
Transcriptional Analysis of Murine Macrophages Infected with Different Strains Identifies Novel Regulation of Host Signaling Pathways
Most isolates of Toxoplasma from Europe and North America fall into one of three genetically distinct clonal lineages, the type I, II and III lineages. However, in South America these strains are rarely isolated and instead a great variety of other strains are found. T. gondii strains differ widely in a number of phenotypes in mice, such as virulence, persistence, oral infectivity, migratory capacity, induction of cytokine expression and modulation of host gene expression. The outcome of toxoplasmosis in patients is also variable and we hypothesize that, besides host and environmental factors, the genotype of the parasite strain plays a major role. The molecular basis for these differences in pathogenesis, especially in strains other than the clonal lineages, remains largely unexplored. Macrophages play an essential role in the early immune response against T. gondii and are also the cell type preferentially infected in vivo. To determine if non-canonical Toxoplasma strains have unique interactions with the host cell, we infected murine macrophages with 29 different Toxoplasma strains, representing global diversity, and used RNA-sequencing to determine host and parasite transcriptomes. We identified large differences between strains in the expression level of known parasite effectors and large chromosomal structural variation in some strains. We also identified novel strain-specifically regulated host pathways, including the regulation of the type I interferon response by some atypical strains. IFNβ production by infected cells was associated with parasite killing, independent of interferon gamma activation, and dependent on endosomal Toll-like receptors in macrophages and the cytoplasmic receptor retinoic acid-inducible gene 1 (RIG-I) in fibroblasts.
Published in the journal:
. PLoS Pathog 9(12): e32767. doi:10.1371/journal.ppat.1003779
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1003779
Summary
Most isolates of Toxoplasma from Europe and North America fall into one of three genetically distinct clonal lineages, the type I, II and III lineages. However, in South America these strains are rarely isolated and instead a great variety of other strains are found. T. gondii strains differ widely in a number of phenotypes in mice, such as virulence, persistence, oral infectivity, migratory capacity, induction of cytokine expression and modulation of host gene expression. The outcome of toxoplasmosis in patients is also variable and we hypothesize that, besides host and environmental factors, the genotype of the parasite strain plays a major role. The molecular basis for these differences in pathogenesis, especially in strains other than the clonal lineages, remains largely unexplored. Macrophages play an essential role in the early immune response against T. gondii and are also the cell type preferentially infected in vivo. To determine if non-canonical Toxoplasma strains have unique interactions with the host cell, we infected murine macrophages with 29 different Toxoplasma strains, representing global diversity, and used RNA-sequencing to determine host and parasite transcriptomes. We identified large differences between strains in the expression level of known parasite effectors and large chromosomal structural variation in some strains. We also identified novel strain-specifically regulated host pathways, including the regulation of the type I interferon response by some atypical strains. IFNβ production by infected cells was associated with parasite killing, independent of interferon gamma activation, and dependent on endosomal Toll-like receptors in macrophages and the cytoplasmic receptor retinoic acid-inducible gene 1 (RIG-I) in fibroblasts.
Introduction
Toxoplasma gondii is a ubiquitous obligate intracellular protozoan parasite that can invade and replicate in almost all cells of a wide range of warm-blooded animals [1]. In humans it is the 2nd most important foodborne pathogen in terms of annual cost of illness and quality adjusted life year loss [2]. Normally a lifelong, largely asymptomatic, infection is established but in immunocompromised individuals and in congenital infections Toxoplasma infection can lead to severe disease and even death. Nevertheless, not all seropositive immunosuppressed patients have reactivating toxoplasmosis [3], and not all congenital infections lead to disease [4]. There is good evidence that both the host genetic background [5]–[7] and the genotype of the infecting strain [8], [9] play a role in the severity of disease.
Despite the existence of a sexual phase in its life cycle, which only occurs in felines, few strains dominate human infections in Europe and North America. Type II strains dominate in Europe, while in North America, types 12, II and III account for the majority of strains isolated from wild-life and patients [9]–[11]. Genotypes not belonging to these lineages are predominant in South America [12]–[14]. A phylogenetic analysis of 956 strains, using single nucleotide polymorphisms (SNPs) identified in five loci, clustered these into 15 haplogroups, including type I, II and III [11], [15]–[17]. Using genome-wide SNPs, it was shown that even within these haplogroups there is often significant diversity and many strains did not fit into the 15 proposed haplogroups. Instead, many strains appear to have formed through recent recombination events [18].
The relationship between strain genotype and virulence in the mouse model is well established; type I isolates, and most South American strains, are highly virulent (LD100 ∼1) [19], whereas type II and III strains are less virulent, with LD50 of ∼103 and 105, respectively [20]. Toxoplasma invasion and modulation of its host cell is mediated by proteins secreted from three secretory organelles, called micronemes, rhoptries and dense granules [21]. Using crosses between type I, II and III it was determined that strain differences in virulence in mice can be largely explained by the exact allelic combination of the Toxoplasma ROP18 and ROP5 genes [19], [20], [22]–[25]. ROP18 and ROP5 are a secreted rhoptry kinase and pseudokinase, respectively, that cooperatively inhibit the murine IFNγ-induced immunity-related GTPases (IRGs) [26]–[29]. These IRGs can vesiculate the parasitophorous vacuole membrane (PVM) which ultimately leads to the destruction of the parasite inside [30]. Toxoplasma polymorphic secreted effectors that modulate host immune signaling pathways also play a role in the differences in virulence between type I, II and III strains. For example, the secreted rhoptry kinase ROP16 from type I and III, but not from type II, is involved in constitutive activation of the STAT transcription factors [22], [31] while the secreted dense granule protein GRA15, from type II, but not from type I and III, mediates high levels of NFκB activation [32]. Expression level differences in ROP38, another secreted rhoptry kinase, mediates strain differences in gene activation along the MAP kinase pathway [33]. Thus, the precise allelic combination of Toxoplasma secreted effectors likely determines how distinct Toxoplasma strains modulate the host immune response. GRA15 and ROP16 effects can be observed in human, mouse and chicken cells [32], [34], suggesting that Toxoplasma effectors that modulate host cell transcription might have evolved to target conserved host proteins.
Part of the variability in disease outcome in human infections may also be tied to strain type. The few severe congenital toxoplasmosis cases in Europe seem to be caused by atypical strains (not type I, II or III) [35]. In North America the incidence of more severe human congenital infections is higher compared to Europe and this seems to be associated with a higher prevalence of non-type II strains [9], most likely type 12 strains as these are also prevalent in North America. North American atypical strains have also been associated with severe ocular disease in non immunosuppressed patients [36]. In South America, where Toxoplasma strain diversity is high, congenital and ocular toxoplasmosis are more prevalent compared to Europe and more often associated with severe symptoms [4], [37]. Some strains from French Guiana can cause severe disease and even death in non immunosuppressed individuals [38]–[41].
Most of the data currently available on Toxoplasma-host cell interactions were obtained using the canonical types I, II or III strains, and little is known about how the atypical strains interact with and modify host cells. Because these atypical strains are correlated with more severe disease manifestations, it is important to determine how they differ from the canonical strains in modulating the host cell. To identify host cell signaling pathways uniquely modulated by atypical Toxoplasma strains we infected macrophages with 29 different strains, representing world-wide diversity, and obtained global transcriptional profiles of both the host and parasites. The parasite transcriptomes showed large differences in gene expression, including expression of host effectors, which correlated with differences in expression of transcriptional regulators such as the Apetala-2 (AP2) factors. Combining genome and transcriptome data also identified that some strains had large structural variations that had an impact on gene expression levels. Analysis of the host cell transcriptomes revealed activation of several signaling pathways in a strain-specific manner. For example, only a few atypical strains induce macrophage type I interferon production, which was associated with parasite killing, independent of interferon gamma, that leads to the release of parasite ligands that trigger intracellular Toll-like receptors and cytoplasmic nucleic acid sensors. This resource provides insight into the host response to a large variety of Toxoplasma strains and, together with the SNPs dataset we recently published for these strains [18], can form the basis for the discovery of novel Toxoplasma effectors.
Results
Co-regulated host and parasite gene clusters are enriched in functional annotation
We used RNA-sequencing (RNA-seq) of poly-adenylated RNA, obtained after infection of primary murine bone-marrow derived macrophages with 29 different Toxoplasma strains for 20 h, to capture the mouse and Toxoplasma transcriptomes. We detected 51% (13,179) of murine genes and 93% (8,138) of Toxoplasma genes (RPKM>1 in at least 1 sample) (Table S1 available online). Overall Toxoplasma induced macrophage gene expression correlated well with Affymetrix microarrays performed on these cells with some of the same strains (Protocols S1–2) but with significantly more genes detected by RNA-seq and a larger dynamic range (Figure S1). We focused on 2,451 differentially expressed Toxoplasma (RPKM>10 in at least 1 sample and at least 4-fold different between 2 samples) and 5,016 differentially regulated host genes (RPKM>5 in at least 1 sample and at least 2-fold different between any 2 samples). To identify co-regulated host and parasite genes we clustered the macrophage and parasite gene expression profiles and also clustered the experiments (strains) to identify parasite strains that modulate host gene expression similarly or that have similar parasite gene expression profiles. Clustering the macrophage and parasite expression data independently into 9–13 major clusters each adequately partitioned the data into co-regulated gene clusters (Figure 1; Figures S2, S3). Most clusters had significant functional enrichment in biological processes and the promoters of the genes in the clusters were significantly enriched for transcription factor binding sites (TFBS) indicating that these clusters represent differential modulation of host/parasite signaling pathways by the different Toxoplasma strains. The induction of host genes in some clusters correlated with strain type: for example, cluster 1 is induced by all type 12 strains, cluster 5 is mainly induced by type II strains or strains that have part of the type II genome and cluster 7 and 9 by type 6 strains, possibly indicating host cells responding to genetic differences between strain types. Genes in the other clusters were differently regulated by strains of the same clonal haplogroup (e.g, ME49 and DEG for cluster 6 and 8) possibly indicating host cells responding to epigenetic differences between strains.

Multiple factors determine gene expression differences among Toxoplasma strains
Gene expression differences among the Toxoplasma strains could have multiple causes. For instance, the specific host cellular state can influence Toxoplasma gene expression [42] and Toxoplasma secreted effectors could modulate host gene expression resulting in an altered host cell environment to which it subsequently responds. Indeed we detected strong correlations between some host and Toxoplasma gene expression clusters (Table S2). For example, host gene cluster 6 and 8, which are negatively correlated (R = −0.93), have a significant negative (R = −0.86) and positive (R = 0.85) correlation with Toxoplasma gene cluster 1, respectively (Figure 1C). Host cluster 6 was enriched for genes and pathways that were previously shown to be modulated in human fibroblasts by the Toxoplasma secreted kinase ROP38 (Figure 1) [33]. Roos and colleagues have shown that ROP38 overexpression has a profound effect on host cell gene expression and modulates host genes enriched in mitochondrial function, metabolic processes and proteasome function and genes modulated by ELK1 [33], a transcriptional activator belonging to the ternary complex factor (Tcf) family. This is consistent with the functional enrichments seen for cluster 6, suggesting that this cluster might be regulated by strain differences in ROP38 expression and that Toxoplasma might be subsequently responding to the different environment established by ROP38. Consistent with this notion the expression level of ROP38 is highly variable among Toxoplasma strains ranging from low expression in RH (RPKM = 1.1) to extremely high expression in CEP (RPKM = 505) and positively correlates with cluster 6 (R = 0.49) and negatively correlates with cluster 9 (R = −0.51).
Differences in expression levels of Toxoplasma transcription factors could also affect a large number of genes. To determine the putative Toxoplasma regulators that could modulate the gene expression clusters we used Genomica [43], which takes a set of putative regulators (Table S1) and assumes that regulators are themselves transcriptionally regulated, so that their expression profiles provide information about their activity level. Although there are many exceptions to this assumption (e.g. activation by phosphorylation of signaling transducers, nuclear translocation of transcription factors upon activation or activation by non-variable polymorphic transcriptional factors), the expression level of specific AP2 factors, which are known to be involved in Toxoplasma gene expression, correlated highly with the expression of Toxoplasma genes in different clusters. For example Toxoplasma cluster 5 and 6 are enriched in genes expressed in the bradyzoite stage and each of these clusters correlate highly with AP2 factors that are known to be upregulated during bradyzoite development [44] (AP2X-10 and AP2Ib-1 for cluster 5 and AP2IX-1 and AP2-IX-9 for cluster 6, Figures 1B; S3 and Table S1). Other non-AP2 putative Toxoplasma gene expression regulators were better predictors of the expression levels of Toxoplasma genes in different clusters (Table S1).
To identify potential large structural variations that also might influence Toxoplasma gene expression levels we made genome-wide graphs of the relative gene expression values for each strain against the position on the genome. This revealed that BOF, B73 and TgCatBr44 each had a large genomic region with on average twice the median expression value (Figure 2A; BOF [chrVIIb; 2.1–2.75 Mb and 3.2–3.52 Mb], B73 [chrX; 5.5–6.2] and TgCatBr44 [chrIX; 0.3–1.28 Mb and 1.7–3.2 Mb]), suggesting a large duplication. We previously sequenced the genome of BOF [18] and we used this data to confirm the putative structural variation identified using relative gene expression levels. To do this we plotted the relative number of reads mapped to chrVIIb averaged over 15 kb intervals. Indeed this identified two regions (2.1–2.75 and 3.2–3.52 Mb) on BOF chrVIIb that have on average twice as many reads compared to the rest of the chromosome and are therefore likely duplicated (Figure 2B). No such duplicated regions were detected in GPHT and FOU, even though these strains are highly related to BOF [18] and together constitute the clonal type 6 haplogroup, showing that even within a clonal lineage there can be significant variation leading to differences in gene expression levels.

Allelic and expression differences in the GRA15 gene determine strain differences in NFκB activation
Mouse gene expression cluster 5 contained 459 genes that were mainly upregulated in cells infected with type II (ME49, DEG and PRU), B41 and B73 strains (Figure 1A). Functional enrichment analysis showed that this cluster was enriched in genes activated by NFκB transcription factors (NFκB1, RelA) and pathway analysis also indicated enrichment in pathways consistent with NFκB activation. Type II, but not type I/III, strains strongly activate NFκB-dependent gene expression, which is due to the polymorphic secreted dense granule protein GRA15 [32]. Indeed, genes in cluster 5 were also highly enriched in genes that are upregulated by GRA15. To investigate the upregulation of NFκB regulated genes and strain type further we sequenced GRA15 from all strains used in this study. This showed that type II, B41 and B73 strains share the same GRA15 allele (GRA15II), which is shorter than the other alleles because of a 255 bp deletion (Figure S4 and S5). As expected, activation and translocation of the NFκB p65 subunit to the host cell nucleus can only be observed in cells infected with parasites strains expressing GRA15II, and is abolished in type II parasites in which GRA15 gene was knocked out (Figure S6).
Nevertheless, we observed that several atypical strains, for example VAND, RAY, WTD3, GUY-KOE, P89, CASTELLS and COUGAR, all of which express a full length copy of GRA15, were also able to induce a less intense but significant NFκB response (Figure 1A), suggesting that other parasite factor(s) may play a role in controlling NFκB activation. To further investigate this, we infected an reporter cell line expressing a luciferase gene under the control of NFκB transcription factor response elements with the different T. gondii strains and observed that strains with higher expression of GRA15, including VAND, WTD3, RAY and GUY-KOE, induced higher activation of the reporter regardless of the GRA15 allele they express (Pearson correlation between GRA15 expression level and luciferase activity = 0.7, Figure 3A). The exception was RH, which expresses a truncated non-functional form of GRA15 due to a premature stop codon [32]. This suggests that expression levels of GRA15 may also be significant in controlling NFκB activation. To test this hypothesis we designed transgenic RH parasites overexpressing either the type II (58 kDa) or type III (66 kDa) GRA15. Levels of transgenic expression of GRA15 were similar in both parasites (Figure 3B). Using immunofluorescence analysis of infected human foreskin fibroblasts we observed that transgenic overexpression of the type III copy of GRA15 in RH parasites also leads to efficient NFκB activation (Figure 3C). Still, not all strains with high expression levels of GRA15 (e.g. BOF) induced a strong pro-inflammatory response (Figure 1A) or are able to induce nuclear translocation of NFκB subunit p65 (Figure S6). Taken together, our data suggest that although GRA15II is the main activator of NFκB, high expression levels of different GRA15 alleles can also induce pro-inflammatory responses in infected host cells, possibly through activation of different NFκB subunits.

The host cell pro-inflammatory response is modulated by multiple Toxoplasma secreted factors
We have previously shown that GRA15 induction of an NFκB-dependent pro-inflammatory response can be antagonized by the activity of Toxoplasma polymorphic rhoptry kinase ROP16 [45]. Recently, ROP38, a rhoptry protein that is highly expressed in type III parasites relative to types I/II, was shown to downregulate several host cell responses, including cytokine production, induced during infection [33]. Indeed, we observed that ROP38 expression was highly variable in the different strains, with the highest expression observed in the type III strain CEP (Figure 4A). We previously observed by microarray analysis that CEP expressing GRA15II induced significantly lower levels of NFκB-regulated genes compared to type I+GRA15II [32]. We therefore wanted to test whether ROP38 can dampen pro-inflammatory responses activated by GRA15II. Indeed, only RH-GRA15II, but not CEP-GRA15II, induced nuclear translocation of NFκB subunit p65 (Figure 3D) even though both transgenic strains expressed similar levels of GRA15 (Figure 3B). To test if the higher expression of ROP38 observed in CEP could be responsible for this phenotype, we engineered type II (PRU) parasites to overexpress a transgenic copy of HA-tagged ROP38 (Figure 4B). Compared to the control type II parasites, activation of the NFκB reporter cell line is markedly reduced in cells infected with parasites overexpressing ROP38 (Figure 4C).

We have also previously shown that GRA15II induces and ROP16I/III represses IL12p40/p70 production by infected macrophages [32], [45]. Because NFkB activation is required for IL12 production we wanted to confirm that other genes, besides GRA15 and ROP16, could play a role in strain differences in IL12 induction. We therefore infected BMDM with F1 progeny derived from IIxIII crosses that have both type II GRA15 and ROP16 alleles (S25, S27, STE7, D3X1) and assessed production of IL12p40 and IL12p70. Even though both the S25 and S27 strains have ROP38 alleles derived from the parental type II strain (data not shown), only the type II and S27 infection led to the production of high levels of IL12p70 (Figures 4D–E), indicating that besides GRA15 and ROP16 other genes play a role in the strain-specific induction of IL12 secretion. Thus, although GRA15II is the main parasite factor controlling NFkB-dependent host cell responses, the expression level of other GRA15 alleles and other Toxoplasma secreted proteins, including ROP16 and ROP38, also affect host cell inflammatory responses. This is reflected in mouse expression clusters 7 and 9, which contain genes that are mainly induced by strains that do not activate a strong inflammatory response (type 6, RH, CAST and CEP).
Effector sequence versus activity correlations can pinpoint critical residues that determine strain differences in activity
Once a Toxoplasma effector that modulates strain-specific regulation of host signaling pathways is identified it is often unclear what exactly determines the strain specificity of that activity. For example type I, II and III all have similar expression levels of the polymorphic rhoptry kinase ROP16 but only ROP16 from types I and III constitutively activate STAT3 and STAT6 [22]. By making chimeric ROP16 from type I and II and subsequent individual substitutions it was determined that a single leucine to serine substitution at position 503 (L→S503) in the kinase domain of type II ROP16 (ROP16II) is responsible for its significantly reduced kinase activity for its STAT3 substrate [22], [46], [47]. To determine whether data from multiple strains could have predicted that the L→S503 is the critical difference that determines strain differences in ROP16 activity we used our RNA-seq data to assemble the ROP16 allele for each strain, perform evolutionary analysis (Figures 5A and S7) and determine for each strain if it could constitutively activate STAT6, a reliable read out for ROP16 activity. All strains with a non-type II ROP16 allele constitutively activated STAT5 and STAT6 (Figures S8, S9). COUGAR, which constitutively activates STAT6, has a ROP16 that only differs at two positions from type II ROP16, it has an alanine at position 502 and a leucine at 503. Thus, sequence versus activity correlations of a polymorphic effector using data from a large collection of strains can directly pinpoint the crucial residue(s) that mediate the strain differences in the activity of the effector. We also observed higher rates of non-synonymous/synonymous (NS/S) polymorphisms at the STAT3-interaction site [22], [46], [47] and part of the kinase domains of ROP16 (Figure 5B). The fact that ROP16 from all non-type II strains is active, with the exception of B73 (Figures S8, S9, data not shown), which is an F1 from a cross between type II and type III [18], suggests that the ROP16II L→S mutation is a recent event that occurred after divergence of type II from the closely related WTD3, B41 and RAY type 12-like strains.

Toxoplasma atypical strains activate type I interferons
Among the novel pathways we identified, we observed that most type 12 Toxoplasma strains, and some other atypical strains, induced a cluster of 543 genes (Cluster 1 in Figure 1) that are enriched in components of the type I interferon signaling pathway, including Ifnb1, the family of RNA helicases Ddx58 (RIG-I), Dhx58 (LGP2) and Ifih1 (MDA5), several interferon inducible proteins, including Ifitm3 and Ifi35, and immunity-related GTPases (IRGs). Upstream regulator analysis indicated that the Interferon Regulatory Factors (IRF1, IRF2, IRF7) were the most likely regulators of this cluster (Figure 1A). Pathway and network analyses of these genes also revealed enrichment for genes involved in IFNβ signaling (Figures 1A and 6A). Another cluster (Cluster 4) containing 399 genes had a high negative correlation with the IFN cluster (R = −0.93). This cluster was highly enriched for genes involved in EIF2 signaling and translation (Figure 1A). It is likely that type I IFN production led to activation of protein kinase R (PKR), which can phosphorylate EIF2A and thereby block translation.

Only a few atypical Toxoplasma strains were capable of inducing high levels of Ifnb and Rsad2 (an IFNβ induced gene also called viperin) expression, the most potent inducers being RUB and COUGAR (Figure 6B). To validate our RNA-seq data, we infected BMDMs with several Toxoplasma strains and measured interferon beta in the supernatants 24 hours post infection. Both IFNβ protein and IFNβ transcripts were produced by cells infected with COUGAR and RUB, but not other canonical or atypical strains tested (Figures 6C–D). Time course experiments showed that IFNβ transcripts can be detected as early as 8 h post infection, peaking at 12 h (Figure 6E and data not shown).
Toxoplasma induction of type I interferon is dependent on IRF3 and endosomal TLRs or RIG-I
Transcriptional regulation of type I interferons by viral pathogens depends on the activity of either IRF3 and/or IRF7 [48]. To investigate whether Toxoplasma induction of IFNβ was also dependent on these host cell transcription factors, we infected immortalized macrophages, derived from wild type or mice deficient for different IRFs, with the ‘activating’ strain COUGAR. Ifnb transcription was completely abolished in infected cells lacking IRF3, but not in cells lacking IRF7 (Figure 6F), showing that the IRF3 transcription factor is essential for parasite-induced IFNβ production. As expected, single IRF knockout cells were still responsive to synthetic double stranded DNA poly d(A)∶d(T), but cells missing both IRF3 and IRF7 were unresponsive (Figure S10A). To test which host cell signaling pathway was being used by atypical Toxoplasma strains to activate IRF3, we infected wild type, Unc93B1 3 d mutant (which are defective in intracellular TLR trafficking and activation) or MyD88 and TRIF double knockout immortalized macrophages with COUGAR and performed qPCR analysis for Rsad2 20 h post infection. Rsad2 transcription was completely abolished in infected MyD88/TRIF dKO macrophages (Figure 6G) and strongly reduced in 3 d cells, suggesting that endosomal Toll-like receptors are essential for parasite-induced IFNβ production in macrophages. Importantly, the absence of TLR signaling did not affect the ability of macrophages to produce IFNβ in response to transfected double stranded DNA (Figure S10B). Nevertheless, we observed that in murine fibroblasts, which express much lower levels of TLRs than macrophages [49], Ifnb transcript levels after both COUGAR infection or poly d(A)∶d(T) treatment are dependent on the cytoplasmic nucleic acid receptor RIG-I (Figure 6H; Figure S10C). Taken together our data show that IFNβ induction by atypical Toxoplasma strains is dependent on activation of different nucleic acid sensing pathways depending on host cell type.
Toxoplasma also induces type I interferon production in human cells
To determine if COUGAR also induced type I IFN production in human cells we performed transcriptional analysis of HFFs infected with either canonical or atypical Toxoplasma strains. We observed that COUGAR could also induce a strong IFNβ signature in human cells, whereas neither the canonical strains nor atypical strains CASTELLS and MAS did (Figure 7A). Since we observed that IFNβ induction is dependent on activation of nucleic acid sensors, we hypothesized that strains capable of inducing IFNβ are somehow being destroyed by the host cells, releasing both parasite DNA and RNA that are then detected by either TLRs or RLRs. To test this, we infected HFFs monolayers with a non-activating type II (PRU strain) or the activating COUGAR strain. As the typical lytic cycle of PRU and COUGAR takes about 72 hours, we chose 24 h and 48 h time points to evaluate parasite growth prior to lysis of host cells. Cells were fixed, stained with antibodies against GRA7 and SAG1 to visualize both parasite vacuoles and parasites respectively. As expected, in monolayers infected with PRU, the number of vacuoles per field remained constant throughout the experiment, while the number of parasites per vacuole increased due to parasite division (Figures 7B–C). In cells infected with COUGAR, we observed a marked reduction in the number of vacuoles per field, suggesting that the host cells were destroying the vacuoles, and the increase in number of parasites per vacuole was negligible (Figures 7B–C).

Previously, we showed that in IFNγ-stimulated MEFs, type II strain vacuoles are rapidly coated with IRGs after which they are destroyed [28], potentially releasing parasite DNA and RNA into the host cell cytoplasm. To test if parasite killing can induce the production of type I IFN, we infected MEFs that were pre-stimulated with IFNγ with a type II strain. Indeed IFNγ-stimulated type II infected cells produced much higher Ifnb levels compared to IFNγ-stimulated or control cells (Figure 7D). Moreover, transfection of Toxoplasma DNA into host cells was sufficient to induce the upregulation of the type I IFN induced gene Rsad2 (Figure 7E).
Our data show that intracellular killing of T. gondii results in the release of parasite nucleic acids that lead to activation of either TLR or RLR receptors, triggering a signaling cascade that activates IRF3 and culminates in the production of IFNβ (Figure 8). This suggests that ‘resting’ cells can eliminate intracellular parasites through a novel, IFNγ-independent killing mechanism, and some atypical strains of Toxoplasma are more susceptible to this killing mechanism, resulting in increased induction of the type I IFN response.
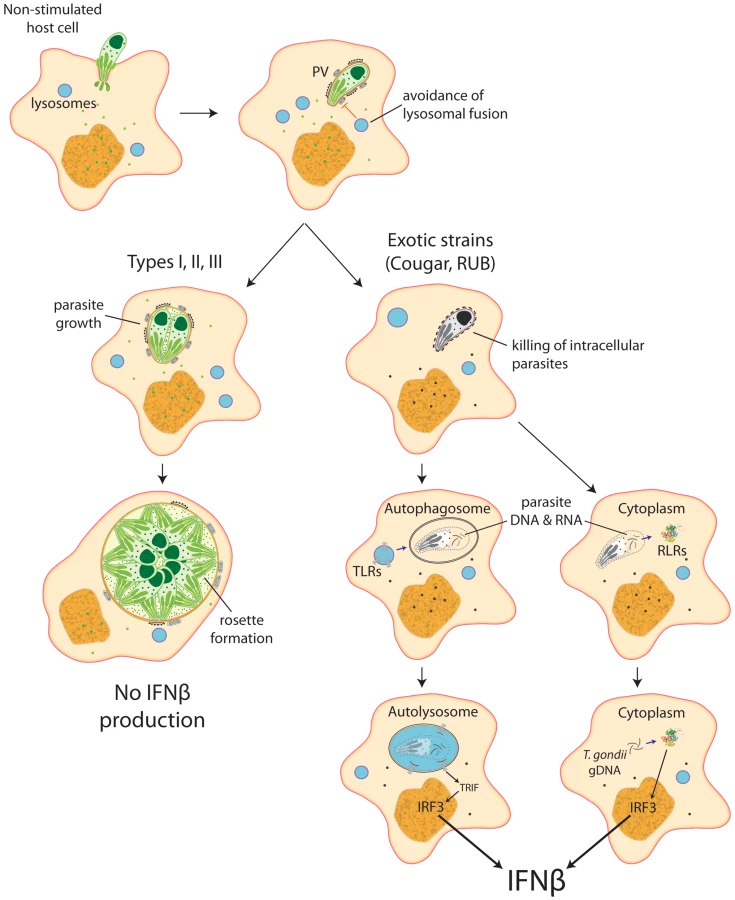
Discussion
In South America both congenital and ocular toxoplasmosis are more prevalent compared to Europe and more often associated with severe symptoms [4], [37]. There is some evidence that this is associated with atypical strains (not type I, II or III), mainly present in South America. Some strains from French Guiana can cause severe disease and even death in non immunosuppressed individuals [38]–[41]. Also in North America atypical (non type I/II/III) strains have been associated with more severe disease symptoms [36]. However, it is currently unknown if these atypical strains differ in their interaction with host cells. Here we provide the complete murine macrophage and Toxoplasma transcriptomes after infection with 29 different Toxoplasma strains. We have identified clusters of co-regulated macrophage and parasite genes that are enriched for functional annotation. We expect that this data will form the basis for exploring the molecular basis for the differential regulation of these gene expression clusters.
As an example we explored a cluster of co-regulated host genes that are enriched in genes regulated by type I IFN and show that only a small number of atypical strains activate the type I IFN response. We show that the type I IFN induction by these strains is associated with the killing of parasites in non-stimulated cells and the recognition of DNA/RNA by endosomal and/or cytoplasmic pattern recognition receptors.
The combination of Toxoplasma secreted effectors that leads to optimal fitness in one host is unlikely to be optimal in all of Toxoplasma's hosts and if strains end up in the wrong host they might cause disease by either causing pathological inflammation or by overwhelming the host. Indeed, different host species display large differences in susceptibility to Toxoplasma and different Toxoplasma strains often differ in virulence in the same host species suggesting that specific Toxoplasma strains might have adapted to certain hosts [50]–[52]. Our data indeed show large differences between strains in the modulation of host genes. One of the most consistent observations is the large difference between Toxoplasma strains in the activation of an inflammatory response. Here we show that this is mediated by differences in the sequence and expression level of GRA15, which can activate the pro-inflammatory transcription factor NFκB, and by differences in the activation of the type I IFN response. Other Toxoplasma effectors, such as the ROP16 and ROP38 protein kinases, can inhibit this inflammatory response. Therefore, the exact sequence and expression levels of GRA15, ROP16 and ROP38, determine the potential of different Toxoplasma strains to induce inflammation. In general the type II, type 12 and COUGAR strains are more pro-inflammatory while type I, type III and type 6 are the least inflammatory. However, even within clonal haplogroups, large differences in the induction of host gene expression exist. In North America, non-type II strains have been associated with more severe toxoplasmosis [9]. Because type 12 strains were recently shown to be prevalent in North America [11], [15]–[17], it is possible that they are the cause of severe disease.
It is interesting that our results indicate that type 12, and the closely related COUGAR strain [18], induce a gene expression profile enriched for type I IFN signaling. For the COUGAR strain, that was also confirmed in human cells. Type I interferons (IFN-α/β) are potent coordinators of antimicrobial responses, controlling both cell autonomous innate responses and T cells memory and effector functions [53], [54]. Besides their role in viral infections, they can also play an important role during parasitic infections by either controlling parasite growth through activation of intracellular killing mechanisms [55]–[60] or enhancing severity of disease caused by Trypanosoma cruzi, Leishmania, and Plasmodium [61]–[64]. Importantly, type I interferons can affect infection persistence [65], [66], which depends on both timing and magnitude of the interferon response [67]. We observed that the exotic strain COUGAR, is more susceptible than type II strains to IFNγ-independent killing, which leads to an early IFNβ production. This strain specific difference in timing and magnitude of type I interferon response may result in different disease outcomes. We have observed that expression of type I interferons by infected host cells is followed by induction of IRGs, which have a major role in controlling parasite growth [30]. Indeed, it has been previously shown that IFNβ can control Toxoplasma growth both in vitro and in vivo [68], [69]. Nevertheless, these studies were performed by pre-treating cells or animals with high doses of recombinant IFNβ, and lack an assessment of the role of type I interferons in physiological conditions. In this regard, type II strains of Toxoplasma were shown to stimulate type I interferon production in vivo by intestinal cells following oral infection with T. gondii cysts [70], [71], however no detailed analysis of the role of this cytokine in disease progression has been performed. Thus, even though induction of IFNβ could potentially be beneficial for the host by controlling intracellular parasite growth, it is striking that in the mouse model COUGAR parasites are significantly more virulent than type II strains [19]. It is currently unclear if this enhanced virulence was due to increased parasite burden or enhanced pathogenesis possibly due to a strong pro-inflammatory response. Because COUGAR is genetically quite different from the type II strain [18] it is possible that its enhanced virulence is unrelated to its induction of IFNβ and due to differences in other polymorphic effectors. However, in the context of recent observations that type I IFN suppresses type II IFN-triggered human anti-mycobacterial responses [72] it is plausible that the type I IFN response induced by some of the Toxoplasma strains could be associated with increased pathogenesis in human patients.
The simultaneous capture of both the parasite and host transcriptomes has also provided us with important insights into the crosstalk between the host and the parasite. We observed significant correlations between the expression of Toxoplasma and host gene clusters. It has been previously observed that the state of the host cell can modulate Toxoplasma gene expression. For example addition of compound 1 to host cells induces differentiation of Toxoplasma into bradyzoites and the knockdown of CDA1, a nucleosome assembly-related protein, demonstrated its involvement in cell cycle arrest and negative control of cell proliferation, mimicking the effects of compound 1 [42]. A potential role for the host cell cycle state in modulating Toxoplasma growth and differentiation has also been observed by others, for example, Toxoplasma spontaneously differentiates into bradyzoites upon invasion of terminally differentiated skeletal muscle and neuronal cells [73], [74]. Recently it was shown that the dense granule protein GRA16 is secreted beyond the PVM and traffics to the host cell nucleus [75], where it could potentially regulate host cell cycle. It was suggested that GRA16 downregulation of cyclin-B1 could be the main cause of the GRA16-induced G2 arrest, which has also been observed by others to be associated with Toxoplasma infection [76], [77]. Many of our strain-specifically regulated host gene clusters are also enriched for processes involved in cell cycle regulation suggesting that this is an important target for Toxoplasma effectors. For example host expression cluster 9 is enriched for host genes modulated by P53 and the CDKN1A kinase. It is also enriched in genes involved in regulation of the host cell cycle, especially G2/M checkpoint regulation. Because these are highly similar to the processes regulated by GRA16 it is tempting to speculate that strain differences in expression of GRA16 and/or sequence of GRA16 might be involved. All together, our study provides global insight of host cells and parasite transcriptional responses upon infection with both canonical and exotic Toxoplasma strains, and allows identification of novel strain specific host cell responses and parasite effectors.
Materials and Methods
Ethics statement
All experiments involving animals were in accordance with guidelines set forth by the American Association for Laboratory Animal Science (AALAS). All protocols used in this work were approved by the Committee on Animal Care (CAC #0611-063-14) at the Massachusetts Institute of Technology.
Reagents
All tissue culture reagents were obtained from Gibco, unless otherwise indicated. Poly(dA∶dT) was obtained from Invivogen. All oligonucleotide primers were obtained from Integrated DNA Technologies.
Mice
C57BL/6J mice were obtained from The Jackson Laboratory. Female, 6–12 weeks old mice were used in all experiments. Mice were housed under specific pathogen free conditions at Massachusetts Institute of Technology animal facility.
Parasites
Toxoplasma was cultured on human foreskin fibroblasts as described previously [32]. The following Toxoplasma strains, with the animal and country it was originally isolated from in brackets, were used in this study: ARI (human, USA), B41 (bear, USA), B73 (bear, USA), BOF (human, Belgium), CAST (human, USA), CASTELLS (sheep, Uruguay), CEP (cat, USA), COUGAR (Cougar, Canada), DEG (human, France), FOU (human, France), GPHT (human, France), GT1 (goat, USA), GUY-DOS (human, French Guiana), GUY-KOE (human, French Guiana), GUY-MAT (human, French Guiana), MAS (human, France), ME49 (sheep, USA), P89 (pig, USA), PRU (human, France), RAY (human, USA), RH (human, USA), ROD (human, USA), RUB (human, French Guiana), TgCatBr5 (cat, Brazil), TgCatBr9 (cat, Brazil), TgCatBr44 (cat, Brazil), VAND (human, French Guiana), VEG (human, USA), WTD3 (white-tailed deer, USA) [16]. The genetic relationship among these strains was previously investigated using genome-wide SNP data [18]. Strains originating from France and French Guiana were provided by the BCR Toxoplasma (http://www.toxocrb.com). To generate PRU (ΔHXGPRT) parasites expressing the type II allele of ROP38, the ROP38 coding region and putative promoter region (1,437 bp upstream of the start codon) were amplified from PRU genomic DNA by PCR (forward, 5′-CGAGAGGGAAGCAACGTTTA-3′, reverse, 5′-TTACGCGTAGTCCGGGACGTCGTACGGGTAAAATTGATGCGTTCTTATCCGACG-3′). Sequence coding for a C terminal HA tag was included in the reverse primer (denoted with italics). ROP38IIHA was then cloned into pTKO-att [32] through LR recombination. The pTKO-att-ROP38IIHA vector was then linearized with NotI (New England Biolabs, Inc.) and transfected into PRUΔHXGPRT by electroporation. Stable integrants were selected in media with 50 µg/ml mycophenolic acid (Axxora) and 50 µg/ml xanthine (Alfa Aesar) and cloned by limiting dilution. Immunofluorescence was used to confirm expression of ROP38II via anti-HA staining. All parasite strains and cell lines were routinely checked for Mycoplasma contamination and it was never detected.
Cells
RIG-I knockout mouse embryonic fibroblasts (MEFs), IRF3, IRF7 and IRF3/7 knockout immortalized mouse macrophages were provided by Katherine Fitzgerald (UMass Medical School). Immortalized macrophages expressing a mutant, non-functional form of Unc93B1, and MyD88/TRIF double-knockout macrophages were provided by Douglas T. Golenbock (UMass Medical School). Human foreskin fibroblasts (HFFs) and MEFs were maintained in DMEM medium supplemented with 10 mM Hepes, 100 U/ml Penicillin-Streptomycin and 10% FCS (PAA Laboratories). Immortalized macrophages were kept in DMEM supplemented with 10 mM Hepes, 100 U/ml Penicillin-Streptomycin, 1 mM Pyruvate, 10% L929 supernatant and 10% FCS.
Bone marrow-derived macrophages infection and RNA isolation
Bone marrow-derived macrophages (BMDMs) were isolated as described [78], and were cultured in RPMI medium supplemented with 25 mM Hepes, 10 mM L-glutamine, 100 U/ml Penicillin-Streptomycin, 50 µM 2-mercaptoethanol and 10% FCS. BMDMs were seeded in 6 well plates at 70% confluency and infected with different strains of T. gondii at three multiplicity of infections (MOIs): 15, 10 and 7.5. At the same time a plaque assay was setup to determine the viability and real MOI of each strain. After 20 h, total RNA was isolated using the RNeasy Plus kit from Qiagen, according to the manufacturer instructions. RNA integrity, size and concentration were verified using the Agilent 2100 Bioanalyzer and samples with similar Toxoplasma infections rates as judged by the Toxoplasma ribosomal band (Figure S11) were selected for RNA-seq.
High-throughput RNA sequencing
The RNA was processed for high-throughput sequencing according to standard Illumina protocols. Briefly, after mRNA pull down from total RNA using Dynabeads mRNA Purification Kit (Invitrogen), mRNA was fragmented into 200–400 base pair-long fragments and reverse transcribed to cDNA. Illumina sequencing adapters were added to each end, and samples were barcoded and multiplexed (four samples per lane) for sequencing on the Illumina HiSeq 2000 machine. On average 68 million 40 bp reads were obtained from each library. Reads in fastq format were mapped to either mouse (build 37.2) or Toxoplasma gondii (strain ME49, version 8.0) genomes using Bowtie 2.0.0 and Tophat 2.0.4. [79]. Transcripts from each sample were assembled, merged, and their expression values in FPKM were calculated using the Cufflinks package, version 2.0.2. [79]. Detailed methodology can be found in the supplemental material, and settings used are included in Table S3A–B.
Clustering and functional annotation of Toxoplasma gondii strains and infected host cells transcriptome
To identify host signaling pathways modulated by Toxoplasma in a strain-specific manner we specifically focused on macrophage genes that had at least a 2-fold difference in expression between macrophages infected with different Toxoplasma strains and had FPKM≥5 in at least 1 strain. To identify differentially regulated Toxoplasma gene clusters we specifically focused on Toxoplasma genes that had at least a 4-fold difference in expression between any two different Toxoplasma strains and had at least a FPKM value of 10 in one strain. We used Genomica [43] to cluster the genes into 10–15 clusters of co-regulated genes. These clusters were then investigated for enrichment for functional annotation, such as enrichment for transcription factor binding sites (TFBS) in their promoters or belonging to a particular pathway using DiRE [80], GSEA [81], and Ingenuity Pathway Analysis (www.ingenuity.com). A more detailed explanation of data analysis can be found in the supplemental methods online (Protocols S1–2).
Identification of structural variation using gene expression levels
We first identified all Toxoplasma protein coding genes with a median expression of >7.5 and subsequently determined the relative expression for each gene for each Toxoplasma strain by dividing the gene expression values, for each strain, by the median gene expression of all strains. We then made genome-wide graphs of these relative expression values for each strain by plotting the average relative expression in steps of 5 genes against the position on the genome (average position for the 5 genes). We then manually inspected the graphs for all 14 Toxoplasma chromosomes for each of the 29 strains for large genomic regions (>0.2 Mb) where a strain had more than 1.5 times the median expression value.
Luciferase assay
HEK293 cells stably expressing a luciferase gene under the control of four consecutive NFκB consensus promoter elements (Systems Biosciences) were infected with the indicated strains of Toxoplasma gondii in 3 MOIs (2.5, 5, 10). At 20 hpi culture supernatants were collected and cells were lysed. Luciferase activity was measured in the lysates using Promega's Luciferase Assay System.
Western blotting
Rabbit polyclonal antibodies to GRA15 were raised against the peptide with the amino acid sequence of GRA15493–510 (CGPPRTENPRQPQVPGENS) and affinity purified with the antigen (YenZym Antibody, San Francisco, CA). 2.5×106 parasites were harvested from T25 flasks with infected HFFs, lysed by addition of lysis buffer, boiled for 5 minutes, placed at 4°C for 5 minutes and centrifuged at 13,000 rpm at 4°C for 15 minutes. Lysates were run on 10% SDS-PAGE protein gel, transferred to a polyvinylidene difluoride membrane and blocked in PBS/0.1% Tween-20/5% nonfat dry milk. The blot was then incubated with primary polyclonal anti-rabbit GRA15 antibodies overnight at room temperature using 1 ug/mL, followed by secondary goat anti-rabbit horseradish peroxidase antibody for 1 hour at room temperature. The blot was then incubated with a luminal based substrate (Immun-Star WesternC, Bio-Rad Laboratories) and chemiluminescence was detected using a charge-coupled device camera (Chemidoc XRS, Bio-Rad Laboratories). Bands were visualized using Quantity One 1-D analysis software.
Immunofluorescence assays
HFFs seeded on coverslips were infected with T. gondii and incubated for different time points. Cells were then fixed with 4% formaldehyde in PBS for 20 min at room temperature, permeabilized and blocked with phosphate-buffered saline (PBS) plus 5% goat serum, 3% serum bovine albumin and 0.2% Triton X-100. Subsequently coverslips were incubated with primary antibody overnight at 4°C, washed twice with PBS, and incubated with fluorescent secondary antibodies and Hoechst to allow visualization of antigen and DNA respectively. Coverslips were mounted on a glass slide with Vectashield (VectorLaboratories). Photographs were taken using NIS-Elements software (Nikon) and a digital camera (CoolSNAP EZ; Roper Industries) connected to an inverted fluorescence microscope (model eclipse Ti-S; Nikon).
Cytokine production
Cell culture supernatants were assayed for cytokines with DuoSet ELISA kits from R&D Systems according to the manufacturer's instructions. IFNβ was quantified from cell culture supernatants using a custom ELISA as described elsewhere [82].
Quantitative PCR
Cells were infected with the indicated strains of Toxoplasma gondii or transfected with parasite DNA using either Xtreme-GENE9 or DOTAP (Roche) transfection reagents according to manufacturer instructions. Eight to 20 hours post infection cells were lysed, total RNA was extracted using the RNeasy Plus kit (Qiagen), and quantified using a NanoDrop spectrophotometer (Thermo Scientific). cDNA was synthetized using the SuperScript III First Strand Synthesis System (Invitrogen), and RT-PCR analysis was performed in a Light Cycler 480 II Real-time PCR machine (Roche) using the SYBR Green I Master kit (Roche) according to the manufacturer's instructions. Primers used are as follows: mIFNβ-Fw: ATAAGCAGCTCCAGCTCCAA; mIFNβ-Rv: CTGTCTGCTGGTGGAGTTCA; mRsad2-Fw: AACCCCCGTGAGTGTCAACTA; mRsad2-Rv: AACCAGCCTGTTTGAGCAGAA; mActin-Fw: TTGAACATGGCATTGTTACCAA; mActin-Rv: TGGCATAGAGGTCTTTACGGA.
Microarrays
Human foreskin fibroblasts grown to confluency into T25 flasks were infected with the indicated strains of T. gondii at 3 different MOIs. At 24 h post infection total RNA was isolated using Trizol (Invitrogen) and cleaned up using RNeasy MinElute kit (Qiagen) following manufacturer's protocols. Subsequently RNA was labeled, hybridized to a human Affymetrix array (Human U133A 2.0) and processed as described elsewhere [32].
Parasite growth
Monolayers of HFFs grown on coverslips were infected with the type II (PA7) strain or COUGAR, fixed at different time points and stained with antibodies against GRA7 and SAG1. Parasite vacuoles (GRA7 positive) per field and the number of parasites per vacuole (SAG1 staining) were counted.
Accession numbers
The sequences reported in this paper have been deposited in Short Read Archive, http://www.ncbi.nlm.nih.gov/sra (accession Nos. SRP008923 and SRP011061)
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. DubremetzJF (1998) Host cell invasion by Toxoplasma gondii. Trends Microbiol 6 : 27–30.
2. HoffmannS, BatzMB, MorrisJGJr (2012) Annual cost of illness and quality-adjusted life year losses in the United States due to 14 foodborne pathogens. J Food Prot 75 : 1292–1302.
3. WalkerM, ZuntJR (2005) Parasitic central nervous system infections in immunocompromised hosts. Clin Infect Dis 40 : 1005–1015.
4. GilbertRE, FreemanK, LagoEG, Bahia-OliveiraLMG, TanHK, et al. (2008) Ocular sequelae of congenital toxoplasmosis in Brazil compared with Europe. PLoS Negl Trop Dis 2: e277.
5. JamiesonSE, Peixoto-RangelAL, HargraveAC, RoubaixL-Ad, MuiEJ, et al. (2010) Evidence for associations between the purinergic receptor P2X(7) (P2RX7) and toxoplasmosis. Genes Immun 11 : 374–383.
6. WitolaWH, MuiE, HargraveA, LiuS, HypoliteM, et al. (2011) NALP1 influences susceptibility to human congenital toxoplasmosis, proinflammatory cytokine response, and fate of Toxoplasma gondii-infected monocytic cells. Infect Immun 79 : 756–766.
7. BelaSR, DutraMS, MuiE, MontpetitA, OliveiraFS, et al. (2012) Impaired innate immunity in mice deficient in interleukin-1 receptor-associated kinase 4 leads to defective type 1 T cell responses, B cell expansion, and enhanced susceptibility to infection with Toxoplasma gondii. Infect Immun 80 : 4298–4308.
8. MeloMB, JensenKD, SaeijJP (2011) Toxoplasma gondii effectors are master regulators of the inflammatory response. Trends Parasitol 27 : 487–495.
9. McLeodR, BoyerKM, LeeD, MuiE, WroblewskiK, et al. (2012) Prematurity and severity are associated with Toxoplasma gondii alleles (NCCCTS, 1981–2009). Clin Infect Dis 54 : 1595–1605.
10. SibleyLD, BoothroydJC (1992) Virulent strains of Toxoplasma gondii comprise a single clonal lineage. Nature 359 : 82–85.
11. KhanA, DubeyJP, SuC, AjiokaJW, RosenthalBM, et al. (2011) Genetic analyses of atypical Toxoplasma gondii strains reveal a fourth clonal lineage in North America. Int J Parasitol 41 : 645–655.
12. DardéM-L (2004) Genetic analysis of the diversity in Toxoplasma gondii. Ann Ist Super Sanita 40 : 57–63.
13. KhanA, JordanC, MuccioliC, VallochiAL, RizzoLV, et al. (2006) Genetic divergence of Toxoplasma gondii strains associated with ocular toxoplasmosis, Brazil. Emerg Infect Dis 12 : 942–949.
14. PenaHFJ, GennariSM, DubeyJP, SuC (2008) Population structure and mouse-virulence of Toxoplasma gondii in Brazil. Int J Parasitol 38 : 561–569.
15. SuC, KhanA, ZhouP, MajumdarD, AjzenbergD, et al. (2012) Globally diverse Toxoplasma gondii isolates comprise six major clades originating from a small number of distinct ancestral lineages. Proc Natl Acad Sci USA 109 : 5844–5849.
16. KhanA, MillerN, RoosDS, DubeyJP, AjzenbergD, et al. (2011) A monomorphic haplotype of chromosome Ia is associated with widespread success in clonal and nonclonal populations of Toxoplasma gondii. mBio 2: e00228–00211.
17. KhanA, FuxB, SuC, DubeyJP, DardeML, et al. (2007) Recent transcontinental sweep of Toxoplasma gondii driven by a single monomorphic chromosome. Proc Natl Acad Sci USA 104 : 14872–14877.
18. MinotS, MeloMB, LiF, LuD, NiedelmanW, et al. (2012) Admixture and recombination among Toxoplasma gondii lineages explain global genome diversity. Proc Natl Acad Sci USA 109 : 13458–13463.
19. KhanA, TaylorS, AjiokaJW, RosenthalBM, SibleyLD (2009) Selection at a single locus leads to widespread expansion of Toxoplasma gondii lineages that are virulent in mice. PLoS Genet 5: e1000404.
20. SaeijJPJ, BoyleJP, CollerS, TaylorS, SibleyLD, et al. (2006) Polymorphic secreted kinases are key virulence factors in toxoplasmosis. Science 314 : 1780–1783.
21. DubeyJP, LindsayDS, SpeerCA (1998) Structures of Toxoplasma gondii tachyzoites, bradyzoites, and sporozoites and biology and development of tissue cysts. Clin Microbiol Rev 11 : 267–299.
22. SaeijJPJ, CollerS, BoyleJP, JeromeME, WhiteMW, et al. (2007) Toxoplasma co-opts host gene expression by injection of a polymorphic kinase homologue. Nature 445 : 324–327.
23. BehnkeMS, KhanA, WoottonJC, DubeyJP, TangK, et al. (2011) Virulence differences in Toxoplasma mediated by amplification of a family of polymorphic pseudokinases. Proc Natl Acad Sci USA 108 : 9631–9636.
24. ReeseML, ZeinerGM, SaeijJP, BoothroydJC, BoyleJP (2011) Polymorphic family of injected pseudokinases is paramount in Toxoplasma virulence. Proc Natl Acad Sci USA 108 : 9625–9630.
25. TaylorS, BarraganA, SuC, FuxB, FentressSJ, et al. (2006) A secreted serine-threonine kinase determines virulence in the eukaryotic pathogen Toxoplasma gondii. Science 314 : 1776–1780.
26. FentressSJ, BehnkeMS, DunayIR, MashayekhiM, RommereimLM, et al. (2010) Phosphorylation of Immunity-Related GTPases by a Toxoplasma gondii-Secreted Kinase Promotes Macrophage Survival and Virulence. Cell Host Microbe 8 : 484–495.
27. SteinfeldtT, Konen-WaismanS, TongL, PawlowskiN, LamkemeyerT, et al. (2010) Phosphorylation of mouse immunity-related GTPase (IRG) resistance proteins is an evasion strategy for virulent Toxoplasma gondii. PLoS Biol 8: e1000576.
28. NiedelmanW, GoldDA, RosowskiEE, SprokholtJK, LimD, et al. (2012) The rhoptry proteins ROP18 and ROP5 mediate Toxoplasma gondii evasion of the murine, but not the human, interferon-gamma response. PLoS Pathog 8: e1002784.
29. BehnkeMS, FentressSJ, MashayekhiM, LiLX, TaylorGA, et al. (2012) The Polymorphic Pseudokinase ROP5 Controls Virulence in Toxoplasma gondii by Regulating the Active Kinase ROP18. PLoS Pathog 8: e1002992.
30. MartensS, ParvanovaI, ZerrahnJ, GriffithsG, SchellG, et al. (2005) Disruption of Toxoplasma gondii parasitophorous vacuoles by the mouse p47-resistance GTPases. PLoS Pathog 1: e24.
31. RosowskiEE, SaeijJP (2012) Toxoplasma gondii Clonal Strains All Inhibit STAT1 Transcriptional Activity but Polymorphic Effectors Differentially Modulate IFNgamma Induced Gene Expression and STAT1 Phosphorylation. PLoS ONE 7: e51448.
32. RosowskiEE, LuD, JulienL, RoddaL, GaiserRA, et al. (2011) Strain-specific activation of the NF-kappaB pathway by GRA15, a novel Toxoplasma gondii dense granule protein. J Exp Med 208 : 195–212.
33. PeixotoL, ChenF, HarbOS, DavisPH, BeitingDP, et al. (2010) Integrative Genomic Approaches Highlight a Family of Parasite-Specific Kinases that Regulate Host Responses. Cell Host Microbe 8 : 208–218.
34. OngYC, BoyleJP, BoothroydJC (2011) Strain-dependent host transcriptional responses to Toxoplasma infection are largely conserved in mammalian and avian hosts. PLoS ONE 6: e26369.
35. AjzenbergD (2012) High burden of congenital toxoplasmosis in the United States: the strain hypothesis? Clin Infect Dis 54 : 1606–1607.
36. GriggME, GanatraJ, BoothroydJC, MargolisTP (2001) Unusual abundance of atypical strains associated with human ocular toxoplasmosis. J Infect Dis 184 : 633–639.
37. AjzenbergD (2011) Unresolved questions about the most successful known parasite. Expert Rev Anti Infect Ther 9 : 169–171.
38. DemarM, HommelD, DjossouF, PeneauC, BoukhariR, et al. (2012) Acute toxoplasmoses in immunocompetent patients hospitalized in an intensive care unit in French Guiana. Clin Microbiol Infect 18: E221–231.
39. CarmeB, DemarM, AjzenbergD, DardeML (2009) Severe acquired toxoplasmosis caused by wild cycle of Toxoplasma gondii, French Guiana. Emerg Infect Dis 15 : 656–658.
40. GrohM, FaussartA, VillenaI, AjzenbergD, CarmeB, et al. (2012) Acute lung, heart, liver, and pancreatic involvements with hyponatremia and retinochoroiditis in a 33-year-old French Guianan patient. PLoS Negl Trop Dis 6: e1802.
41. DardéML, VillenaI, PinonJM, BeguinotI (1998) Severe toxoplasmosis caused by a Toxoplasma gondii strain with a new isoenzyme type acquired in French Guyana. J Clin Microbiol 36 : 324.
42. RadkeJR, DonaldRG, EibsA, JeromeME, BehnkeMS, et al. (2006) Changes in the expression of human cell division autoantigen-1 influence Toxoplasma gondii growth and development. PLoS Pathog 2: e105.
43. SegalE, FriedmanN, KollerD, RegevA (2004) A module map showing conditional activity of expression modules in cancer. Nat Genet 36 : 1090–1098.
44. BehnkeMS, WoottonJC, LehmannMM, RadkeJB, LucasO, et al. (2010) Coordinated progression through two subtranscriptomes underlies the tachyzoite cycle of Toxoplasma gondii. PLoS ONE 5: e12354.
45. JensenKD, WangY, WojnoED, ShastriAJ, HuK, et al. (2011) Toxoplasma polymorphic effectors determine macrophage polarization and intestinal inflammation. Cell Host Microbe 9 : 472–483.
46. YamamotoM, StandleyD, TakashimaS, SaigaH, OkuyamaM, et al. (2009) A single polymorphic amino acid on Toxoplasma gondii kinase ROP16 determines the direct and strain-specific activation of Stat3. J Exp Med
47. OngY-C, ReeseML, BoothroydJC (2010) Toxoplasma Rhoptry Protein 16 (ROP16) Subverts Host Function by Direct Tyrosine Phosphorylation of STAT6. J Biol Chem 285 : 28731–28740.
48. TakeuchiO, AkiraS (2009) Innate immunity to virus infection. Immunol Rev 227 : 75–86.
49. SuAI, CookeMP, ChingKA, HakakY, WalkerJR, et al. (2002) Large-scale analysis of the human and mouse transcriptomes. Proc Natl Acad Sci USA 99 : 4465–4470.
50. SaeijJP, BoyleJP, BoothroydJC (2005) Differences among the three major strains of Toxoplasma gondii and their specific interactions with the infected host. Trends Parasitol 21 : 476–481.
51. ElmoreSA, JonesJL, ConradPA, PattonS, LindsayDS, et al. (2010) Toxoplasma gondii: epidemiology, feline clinical aspects, and prevention. Trends Parasitol 26 : 190–196.
52. SergentV, CautainB, KhalifeJ, DesléeD, BastienP, et al. (2005) Innate refractoriness of the Lewis rat to toxoplasmosis is a dominant trait that is intrinsic to bone marrow-derived cells. Infect Immun 73 : 6990–6997.
53. HuberJP, FarrarJD (2011) Regulation of effector and memory T-cell functions by type I interferon. Immunology 132 : 466–474.
54. MacMickingJD (2012) Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat Rev Immunol 12 : 367–382.
55. CostaVM, TorresKC, MendoncaRZ, GresserI, GollobKJ, et al. (2006) Type I IFNs stimulate nitric oxide production and resistance to Trypanosoma cruzi infection. J Immunol 177 : 3193–3200.
56. KogaR, HamanoS, KuwataH, AtarashiK, OgawaM, et al. (2006) TLR-dependent induction of IFN-beta mediates host defense against Trypanosoma cruzi. J Immunol 177 : 7059–7066.
57. MattnerJ, Wandersee-SteinhauserA, PahlA, RollinghoffM, MajeauGR, et al. (2004) Protection against progressive leishmaniasis by IFN-beta. J Immunol 172 : 7574–7582.
58. MorrellCN, SrivastavaK, SwaimA, LeeMT, ChenJ, et al. (2011) Beta interferon suppresses the development of experimental cerebral malaria. Infect Immun 79 : 1750–1758.
59. VigarioAM, BelnoueE, GrunerAC, MauduitM, KayibandaM, et al. (2007) Recombinant human IFN-alpha inhibits cerebral malaria and reduces parasite burden in mice. J Immunol 178 : 6416–6425.
60. PichyangkulS, YongvanitchitK, Kum-arbU, HemmiH, AkiraS, et al. (2004) Malaria blood stage parasites activate human plasmacytoid dendritic cells and murine dendritic cells through a Toll-like receptor 9-dependent pathway. J Immunol 172 : 4926–4933.
61. ChesslerAD, CaradonnaKL, Da'daraA, BurleighBA (2011) Type I interferons increase host susceptibility to Trypanosoma cruzi infection. Infect Immun 79 : 2112–2119.
62. KhouriR, BaficaA, Silva MdaP, NoronhaA, KolbJP, et al. (2009) IFN-beta impairs superoxide-dependent parasite killing in human macrophages: evidence for a deleterious role of SOD1 in cutaneous leishmaniasis. J Immunol 182 : 2525–2531.
63. SharmaS, DeOliveiraRB, KalantariP, ParrocheP, GoutagnyN, et al. (2011) Innate immune recognition of an AT-rich stem-loop DNA motif in the Plasmodium falciparum genome. Immunity 35 : 194–207.
64. HaqueA, BestSE, AmmerdorfferA, DesbarrieresL, de OcaMM, et al. (2011) Type I interferons suppress CD4(+) T-cell-dependent parasite control during blood-stage Plasmodium infection. Eur J Immunol 41 : 2688–2698.
65. TeijaroJR, NgC, LeeAM, SullivanBM, SheehanKC, et al. (2013) Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 340 : 207–211.
66. WilsonEB, YamadaDH, ElsaesserH, HerskovitzJ, DengJ, et al. (2013) Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 340 : 202–207.
67. WangY, SwieckiM, CellaM, AlberG, SchreiberRD, et al. (2012) Timing and magnitude of type I interferon responses by distinct sensors impact CD8 T cell exhaustion and chronic viral infection. Cell Host Microbe 11 : 631–642.
68. OrellanaMA, SuzukiY, AraujoF, RemingtonJS (1991) Role of beta interferon in resistance to Toxoplasma gondii infection. Infect Immun 59 : 3287–3290.
69. SchmitzJL, CarlinJM, BordenEC, ByrneGI (1989) Beta interferon inhibits Toxoplasma gondii growth in human monocyte-derived macrophages. Infect Immun 57 : 3254–3256.
70. MinnsLA, MenardLC, FoureauDM, DarcheS, RonetC, et al. (2006) TLR9 is required for the gut-associated lymphoid tissue response following oral infection of Toxoplasma gondii. J Immunol 176 : 7589–7597.
71. FoureauDM, MielcarzDW, MenardLC, SchulthessJ, WertsC, et al. (2010) TLR9-dependent induction of intestinal alpha-defensins by Toxoplasma gondii. J Immunol 184 : 7022–7029.
72. TelesRM, GraeberTG, KrutzikSR, MontoyaD, SchenkM, et al. (2013) Type I interferon suppresses type II interferon-triggered human anti-mycobacterial responses. Science 339 : 1448–1453.
73. Ferreira da Silva MdaF, BarbosaHS, GrossU, LuderCG (2008) Stress-related and spontaneous stage differentiation of Toxoplasma gondii. Mol Biosyst 4 : 824–834.
74. JonesTC, BienzKA, ErbP (1986) In vitro cultivation of Toxoplasma gondii cysts in astrocytes in the presence of gamma interferon. Infect Immun 51 : 147–156.
75. BougdourA, DurandauE, Brenier-PinchartMP, OrtetP, BarakatM, et al. (2013) Host cell subversion by Toxoplasma GRA16, an exported dense granule protein that targets the host cell nucleus and alters gene expression. Cell Host Microbe 13 : 489–500.
76. BrunetJ, PfaffAW, AbidiA, UnokiM, NakamuraY, et al. (2008) Toxoplasma gondii exploits UHRF1 and induces host cell cycle arrest at G2 to enable its proliferation. Cell Microbiol 10 : 908–920.
77. MolestinaRE, El-GuendyN, SinaiAP (2008) Infection with Toxoplasma gondii results in dysregulation of the host cell cycle. Cell Microbiol 10 : 1153–1165.
78. AustinP, McCullochE, TillJ (1971) Characterization of the factor in L-cell conditioned medium capable of stimulating colony formation by mouse marrow cells in culture. J Cell Physiol 77 : 121–134.
79. TrapnellC, RobertsA, GoffL, PerteaG, KimD, et al. (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7 : 562–578.
80. GoteaV, OvcharenkoI (2008) DiRE: identifying distant regulatory elements of co-expressed genes. Nucleic Acids Res 36: W133–139.
81. SubramanianA, TamayoP, MoothaVK, MukherjeeS, EbertBL, et al. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102 : 15545–15550.
82. RobertsZJ, GoutagnyN, PereraPY, KatoH, KumarH, et al. (2007) The chemotherapeutic agent DMXAA potently and specifically activates the TBK1-IRF-3 signaling axis. J Exp Med 204 : 1559–1569.
83. Korber B (2000) HIV Signature and sequence variation analysis. . In: Rodrigo AG, Learn GH, editors. Computational analysis of HIV molecular sequences. Dordrecht, Netherlands: Kluwer Academic Publishers. pp. 55–72.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2013 Číslo 12
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Influence of Mast Cells on Dengue Protective Immunity and Immune Pathology
- Host Defense via Symbiosis in
- Coronaviruses as DNA Wannabes: A New Model for the Regulation of RNA Virus Replication Fidelity
- Myeloid Dendritic Cells Induce HIV-1 Latency in Non-proliferating CD4 T Cells
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy