
The Molecular Basis for Control of ETEC Enterotoxin Expression in Response to Environment and Host
Diarrheagenic illness remains a major disease burden in the developing world. Enterotoxigenic Escherichia coli (ETEC) are the leading bacterial cause of such disease; hundreds of millions of cases occur every year. The severe watery diarrhoea associated with ETEC infections results from the action of enterotoxins. The toxins target human gut epithelial cells and trigger the loss of water and electrolytes into the gut lumen. Oral rehydration therapy can counteract this process. Hence, glucose and salt solutions promote rehydration of the patient. In this work we show that the gene regulatory mechanisms controlling toxin expression respond directly to sugar and salt. Furthermore, we describe a molecular mechanism to explain these effects. Hence, we provide a starting point for the optimisation of oral rehydration solutions to reduce toxin expression over the course of an ETEC infection.
Published in the journal:
. PLoS Pathog 11(1): e32767. doi:10.1371/journal.ppat.1004605
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004605
Summary
Diarrheagenic illness remains a major disease burden in the developing world. Enterotoxigenic Escherichia coli (ETEC) are the leading bacterial cause of such disease; hundreds of millions of cases occur every year. The severe watery diarrhoea associated with ETEC infections results from the action of enterotoxins. The toxins target human gut epithelial cells and trigger the loss of water and electrolytes into the gut lumen. Oral rehydration therapy can counteract this process. Hence, glucose and salt solutions promote rehydration of the patient. In this work we show that the gene regulatory mechanisms controlling toxin expression respond directly to sugar and salt. Furthermore, we describe a molecular mechanism to explain these effects. Hence, we provide a starting point for the optimisation of oral rehydration solutions to reduce toxin expression over the course of an ETEC infection.
Introduction
ETEC are Gram negative bacteria that cause severe diarrhoea, known as non-vibrio cholera, in humans [1], [2]. First isolated in 1971, ETEC are responsible for 210 million infections annually, mostly in developing countries, leading to 380,000 deaths [3]. Disease results primarily from the action of two enterotoxins. The heat-labile toxin (LT) is similar in structure and function to cholera toxin [4], [5]. The heat-stable toxin (ST) mimics the human hormone guanylin [6]. Both toxins are secreted by ETEC during infection. Made up of two subunits, encoded by the eltAB operon, LT has the configuration AB5 [5], [7]. In the gut, LT binds to host cell GM1 gangliosides and is endocytosed [8], [9]. This triggers constitutive cAMP production in the affected cell [8]. The ST toxin, encoded by the estA gene, also interferes with cell signalling [6]. Hence, ST binds to the guanylate cyclase C receptor and stimulates overproduction of cGMP. The combined actions of LT and ST cause loss of H2O, and electrolytes, from epithelial cells into the gut lumen [4]. Oral Rehydration Therapy (ORT) is used to redress the resulting electrolyte imbalance and rehydrate the patient [10]. In its most simple form, ORT requires only an aqueous solution of glucose and salt. Hence, the availability of metabolites and cations are a central theme of ETEC mediated disease. The effect of ORT on human physiology is well understood: glucose and Na2+ are transported across the epithelial membrane, along with water, to promote rehydration [11]. Surprisingly, despite the existence of molecular mechanisms that allow bacteria to respond to these signals, the consequences for ETEC are unknown.
In E. coli, the transcriptional response to glucose is controlled by cAMP receptor protein (CRP) [12]. In the absence of glucose, intracellular cAMP levels increase and CRP binds DNA targets with the consensus sequence 5′-TGTGA-n6-TCACA-3′ [13]. Subsequently, gene expression is reprogrammed to make use of alternative carbon sources [14]. Note that the gene regulatory network managed by CRP includes many indirect pathways [14], [15]. Hence, CRP is also a pleiotropic regulator of transcription. Whilst indirect regulatory effects are difficult to characterise, genes that are directly controlled by CRP can be divided into distinct classes [12]. At Class II targets, CRP binds to a site overlapping the promoter -35 element and interacts directly with both the N-terminal and C-terminal domains of the RNA polymerase α subunit (αNTD and αCTD). At Class I targets, CRP binds further upstream and interacts only with αCTD. This interaction can be further stabilised by UP-elements, AT-rich DNA sequences, adjacent to the CRP site, that facilitate αCTD-DNA interactions [12]. At both classes of promoter, the various contacts enhance gene expression by stabilising the transcription initiation complex. Unsurprisingly, most genes regulated by CRP encode proteins involved in metabolism. However, in some bacteria, CRP has been co-opted as a virulence regulator [16].
The Histone-like Nucleoid Structuring (H-NS) factor is a component of bacterial nucleoprotein. Consequently, H-NS also influences gene expression on a global scale [17]. Briefly, H-NS targets sections of the genome with a low GC content [17]. Depending on H-NS conformation, the resulting nucleoprotein complexes can be filamentous or bridged in organisation [18]. Filamentous complexes favour gene regulation by excluding RNA polymerase, and transcriptional regulators, from their targets [19], [20]. Bridged complexes favour RNA polymerase trapping [21]. In all scenarios, it is thought that H-NS acts primarily to silence transcription [22]. The conformation of H-NS, and hence the way in which it modulates DNA topology, can be controlled by divalent cations. Consequently, H-NS mediated repression can be relieved by increased osmolarity [23]. Like CRP, H-NS has been incorporated into the virulence gene regulatory networks of many bacteria [17].
In this work we define the molecular trigger that controls toxin expression in ETEC. We show that CRP and H-NS are key regulatory factors. Strikingly, this allows ETEC to integrate extracellular signals of osmolarity and metabolism to control toxin production. Hence, we propose that ETEC toxicity responds directly to osmo-metabolic flux. Interestingly, the precise regulatory settings are different for each toxin encoding gene. The differences result from i) varying promoter configurations and ii) competition between CRP and H-NS for overlapping DNA targets. This is significant since fluctuations in osmolarity, and changes in the availability of metabolites, are central to ETEC infection and its treatment.
Results
Binding of CRP and H-NS across the ETEC H10407 genome
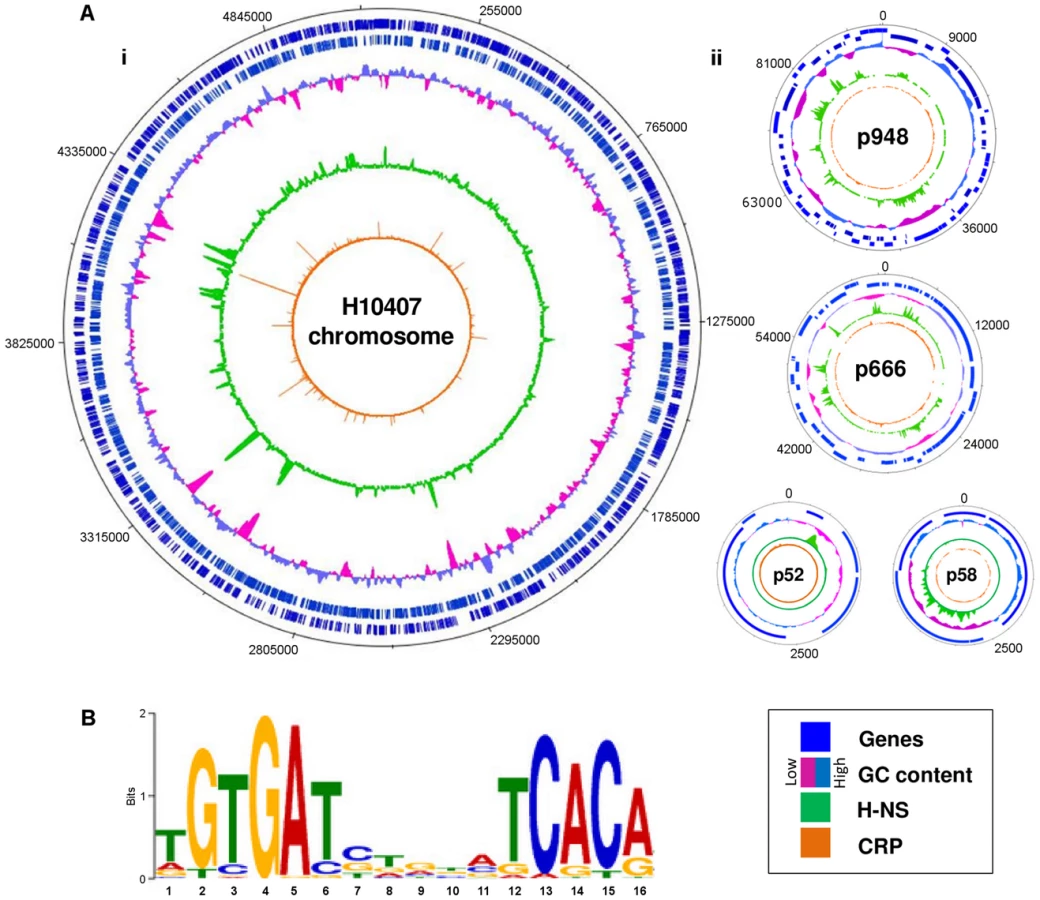
The prototypical ETEC strain H10407 reproducibly elicits diarrhoea in human volunteers and has a well-defined genome that shares 3,766 genes with E. coli K-12 [1]. Pathogenicity arises from 599 ancillary genes encoded by 25 discrete chromosomal loci and 4 plasmids. The plasmids, named p948, p666, p58 and p52, encode the enterotoxins. Derivatives of the estA gene are found on plasmids p666 (estA1) and p948 (estA2). A single copy of the eltAB operon is encoded by plasmid p666. We used Chromatin Immunoprecipitation (ChIP) coupled with next-generation DNA sequencing (ChIP-seq) to map CRP and H-NS targets across the ETEC H10407 genome. The binding profiles are shown in Fig. 1A. In each plot genes are illustrated by blue lines (tracks 1 and 2), DNA G/C content by a cyan and pink graph (track 3), H-NS binding is in green (track 4) and CRP binding is shown in orange (track 5). As expected, H-NS binding is inversely correlated with DNA G/C content (compare tracks 3 and 4). Similarly, CRP binding occurs in expected locations; 96% of the CRP binding sites are associated with the DNA logo shown in Fig. 1B (i.e. the known CRP consensus sequence (13–15)). We identified a total of 111 high-confidence CRP targets (Table 1). Of these targets 93% were present in the genome sequences of both ETEC H10407 and E. coli K-12. The most common location for CRP sites was in intergenic regions (66% of targets) whilst a smaller number of targets were found within genes (34%). Consistent with expectations, CRP sites were most frequently located ∼40.5 bp, or ∼92.5 bp, upstream of experimentally determined transcription start sites (TSSs). Surprisingly, CRP binding was restricted to the ETEC chromosome (Fig. 1Ai). Conversely, H-NS bound to chromosomal and plasmid loci (Fig. 1Ai), including all toxin encoding genes (Fig. 1Aii).

Unoccupied high-affinity CRP binding targets on p948 and p666 are bound by H-NS
To better understand the lack of CRP binding to p948 and p666 we took a bioinformatic approach. CRP targets were aligned to generate a position weight matrix (PWM). The PWM was then used to search p948 and p666 for CRP sites. A continuum of over 100 potential CRP targets was identified. However, we recognise that the vast majority of these are likely to be false positives. Hence, we next sought to differentiate between genuine CRP sites and spurious predictions. To do this, predicted sites were scored, grouped, and ranked on the basis of their match to the PWM (Fig. 2A, S1 Table). Electrophoretic mobility shift assays (EMSA) were then used to measure binding of CRP to a target from each group so that a meaningful cut-off could be established. The result is illustrated graphically in Fig. 2B. The raw data are shown in S1A Fig. We found that predicted sites with a score<10 did not bind CRP. To assess the affinity of CRP for all predicted targets scoring >10 a second set of EMSA experiments was done (S1B Fig.). Hence, we identified a total of 5 potential CRP targets on p666 and p948. Interestingly, the estA1 and estA2 genes, which both encode ST, were amongst the 5 targets (Fig. 2C). Remarkably, all 5 of the plasmid borne CRP targets identified in silico, and bound tightly by CRP in vitro, were occupied by H-NS in vivo (Fig. 2C).

The estA2 gene is transcribed from a Class I CRP dependent promoter
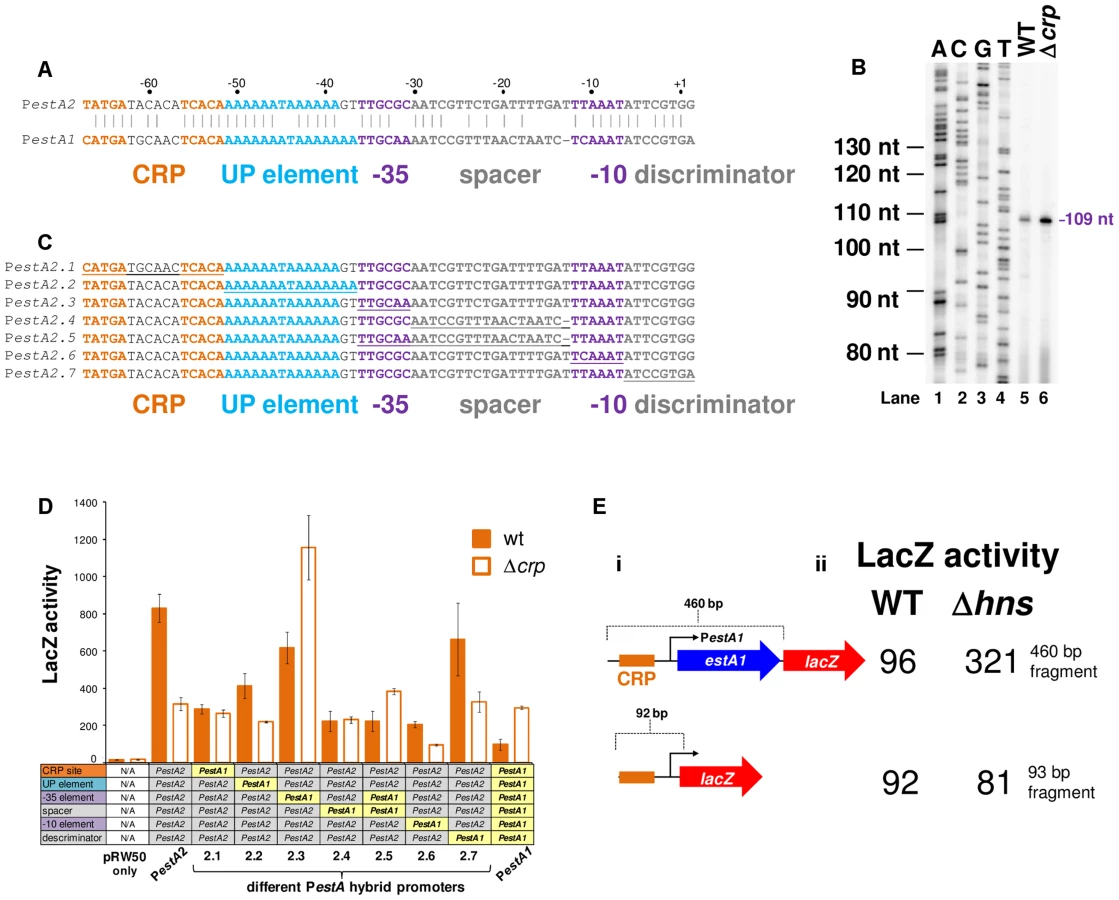
To understand if CRP could regulate ST production we focused first on estA2. This derivative of the toxin is more commonly associated with human disease and ETEC H10407 is somewhat unusual in also encoding estA1 [24]. The sequence of the estA2 regulatory region is shown in Fig. 3A. A 93 bp DNA fragment, containing the regulatory region, was cloned into the lacZ reporter plasmid pRW50 to generate a lacZ fusion (S2A Fig.). The estA2 TSS was then determined using mRNA primer extension analysis. We detected a single extension product, of 109 nucleotides (nt) in length (Fig. 3B). The position of the TSS is labelled “+1” in Fig. 3A. Promoter -10 (5′-TTAAAT-3′) and -35 (5′-TTGCGC-3′) elements were observed at the expected positions upstream of the TSS. Throughout this work we refer to this promoter, highlighted purple in Fig. 3A, as PestA2. To confirm CRP binding at the predicted site we used DNase I footprinting (Fig. 3C). As expected, CRP protected the predicted target from digestion. Additionally, CRP induced DNase I hypersensitivity in the centre of the site. Note that the CRP site is centred 59.5 bp upstream of the TSS and adjacent to an AT-rich sequence that may be an UP element (Fig. 3A). Thus, we hypothesised that PestA2 is a class I CRP activated promoter. To test our hypothesis we first determined whether CRP could indeed activate PestA2. To do this, we compared LacZ expression in M182Δlac and M182ΔlacΔcrp cells carrying the PestA2::lacZ fusion. The data show that loss of CRP results in a 3-fold decrease in LacZ expression from PestA2 (Fig. 3D). We next tested the ability of CRP to activate PestA2 in vitro. The 93 bp DNA fragment was cloned upstream of the λoop terminator in plasmid pSR. In the context of this construct a 112 nt transcript is generated by RNA polymerase from PestA2 in vitro. The amount of transcript can then be quantified by electrophoresis. The result of the analysis, with and without CRP, is shown in Fig. 3E. As expected, an intense band corresponding to the 112 nt transcript was observed. Production of the transcript was stimulated by CRP. Note that CRP had no effect on production of the 108 nt control RNAI transcript from the plasmid replication origin. Finally, we examined the AT-rich DNA sequence (highlighted blue in Fig. 3A) located between the CRP site and the promoter -35 element. We found that increasing the GC content of the putative UP-element altered migration of the 93 bp DNA fragment on an agarose gel, consistent with a change in DNA topology (S3A Fig.). Moreover, these changes to the UP-element rendered PestA2 insensitive to CRP in vivo and in vitro (S3B Fig.).

H-NS excludes CRP from the estA2 promoter and represses estA2 transcription
Promoters can be liberated from H-NS repression if separated from flanking, H-NS bound, DNA [25]. We reasoned that this might be why, when isolated on the 93 bp fragment, PestA2 was active and dependent on CRP. To test this logic we generated a further two PestA2::lacZ fusions using the pRW50 plasmid system. The additional PestA2 DNA fragments were both 460 bp in length and include the full estA2 gene that was entirely bound by H-NS in our ChIP-seq assay (Fig. 2C). The CRP site was ablated in one of the additional fragments by introducing point mutations that are predicted to disrupt CRP binding. The sequence of the DNA fragments is shown in S2A Fig. The lacZ fusions are illustrated graphically in Fig. 4A. Our expectation was that the longer 460 bp fragment would bind H-NS whilst the starting 93 bp fragment would not. To test this prediction we used ChIP. Thus, we compared H-NS binding to the different PestA2 containing fragments in vivo. Fig. 4B shows results of a PCR analysis to measure enrichment of the PestA2 locus. As expected, PestA2 was only enriched in anti-H-NS immunoprecipitates when in the context of the 460 bp fragment. Crucially, enrichment is specific because, in a set of control PCR reactions, there was no enrichment of the yabN locus in any immunoprecipitate.

Our ChIP analysis suggests that the 460 bp fragment containing PestA2 is subject to regulation by H-NS. To confirm that this was the case, the various pRW50 derivatives were used to transform M182Δlac and M182ΔlacΔhns cells. We then measured LacZ activity, driven by PestA2, in the transformants. Consistent with our expectations the data show that PestA2 is repressed 5-fold by H-NS only in the context of the 460 bp DNA fragment (Fig. 4C). Importantly, mutations in the CRP binding site abolish PestA2 activity in the absence of H-NS. Hence, the measured LacZ expression must be driven by PestA2 rather than any spurious promoters located within the estA2 gene. Taken together our ChIP-seq and LacZ activity data show that H-NS prevents CRP from activating PestA2.
The estA2 and estA1 promoters are differently regulated by CRP but similarly regulated by H-NS
The estA1 regulatory region, located on plasmid p666, contains a sequence similar to PestA2 (Fig. 5A). We expected that this sequence would be the estA1 promoter (PestA1). To test this expectation we created a 92 bp PestA1::lacZ fusion, equivalent to the 93 bp PestA2::lacZ fusion described above, and mapped the 5′ end of the resulting mRNA. As expected, the primer extension product was 109 nt in length (Fig. 5B). Hence, PestA1 and PestA2 use equivalent TSSs. However, we were surprised that the intensity of the PestA1 primer extension product increased in cells lacking CRP (Fig. 5B). Closer examination of the alignment in Fig. 5A shows that, whilst PestA1 and PestA2 are similar, there are differences in the sequence and position of key promoter elements. To try and understand which changes result in the aberrant behaviour of PestA1 we made a set of hybrid promoters. The hybrid constructs are derived from the CRP-activated estA2 promoter. In each hybrid, named PestA2.1 through PestA2.7, a region of PestA2 was replaced with the equivalent region from PestA1 (see underlined sequences in Fig. 5C). The ability of the different hybrid promoters to drive lacZ expression, with and without CRP, was then tested. The results are shown in Fig. 5D. Note that, in Fig. 5D, the composition of each hybrid promoter is indicated in the grid below the graph. For example, PestA2.1 is derived from PestA2 but contains the PestA1 CRP site. As expected, both PestA1 and PestA2 were able to drive lacZ expression but CRP had opposite effects. Moreover, maximal expression from PestA1 was 3-fold lower than from PestA2. Only PestA2.3 and PestA2.5, which both carried the same changes in the promoter -35 element, exhibited a reversed dependence on CRP. Hence, the PestA1 -35 element must be responsible for the altered CRP dependence. All other hybrid promoters exhibited an overall reduction in activity compared to the parent PestA2 construct. We conclude that this combination of changes results in the lower activity of PestA1. Note that both PestA1 and PestA2 were bound by H-NS in our ChIP-seq analysis (Fig. 2C). We reasoned that cloning PestA1, with flanking DNA, would reveal H-NS mediated repression. We generated a derivative of the PestA1::lacZ fusion where the downstream boundary was extended to include the entire estA1 gene (S2B Fig., Fig. 5Ei). As expected, transcription from PestA1 was repressed by H-NS in the presence of downstream DNA (Fig. 5Eii).
The eltAB operon is indirectly repressed by CRP and directly repressed by H-NS
We next turned our attention to the LT toxin promoter (PeltAB) [26], [27]. Previously, Bodero and Munson [27] showed that transcription from this promoter was repressed by CRP. A mechanism for repression was proposed whereby CRP acted directly by binding three DNA targets overlapping PeltAB [27]. Even so, no CRP binding at PeltAB was identified by our ChIP-seq analysis (Fig. 6A). It is possible that this is because H-NS also excludes CRP from this locus (Fig. 6A). However, we also failed to identify CRP targets at PeltAB in our bioinformatic screen, even below the stringent cut-off (Fig. 2, S1 Table). In retrospect, this appears to be because all of three PeltAB CRP binding sites contain at least 4 mismatches to the consensus for CRP binding (Fig. 6A). Hence, we measured the affinity of CRP for PeltAB using EMSA assays. In parallel, we tested CRP binding to PestA2 as a control. As expected, CRP bound tightly to PestA2 at low concentrations (Fig. 6B, lanes 1–6). At high CRP concentrations further non-specific binding was observed (evidenced by a conspicuous “smear” in DNA migration in lane 7). In the equivalent experiment, with PeltAB, no specific binding of CRP was observed (lanes 8–13). However, non-specific CRP binding was again detectable at high protein concentrations (lane 14). Hence, CRP does not bind specifically to PeltAB. We hypothesised that previously observed changes in PeltAB activity, in cells lacking CRP, may occur indirectly. To test this, we cloned a 359 bp DNA fragment, containing PeltAB, into our pRW50 lacZ expression system. We also made a truncated 118 bp derivative of this construct where two of the three putative CRP targets were removed. A derivative of the truncated 118 bp construct, where the remaining CRP site was completely ablated by point mutations, was also made. The DNA sequences of the different constructs are shown in S2C Fig. They are illustrated graphically in Fig. 6Ci. Consistent with previous measurements, we found that transcription from PeltAB increased 2.5 fold in the absence of CRP. However, the response of PeltAB was identical when the CRP binding sites were removed (Fig. 6Cii). Hence, although CRP represses transcription from PeltAB, this must occur indirectly.

Given the configuration of H-NS binding at the eltAB locus (Fig. 6A) we reasoned that PeltAB would be repressed by H-NS in the presence of sufficient flanking DNA. As we had done previously for PestA1 and PestA2, we compared the binding of H-NS to PeltAB in the presence and absence of the downstream flanking sequence. The different DNA constructs are illustrated in Fig. 7A and results of ChIP experiments to measure H-NS binding are shown in Fig. 7B. As predicted, enrichment of PeltAB, in immunoprecipitations with anti-H-NS, was only observed in the presence of downstream DNA. Importantly, this enrichment was specific to PeltAB and not observed for the control locus yabN. Corresponding LacZ activities, for the different DNA constructs, measured in M182 or the Δhns derivative, are shown in Fig. 7C. Incorporation of flanking DNA downstream of PeltAB resulted in a 15-fold reduction in LacZ activity that was largely relieved in the absence of H-NS.

CRP and H-NS allow the estA1, estA2 and eltAB promoters to respond to glucose and salt
Given the established regulatory connections between CRP and glucose, and between H-NS and salt, we next measured changes in the activity of PestA1, PestA2 and PeltAB in response to glucose and salt. A complete description of assay conditions is provided in the Materials and Methods section. Briefly, to establish the range of conditions across which the promoters were able to respond, we examined the effect of titrating glucose or salt into the growth medium individually. In all experiments, we used the promoter::lacZ fusions that included downstream flanking DNA. This was to ensure that signals sensed by both CRP and H-NS could be integrated. As expected, the activity of PestA1 was low. Consequently, the effects of glucose and salt were negligible (S4A Fig.). Conversely, the activity of PestA2 was sensitive to both glucose and salt (S4B Fig.). Thus, lacZ expression driven by PestA2 was repressed by glucose (orange line) and enhanced by salt (green line). As expected, PeltAB activity increased in the presence of both salt and glucose, but induction by salt was more prominent (S4C Fig.). We hypothesised that, for PestA2, the inhibitory effect of glucose should override the stimulatory effect of salt. Our reasoning was that, although H-NS can repress PestA2, the promoter is ultimately dependent on CRP for activity. Hence, we examined the effect of adding salt and glucose, to cells carrying the PestA2::lacZ fusion, separately and in combination (Fig. 8A). As predicted, the inhibitory effect of glucose was dominant (Fig. 8Ai) and was still observed in the absence of H-NS (Fig. 8Aii). Conversely, the stimulatory effect of salt required H-NS (compare green bars in Fig. 8). Importantly, in a separate experiment, we also showed that the effect of glucose on PestA2 activity requires that the CRP site is intact (S4D Fig.). The combined effect of salt and glucose on PeltAB was more difficult to predict because CRP acts via an undefined, and indirect, mechanism. The result of the analysis (Fig. 8B) shows that the stimulatory effects of salt and glucose on transcription from PeltAB are not additive. Moreover, the stimulatory effect of glucose requires H-NS.

The response of PeltAB and PestA2 to CRP and H-NS is conserved in other ETEC isolates and during host cell attachment
Examination of all sequenced ETEC genomes reveals slight variations in the sequence of the eltAB and estA2 promoter sequences (recall that ETEC H10407 is somewhat anomalous in also encoding estA1). Thus, we next sought to understand if our model for regulation of LT and ST expression was broadly applicable. We focused our efforts on ETEC E24377A since i) the genome has been sequenced and ii) a vast array of independently generated transcriptomic data are available for this organism [28], [29]. Using ETEC E24377A DNA as a template, we generated a 460 bp PestA2, and 1126 bp PeltAB DNA fragment. The sequences are shown in S2D Fig. The DNA fragments were cloned into pRW50 and the ability of the promoters to drive lacZ expression in response to CRP and H-NS was measured. As expected, transcription from PestA2 was repressed by H-NS and activated by CRP whilst PeltAB was repressed by H-NS (Fig. 9A). We observed no effect of CRP on transcription from PeltAB in the context of the 1126 bp ETEC E24377A fragment. This is not unexpected because CRP acts indirectly and these indirect CRP effects have only previously been observed in the context of short DNA fragments containing PeltAB that are not subject to direct repression by H-NS. We note that Sahl and Rasko previously examined the global transcriptome response of E24377A to glucose levels and bile salts [28]. In exact agreement with our model for toxin regulation, and the data in Fig. 9A, this study confirmed that i) salt induced expression of both toxins and ii) glucose inhibited expression of estA2 [28]. Fortuitously, changes in the ETEC E24377A transcriptome, prompted by ETEC attachment to human gut epithelial cells, have also been quantified comprehensively [29]. Briefly, in these experiments, ETEC were added to sets of Caco-2 intestinal epithelial cell tissue cultures. Over a time course, ETEC that had adhered to host cells were separated from non-adhered ETEC. The transcriptomes of adhered and non-adhered ETEC were then compared. By mining these data, we next sought to determine if our model was consistent with observed changes in the transcription of crp, hns, eltA and estA during host cell attachment. Briefly, our data predict that changes in estA expression should be directly correlated to changes in the level of CRP and inversely correlated with changes in levels of H-NS. Conversely, levels of eltA expression should be inversely correlated with levels of H-NS. The result of the analysis is illustrated in Fig. 9B. The data show that the relative levels of crp transcription in attached and unattached cells are similar (orange line). However, levels of hns transcription change dramatically (green line) 60 minutes after host cell attachment. As predicted by our model, levels of estA2 and eltA transcription (dashed lines) inversely track changes hns transcript levels. When undertaking this analysis we noticed that, although there was little change in the relative level of crp mRNA between attached and unattached ETEC cells, the absolute level of crp mRNA did fluctuate across the time course of the experiment and between biological replicates. Strikingly, when these absolute mRNA levels are compared there is a clear linear relationship between crp and estA2 expression (Fig. 9C). Note that in Fig. 9C the absolute level of hns mRNA has been added in parenthesis for each data point. Remarkably, the only two outlying data points in this plot correspond to the two samples with increased hns expression. We conclude that regulation of estA2 and eltA by CRP and H-NS is important during the attachment of ETEC to human intestinal epithelial cells, and that the regulatory control of ETEC toxins is conserved across different strains.

Disrupting the regulatory switch attenuates ETEC virulence
Taken together, our data suggest that CRP and H-NS form a regulatory switch that controls ETEC toxicity. We next sought to examine the effect of disabling the switch on virulence. This is not straightforward because no animal model faithfully mimics the disease caused by ETEC in humans. However, intranasal mouse models have been used as a proxy for measuring E. coli pathogenicity [30]. Importantly, pathogenic E. coli cause more severe disease in this model than non-pathogenic strains [30]. Furthermore, ETEC strains lacking genes encoding toxins and known colonisation factors are less virulent in this model [31]. We opted to disrupt the regulatory switch by removing the crp rather than the hns gene. This was a deliberate decision since E. coli strains lacking hns are severely attenuated for growth in laboratory conditions. Conversely, the crp null derivative of ETEC H10407 was only mildly compromised for growth in liquid culture. Hence, we compared pathogenicity of ETEC H10407, and the crp derivative, using the intranasal mouse model [30]. Note that the outcome of this experiment is difficult to predict since the effects of CRP on pathogenicity likely go far beyond the control of toxin expression. However, it is reasonable to assume that ETEC virulence should differ in cells lacking crp. The median survival of mice challenged with wild type ETEC was 53 hours and the mortality rate was 100%. Conversely, the median survival of mice challenged with Δcrp ETEC was 72 h and 20% of the mice survived (Fig. 9D). Thus, whilst the full extent to which CRP co-ordinates the ETEC virulence programme remains to be determined, CRP is clearly central to the pathogenic response.
Discussion
A complex hierarchy of salt and glucose-dependent regulation controls toxin expression
We propose that toxin expression in ETEC can be controlled by osmo-metabolic flux. This is relevant to conditions in the small intestine (osmolarity equivalent to 300 mM NaCl) disease symptoms (the extrusion of cations and cAMP into the gut lumen) and treatment (the ingestion of solutions containing glucose and salt) [7]–[11], [32]. A molecular model, describing how the different signals are integrated, is illustrated in Fig. 10. Two gene regulatory proteins, CRP and H-NS, are central to our model. Hence, H-NS directly represses the expression of eltAB, estA1 and estA2 (pathways “a” and “b” in Fig. 10). For estA2 and eltAB this repression can be relieved, in an H-NS dependent manner, by increased osmolarity. At PestA2 CRP directly activates transcription by a Class I mechanism (pathway “c”). H-NS can interfere with this process by competing with CRP for binding at PestA2 (pathway “d”). Finally, CRP can indirectly repress expression of eltAB via an unknown pathway that is influenced by H-NS (“e”). Both pathways “c” and “e” are sensitive to glucose availability because of their dependence on CRP. We speculate that pathway “e” may include H-NS since the effects of salt and sugar on eltAB expression were epistatic (Fig. 8). Our model for H-NS repression of eltAB is consistent with previous work [26]. However, our conclusion that eltAB is indirectly repressed by CRP disagrees with a previous study [27]. Even so, we were able to faithfully reproduce most of the observations previously described by Bodero and Munson [27]. We note that Bodero and Munson previously suggested that CRP may bind targets at PeltAB with a 7, rather than 6, base pair spacer between the two CRP half sites. Such CRP targets have never been described amongst hundreds of known CRP regulated promoters. Furthermore, we found no such CRP sites in our ChIP-seq analysis. Given that these DNA sequences can be deleted, without negating the effect of CRP on PeltAB activity, the regulatory effect of CRP must be indirect.

Oral Rehydration Therapy is likely to impact on toxin expression
Our model for regulation of ST and LT expression is pertinent to both ETEC mediated disease and its treatment. ST and LT trigger the extrusion of H2O, cations, and cAMP (the cofactor for CRP) from the small intestine into the gut lumen [4]–[9]. Furthermore, solutions of salt and glucose are consumed by patients to reverse this process [10], [11]. We speculate that, during infection, extrusion of electrolytes and cAMP into the gut lumen could create a positive feedback loop to drive toxin expression. Importantly, our model also suggests that ORT may provide benefits beyond stimulating rehydration of the patient. The concentration of glucose used in ORT is ∼10-fold higher than is required to repress estA2 expression. Hence, even if 90% of glucose present in ORT solutions is absorbed before reaching the site of infection, sufficient glucose should be present to down regulate toxin expression. Furthermore, even though salt is able to induce expression of estA2 and eltAB, the effect is only observed at concentrations far higher than those found in ORT solutions.
Differential regulation of estA1 and estA2 by CRP
Our observation that estA1 and estA2 are oppositely regulated by CRP is intriguing given the similarities between the promoter sequences of these genes. Differential regulation is dependent on the promoter -35 element (Fig. 5). At Class I CRP regulated promoters an αCTD protomer sits between CRP and domain 4 of the RNA polymerase σ subunit, which is bound to the promoter -35 element [12]. Thus, one possible explanation is that changes in the -35 element result in subtle repositioning of σ. This could result in unproductive interactions between αCTD and σ when CRP is present.
H-NS prevents CRP regulation of select target genes
Our data indicate that several strong CRP binding sites in the H10407 genome are occluded by H-NS. This strongly suggests that the CRP regulon has evolved to incorporate additional environmental signals through the action of H-NS. The repressive effect of H-NS on transcription has been widely described [23]. H-NS represses transcription predominantly by occluding the binding of RNAP or by trapping RNAP at promoters [20]. Recently, it was shown that H-NS occludes many binding sites for the CRP homologue, FNR, in E. coli [21]. Thus, occlusion of transcription factor binding sites appears to be a major function of H-NS, especially for CRP family proteins. Note that, in order to exclude CRP from target promoters, sites of H-NS nucleation and CRP binding need not overlap precisely. For example, at both estA1 and estA2, maximal H-NS binding is observed within the coding sequence of the gene (Fig. 2C). Despite this, H-NS oligomerisation across adjacent DNA is sufficient to prevent CRP binding.
Conclusions
In summary, our model provides a framework for better understanding ETEC mediated disease and its treatment. Moreover, our catalogue of CRP and H-NS binding targets provide a useful community resource for further studies of all E. coli strains. In particular, our ChIP-seq data for CRP report >50 targets not identified previously in E. coli K-12 and 8 ETEC-specific targets. Finally, our data show how very small changes in the organisation of gene regulatory regions can have major effects on gene expression, such that transcription responds differently to the same environmental cues.
Materials and Methods
Strains, plasmids and oligonucleotides
ETEC strain H10407 is described by Crossman et al. [1]. The C-terminal crp-3×FLAG tag was introduced into the H10407 chromosome using the recombineering method of Stringer et al. [33]. Wild type E. coli K-12 strains JCB387 and M182 have been described previously [34], [35]. The Δhns M182 derivative was generated by P1 transduction of hns::kan from E. coli K12 derivative YN3144 (a gift from Ding Jin). Plasmids pRW50 and pSR are described by Lodge et al. [36] and Kolb et al. [37]. More detailed descriptions of strains and plasmids, along with the sequences of oligonucleotides, are provided in S2 Table.
ChIP-seq
Cultures were grown to mid-log phase in M9 minimal medium with 1% (w/v) fructose at 37°C. Targeted ChIP experiments (Fig. 4 and 6) were done exactly as described by Singh and Grainger [38] using PestA2 or PeltAB fragments cloned in pRW50 carried in strain M182. The ChIP-seq was done as described extensively by Singh et al. [25] using strain H10407. Briefly, H-NS and CRP-3×FLAG were immunoprecipitated using protein A sepharose (GE Healthcare) in combination with 2 µL of anti-H-NS or 2 µl of anti-FLAG respectively. After immunoprecipitation and washing, beads were resupended in 100 µL 1× Quick Blunting Buffer (NEB) with dNTPs (as specified by the manufacturer) and 2 µL Quick Blunting Enzyme Mix, and incubated for 30 minutes at 24°C with gentle mixing. After being collected by centrifugation, the beads were again washed and the associated DNA was A-tailed by resuspension of beads in 100 µL 1× NEB buffer #2 supplemented with 2 mM dATP and 10 units of Klenow Fragment (3′→5′ exo-; NEB). Following incubation for 30 minutes at 37°C, with gentle mixing, the beads were again collected and washed. Illumina adapters (1 µl NEXTflex ChIP-seq barcoded adapters; BioO Scientific) were added to beads resuspended in 100 µL 1× Quick Ligation reaction buffer and 4 µL Quick T4 DNA Ligase (NEB), and incubated for 15 minutes at 24°C with gentle mixing. After washing the beads, the DNA was the eluted into a fresh tube by addition of 100 µL ChIP elution buffer (50 mM Tris–HCl, pH 7.5, 10 mM EDTA, 1% SDS) and incubation at 65°C for 10 minutes. The eluate was collected by centrifugation for one minute at 4000 rpm. Crosslinks were reversed by incubation for 10 minutes at 100°C. Samples were purified by phenol extraction and precipitated with ethanol, 40 µg glycogen and 8.3 mM sodium acetate. DNA was pelleted for 15 minutes at 4°C at top speed in a microcentrifuge, washed with 70% ethanol, dried and resuspended in 11 µL H2O. After quantification by PCR each library was amplified, purified and resuspended in 20 µL H2O. Libraries were the sequenced using a HiSeq 2000 sequencer (Illumina; University at Buffalo Next Generation Sequencing Core Facility). Sequence reads were aligned to non-repetitive sequences in the E. coli H10407 genome using CLC Genomics Workbench and overall coverage was determined using custom Python scripts. Sequence reads have been submitted to the EBI ArrayExpress database and can be accessed using accession number E-MTAB-2917.
Bioinformatics
ChIP-seq peaks were identified as described previously [25]. We refer to these peaks as “high stringency” peaks. A second round of peak calling was performed in which the sequence read threshold values (i.e. the minimum number of sequence reads at a given genomic position that is required for a peak to be called) was reduced by 20%. We refer to these peaks as “low stringency” peaks. MEME [39] was used to identify enriched sequence motifs in the sequences from 50 bp upstream to 50 bp downstream of the high stringency peak centres. Thus, we identified a motif closely resembling the known CRP consensus site in many of the regions surrounding high stringency ChIP-seq peaks. These CRP site sequences are included in Table 1. Those high stringency peaks for which MEME did not identify a motif were used for a second round of analysis using MEME. This also identified a motif closely resembling the known CRP consensus site. These CRP site sequences are also included in Table 1. We used MEME to identify enriched sequence motifs in the low stringency peak list. This also identified a motif closely resembling the known CRP consensus site. These CRP site sequences are also included in Table 1. “High-confidence” ChIP-seq peaks listed in Table 1 include all the high stringency peaks but only those low stringency peaks for which we identified a motif using MEME. A complete list of all peaks, including low stringency peaks for which a motif was not identified by MEME, is provided in S3 Table. In order to assess the location of CRP sites with respect to TSSs we used the targets listed in Table 1. For each target the predicted sequence from MEME was used in a BLAST search against the E. coli K-12 MG1655 genome. All but 11 CRP sites in ETEC had a single perfect match in the E. coli K-12 chromosome. For each perfect match the distance from the centre of the CRP site to all transcription start sites was calculated. Transcription start site coordinates are from Kim et al. [40] and Cho et al. [41]. Distances between −200 and +100 were selected and all other distances were discarded. Distances were then grouped in bins of 5 bp each and the most common distance bins were identified. Note that, because the position of the CRP site was transposed onto the E. coli K-12 genome, the distance between CRP sites and TSSs
The PWM describing CRP binding sites was generated using the PREDetector software package and our previous list of 68 CRP binding sites in the E. coli K-12 genome [15], [42]. Subsequent bioinformatic screens of plasmids p666 and p948 were done by importing the relevant genbank files into PREDetector and running a binding site search with a cut-off of 7 using settings that did not exclude CRP sites within genes. The “score” for each site predicted by PREDetector increases if a closer match to the PWM is found. To generate the chromosome and plasmid maps shown in Fig. 1 we used DNA plotter software [43].
Data shown in Fig. 9B–C were extracted from the publically available datasets of Kansal et al. [29] that measure changes in the ETEC E24377A transcriptome upon contact with Caco-2 intestinal epithelial cells. The data are hosted under the GEO accession code GSE40427. For each assay condition (planktonic and attached ETEC cells) we extracted the signal intensity for microarray probe sets A1527 (crp), UTI189_C1433 (hns), D4754 (eltA) and D4048 (estA). The average signal intensity was calculated and the fold change in transcription in attached compared to planctonic ETEC cells was determined for each time point. The data in Fig. 9C show a comparison of absolute signal intensities for probe sets A1527 (crp) and D4048 (estA) compared for each of the two replicates obtained at 30, 60 or 120 minutes after attachment to host cells. Signal intensities obtained after 30 minutes growth in LB medium (three replicates) are also included in this analysis.
Proteins
The CRP and σ70 purification was done exactly as described previously [44], [45]. RNA polymerase core enzyme was purchased from Epicenter. RNA polymerase holoenzyme was generated by incubating the core enzyme with an equimolar concentration of σ70 at room temperature for 20 minutes prior to use. H-NS was overexpressed in T7 express cells from plasmid pJ414hns. After overexpressing H-NS, cells were collected from the culture by centrifugation and resuspended in buffer A (20 mM Tris-HCl pH 7.2, 1 mM EDTA and 10% (v/v) glycerol) containing 100 mg/ml PMSF. Cells were lysed by sonication and the sample was cleared by centrifugation. The supernatent was loaded directly onto a Heparin column (Amersham) pre-equilibrated with buffer A. A linear NaCl gradient was applied and H-NS was found to elute at approximately 500 mM NaCl. The peak fractions were pooled and diluted 3-fold with buffer A. The sample was then loaded onto an S-FF column (Amersham) pre-equilibrated with Buffer A. A NaCl gradient was applied and H-NS eluted at approximately 550 mM NaCl. The H-NS containing fractions were then dialysed against a buffer containing 20 mM Tris HCl (pH 7.2), 300 mM KCl and 10% Glycerol (v/v)for storage at −80°C.
DNAse I footprinting and Electrophoretic Mobility Shift Assays
DNA fragments for DNAse I footprinting or EMSA assays were excised from pSR by sequential digestion with HindIII and then AatII. After digestion, fragments were labelled at the HindIII end using [γ-32P]-ATP and T4 polynucleotide kinase. DNAse I footprints and EMSA experiments were then done as described by Grainger et al. [45] except that cAMP was added to reactions at a concentration of 0.2 mM. Radio-labelled DNA fragments were used at a final concentration of ∼10 nM. Note that all in vitro DNA binding reactions contained a vast excess (12.5 µg ml−1) of Herring sperm DNA as a non-specific competitor. Footprints were analysed on a 6% DNA sequencing gel (molecular dynamics). The results of all footprints and EMSA experiments were visualized using a Fuji phosphor screen and Bio-Rad Molecular Imager FX.
Primer extension assays
Transcript start sites were mapped by primer extension, as described in Lloyd et al. [46] using RNA purified from strains carrying the 92 bp PestA1 or 93 bp PestA2 fragment cloned in pRW50. The 5′ end-labelled primer D49724, which anneals downstream of the HindIII site in pRW50, was used in all experiments. Primer extension products were analysed on denaturing 6% polyacrylamide gels, calibrated with size standards, and visualized using a Fuji phosphor screen and Bio-Rad Molecular Imager FX.
In vitro transcription assays
The in vitro transcription experiments were performed as described previously Savery et al. [35] using the system of Kolb et al. [38]. A Qiagen maxiprep kit was used to purify supercoiled pSR plasmid carrying the different promoter inserts. This template (∼16 µg ml−1) was pre-incubated with purified CRP in buffer containing 0.2 mM cAMP, 20 mM Tris pH 7.9, 5 mM MgCl2, 500 µM DTT, 50 mM KCl, 100 µg ml−1 BSA, 200 µM ATP, 200 µM GTP, 200 µM CTP, 10 µM UTP with 5 µCi [α-32P]-UTP. The reaction was started by adding purified E. coli RNA polymerase. Labelled RNA products were analysed on a denaturing polyacrylamide gel.
β-galactosidase assays and addition of glucose and salt to growth medium
β-Galactosidase assays were done using the protocol of Miller [47]. All assay values are the mean of three independent experiments with a standard deviation <10% of the mean. Cells were grown aerobically at 37°C to mid-log phase in LB medium unless stated otherwise. For all experiments investigating the effects of glucose and salt M9 minimal medium was used so that the glucose and salt concentrations could be controlled more accurately. The amount of glucose is shown as percentage w/v. The addition of “salt” refers to a 3∶1 molar ration of NaCl to KCl. We have arbitrarily described 30 mM NaCl and 10 mM KCl as being a “1%” salt solution.
Intranasal mouse infection model assays
Strains of ETEC were grown in Luria Broth (LB) to an OD600 of 1.0. Groups of 10 mice (8–10 week old BALB/c) were infected intranasally with approximately 1×109 colony forming units of bacteria in 100 µl of inoculums according to Byrd et al. [30]. Mice were monitored daily for 6 days post-infection for weight and morbidity.
Ethics statement
The protocol 12-02-015IBT “Oral Immunization of Mice with Enterotoxigenic: E coli (ETEC)” has been approved by the Noble Life Sciences IACUC committee. All animal care and use procedures adhere to the guidelines set by the Public Health Service Policy, U.S. Dept. of Agriculture (USDA) and the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.
Supporting Information
Zdroje
1. CrossmanLC, ChaudhuriRR, BeatsonSA, WellsTJ, DesvauxM, et al. (2010) A commensal gone bad: complete genome sequence of the prototypical enterotoxigenic Escherichia coli strain H10407. J Bacteriol 192 : 5822–5831.
2. SackRB (2011) The discovery of cholera - like enterotoxins produced by Escherichia coli causing secretory diarrhoea in humans. Indian J Med Res 133 : 171–80.
3. GuptaSK, KeckJ, RamPK, CrumpJA, MillerMA, et al. (2008) Analysis of Data Gaps Pertaining to Enterotoxigenic Escherichia coli Infections in Low and Medium Human Development Index Countries, 1984–2005. Epidemiology and Infection 136 : 721–738.
4. de HaanL, HirstTR (2004) Cholera toxin: a paradigm for multi-functional engagement of cellular mechanisms. Mol Membr Biol 21 : 77–92.
5. ZhangRG, ScottDL, WestbrookML, NanceS, SpanglerBD, et al. (1995) The three-dimensional crystal structure of cholera toxin. J Mol Biol 251 : 563–73.
6. TaxtA, AaslandR, SommerfeltH, NataroJ, PuntervollP (2010) Heat-stable enterotoxin of enterotoxigenic Escherichia coli as a vaccine target. Infect Immun 78 : 1824–31.
7. YamamotoT, TamuraT, YokotaT (1984) Primary structure of heat-labile enterotoxin produced by Escherichia coli pathogenic for humans. J Biol Chem 259 : 5037–44.
8. de HaanL, VerweijWR, FeilIK, HoltropM, HolWG, et al. (1998) Role of GM1 binding in the mucosal immunogenicity and adjuvant activity of the Escherichia coli heat-labile enterotoxin and its B subunit. Immunology 94 : 424–430.
9. SaslowskyDE, te WelscherYM, ChinnapenDJ, WagnerJS, WanJ, et al. (2013) Ganglioside GM1-mediated transcytosis of cholera toxin bypasses the retrograde pathway and depends on the structure of the ceramide domain. J Biol Chem 288 : 25804–9.
10. NalinDR, CashRA, IslamR, MollaM, PhillipsRA (1968) Oral maintenance therapy for cholera in adults. Lancet 2 : 370–3.
11. GuerrantRL, Carneiro-FilhoBA, DillinghamRA (2003) Cholera, diarrhea, and oral rehydration therapy: triumph and indictment. Clin Infect Dis 37 : 398–405.
12. BusbyS, EbrightRH (1999) Transcription activation by catabolite activator protein (CAP). J Mol Biol 293 : 199–213.
13. ParkinsonG, WilsonC, GunasekeraA, EbrightYW, EbrightRH, et al. (1996) Structure of the CAP-DNA complex at 2.5 angstroms resolution: a complete picture of the protein-DNA interface. J Mol Biol 260 : 395–408.
14. ZhengD, ConstantinidouC, HobmanJL, MinchinSD (2004) Identification of the CRP regulon using in vitro and in vivo transcriptional profiling. Nucleic Acids Res 32 : 5874–93.
15. GraingerDC, HurdD, HarrisonM, HoldstockJ, BusbySJ (2005) Studies of the distribution of Escherichia coli cAMP-receptor protein and RNA polymerase along the E. coli chromosome. Proc Natl Acad Sci U S A 102 : 17693–8.
16. RossiterAE, BrowningDF, LeytonDL, JohnsonMD, GodfreyRE, et al. (2011) Transcription of the plasmid-encoded toxin gene from enteroaggregative Escherichia coli is regulated by a novel co-activation mechanism involving CRP and Fis. Mol Microbiol 81 : 179–91.
17. NavarreWW, PorwollikS, WangY, McClellandM, RosenH, et al. (2006) Selective silencing of foreign DNA with low GC content by the H-NS protein in Salmonella. Science 313 : 236–8.
18. LiuY, ChenH, KenneyLJ, YanJ (2010) A divalent switch drives H-NS/DNA-binding conformations between stiffening and bridging modes. Genes Dev 24 : 339–44.
19. LimCJ, LeeSY, KenneyLJ, YanJ (2012) Nucleoprotein filament formation is the structural basis for bacterial protein H-NS gene silencing. Sci Rep 2 : 509.
20. MyersKS, YanH, OngIM, ChungD, LiangK, et al. (2013) Genome-scale analysis of Escherichia coli FNR reveals complex features of transcription factor binding. PLoS Genet 9 (6) e1003565.
21. DameRT, WymanC, WurmR, WagnerR, GoosenN (2002) Structural basis for H-NS-mediated trapping of RNA polymerase in the open initiation complex at the rrnB P1. J Biol Chem 277 : 2146–50.
22. DormanCJ (2007) H-NS, the genome sentinel. Nat Rev Microbiol 5 : 157–61.
23. AtlungT, IngmerH (1997) H-NS: a modulator of environmentally regulated gene expression. Mol Microbiol 24 : 7–17.
24. SteinslandH1, Valentiner-BranthP, PerchM, DiasF, FischerTK, AabyP, MølbakK, SommerfeltH (2002) Enterotoxigenic Escherichia coli infections and diarrhea in a cohort of young children in Guinea-Bissau. J Infect Dis 186 : 1740–7.
25. SinghSS, SinghN, BonocoraRP, FitzgeraldDM, WadeJT, et al. (2014) Widespread suppression of intragenic transcription initiation by H-NS. Genes Dev 28 : 214–219.
26. YangJ, TauschekM, StrugnellR, Robins-BrowneRM (2005) The H-NS protein represses transcription of the eltAB operon, which encodes heat-labile enterotoxin in enterotoxigenic Escherichia coli, by binding to regions downstream of the promoter. Microbiology 151 : 1199–1208.
27. BoderoMD, MunsonGP (2009) Cyclic AMP receptor protein-dependent repression of heat-labile enterotoxin. Infect Immun 77 : 791–8.
28. SahlJW, RaskoDA (2012) Analysis of global transcriptional profiles of enterotoxigenic Escherichia coli isolate E24377A. Infect Immun 80 : 1232–1242.
29. KansalR, RaskoDA, SahlJW, MunsonGP, RoyK (2013) Transcriptional modulation of enterotoxigenic Escherichia coli virulence genes in response to epithelial cell interactions. Infect Immun 81 : 259–70.
30. ByrdW, MogSR, CasselsFJ (2003) Pathogenicity and immune response measured in mice following intranasal challenge with enterotoxigenic Escherichia coli strains H10407 and B7A. Infect Immun 71 : 13–21.
31. ByrdW, BoedekerEC (2013) Attenuated Escherichia coli strains expressing the colonization factor antigen I (CFA/I) and a detoxified heat-labile enterotoxin (LThK63) enhance clearance of ETEC from the lungs of mice and protect mice from intestinal ETEC colonization and LT-induced fluid accumulation. Vet Immunol Immunopathol 152 : 57–67.
32. GuptaS, ChowdhuryR (1997) Bile affects production of virulence factors and motility of Vibrio cholerae. Infect Immun 65 : 1131–1134.
33. StringerAM, SinghN, YermakovaA, PetroneBL, AmarasingheJJ, et al. (2012) FRUIT, a scar-free system for targeted chromosomal mutagenesis, epitope tagging, and promoter replacement in Escherichia coli and Salmonella enterica. PLoS One 7: e44841.
34. PageL, GriffithsL, ColeJA (1990) Different physiological roles of two independent pathways for nitrite reduction to ammonia by enteric bacteria. Arch Microbiol 154 : 349–54.
35. BusbyS, KotlarzD, BucH (1983) Deletion mutagenesis of the Escherichia coli galactose operon promoter region. J Mol Biol 167 : 259–274.
36. LodgeJ, FearJ, BusbyS, GunasekaranP, KaminiNR (1992) Broad host range plasmids carrying the Escherichia coli lactose and galactose operons. FEMS Microbiol Lett 74 : 271–6.
37. KolbA, KotlarzD, KusanoS, IshihamaA (1995) Selectivity of the Escherichia coli RNA polymerase E sigma 38 for overlapping promoters and ability to support CRP activation. Nucleic Acids Res 23 : 819–26.
38. SinghSS, GraingerDC (2013) H-NS can facilitate specific DNA-binding by RNA polymerase in AT-rich gene regulatory regions. PLoS Genet 9: e1003589.
39. BaileyTL, BodenM, BuskeFA, FrithM, GrantCE, et al. (2009) MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37 (Web Server issue) W202–8.
40. KimD, HongJS, QiuY, NagarajanH, SeoJH, et al. (2012) Comparative analysis of regulatory elements between Escherichia coli and Klebsiella pneumoniae by genome-wide transcription start site profiling. PLoS Genet 8 (8) e1002867.
41. ChoBK, KimD, KnightEM, ZenglerK, PalssonBØ (2014) Genome-scale reconstruction of the sigma factor network in Escherichia coli: topology and functional states. BMC Biol 12 : 4.
42. HiardS, MareeR, ColsonS, HoskissonPA, TitgemeyerF, et al. (2007) PREDetector: A new tool to identify regulatory elements in bacterial genomes. Biochem Biophys Res Commun 357 (4) 861–4.
43. CarverT, ThomsonN, BleasbyA, BerrimanM, ParkhillJ (2009) DNAPlotter: circular and linear interactive genome visualization. Bioinformatics 25 : 119–20.
44. SaveryNJ, LloydGS, KainzM, GaalT, RossW, et al. (1998) Transcription activation at Class II CRP-dependent promoters: identification of determinants in the C-terminal domain of the RNA polymerase alpha subunit. EMBO J 17 : 3439–3447.
45. GraingerDC, GoldbergMD, LeeDJ, BusbySJ (2008) Selective repression by Fis and H-NS at the Escherichia coli dps promoter. Mol Microbiol 68 : 1366–1377.
46. LloydGS, HollandsK, GodfreyRE, BusbySJ (2008) Transcription initiation in the Escherichia coli K-12 malI-malX intergenic region and the role of the cyclic AMP receptor protein. FEMS Microbiol Lett 288 : 250–7.
47. Miller J (1972) Experiments in Molecular Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 1
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Infections in Humans and Animals: Pathophysiology, Detection, and Treatment
- : Trypanosomatids Adapted to Plant Environments
- Environmental Drivers of the Spatiotemporal Dynamics of Respiratory Syncytial Virus in the United States
- Dengue Virus RNA Structure Specialization Facilitates Host Adaptation
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy