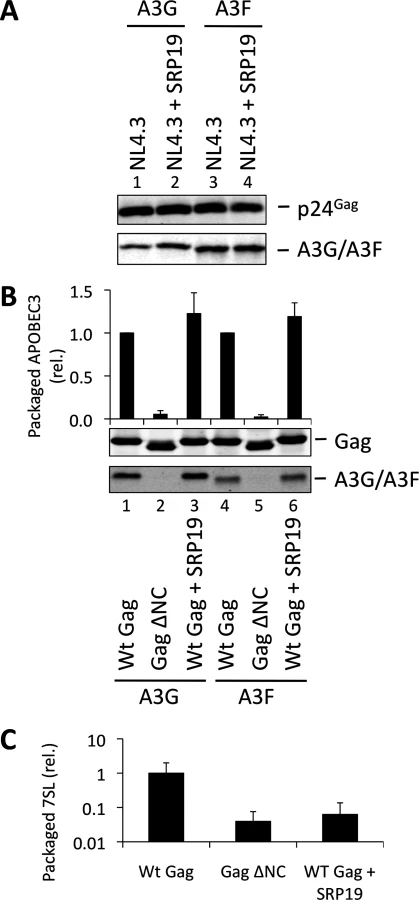
To investigate a potential redundancy between HIV-1 gRNA and host 7SL RNA, we next transfected 293T cells expressing A3G or A3F with a vector encoding SRP19 to inhibit 7SL packaging, as well as the aforementioned ΔΨ construct to generate VLPs depleted of gRNA. We observed that A3G and A3F were both packaged with normal efficiencies irrespective of SRP19 over-expression and/or the prevention of gRNA packaging (Fig. 4). Our observations imply that APOBEC3 proteins are packaged into these particles through the action of other RNAs.

Gag fusion proteins demonstrate that 7SL RNA and Y RNAs can promote APOBEC3 protein packaging
Our data suggested that a variety of host cell RNAs might be involved in APOBEC3 protein packaging into virions. To investigate this further, we next devised an alternative experimental approach whereby the packaging of RNA was dictated by heterologous RNA binding proteins (or domains thereof) rather than by the product of binding to NC and intracellular abundance. To do so, RNA binding domains were genetically fused to the carboxy-terminus of Gag ΔNC, and these proteins were used to generate VLPs in the presence or absence of A3G or A3F. In case of poor virion production, Gag ΔNC was co-expressed to ensure efficient VLP production [52,53].
To verify the experimental system, we initially expressed SRP19 fused to Gag ΔNC, anticipating that SRP19 would bind 7SL RNA and recruit it into VLPs together with A3G or A3F. Fig. 5A demonstrates that the addition of 6 amino acids to the C-terminus of Gag ΔNC, creating convenient restriction sites to allow fusions to Gag ΔNC (lanes 3 and 7), does not mediate APOBEC3 packaging. Also, A3G/F packaging was not mediated by the fusion of Gag to GFP, a protein that does not specifically bind to RNA (lanes 4 and 8). Although SRP19 was cleaved from Gag (lanes 5 and 10), the produced particles still contained intact Gag ΔNC-SRP19. Quantification of the amount of 7SL RNA in these VLPs demonstrated that the presence of SRP19 enables the efficient packaging of 7SL RNA (Fig. 5B). Importantly, these Gag ΔNC-SRP19 particles were able to package A3G efficiently, but not A3F. This observation is in agreement with the iCLIP data, which show that A3F does not preferentially bind to 7SL. Thus, while 7SL RNA is not required for the packaging of A3G into HIV-1 particles with an intact NC domain (Fig. 3), it is evidently able to promote packaging of A3G when selectively captured by VLPs.

Our iCLIP data suggested that both A3G as well as A3F can bind to Y RNAs, small non-coding RNAs of ∼100 nucleotides that are components of RoRNPs [54,55] (Fig. 1D). These RNAs are also incorporated into HIV-1 particles [35,49]. To investigate their role in A3G/F packaging, we first tried to “knock down” their levels using siRNA mediated silencing. Although we could reduce cellular Y RNA concentrations, the levels that were encapsidated remained unaltered, in line with previous findings where cellular Y RNAs were reduced following RNAi-mediated Ro60 depletion but packaging into MLV particles was unchanged [56].
We then used our established Gag ΔNC fusion system to ask if Y RNAs would be able to recruit A3G or A3F into viral particles. Ro60 was fused to Gag and 293T cells were co-transfected to express HA tagged A3G or A3F with wild type Gag, Gag ΔNC, Gag ΔNC-Ro60, Gag ΔNC-Ro60 together with wild type Gag or with Gag ΔNC. VLPs were produced and immunoblotting showed that, although Gag fused to Ro60 alone did not produce detectable quantities of particles, mixed particles containing the Gag-Ro60 fusion protein and either Wt Gag or Gag ΔNC were formed (Fig. 5C). Quantitative RT-PCR specific for Y3 RNA was performed on RNA extracted from these particles; both mixed Gag ΔNC + Gag ΔNC-Ro60 and wild type Gag particles each contained ∼7-fold more Y3 RNA compared to Gag ΔNC VLPs (Fig. 5D), thus validating our approach. Analysis of the A3G/F contents demonstrated a strong restoration of packaging for the Gag ΔNC + Gag ΔNC-Ro60 mixed particles, though the levels did not match those noted with wild type Gag VLPs (Fig. 5C). These results indicate that controlled packaging of specific RNA ligands of A3G or A3F can determine their encapsidation, presumably by bridging between assembling Gag fusion proteins and APOBEC3 proteins, further demonstrating that specific RNAs can promote the encapsidation of APOBEC3 proteins.
Diverse RNA-binding domains are able to promote the packaging of A3G and A3F
Having shown that specific RNAs can recruit APOBEC3 proteins into assembling HIV-1 particles, we next asked whether this was a general property of packaged RNAs. To address this, a series of RNA binding domains (RBDs) of cell-encoded RNA binding proteins known to possess broad RNA binding capabilities were fused to Gag ΔNC as above. Specifically, we used the RBDs from two heterogeneous ribonucleoprotein (hnRNP) proteins: hnRNP C1, that binds to uridine tracts of RNAs [39], and hnRNP K, which is the prototypic protein for the KH RNA binding motif and has high affinity to poly(C) RNA [57,58]. We also fused the splicing factor SRSF2 that has a degenerate RNA binding sequence motif [59] and the double stranded RNA binding protein Staufen-1 [60,61].
These expression constructs were co-transfected with A3G/F into 293T cells together with vectors for wild type Gag or Gag ΔNC, and VLPs analysed by immunoblot (Fig. 6). Remarkably, all four RBDs readily rescued packaging of A3G and A3F in mixed virions with Gag ΔNC with (generally) similar efficiency as the wild type Gag or NC itself when reconnected to Gag ΔNC (compare lanes 6, 8, 10 and 12 with 1, 2 and 4; and 18, 20, 22 and 24 with 13, 14 and 16). Given that A3G and A3F exhibit very broad RNA binding characteristics (Fig. 1), we conclude that a multitude of such RNA substrates, if packaged, can serve to draw A3G/F into VLPs.

A3G and A3F do not preferentially bind to packaged RNAs
Fig. 6 shows that A3G and A3F can be incorporated into VLPs when diverse RBDs are fused to Gag. This is consistent with our iCLIP data, demonstrating that A3G and A3F are able to bind to multiple diverse RNAs. One obvious question that is raised by these observations is whether A3G/F are preferentially encapsidated by assembling HIV-1 VLPs, or whether they are inevitably packaged as a natural consequence of their promiscuous RNA binding capabilities. To address this directly, we carefully quantified the ratios of A3G and A3F to RNA in the lysates of virus producing cells and the matching particle preparations (Fig. 7). Accordingly, 293T cells were transiently transfected with vectors expressing T7-tagged versions of A3G or A3F, as well as the wild type Gag expression vector. Cells were also transfected with an irrelevant plasmid to serve as a negative control. VLPs and cell lysates were collected at 48 h post transfection. VLPs were isolated through a continuous sucrose gradient and the fractions containing VLPs were identified by immunoblot (S6 Fig.). Protein quantities were then determined against a standard curve of recombinant T7-A3G (Fig. 7A and 7B), and RNA in cell lysates and VLPs were extracted and quantified by Qubit (Fig. 7C). Culture supernatant from cells transfected with an irrelevant plasmid was used to assess background, and RNA was not detected in these samples (threshold of detection, 20 pg/ml). Interestingly, the calculated ratios of A3G/F to RNA were similar in cells and in virus particles (Fig. 7D, mean of 4 independent experiments). In other words, there is no evident enrichment of A3G or A3F in virions relative to virus-producing cells. Taking all our findings together leads us to conclude that the packaging of A3G and A3F into HIV-1 particles is driven by RNA binding, and that multiple/diverse RNAs can fulfil this role provided they are themselves packaged.

Discussion
A3G and A3F are antiviral proteins that have to be packaged into newly synthesised retrovirus particles to exert their activity. However, the details of the packaging mechanism remain incompletely understood, though a preference for encapsidating newly synthesised A3G has been established [29–35,47,48,51]. Here, we used for the first time a high throughput method to identify the RNAs that these two proteins bind to in living cells productively infected with vif-deficient HIV-1 and in cell-free viral particles. We then systematically addressed which RNAs are either necessary or sufficient for A3G and A3F incorporation into budding virions.
An important innovation with our study was the employment of a reference (“non-RNA binding”) protein during the iCLIP procedure, in this case GFP. This enabled us to identify A3G/F RNA ligands that are enriched over background RNA associations that are a presumed property of any protein (Fig. 1; exemplified here by the generation of iCLIP libraries from GFP-containing cells). While some preferential binding to certain classes of RNAs was apparent, it was nonetheless evident that the patterns of A3G/F binding were mostly non-discriminatory. We speculate that such promiscuity in RNA binding could be explained by the capacity of A3G/F to interact with myriad RNA sequences that are available for binding simply because they are not already occupied by other proteins.
We investigated in detail some specific RNAs that are found in retroviral particles [37]. We found that A3G and A3F do not require HIV-1 gRNA or 7SL for packaging into HIV-1 particles (Fig. 2, 3 and 4). However, using our Gag ΔNC fusion assay, we could show that A3G (but not A3F) can utilise 7SL to be incorporated into viral particles (Fig. 5). These observations correlated well with our iCLIP data, where it was shown that A3F does not bind to 7SL.
In accordance with previous studies [30–33], we observed that A3G and A3F are not packaged into Gag ΔNC VLPs. Importantly, these particles can clearly incorporate APOBEC3 proteins when a variety of unrelated RNAs are recruited into assembling virions via fusions of Gag ΔNC to a series of unrelated RBDs (Fig. 5 and 6). In other words, under experimental conditions, A3G and A3F can be packaged into HIV-1 particles via interactions with diverse and unrelated RNAs. Our data also show that 80% of the RNA sequences that A3G/F bind to inside vif-deficient (but otherwise normal) viral particles are of viral origin. If we assume that this distribution correlates with the binding of A3G/F to RNAs that are being packaged during particle production, our data imply that viral genomic RNA ordinarily mediates A3G/F packaging. An alternative possibility is that non-viral RNAs recruit A3G/F into particles, but that A3G/F then release these RNAs and bind to gRNA following particle formation and release.
Lastly, fastidious quantification of A3G/F and total RNA levels in cells and budded virions revealed that viral particles are not enriched for APOBEC3 protein content (Fig. 7). While RNA binding is clearly required for APOBEC3 protein packaging, these observations indicate that there is no selectivity for engaging RNAs that are destined for incorporation into assembling viruses. Accordingly, we speculate that cytoplasmic APOBEC3 proteins exploit their relatively non-specific RNA binding capabilities to patrol the cytosol and bind to unoccupied sites on RNAs. In the context of cellular RNAs, this may account for the pronounced accumulation of APOBEC3 proteins in RNA-rich microdomains such as P-bodies and stress granules [23–26]. For viruses with RNA genomes, such as retroviruses, this can result in encapsidation into newly formed viral particles. Given the DNA editing function of APOBEC3 proteins, viruses that have RNA genomes and replicate via DNA intermediates—namely retroviruses and hepadnaviruses—will be susceptible to hypermutation and inhibition [4,5,9]. Indeed, we propose that the relatively non-specific RNA binding characteristics of A3G/F render these proteins well suited to the inhibition of a wide variety of viral and transposon targets. Moreover, this feature may further ensure that viral sequence variation, a noted hallmark of HIV-1, will not afford a means of escape from APOBEC3-mediated restriction: perhaps this underlies the evolution of an entirely different (protein-based) evasion mechanism, namely Vif-induced protein degradation?
Materials and Methods
Plasmids and cells
cDNAs encoding SRP19, GFP, A3G or A3F were cloned between the XbaI and BamHI sites of pCGTHCFFLT7 [62]. DNA fragments encoding T7-tagged derivatives of GFP, A3G or A3F were cloned between the XhoI and EcoRI sites of the retrovirus vector, pCMS28 [26]. A GFP-containing fragment with a GST linker sequence at its 3’-end was cloned between the EcoRI and XhoI restriction sites of pCMS28, and A3G or A3F cDNAs were then inserted using the NotI and XhoI sites. Plasmids expressing HA-tagged A3G and A3F were previously described [50]. vif-deficient HIV-1NL4-3 [27] and HIV-1IIIB [63] strains were used where indicated. The wild type Gag-Pol vector, pCMS446, was generated by inserting the HIV-1 5’ UTR-Gag-Pol, Protease activity inactivated (nucleotides 455–5096 from the HV-1HXB2 isolate [GenBank: K03455.1] [64]) fragment into pcDNA3.1 (Invitrogen) that contains the HIV-1 RRE [65]. pGag was generated by deleting the Pol sequence 3’ to the Gag stop codon. pGag ΔNC was generated by cloning the analogous 5’ UTR-Gag-Pol fragment from the Zwt-p6 proviral construct [45] into the same RRE-containing vector. SL2 and 3 were deleted from pGag by overlapping PCR using the primers 5′-AGGGGCGGCGACTGGTGAGAGATGGGTGCGAGAGCGTCAGTATTAAGC-3′ and 5′-TGACGCTCTCGCACCCATCTCTCACCAGTCGCCGCCCCTCGCCTCTTGC-3′ to generate pGag ΔΨ [46]. pGag ΔNC NB was created by inserting NheI and BamHI restriction sites in frame 5’ to the stop codon of Gag in pGag ΔNC. NC (HIV-1HXB2 strain), GFP, SRP19, Ro60, hnRNP C1 (amino acids 1–104), hnRNP K (amino acids 38 to 464 with amino acids 323–338 deleted), SRSF2 (amino acids 2–93) and Staufen 1 cDNAs were inserted into pGag ΔNC NB using the NheI and BamHI sites.
293T and HeLa cells were obtained from the American Tissue Culture Collection (ATCC). TZM-bl cells were obtained through the NIH AIDS Reagents Repository Program (ARRP). These cell lines were cultured in Dulbecco’s modified Eagle’s medium (Invitrogen, UK) supplemented with 10% foetal bovine serum and 1% penicillin/streptomycin. CEM-SS T cells, from ARRP, were cultured in Roswell Park Memorial Institute 1640 medium (Invitrogen, UK) supplemented with 10% foetal bovine serum and 1% penicillin/streptomycin. Stable CEM-SS T cell lines were generated by standard retroviral transduction using MLV-based vectors expressing GFP, GFP-A3G GFP-A3F, T7-GFP, T7-A3G or T7-A3F and selected with 1 μg/ml puromycin. 293T cells stably expressing HA-tagged A3G or A3F were generated by transduction with MLV based vectors expressing the proteins of interest and selection with 1 μg/ml puromycin. Expression levels of the A3 proteins were assessed by immunoblot using rabbit polyclonal sera specific for A3G [66] or A3F [67] for primary detection.
Individual-nucleotide resolution UV cross-linking and immunoprecipitation
iCLIP has been described in detail previously [39]. Briefly, CEM-SS T cells stably expressing the proteins of interest were infected with vif-deficient HIVIIIB. 48 h later, a sample was collected to assess infection by intracellular p24Gag staining and flow cytometry, confirming that at least 80% of cells were productively infected. The supernatant was collected, filtered through a 0.45 μm pore filter, and viruses isolated through a 20% sucrose cushion (wt/vol) at 21000 × g for 2 h at 4°C and resuspended in PBS. Cells were collected, washed 6 times with PBS and resuspended in PBS. Cells and viruses were then radiated with 400 mJ/cm2 using a Stratlinker 2400. Cells were pelleted by centrifugation and the supernatant discarded. Cells and viruses were then resuspended in 1 ml of lysis buffer (50 mM Tris-HCL, pH 7.4; 100 mM NaCl; 1% NP-40; 0.1% SDS; 0.5% sodium deoxycholate and protease inhibitor) and sonicated. 0.16 μg of RNase A were added to High RNase samples and 0.04 ng to the other samples. Tubes were incubated at 37°C for 3 min and added to protein G dynabeads previously incubated with anti-T7 antibody (Novagen) or anti-GFP antibody (Roche). The RNAs were dephosphorylated using Shrimp alkaline phosphatase (Promega) and a pre-adenylated linker was ligated to the 3’-end of RNAs on beads. RNAs were radiolabeled with P32-ϒ-ATP and separated using SDS-polyacrylamide gel electrophoresis, electrophoretically transferred to nitrocellulose and visualised on film. Pieces of the membrane containing the RNAs of interest were excised and resuspended in PK buffer (100 mM Tris-HCl pH 7.5, 50 mM NaCl, 10 mM EDTA) containing 2 mg/ml proteinase K. RNAs were isolated with phenol/chloroform (Ambion) and precipitated with 2.5 volumes of 100% ethanol, 0.1 volumes of sodium acetate (3 M, pH 5.5) and 0.5 μl of glycoblue. RNAs were then pelleted and reverse transcribed using barcoded primers. cDNAs were run on a TBE-urea polyacrylamide gel and products ranging from 70–85, 85–110 and >110 base pairs were excised. Nucleic acids were extracted and circularised. A primer that anneals to the linker previously ligated to the RNAs was used to create a double stranded region and this DNA was digested with BamHI. cDNA was then amplified by PCR and sequenced on one lane of an Illumina GA2 flow cell with 50 nucleotides run length. Data is available at ArrayExpress with the accession number E-MTAB-2700.
Before mapping reads, adapter sequences were removed, and the barcodes for each sample within each library were used to identify which sequence was immunoprecipitated from each protein. Mapping of the reads was performed against the human genome (version Hg19/ GRCh37) and HIV-1IIIB (GenBank ID EU541617) genome using bowtie [41]. Reads that aligned to a single position on the human genome or HIV-1 genome with at most two mismatches were considered for analysis. Genomic annotations were then assigned based on gene annotations provided by Ensembl (v59). Reads were also aligned to human repeat sequences using repeat masker [42] and a database of consensus sequences provided by the software. A maximum of 3 mismatches were allowed.
Viral particle production, packaging assays and RNA and protein quantifications
HIV-1 virions were produced by transfecting 293T cells with vif-deficient pNL4-3 or pIIIB using polyethyleneimine (PEI). Virus were then harvested 48 h later and filtered through a 0.45 μm pore filter. Lentiviral vectors were produced in 293T cells by transfecting the p8.91 packaging plasmid, a lentiviral vector and the VSVg envelope plasmid pMDG2.1 [43] at a ratio of 2 : 2:1 using PEI. Vectors were harvested 48 h after transfection and filtered. Viral particles were quantified by p24Gag enzyme linked immunosorbent assay (Perkin Elmer). VLPs were produced by transfecting the packaging plasmid of interest and the Rev expression vector, at a ratio of 2 : 1. Wherever stated, HA - or T7-tagged GFP, A3G or A3F vectors were co-transfected at a ratio of 1 : 5 to the packaging plasmid. The supernatant was collected 48 h later, filtered through a 0.45μm pore filter and isolated through a 20% sucrose cushion (wt/vol) at 21000 × g for 2 h at 4°C. Cells and viral pellets were lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl pH 7.4, 100 mM NaCl, 1 mM MgCl2, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate). T7-tagged A3G was purified as described before [68]. Expression, VLP production and packaging of APOBEC3 proteins were assessed by standard immunoblot using anti-HA (mouse monoclonal; 12CA5) or anti-T7 (Novagen) and anti-p24Gag (mouse monoclonal; p24-2 [69]) antibodies and detected and quantified by Li-cor Odyssey infrared imaging using IRDye800CW or IRDye680LT-labeled secondary antibodies. VLPs produced for RNA and protein quantification were filtered, layered over a continuous sucrose gradient (60–20%) and centrifugated at 150,000 × g for 1h15min at 4°C. 1mL fractions were collected and centrifugated at 21000 × g for 2 h at 4°C. The supernatant was removed and the pellet resuspended in RIPA buffer. The fractions containing p24Gag were identified by immunoblot. The fraction with highest level of p24Gag was then used to quantify the packaged A3G/F by immunoblot using a standard curve of purified T7-A3G. The remainder of the fraction was used to extract RNA using a microRNA extraction kit (Promega). Total RNA was then quantified using the Qubit RNA HS assay kit (Life Technologies), following the manufacturer’s instructions.
Reverse transcription and quantitative PCR:
RNA was extracted from viral particles using Tri Reagent LS (Sigma) according to the manufacturer’s instructions. 0.5 μg of total RNA was used to synthesise cDNA with the high-capacity cDNA reverse transcription kit (Applied Biosystems) and random primers using the manufacturer’s protocol. qPCR was then performed using the primers 5′-TAACTAGGGAACCCACTGC-3′, 5′-GCTAGAGATTTTCCACACTG-3′ and the probe 5′-6-carboxyfluorescein [FAM]-ACACAACAGACGGGCACACACTA-6-carboxytetramethylrhodamine [TAMRA]-3′ to detect HIV-1 gRNA; SYBR Green (Applied Biosystems) was used to detect 7SL with the primers 5′-GGGCTGTAGTGCGCTATGC-3′ and 5′-CCCGGGAGGTCACCATATT-3′, and to quantify hY3 RNA with the primers 5′-GGCTGGTCCGAGTGCAGTG-3′ and 5′-AAAGGCTAGTCAAGTGAAGCAGTGG-3′.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Holmes RK, Malim MH, Bishop KN (2007) APOBEC-mediated viral restriction: not simply editing? Trends Biochem Sci 32 : 118–128. 17303427
2. Desimmie B a, Delviks-Frankenberrry K a, Burdick RC, Qi D, Izumi T, et al. (2013) Multiple APOBEC3 Restriction Factors for HIV-1 and One Vif to Rule Them All. J Mol Biol: 1–26.
3. LaRue RS, Andrésdóttir V, Blanchard Y, Conticello SG, Derse D, et al. (2009) Guidelines for naming nonprimate APOBEC3 genes and proteins. J Virol 83 : 494–497. doi: 10.1128/JVI.01976-08 18987154
4. Sheehy AM, Gaddis NC, Choi JD, Malim MH (2002) Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418 : 4–8. 12167863
5. Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, et al. (2003) Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 424 : 99–103. 12808466
6. Gaddis NC, Sheehy AM, Ahmad KM, Swanson M, Bishop KN, et al. (2004) Further Investigation of Simian Immunodeficiency Virus Vif Function in Human Cells. J Virol 78 : 12041–12046. 15479843
7. Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-mahrt SK, et al. (2003) DNA deamination mediates innate immunity to retroviral infection. Cell 113 : 803–809. 12809610
8. Okeoma CM, Lovsin N, Peterlin BM, Ross SR (2007) APOBEC3 inhibits mouse mammary tumour virus replication in vivo. Nature 445 : 927–930. 17259974
9. Turelli P, Mangeat B, Jost S, Vianin S, Trono D (2004) Inhibition of hepatitis B virus replication by APOBEC3G. Science 303 : 1829.
10. Narvaiza I, Linfesty DC, Greener BN, Hakata Y, Pintel DJ, et al. (2009) Deaminase-independent inhibition of parvoviruses by the APOBEC3A cytidine deaminase. PLoS Pathog 5: e1000439. doi: 10.1371/journal.ppat.1000439 19461882
11. Esnault C, Heidmann O, Delebecque F, Dewannieux M, Ribet D, et al. (2005) APOBEC3G cytidine deaminase inhibits retrotransposition of endogenous retroviruses. Nature 433 : 430–433. 15674295
12. Marin M, Rose KM, Kozak SL, Kabat D (2003) HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat Med 9 : 1398–1403. 14528301
13. Yu X, Yu Y, Liu B, Luo K, Kong W, et al. (2003) Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 302 : 1056–1060. 14564014
14. Sheehy AM, Gaddis NC, Malim MH (2003) The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat Med 9 : 1404–1407. 14528300
15. Guo Y, Dong L, Qiu X, Wang Y, Zhang B, et al. (2014) Structural basis for hijacking CBF-β and CUL5 E3 ligase complex by HIV-1 Vif. Nature 505 : 229–233. doi: 10.1038/nature12884 24402281
16. Zhang H, Yang B, Pomerantz RJ, Zhang C, Arunachalam SC, et al. (2003) The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 424 : 94–98. 12808465
17. Bishop KN, Verma M, Kim E-Y, Wolinsky SM, Malim MH (2008) APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog 4: e1000231. 19057663
18. Iwatani Y, Chan DSB, Wang F, Maynard KS, Sugiura W, et al. (2007) Deaminase-independent inhibition of HIV-1 reverse transcription by APOBEC3G. Nucleic Acids Res 35 : 7096–7108. 17942420
19. Gillick K, Pollpeter D, Phalora P, Kim E-Y, Wolinsky SM, et al. (2013) Suppression of HIV-1 infection by APOBEC3 proteins in primary human CD4(+) T cells is associated with inhibition of processive reverse transcription as well as excessive cytidine deamination. J Virol 87 : 1508–1517. doi: 10.1128/JVI.02587-12 23152537
20. Navarro F, Bollman B, Chen H, König R, Yu Q, et al. (2005) Complementary function of the two catalytic domains of APOBEC3G. Virology 333 : 374–386. 15721369
21. Iwatani Y, Takeuchi H, Strebel K, Levin JG (2006) Biochemical activities of highly purified, catalytically active human APOBEC3G: correlation with antiviral effect. J Virol 80 : 5992–6002. 16731938
22. Huthoff H, Autore F, Gallois-Montbrun S, Fraternali F, Malim MH (2009) RNA-dependent oligomerization of APOBEC3G is required for restriction of HIV-1. PLoS Pathog 5: e1000330. 19266078
23. Chiu Y-L, Soros VB, Kreisberg JF, Stopak K, Yonemoto W, et al. (2005) Cellular APOBEC3G restricts HIV-1 infection in resting CD4+ T cells. Nature 435 : 108–114. 15829920
24. Kozak SL, Marin M, Rose KM, Bystrom C, Kabat D (2006) The anti-HIV-1 editing enzyme APOBEC3G binds HIV-1 RNA and messenger RNAs that shuttle between polysomes and stress granules. J Biol Chem 281 : 29105–29119. 16887808
25. Wichroski MJ, Robb GB, Rana TM (2006) Human retroviral host restriction factors APOBEC3G and APOBEC3F localize to mRNA processing bodies. PLoS Pathog 2: e41. 16699599
26. Gallois-Montbrun S, Kramer B, Swanson CM, Byers H, Lynham S, et al. (2007) Antiviral protein APOBEC3G localizes to ribonucleoprotein complexes found in P bodies and stress granules. J Virol 81 : 2165–2178. 17166910
27. Phalora PK, Sherer NM, Wolinsky SM, Swanson CM, Malim MH (2012) HIV-1 replication and APOBEC3 antiviral activity are not regulated by P bodies. J Virol 86 : 11712–11724. 22915799
28. Izumi T, Burdick R, Shigemi M, Plisov S, Hu W-S, et al. (2013) Mov10 and APOBEC3G localization to processing bodies is not required for virion incorporation and antiviral activity. J Virol 87 : 11047–11062. doi: 10.1128/JVI.02070-13 23926332
29. Soros VB, Yonemoto W, Greene WC (2007) Newly synthesized APOBEC3G is incorporated into HIV virions, inhibited by HIV RNA, and subsequently activated by RNase H. PLoS Pathog 3: e15. 17291161
30. Luo K, Liu B, Xiao Z, Yu Y, Gorelick R, et al. (2004) Amino-Terminal Region of the Human Immunodeficiency Virus Type 1 Nucleocapsid Is Required for Human APOBEC3G Packaging. J Virol 78 : 11841–11852. 15479826
31. Schäfer A, Bogerd HP, Cullen BR (2004) Specific packaging of APOBEC3G into HIV-1 virions is mediated by the nucleocapsid domain of the gag polyprotein precursor. Virology 328 : 163–168. 15464836
32. Svarovskaia ES, Xu H, Mbisa JL, Barr R, Gorelick RJ, et al. (2004) Human apolipoprotein B mRNA-editing enzyme-catalytic polypeptide-like 3G (APOBEC3G) is incorporated into HIV-1 virions through interactions with viral and nonviral RNAs. J Biol Chem 279 : 35822–35828. 15210704
33. Zennou V, Perez-caballero D, Bieniasz PD (2004) APOBEC3G Incorporation into Human Immunodeficiency Virus Type 1 Particles. J Virol 78 : 12058–12061. 15479846
34. Khan MA, Kao S, Miyagi E, Goila-gaur R, Opi S, et al. (2005) Viral RNA Is Required for the Association of APOBEC3G with Human Immunodeficiency Virus Type 1 Nucleoprotein Complexes. J Virol 79 : 5870–5874. 15827203
35. Wang T, Tian C, Zhang W, Luo K, Sarkis PTN, et al. (2007) 7SL RNA mediates virion packaging of the antiviral cytidine deaminase APOBEC3G. J Virol 81 : 13112–13124. 17881443
36. Chiu Y, Witkowska HE, Hall SC, Santiago M, Soros VB, et al. (2006) High-molecular-mass APOBEC3G complexes restrict Alu retrotransposition. Proc Natl Acad Sci USA 103 : 15588–15593. 17030807
37. Berkowitz R, Fisher J, Goff SP (1996) RNA Packaging. In: Kräusslich H-G, editor. Morphogenesis and Maturation of Retroviruses SE - 6. Springer Berlin Heidelberg, Vol. 214. pp. 177–218.
38. Ule J, Jensen KB, Ruggiu M, Mele A, Ule A, et al. (2003) CLIP identifies Nova-regulated RNA networks in the brain. Science 302 : 1212–1215. 14615540
39. König J, Zarnack K, Rot G, Curk T, Kayikci M, et al. (2010) iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol 17 : 909–915.
40. Chi SW, Zang JB, Mele A, Darnell RB (2009) Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature 460 : 479–486. doi: 10.1038/nature08170 19536157
41. Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. doi: 10.1186/gb-2009-10-3-r25 19261174
42. Smit Afa Green (n.d.) Repeat Masker. Available: http://repeatmasker.org.
43. Zufferey R, Nagy D, Mandel R, Naldini L, Trono D (1997) Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol 15 : 871–875. 9306402
44. Demaison C, Parsley K, Brouns G, Scherr M, Battmer K, et al. (2002) High-level transduction and gene expression in hematopoietic repopulating cells using a human immunodeficiency virus type 1-based lentiviral vector containing an internal spleen focus forming virus promoter. Hum Gene Ther 13 : 803–813. 11975847
45. Accola MA, Strack B, Göttlinger HG (2000) Efficient Particle Production by Minimal Gag Constructs Which Retain the Carboxy-Terminal Domain of Human Immunodeficiency Virus Type 1 Capsid-p2 and a Late Assembly Domain. J Virol 74 : 5395–5402. 10823843
46. Luban J, Goff SP (1994) Mutational Analysis of cis-Acting Packaging Signals in Human Immunodeficiency Virus Type 1 RNA. J Virol 68 : 3784–3793. 8189516
47. Onafuwa-Nuga A a, Telesnitsky A, King SR (2006) 7SL RNA, but not the 54-kd signal recognition particle protein, is an abundant component of both infectious HIV-1 and minimal virus-like particles. RNA 12 : 542–546. 16489186
48. Khan M a, Goila-Gaur R, Opi S, Miyagi E, Takeuchi H, et al. (2007) Analysis of the contribution of cellular and viral RNA to the packaging of APOBEC3G into HIV-1 virions. Retrovirology 4 : 48. 17631688
49. Onafuwa-nuga AA, King SR, Telesnitsky A (2005) Nonrandom Packaging of Host RNAs in Moloney Murine Leukemia Virus. J Virol 79 : 13528–13537. 16227273
50. Gallois-Montbrun S, Holmes RK, Swanson CM, Fernández-Ocaña M, Byers HL, et al. (2008) Comparison of cellular ribonucleoprotein complexes associated with the APOBEC3F and APOBEC3G antiviral proteins. J Virol 82 : 5636–5642. doi: 10.1128/JVI.00287-08 18367521
51. Bach D, Peddi S, Mangeat B, Lakkaraju A, Strub K, et al. (2008) Characterization of APOBEC3G binding to 7SL RNA. Retrovirology 5 : 54. doi: 10.1186/1742-4690-5-54 18597676
52. Sherer NM, Lehmann MJ, Jimenez-Soto LF, Ingmundson A, Horner SM, et al. (2003) Visualization of retroviral replication in living cells reveals budding into multivesicular bodies. Traffic 4 : 785–801. 14617360
53. Müller B, Daecke J, Fackler OT, Dittmar T, Zentgraf H, et al. (2004) Construction and Characterization of a Fluorescently Labeled Infectious Human Immunodeficiency Virus Type 1 Derivative. J Virol 78 : 10803–10813.
54. Lerner MR, Boyle JA, Hardin JA, Steitz JA, Science S, et al. (1981) Two Novel Classes of Small Ribonucleoproteins Detected by Antibodies Associated with Lupus Erythematosus. Science (80) 211 : 400–402. 6164096
55. Hendrick JP, Wolin SL, Rinke J, Steitz JA (1981) Ro small cytoplasmic ribonucleoproteins are a subclass of La ribonucleoproteins : further characterization of the Ro and La Small ribonucleoproteins from uninfected mammalian cells. Mol Cell Biol 12 : 1138–1149. 6180298
56. Garcia EL, Onafuwa-Nuga A, Sim S, King SR, Wolin SL, et al. (2009) Packaging of host mY RNAs by murine leukemia virus may occur early in Y RNA biogenesis. J Virol 83 : 12526–12534. doi: 10.1128/JVI.01219-09 19776129
57. Swanson MS, Dreyfuss G (1988) heterogeneous nuclear ribonucleoprotein Classification and Purification of Proteins of Heterogeneous Nuclear Ribonucleoprotein Particles by RNA-Binding Specificities. Mol Cell Biol 8 : 2237–2241.
58. Makeyev A V, Liebhaber SA (2002) The poly ( C ) -binding proteins: a multiplicity of functions and a search for mechanisms. RNA 8 : 265–278. 12003487
59. Pandit S, Zhou Y, Shiue L, Coutinho-Mansfield G, Li H, et al. (2013) Genome-wide analysis reveals SR protein cooperation and competition in regulated splicing. Mol Cell 50 : 223–235. doi: 10.1016/j.molcel.2013.03.001 23562324
60. Saunders LR, Barber GN (2003) The dsRNA binding protein family: critical roles, diverse cellular functions. FASEB J 17 : 961–983. 12773480
61. De Lucas S, Oliveros JC, Chagoyen M, Ortín J (2014) Functional signature for the recognition of specific target mRNAs by human Staufen1 protein. Nucleic Acids Res 42 : 4516–4526. doi: 10.1093/nar/gku073 24470147
62. Wilson a C Peterson MG, Herr W (1995) The HCF repeat is an unusual proteolytic cleavage signal. Genes Dev 9 : 2445–2458. 7590226
63. Simon JH, Southerling TE, Peterson JC, Meyer BE, Malim MH (1995) Complementation of vif-defective human immunodeficiency virus type 1 by primate, but not nonprimate, lentivirus vif genes. J Virol 69 : 4166–4172. 7769676
64. Ratner L, Haseltine W, Patarca R, Livak KJ, Starcich B, et al. (1985) Complete nucleotide sequence of the AIDS virus, HTLV-III. Nature 313 : 277–284. 2578615
65. Swanson CM, Puffer B a, Ahmad KM, Doms RW, Malim MH (2004) Retroviral mRNA nuclear export elements regulate protein function and virion assembly. EMBO J 23 : 2632–2640. 15201866
66. Newman ENC, Holmes RK, Craig HM, Klein KC, Lingappa JR, et al. (2005) Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr Biol 15 : 166–170. 15668174
67. Holmes RK, Koning FA, Bishop KN, Malim MH (2007) APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation: Comparisons with APOBEC3G. J Biol Chem 282 : 2587–2595. 17121840
68. Shlyakhtenko LS, Lushnikov AY, Li M, Lackey L, Harris RS, et al. (2011) Atomic force microscopy studies provide direct evidence for dimerization of the HIV restriction factor APOBEC3G. J Biol Chem 286 : 3387–3395. doi: 10.1074/jbc.M110.195685 21123176
69. Gaddis NC, Chertova E, Sheehy AM, Henderson LE, Malim MH (2003) Comprehensive investigation of the molecular defect in vif-deficient human immunodeficiency virus type 1 virions. J Virol 77 : 5810–5820. 12719574
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 1
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Infections in Humans and Animals: Pathophysiology, Detection, and Treatment
- : Trypanosomatids Adapted to Plant Environments
- Environmental Drivers of the Spatiotemporal Dynamics of Respiratory Syncytial Virus in the United States
- Dengue Virus RNA Structure Specialization Facilitates Host Adaptation
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy