
Genome-Wide RNAi Screen Identifies Novel Host Proteins Required for Alphavirus Entry
The enveloped alphaviruses include important and emerging human pathogens such as Chikungunya virus and Eastern equine encephalitis virus. Alphaviruses enter cells by clathrin-mediated endocytosis, and exit by budding from the plasma membrane. While there has been considerable progress in defining the structure and function of the viral proteins, relatively little is known about the host factors involved in alphavirus infection. We used a genome-wide siRNA screen to identify host factors that promote or inhibit alphavirus infection in human cells. Fuzzy homologue (FUZ), a protein with reported roles in planar cell polarity and cilia biogenesis, was required for the clathrin-dependent internalization of both alphaviruses and the classical endocytic ligand transferrin. The tetraspanin membrane protein TSPAN9 was critical for the efficient fusion of low pH-triggered virus with the endosome membrane. FUZ and TSPAN9 were broadly required for infection by the alphaviruses Sindbis virus, Semliki Forest virus, and Chikungunya virus, but were not required by the structurally-related flavivirus Dengue virus. Our results highlight the unanticipated functions of FUZ and TSPAN9 in distinct steps of alphavirus entry and suggest novel host proteins that may serve as targets for antiviral therapy.
Published in the journal:
. PLoS Pathog 9(12): e32767. doi:10.1371/journal.ppat.1003835
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1003835
Summary
The enveloped alphaviruses include important and emerging human pathogens such as Chikungunya virus and Eastern equine encephalitis virus. Alphaviruses enter cells by clathrin-mediated endocytosis, and exit by budding from the plasma membrane. While there has been considerable progress in defining the structure and function of the viral proteins, relatively little is known about the host factors involved in alphavirus infection. We used a genome-wide siRNA screen to identify host factors that promote or inhibit alphavirus infection in human cells. Fuzzy homologue (FUZ), a protein with reported roles in planar cell polarity and cilia biogenesis, was required for the clathrin-dependent internalization of both alphaviruses and the classical endocytic ligand transferrin. The tetraspanin membrane protein TSPAN9 was critical for the efficient fusion of low pH-triggered virus with the endosome membrane. FUZ and TSPAN9 were broadly required for infection by the alphaviruses Sindbis virus, Semliki Forest virus, and Chikungunya virus, but were not required by the structurally-related flavivirus Dengue virus. Our results highlight the unanticipated functions of FUZ and TSPAN9 in distinct steps of alphavirus entry and suggest novel host proteins that may serve as targets for antiviral therapy.
Introduction
Alphaviruses are small enveloped plus-sense RNA viruses that are transmitted in nature by mosquito vectors [1]. These viruses can cause debilitating illness, severe arthritis, and encephalitis in humans, as in the case of the recently emerged alphavirus Chikungunya virus and the encephalitic alphaviruses Eastern, Western, and Venezuelan equine encephalitis virus [2], [3].
Alphaviruses have highly organized structures, with an internal nucleocapsid core surrounded by a viral membrane containing a lattice of the E2 and E1 proteins [4], [5]. They infect cells via binding to receptors on the plasma membrane and internalization by clathrin-mediated endocytosis [1], [6], [7]. The E1 envelope protein then mediates low pH-dependent fusion of the virus and endosome membranes, releasing the nucleocapsid into the cytoplasm. Virus replication occurs in the cytoplasm and the envelope proteins are transported via the host secretory pathway to the cell surface where new virus particles bud [1].
While there has been considerable progress in defining the structure and function of the viral proteins, relatively little is known about the host factors involved in alphavirus infection. Here we used a genome-wide siRNA screen to identify such host factors in human cells, and defined the mechanisms of several novel host proteins that promote alphavirus infection. These factors were involved in distinct steps of the virus entry pathway: the protein FUZ promoted endocytic uptake and the protein TSPAN9 promoted fusion of internalized, low pH-triggered virus in the endosome.
Results
siRNA screen
To identify novel host proteins involved in various aspects of alphavirus infection, we performed a genome-wide RNA interference screen using the prototype alphavirus Sindbis virus, engineered to express firefly luciferase in the cytoplasm of infected cells (SINV-Luc). Conditions were optimized to detect multiple cycles of virus infection, which were dependent on both clathrin and endosomal acidification (Fig. S1A, B). A total of 21,687 genes were targeted in human U-2 OS cells using pools of 3 siRNAs per gene, and screened for effects on SINV-Luc infection (Figs. 1A and S1C, D) (Table S1).

Analysis of robust z-scores and comparison with the positive and negative controls on each plate were used to identify 400 genes that may promote alphavirus infection and 59 genes that potentially encode antiviral factors (Materials and Methods, Fig. 1A). The siRNAs targeting these 459 genes were then tested to confirm effects on multi-cycle infection, to determine effects on single cycle infection (Fig. S1E), and to eliminate cytotoxic siRNA pools. 356 of the 400 siRNA pools that inhibited virus infection showed the same phenotype upon retesting, and 83 reproduced this effect with multiple (2–3) siRNAs. All of the 59 siRNA pools that promoted virus infection were positive upon retesting, and 56 out of 59 showed this phenotype with multiple siRNAs. After the elimination of siRNA pools in which only 1 siRNA reproduced the original phenotype and pools that decreased cell viability, we identified 118 SINV host factors: 62 factors that promote SINV infection (Table S2), and 56 factors that inhibit SINV infection (Table S3). Comparison of multi-cycle and single cycle infection assays suggested that the majority of these host factors are involved in initial SINV entry and replication (Fig. 1A, last column, and Tables S2 and S3). Comparison with screens for proteins involved in transferrin and epidermal growth factor entry [8] and clathrin-coated vesicle function [9] showed that ∼37% of our 118 SINV host factors are implicated in some aspect of endocytosis (Table S4). About 20% of our SINV host factors were identified in screens for host genes involved in infection by other animal viruses (Table S5).
The 62 host factors that promote SINV infection were grouped into functions with p values indicating the significance of their over-representation vs. the original siRNA library (Fig. 1B). They include host proteins known to be involved in alphavirus endocytic entry, such as vacuolar ATPase subunit ATP6V0C and dynamin 2, and NDST1, which is involved in sulfated proteoglycan synthesis and would thus promote SINV-Luc attachment to host cells [6], [10]. All three of these proteins were also identified in endocytosis screens and tests of other viruses (Tables S4 and S5). Our initial screen also ranked vATPase subunits ATP6AP2, ATP6V1B1 and ATP60E within the top 1000 proviral hits (Table S1). The 56 host factors that inhibit SINV infection include MAP1LC3B, which is involved in cargo recognition during selective autophagy, a pathway that protects against SINV infection [11]. Our screen did not identify NRAMP2, the iron transporter that acts as a receptor for SINV [7], as a host factor, perhaps due to its genetic redundancy in mammalian cells [7] or to differences in the libraries, host cells, etc. However, the iron-sensing protein FBXL5 was identified as an inhibitor of SINV infection (Table S3). FBXL5 promotes the degradation of the iron regulatory proteins that stabilize the mRNA for NRAMP2 [12]. Thus, our screen identified a number of factors with clearly identifiable functions in either promoting or inhibiting SINV infection.
Defining inhibition by ARCN1, FUZ and TSPAN9 depletion
We chose three host factors that promote SINV infection for further study, based on the strength of their phenotypes and their possible novel roles in alphavirus infection. Archain1 (ARCN1) is the delta subunit of the COPI coatomer complex, which mediates vesicular traffic in both the secretory and endocytic pathways [13] (Table S4). COPI proteins can play roles in the internalization, RNA replication, and morphogenesis of a variety of viruses [14]–[16] (Table S5), but their role in alphavirus infection has not been defined [17]. Our initial screen also ranked the COPI coatomer proteins COPA, COPB, COPB2 and COPG within the top 1000 proviral hits (Table S1). Fuzzy homologue (FUZ) is an effector protein involved in planar cell polarity, ciliogenesis, and mammalian embryonic development [18]–[21]. FUZ has no reported roles in virus infection or endocytosis. TSPAN9 [22] is a little characterized member of the tetraspanin family of membrane proteins, which have a wide variety of reported functions including acting as membrane organizers, modulating effectors and signaling complexes, and affecting cellular adhesion, motility and membrane fusion [23]. TSPAN9 was identified in a broad endocytosis screen (Table S4) but has no known roles in promoting virus infection (Table S5).
For each of these factors, we confirmed that transfection of the 3 individual siRNAs into U-2 OS cells resulted in inhibition of SINV-Luc infection (Fig. 2A). Quantigene analysis of U-2 OS cells demonstrated that target gene mRNA levels were reduced to <15% of the control by all 3 ARCN1 and FUZ siRNAs and for TSPAN9-1 siRNA, while TSPAN9-2 and -3 siRNAs caused reductions to 22–38 % of control (Fig. 2B). siRNA transfection of HeLa cells produced similar inhibition of infection (Fig. 3A), confirming that the effect is not cell-type specific. We also confirmed both inhibition of virus infection and decreased mRNA levels using either an additional shRNA or an esiRNA (Fig. S2). We selected the individual siRNAs ARCN1-1, FUZ-1, and TSPAN9-1 for further studies. Infection by the alphaviruses SINV-GFP, SINV-pE2S1, Semliki Forest virus (SFV), and Chikungunya virus (CHIKV) was inhibited by these siRNAs to a level similar to the inhibition observed for SINV-Luc (Fig. 3B). SINV-pE2S1 is a strain of SINV whose infection and attachment are independent of heparan sulfate proteoglycans [10]. Thus, the requirement for ARCN1, FUZ, and TSPAN9 is conserved among several different alphaviruses. Infection by the rhabdovirus vesicular stomatitis virus (VSV) was similarly inhibited. In contrast, while infection by the flavivirus Dengue virus 2 (DENV2) was inhibited by clathrin or ARCN1 depletion (Fig. 3B) and was dependent on endosomal acidification (data not shown), it was resistant to depletion of either FUZ or TSPAN9 (Fig. 3B). Human umbilical vein endothelial cells (HUVEC) were transfected with the indicated siRNAs and tested for infection with SFV and CHIKV (Fig. 3C). Depletion of clathrin, ARCN1, FUZ or TSPAN9 significantly inhibited infection by both viruses, thus demonstrating a requirement for these proteins during alphavirus infection of primary human cells.


Depletion of ARCN1, FUZ or TSPAN9 inhibited single cycle SINV infection (Fig. 1A and Table S2). We therefore analyzed the initial stage(s) of the alphavirus lifecycle that might be dependent on these proteins. Fusion of prebound SINV-GFP or SFV with the plasma membrane bypassed the requirement for clathrin, FUZ and TSPAN9 in infection (Fig. 4A), suggesting that like clathrin, FUZ and TSPAN9 are involved in virus entry steps. In contrast, the requirement for ARCN1 was not bypassed (Fig. 4A). Direct binding experiments showed a decrease of >80% in SINV or SFV binding to ARCN1-depleted cells but no decrease with FUZ or TSPAN9-depleted cells (Figs. 4B and S3A). Viral RNA transfection experiments showed a moderate but statistically significant decrease in SINV and SFV infection of ARCN1-depleted cells, most likely through effects on viral RNA transcription/replication (Fig. S3B, C). Thus, ARCN1 depletion most significantly affected alphavirus infection by a strong inhibition of cell surface binding, presumably due to reduced receptor delivery to the plasma membrane. Inhibition was observed for both SINV, which uses NRAMP2 as a receptor [7], and SFV, for which the receptor has not been identified. Decreased virus-cell binding is a mechanism of inhibition that has not been previously described in other studies of the role of the COPI complex in virus infection. While COPI depletion has been reported to inhibit endocytic uptake and traffic [15], [17], its potential effects on alphavirus endocytosis would not be detectable in our system due to the upstream block in alphavirus attachment.

FUZ depletion inhibits virus uptake
To define the roles of FUZ and TSPAN9 in the virus entry pathway, we took advantage of our specific reagents for the steps in SFV entry. We first tested whether depletion of either protein inhibited SFV endocytosis. FUZ-depleted cells showed a striking reduction in SFV internalization, to ∼20% of the level in control cells (Fig. 5A). TSPAN9-depleted cells showed a small decrease in SFV internalization, to ∼80% of control cells (Fig. 5A). We then tested the uptake of transferrin, a ligand that is internalized by clathrin-mediated endocytosis, accumulated in early endosome compartments, and recycled to the plasma membrane [24]. Labeling of FUZ-depleted cells by internalized transferrin was reduced to ∼10% of the level in control cells, while labeling of TSPAN9-depleted cells showed a modest decrease that was not statistically significant (Fig. 5B). Uptake of LDL, which is also internalized by clathrin-mediated endocytosis, was also decreased in the FUZ-depleted cells (Fig. S4). In contrast, the fluid-phase endocytic marker fluorescent dextran produced equivalent labeling of control, TSPAN9-depleted and FUZ-depleted cells (Fig. 5C). Lysotracker staining indicated that general endosome/lysosome acidification was also unimpaired by FUZ or TSPAN9 depletion (data not shown).
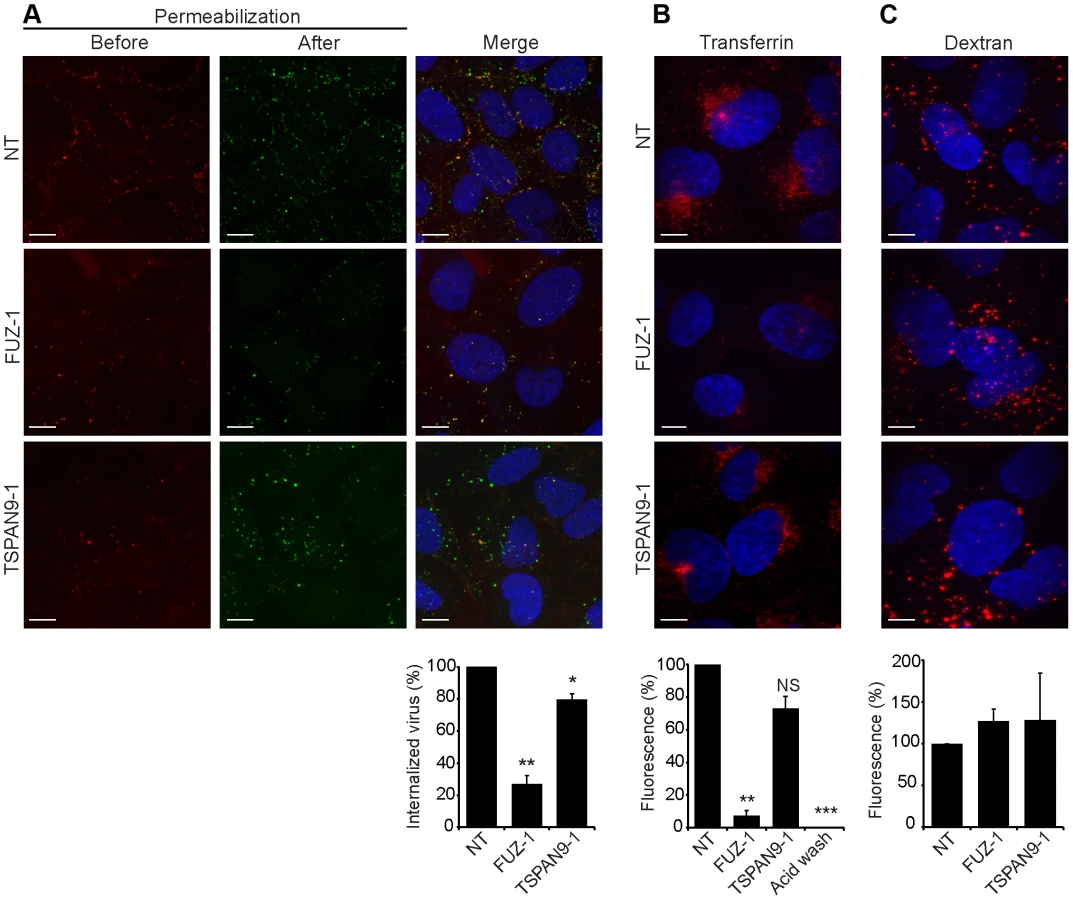
TSPAN9 depletion inhibits intracellular virus fusion
Upon exposure to the low pH environment of the early endosome, the alphavirus fusion protein E1 undergoes acid-induced conformational changes that can be detected by the conformation-specific SFV monoclonal antibody E1a-1 [25]. Since TSPAN9 depletion did not strongly inhibit SFV uptake, we tested if the depleted cells could trigger the E1 conformational change (Fig. 6A). Control cells showed abundant E1a-1staining, and NH4Cl treatment confirmed that staining was dependent on endosomal acidification. Little E1a-1 staining (<10% of control) was observed in FUZ-depleted cells, in keeping with their strong decrease in SFV endocytosis. TSPAN9-depleted cells supported the E1 conformational change, showing a small decrease in E1a-1 staining comparable to the reduction in virus endocytosis. These results suggested that the block in TSPAN9-depleted cells was at a step after the acid-induced conformational change in E1. We directly assayed intracellular virus fusion using SFV labeled with a self-quenching concentration of the lipophilic fluorescent dye DiD, and using E1a-1 staining to correct to equivalent levels of internalized, acid-triggered virus. Membrane fusion of E1a-1-positive SFV was decreased by ∼80% in TSPAN9-depleted cells (Fig. 6B).

Discussion
Our data demonstrated that ARCN1 depletion blocked alphavirus-cell binding, and identified two novel host factors, FUZ and TSPAN9, that promote alphavirus entry and infection. FUZ is a cytoplasmic protein of ∼46 kDa, and was originally identified in Drosophila as Fuzzy, an effector of planar cell polarity [26]. FUZ knockouts in mice cause embryonic lethality and neural tube defects [18], [19], and human FUZ mutations are associated with neural tube defects [27]. While these developmental phenotypes are complex, defects in FUZ act at least in part through their effects on formation of the primary cilium, a structure with roles in signaling pathways [28], [29]. FUZ promotes ciliogenesis, and has been shown to affect the intraflagellar transport system and the localization of the small GTPase rab8 [20], [21]. The relative importance of FUZ in trafficking of internal versus membrane components of the cilium and the potential roles of FUZ in other aspects of membrane traffic remain unclear [29].
Our data indicate that, in addition to its functions in ciliogenesis and development, FUZ plays an important role in clathrin-mediated endocytosis. FUZ depletion blocked alphavirus endocytic uptake and inhibited intracellular transferrin and LDL accumulation. While infection by a panel of alphaviruses and VSV was inhibited by FUZ depletion, both fluid phase uptake and DENV infection were unaffected. This may reflect differences in the entry pathway of DENV versus the pathways of alphaviruses and VSV. All of these viruses are dependent on clathrin, but only DENV fusion occurs in late endosomes [30] and is promoted by acidic phospholipids [31], suggesting a parallel clathrin pathway that is not dependent on FUZ. FUZ mRNA was reported to be expressed in all assayed human tissues [32], suggesting that FUZ could play a role in clathrin-mediated endocytosis in a wide variety of cell types. Although FUZ is essential for mouse development, its depletion by siRNA targeting in primary cells and cell lines did not cause cytotoxic effects within the time scale of our experiments. Given that cells can adapt to the depletion or inhibition of specific endocytic routes by up-regulation of alternative pathways [33], [34], the acute depletion of FUZ will be an important tool to define its mechanism.
TSPAN9 is a 4-pass membrane protein of ∼27 kDa with the N - and C-termini located in the cytoplasm. Its large extracellular loop contains a glycosylation site and 6 cysteine residues, placing it in group 2b of the tetraspanin superfamily [23]. TSPAN9 mRNA is expressed at similar levels in all assayed human tissues [32] and is also expressed in a wide variety of cell lines [22]. Immunolocalization studies in U-2 OS cells stably overexpressing TSPAN9 showed that it is localized at the plasma membrane and on vesicular structures in the cytoplasm (Fig. S5A). TSPAN9 was detected in a systems survey that identified ∼4600 factors involved in transferrin and epidermal growth factor endocytosis [8]. While our data document some reduction in both transferrin and alphavirus endocytosis, this was a relatively minor effect of TSPAN9 depletion. While SFV binding, endocytosis, and low pH-triggered E1 conformational changes occurred quite efficiently in depleted cells, the fusion of internalized virus was strongly blocked. Overexpression of TSPAN9 in U-2 OS cells increased the efficiency of SFV infection compared to that of control cells (Fig. S5B). Together our data support a role for TSPAN9 in a late step of alphavirus entry. TSPAN9 could be required for the correct routing of the virus to the early endosome compartment, or for maintaining the early endosome membrane in a fusion-permissive state. Such models could explain why DENV, which fuses in the late endosome, is resistant to TSPAN9 depletion.
In conclusion, our study reveals distinct and unexpected roles for FUZ and TSPAN9 in alphavirus entry. FUZ and TSPAN9 are conserved from mosquitoes to humans, and thus our results suggest that these proteins play important roles during alphavirus infection of both the mosquito vector and the mammalian host. Defining these roles will be important to understanding the early steps of alphavirus infection and the utility of FUZ and TSPAN9 as novel antiviral targets.
Materials and Methods
Cell lines
U-2 OS cells were maintained in McCoy's 5A medium with 10% fetal bovine serum (FBS), 100 U penicillin/ml, 100 µg streptomycin/ml, and L-glutamine at 37°C. HeLa cells were maintained in DMEM with 10% FBS, 100 U penicillin/ml, 100 µg streptomycin/ml, and L-glutamine at 37°C. To make cell stocks for transfection, U-2 OS or HeLa cells were split 4 days prior to siRNA transfection and seeded at 1×106 in 75 cm2 flasks.
Sindbis test viruses
The pE2R1-GFP/2A Sindbis virus infectious clone (here referred to as SINV-GFP) expressing green fluorescent protein in the cytoplasm [35] was a kind gift from Dr. Hans Heidner. This infectious clone was used to generate a luciferase-expressing virus (SINV-Luc) by replacing GFP with luciferase using overlap PCR. Virus stocks were generated by in vitro RNA transcription and electroporation of BHK-21 cells as previously described [36], and titers were determined on BHK-21 cells.
Initial multi-cycle RNAi screen
Screening was performed at the RNAi core at New York University Langone Medical Center using an arrayed library targeting 21,687 human genes in pools of 3 siRNAs per gene (Ambion Silencer® human genome siRNA library V3). RNAi pools were pre-arrayed in duplicate on Corning white polystyrene 384 well plates, and used to reverse-transfect U-2 OS cells (3000 cells/well) at a final concentration of 37.5 nM siRNA, 0.375% Lipofectamine RNAiMAX (Invitrogen) in medium without antibiotics. Six wells of each of the following control siRNAs were included on each plate: a nontargeting (NT) siRNA (Ambion Silencer® negative control #7), an siRNA targeting the clathrin heavy chain (CLTC) (Dharmacon clathrin heavy chain oligo I) [37], and an siRNA targeting RPL27A (Ambion siRNA ID#: 9257), which induces cell death by targeting a protein required for ribosome biogenesis [38], and which served as a control for transfection efficiency. Transfected cells were incubated for 48 h at 37°C, infected with SINV-Luc at a multiplicity of infection (MOI) of 1, and incubated for 24 h at 37°C, conditions optimized for multiple rounds of infection and the linear range of the assay (Figs. S1A and B). The SteadyGlo Luciferase Assay System reagent (Promega) was added to a final concentration of 25% and plates were read by an EnVision Multilabel plate reader (Perkin Elmer) using the luminescence aperture.
Data analysis
Wells of interest from the initial screen were selected by analysis of the robust z-score and of the controls on each plate. Using CellHTS2 (software package implemented in Bioconductor/R), the raw data were first transformed in log-2 space and normalized to the per-plate median to remove systematic plate-to-plate variation. The robust z-score of each well was then calculated based on the median and median absolute deviation [39]. We also compared the experimental wells to the NT and clathrin siRNA controls on each plate, selecting those genes with lower luciferase activity than clathrin siRNA in both replicates, thereby reducing the likelihood of false positives. The ranked list resulting from these two criteria was manually curated to remove genes in which knockdown was previously reported to cause cell death, or in which luciferase levels were at background, indicative of cell death. We also eliminated genes that appeared to indirectly affect virus infection (e.g., ribosomal proteins). A final list of 400 genes in which infection was reduced by siRNA (corresponding to robust z-scores<−3) and 59 genes in which infection was promoted by siRNA (corresponding to robust z-scores>2) was selected for follow-up screening.
Microsoft EXCEL was used to calculate statistical significance by two-tailed unpaired Student's t-test.
Follow-up screening and confirmation
siRNAs targeting the identified 459 genes were further tested individually for their effects on multi-cycle virus infection, and tested as pools for effects on cell viability and single cycle infection. Selected genes were also targeted by additional siRNAs or shRNAs, tested in HeLa and primary human endothelial cells, and evaluated for effects on other alphaviruses including SFV and CHIKV, and on VSV and DENV. These methods are described in detail in the supplementary information.
Assays of steps in the alphavirus lifecycle
Assay for low pH-dependent fusion at the plasma membrane
U-2 OS cells were transfected as above with the indicated siRNAs in 384 well plates, and cultured for 48 h. Virus was pre-bound to cells on ice for 90 min and then pulsed with pH 5.5 medium for 1 min at 37°C to trigger fusion of virus with the cell plasma membrane [40]. Cells were cultured overnight at 28°C in the presence of 20 mM NH4Cl to prevent secondary infection, and infected cells scored by GFP expression or immunofluorescence. Fluorescence images were captured on a Zeiss Axiovert 200M microscope at 5X magnification. Infected cells were quantitated using Cell Profiler v. 2.0 r11710 cell image analysis software (Broad Institute) [41].
Viral RNA transfection
U-2 OS cells were transfected as above with the indicated siRNAs in 384 well plates, and cultured for 48 h. To test effects on viral RNA transcription/translation, cells were then transfected with SINV-mCherry RNA (a kind gift from Dr. G. Martinez in our lab) or SFV RNA using lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Cells were incubated at 28°C for 16 h in the presence of 20 mM NH4Cl and infected cells were quantitated by fluorescence microscopy as described above.
Virus binding
U-2 OS cells seeded at 4×104 cells/well on 8 well Lab-Tek chambered cover glass (Thermo Scientific) were transfected with 22.5 nM siRNA and 0.15% RNAiMax, and cultured for 48 h. To quantitate virus-cell surface binding, U-2 OS cells were washed 2X with Rmed (RPMI without bicarbonate, plus 10 mM HEPES and 0.2% BSA, pH 6.8). SINV-Luc (MOI = 10) or purified SFV (0.25 µg/well) in Rmed were pre-bound to U-2 OS cells on ice. Cells were then washed once with Rmed, fixed with 3% paraformaldehyde (PFA) in PBS, and bound virus was detected by staining with anti-SINV mAbs R2 and R6 or anti-SFV E1/E2 pAb followed by goat anti-mouse or goat anti-rabbit Ig Alexa fluor 488, respectively. Z-stack images were captured using a Leica SP2 confocal microscope, and brightfield images were used to determine the cell boundaries. Bound virus was quantified using Image J to determine the area of the virus particles bound per 200 cells, and normalized to the value obtained with the NT control. Commercially available antibodies against NRAMP2 were not able to detect cell surface expression of NRAMP2 in U-2 OS or HeLa cells (data not shown), precluding direct tests of receptor levels.
Virus endocytosis
U-2 OS cells were transfected as described for the virus binding assay. Purified SFV (0.25 µg/well) in Rmed pH 6.8 was bound to U-2 OS cells for 90 minutes on ice. The cells were incubated at 37°C in Rmed pH 7.4 for 30 min to permit endocytosis, washed on ice 3X with PBS and fixed with 3% PFA. Cell surface virus was stained with anti-SFV E1/E2 pAb and goat anti-rabbit Ig Alexa fluor 568. Cells were then permeabilized with 0.1% Triton X-100 and intracellular virus was stained with anti-SFV E1/E2 rabbit pAb and goat anti-rabbit Alexa fluor 488. Fluorescence microscopy was performed as described for the binding assay. Endocytosed particles containing only green stain were counted manually in 200 cells, and data normalized to the value obtained for the NT control.
Assay for conversion of E1 to the acid-induced conformation
U-2 OS cells were transfected as described for the virus binding assay. Purified SFV (0.25 µg/well) in Rmed pH 6.8 was bound to cells for 90 min on ice. The cells were then incubated at 37°C for 20 min to permit endocytosis of bound virus, washed 3X with ice-cold PBS and fixed with 3% PFA. Cells were permeabilized with 0.1% Triton X-100 and stained with mAb E1a-1, a previously described monoclonal antibody to the acid conformation of SFV E1 [25], followed by anti-mouse Ig Alexa fluor 568. Virus particles were detected and quantified as described for the binding assay, counting 200 cells/condition.
Lipid mixing virus fusion assay
Purified SFV (25 µg) was labeled by incubating with a final concentration of 7.5 µM 1,1′-Dioctadecyl-3,3,3′,3′-Tetramethylindodicarbocyanine, 4-Chlorobenzenesulfonate (DiD) in 50 µl HNE buffer (5 mM HEPES, 150 mM NaCl, 0.1 mM EDTA, pH 7.0) for 10 min at RT with intermittant vortexing. DiD incorporation was essentially complete at this concentration of dye, and no difference in the results was observed when labeled virus was passed over a desalting column. Labeled virus was therefore used without further purification. U-2 OS cells were incubated for 90 min on ice with 1 µg/well of DiD-labeled SFV in Rmed pH 6.8. The medium was changed to Rmed pH 7.4 and the samples warmed to 37°C for 20 min to permit endocytic uptake of bound virus. Cells were washed with PBS, fixed with 3% PFA, and evaluated for dequenching of DiD as described [42] for the bulk fusion assay. In brief, Z-stacks of 10 fields of cells per experiment were imaged using the 633 nm helium-neon laser on a Leica SP2 or SP5 confocal microscope, and the total fluorescent signal per imaging field was quantitated using Image J software, and normalized for cell number. From 400–600 cells were quantitated per condition. Cells were then permeabilized and stained with mAb E1a-1 as above to detect the low pH conformation of E1. The level of DiD staining was corrected by using mAb E1a-1 staining to adjust to equivalent levels of internalized, acid-triggered virus in each sample. Fusion values were then compared to those obtained for the NT control. Negligible fusion was detected in parallel NT samples treated with 20 mM NH4Cl to inhibit endosomal acidification (data not shown).
Assays of the cellular endocytic pathway
U-2 OS cells were transfected on 8 well chambered cover glass as described for the virus binding assay and then treated as follows:
Transferrin uptake
Cells were prebound on ice for 1 h with 20 µg/ml Alexa fluor 568-conjugated human transferrin in McCoy's 5A medium without serum, incubated for 30 min at 37°C, washed once with 0.5M NaCl, 0.2M acetic acid, pH 2.5, then 2X with PBS, and fixed with 3% PFA. Internalized transferrin, LDL, and dextran (described below) were quantitated with Image J as described for the virus binding assay.
LDL uptake
Cells were incubated in McCoy's 5A medium with 0.1% BSA without serum overnight to induce LDL-receptor expression, and then were prebound on ice for 1 h with 10 µg/ml of DiI-labeled human LDL in McCoy's 5A in the absence of serum. The cells were then incubated for 1 h at 37°C, washed 2X with 4 mg/ml dextran sulfate pH 6.5, then 1x with PBS, and fixed with 3% PFA.
Dextran uptake
Cells were incubated with 0.5 mg/ml Alexa fluor 568-conjugated dextran (10,000 MW) in U-2 OS medium for 3 h at 37°C, washed, cultured in dextran-free U-2 OS medium for 2 h, washed 3X with PBS and fixed with 3% PFA.
Lysotracker assay
U-2 OS cells on 384 wells plates were transfected with siRNAs as described previously. Cells were incubated with 100 nM lysotracker red DND-99 in U-2 OS medium for 1 h at 37°C, transferred to U-2 OS medium without lysotracker, and imaged immediately at 20x magnification using the Zeiss Axiovert 200M microscope. All probes were from Invitrogen/Life Technologies.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Kuhn RJ (2007) Togaviridae: The Viruses and Their Replication. In: Knipe DM, Howley PM, editors. Fields Virology. Fifth ed. Philadelphia, PA: Lippincott, Williams and Wilkins. pp. 1001–1022.
2. EnserinkM (2007) Infectious diseases. Chikungunya: no longer a third world disease. Science 318 : 1860–1861.
3. SchwartzO, AlbertML (2010) Biology and pathogenesis of chikungunya virus. Nat Rev Micro 8 : 491–500.
4. JoseJ, SnyderJE, KuhnRJ (2009) A structural and functional perspective of alphavirus replication and assembly. Future Microbiol 4 : 837–856.
5. VaneyMC, DuquerroyS, ReyFA (2013) Alphavirus structure: activation for entry at the target cell surface. Curr Opin Virol 3 : 151–158.
6. KielianM, Chanel-VosC, LiaoM (2010) Alphavirus entry and membrane fusion. Viruses 2 : 796–825.
7. RosePP, HannaSL, SpiridigliozziA, WannissornN, BeitingDP, et al. (2011) Natural resistance-associated macrophage protein is a cellular receptor for sindbis virus in both insect and mammlian hosts. Cell Host Microbe 10 : 97–104.
8. CollinetC, StoterM, BradshawCR, SamusikN, RinkJC, et al. (2010) Systems survey of endocytosis by multiparametric image analysis. Nature 464 : 243–249.
9. BornerGH, AntrobusR, HirstJ, BhumbraGS, KozikP, et al. (2012) Multivariate proteomic profiling identifies novel accessory proteins of coated vesicles. J Cell Biol 197 : 141–160.
10. KlimstraWB, HeidnerHW, JohnstonRE (1999) The furin protease cleavage recognition sequence of sindbis virus PE2 can mediate virion attachment to cell surface heparan sulfate. J Virol 73 : 6299–6306.
11. OrvedahlA, SumpterRJr, XiaoG, NgA, ZouZ, et al. (2011) Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 480 : 113–117.
12. ThompsonJW, BruickRK (2012) Protein degradation and iron homeostasis. Biochim Biophys Acta 1823 : 1484–1490.
13. BethuneJ, WielandF, MoellekenJ (2006) COPI-mediated transport. J Membr Biol 211 : 65–79.
14. CherryS, KunteA, WangH, CoyneC, RawsonRB, et al. (2006) COPI activity coupled with fatty acid biosynthesis is required for viral replication. PLoS Pathog 2: e102.
15. CuretonDK, Burdeinick-KerrR, WhelanSP (2012) Genetic inactivation of COPI coatomer separately inhibits vesicular stomatitis virus entry and gene expression. J Virol 86 : 655–666.
16. ZhangL, LeeSY, BeznoussenkoGV, PetersPJ, YangJS, et al. (2009) A role for the host coatomer and KDEL receptor in early vaccinia biogenesis. Proc Natl Acad Sci USA 106 : 163–168.
17. DaroE, SheffD, GomezM, KreisT, MellmanI (1997) Inhibition of endosome function in CHO cells bearing a temperature-sensitive defect in the coatomer (COPI) component epsilon-COP. J Cell Biol 139 : 1747–1759.
18. GrayRS, AbituaPB, WlodarczykBJ, Szabo-RogersHL, BlanchardO, et al. (2009) The planar cell polarity effector Fuz is essential for targeted membrane trafficking, ciliogenesis and mouse embryonic development. Nature Cell Biol 11 : 1225–1232.
19. HeydeckW, ZengH, LiuA (2009) Planar cell polarity effector gene Fuzzy regulates cilia formation and Hedgehog signal transduction in mouse. Dev Dyn 238 : 3035–3042.
20. BrooksER, WallingfordJB (2012) Control of vertebrate intraflagellar transport by the planar cell polarity effector Fuz. J Cell Biol 198 : 37–45.
21. ZilberY, BabayevaS, SeoJH, LiuJJ, MootinS, et al. (2013) The PCP effector Fuzzy controls cilial assembly and signaling by recruiting Rab8 and Dishevelled to the primary cilium. Mol Biol Cell 24 : 555–565.
22. SerruV, DessenP, BoucheixC, RubinsteinE (2000) Sequence and expression of seven new tetraspans. Biochim Biophys Acta 1478 : 159–163.
23. LevyS, ShohamT (2005) The tetraspanin web modulates immune-signalling complexes. Nat Rev Immunol 5 : 136–148.
24. EnnsCA, RutledgeEA, WilliamsAM (1996) The transferrin receptor. Biomembranes 4 : 255–287.
25. KielianM, JungerwirthS, SayadKU, DeCandidoS (1990) Biosynthesis, maturation, and acid-activation of the Semliki Forest virus fusion protein. J Virol 64 : 4614–4624.
26. CollierS, GubbD (1997) Drosophila tissue polarity requires the cell-autonomous activity of the fuzzy gene, which encodes a novel transmembrane protein. Development 124 : 4029–4037.
27. SeoJH, ZilberY, BabayevaS, LiuJ, KyriakopoulosP, et al. (2011) Mutations in the planar cell polarity gene, Fuzzy, are associated with neural tube defects in humans. Hum Mol Genet 20 : 4324–4333.
28. GerdesJM, DavisEE, KatsanisN (2009) The vertebrate primary cilium in development, homeostasis, and disease. Cell 137 : 32–45.
29. WallingfordJB, MitchellB (2011) Strange as it may seem: the many links between Wnt signaling, planar cell polarity, and cilia. Genes Dev 25 : 201–213.
30. van der SchaarHM, RustMJ, WaartsBL, van der Ende-MetselaarH, KuhnRJ, et al. (2007) Characterization of the early events in dengue virus cell entry by biochemical assays and single-virus tracking. J Virol 81 : 12019–12028.
31. ZaitsevaE, YangST, MelikovK, PourmalS, ChernomordikLV (2010) Dengue virus ensures its fusion in late endosomes using compartment-specific lipids. PLoS Pathog 6: e1001131.
32. WuC, MacleodI, SuAI (2013) BioGPS and MyGene.info: organizing online, gene-centric information. Nucleic Acids Res 41: D561–565.
33. MercerJ, SchelhaasM, HeleniusA (2010) Virus entry by endocytosis. Annu Rev Biochem 79 : 803–833.
34. SunE, HeJ, ZhuangX (2013) Live cell imaging of viral entry. Curr Opin Virol 3 : 34–43.
35. ThomasJM, KlimstraWB, RymanKD, HeidnerHW (2003) Sindbis virus vectors designed to express a foreign protein as a cleavable component of the viral structural polyprotein. J Virol 77 : 5598–5606.
36. LiljeströmP, LusaS, HuylebroeckD, GaroffH (1991) In vitro mutagenesis of a full-length cDNA clone of Semliki Forest virus: the small 6,000-molecular-weight membrane protein modulates virus release. J Virol 65 : 4107–4113.
37. HinrichsenL, HarborthJ, AndreesL, WeberK, UngewickellEJ (2003) Effect of clathrin heavy chain - and alpha-adaptin-specific small inhibitory RNAs on endocytic accessory proteins and receptor trafficking in HeLa cells. J Biol Chem 278 : 45160–45170.
38. WildT, HorvathP, WylerE, WidmannB, BadertscherL, et al. (2010) A protein inventory of human ribosome biogenesis reveals an essential function of exportin 5 in 60S subunit export. PLoS Biol 8: e1000522.
39. BirminghamA, SelforsLM, ForsterT, WrobelD, KennedyCJ, et al. (2009) Statistical methods for analysis of high-throughput RNA interference screens. Nat Methods 6 : 569–575.
40. LiaoM, KielianM (2005) Domain III from class II fusion proteins functions as a dominant-negative inhibitor of virus-membrane fusion. J Cell Biol 171 : 111–120.
41. KamentskyL, JonesTR, FraserA, BrayMA, LoganDJ, et al. (2011) Improved structure, function and compatibility for CellProfiler: modular high-throughput image analysis software. Bioinformatics 27 : 1179–1180.
42. Ayala-NunezNV, WilschutJ, SmitJM (2011) Monitoring virus entry into living cells using DiD-labeled dengue virus particles. Methods 55 : 137–143.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2013 Číslo 12
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Influence of Mast Cells on Dengue Protective Immunity and Immune Pathology
- Host Defense via Symbiosis in
- Coronaviruses as DNA Wannabes: A New Model for the Regulation of RNA Virus Replication Fidelity
- Myeloid Dendritic Cells Induce HIV-1 Latency in Non-proliferating CD4 T Cells
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy