
-Induced Activation of EGFR Prevents Autophagy Protein-Mediated Killing of the Parasite
Toxoplasma gondii resides in an intracellular compartment (parasitophorous vacuole) that excludes transmembrane molecules required for endosome - lysosome recruitment. Thus, the parasite survives by avoiding lysosomal degradation. However, autophagy can re-route the parasitophorous vacuole to the lysosomes and cause parasite killing. This raises the possibility that T. gondii may deploy a strategy to prevent autophagic targeting to maintain the non-fusogenic nature of the vacuole. We report that T. gondii activated EGFR in endothelial cells, retinal pigment epithelial cells and microglia. Blockade of EGFR or its downstream molecule, Akt, caused targeting of the parasite by LC3+ structures, vacuole-lysosomal fusion, lysosomal degradation and killing of the parasite that were dependent on the autophagy proteins Atg7 and Beclin 1. Disassembly of GPCR or inhibition of metalloproteinases did not prevent EGFR-Akt activation. T. gondii micronemal proteins (MICs) containing EGF domains (EGF-MICs; MIC3 and MIC6) appeared to promote EGFR activation. Parasites defective in EGF-MICs (MIC1 ko, deficient in MIC1 and secretion of MIC6; MIC3 ko, deficient in MIC3; and MIC1-3 ko, deficient in MIC1, MIC3 and secretion of MIC6) caused impaired EGFR-Akt activation and recombinant EGF-MICs (MIC3 and MIC6) caused EGFR-Akt activation. In cells treated with autophagy stimulators (CD154, rapamycin) EGFR signaling inhibited LC3 accumulation around the parasite. Moreover, increased LC3 accumulation and parasite killing were noted in CD154-activated cells infected with MIC1-3 ko parasites. Finally, recombinant MIC3 and MIC6 inhibited parasite killing triggered by CD154 particularly against MIC1-3 ko parasites. Thus, our findings identified EGFR activation as a strategy used by T. gondii to maintain the non-fusogenic nature of the parasitophorous vacuole and suggest that EGF-MICs have a novel role in affecting signaling in host cells to promote parasite survival.
Published in the journal:
. PLoS Pathog 9(12): e32767. doi:10.1371/journal.ppat.1003809
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1003809
Summary
Toxoplasma gondii resides in an intracellular compartment (parasitophorous vacuole) that excludes transmembrane molecules required for endosome - lysosome recruitment. Thus, the parasite survives by avoiding lysosomal degradation. However, autophagy can re-route the parasitophorous vacuole to the lysosomes and cause parasite killing. This raises the possibility that T. gondii may deploy a strategy to prevent autophagic targeting to maintain the non-fusogenic nature of the vacuole. We report that T. gondii activated EGFR in endothelial cells, retinal pigment epithelial cells and microglia. Blockade of EGFR or its downstream molecule, Akt, caused targeting of the parasite by LC3+ structures, vacuole-lysosomal fusion, lysosomal degradation and killing of the parasite that were dependent on the autophagy proteins Atg7 and Beclin 1. Disassembly of GPCR or inhibition of metalloproteinases did not prevent EGFR-Akt activation. T. gondii micronemal proteins (MICs) containing EGF domains (EGF-MICs; MIC3 and MIC6) appeared to promote EGFR activation. Parasites defective in EGF-MICs (MIC1 ko, deficient in MIC1 and secretion of MIC6; MIC3 ko, deficient in MIC3; and MIC1-3 ko, deficient in MIC1, MIC3 and secretion of MIC6) caused impaired EGFR-Akt activation and recombinant EGF-MICs (MIC3 and MIC6) caused EGFR-Akt activation. In cells treated with autophagy stimulators (CD154, rapamycin) EGFR signaling inhibited LC3 accumulation around the parasite. Moreover, increased LC3 accumulation and parasite killing were noted in CD154-activated cells infected with MIC1-3 ko parasites. Finally, recombinant MIC3 and MIC6 inhibited parasite killing triggered by CD154 particularly against MIC1-3 ko parasites. Thus, our findings identified EGFR activation as a strategy used by T. gondii to maintain the non-fusogenic nature of the parasitophorous vacuole and suggest that EGF-MICs have a novel role in affecting signaling in host cells to promote parasite survival.
Introduction
Toxoplasma gondii is an obligate intracellular protozoan parasite that infects around a third of the human population worldwide. T. gondii is of clinical importance because it causes encephalitis in immunocompromised individuals and retino-choroiditis in immunocompetent and immunosuppressed patients. T. gondii can also cause congenital infection that may result in cerebral and ocular disease. Tachyzoites of T. gondii infect virtually any nucleated cell through active invasion. This process is dependent on the parasite actin-myosin motor and sequential secretion of proteins from micronemes and rhoptries, specialized organelles present in the apical end of the parasite [1]. Once secreted, T. gondii micronemal proteins (MICs) are expressed at the parasite surface membrane and they interact with host cell receptors [2]. MICs contain adhesive domains such as type I thrombospondin repeats, apple domains, EGF repeats and integrin A domains [3], [4]. The connection between transmembrane MICs to the actin-myosin motor (glideosome) of the parasite together with the binding of host cell receptors by MICs is considered to enable the organism to penetrate host cells [5], [6]. Following the release of MICs, rhoptries secrete rhoptry neck proteins (RONs) that are critical for the formation of a structure called the moving junction (MJ) [7], [8]. The MJ anchors the parasite to the host cell while the parasite penetrates it. The MJ is also believed to function as a sieve that excludes host type I transmembrane proteins from entering the PV membrane (PVM) [8], [9]. The end result is the formation of a parasitophorous vacuole that is devoid of host proteins required for recruitment of endosomes and lysosomes [10].
T. gondii cannot withstand the lysosomal environment. Thus, the non-fusogenic nature of the PV is critical since it allows the parasite to survive and replicate. The immune system can deprive the parasite from this niche by disrupting the PVM through the effects of IFN-γ/Immunity related GTPases (IRG) [11], [12] and by making the PV fusogenic through the effects of CD40 ligation [13]–[15]. CD40 re-routes the PV to the lysosomes through the autophagy machinery [13]–[15].
Autophagy is a conserved cellular mechanism of lysosomal degradation. During autophagy, portions of the cytosol or organelles are encircled by an isolation membrane [16]. The expansion of the isolation membrane results in the formation of a double membrane structure called autophagosome that delivers its contents to the lysosomes for degradation [16]. Autophagy is recognized as a mechanism stimulated by innate and adaptive immune mechanisms to degrade numerous intracellular pathogens [17]. However, various bacteria and viruses have evolved mechanisms to prevent autophagic degradation by targeting autophagy proteins to avoid recognition by the autophagy machinery or prevent the initiation and maturation of autophagosomes [18]–[24]. Much less is known regarding whether pathogens manipulate signaling cascades that regulate autophagy to prevent their degradation. HIV-1 envelope can activate the negative regulator of autophagy mTOR and it has been proposed that this would prevent lysosomal degradation of virions [25]. Bioinformatic analysis of human THP-1 cells infected with Mycobacterium tuberculosis suggested that the pathogen activates host signaling cascades that impair autophagy [26].
The highly successful nature of T. gondii as a pathogen together with evidence that the autophagy pathway can trigger lysosomal killing of the pathogen raise the possibility that T. gondii prevents autophagic targeting of the PV to maintain the non-fusogenic nature of the PV. Moreover, approximately 25–35% of various CD40+ cells subjected to CD40 ligation are unable to kill T. gondii further suggesting that the parasite may utilize mechanism(s) to prevent induction of autophagic killing. Here we report that maintenance of the non-fusogenic nature of the PV requires T. gondii-induced activation of EGFR-Akt, a signaling cascade that prevents autophagy protein-dependent vacuole-lysosomal fusion, lysosomal degradation and killing of the parasite. Blockade of EGFR-Akt may prove of therapeutic benefit for toxoplasmosis since it is sufficient to induce killing of the parasite without the need for immune-induced activation of host cells.
Results
T. gondii induces rapid Akt activation in non-hematopoietic cells through phosphatidylinositol 3-kinase (PI3K)
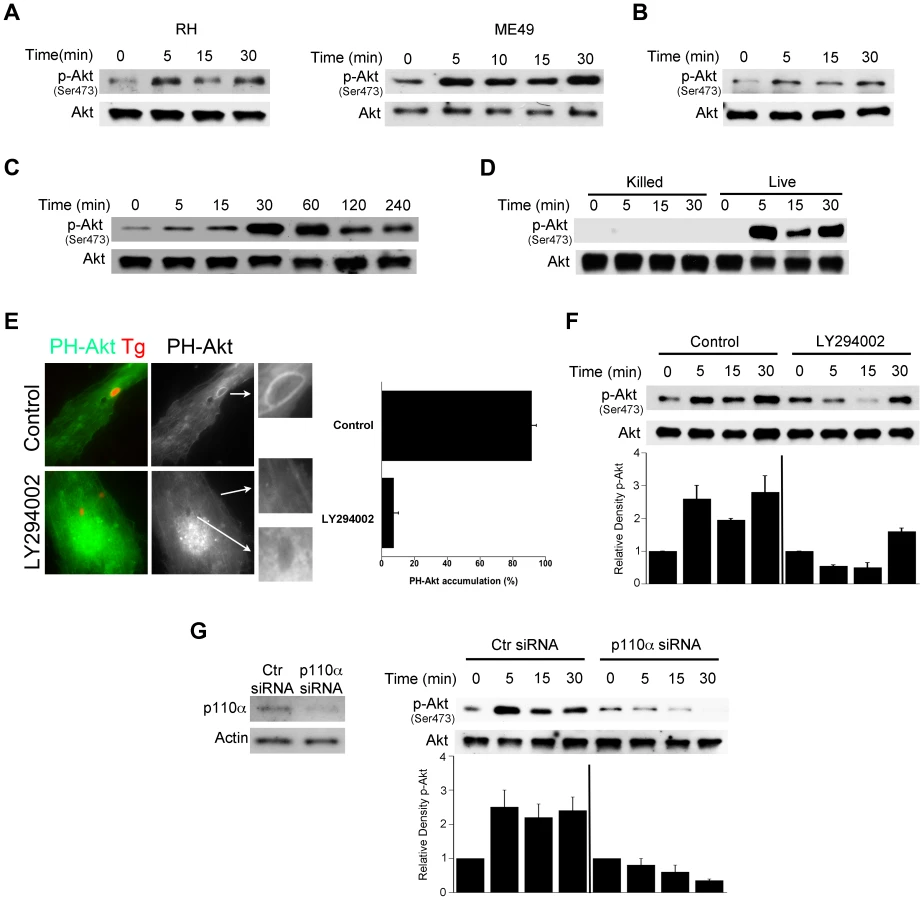
We determined whether Akt is quickly activated by T. gondii during infection of various non-hematopoietic cells. Activation of Akt is a multistep process where phosphorylation of Serine 473 results in full activation of the molecule [27]. Primary human brain microvascular endothelial cells (HBMEC) were infected with either type I (RH) or type II (ME49) strains of T. gondii under conditions that caused synchronized infection. T. gondii infection resulted in an enhanced phosphorylation of Akt Serine 473 as assessed by immunoblot (Figure 1A). Similar results were obtained with a mouse endothelial cell line mHEVc (Figure 1B). T. gondii also caused Akt phosphorylation in a human retinal pigment epithelial (RPE) cell line, an effect that decreased at later time points post-infection (Figure 1C). We assessed whether viable parasites are required to induce activation of Akt. HBMEC were challenged with live or killed parasites followed by determination of Akt activation. Viable but not killed tachyzoites induced Akt phosphorylation (Figure 1D). Activation of phosphatidylinositol 3-kinase (PI3K) with resulting production of phosphatidylinositol 3,4,5 trisphosphate (PIP3) production is a major trigger of Akt activation [28]. The amino-terminal pleckstrin homology (PH) domain of Akt mediates recruitment of this molecule to plasma membrane containing increased PI(3,4,5)P3 or PI(3,4)P2 [29]. Indeed, the PH domain of Akt fused to GFP (PH-Akt-GFP) has been used as a probe to examine sites of PIP3 accumulation [30]. HBMEC were transiently transfected with a plasmid encoding PH-Akt-GFP followed by challenge with RH T. gondii that express cytoplasmic RFP (T. gondii-RFP). T. gondii-infected cells exhibited accumulation of PH-Akt-GFP around the parasite (Figure 1E). To examine the role of PI3K in this process, HBMEC were incubated with or without LY294002, a specific PI3K inhibitor, followed by challenge with the parasite. LY294002 did not affect the percentage of infected cells (not shown). Accumulation of PH-Akt-GFP around T. gondii was ablated by LY294002 (p<0.01) (Figure 1E). Moreover, incubation with LY294002 impaired the upregulation of Akt phosphorylation induced by T. gondii, especially in the earlier time points post-infection (Figure 1F). Similarly, Akt phosphorylation during T. gondii infection was impaired in HBMEC transfected with siRNA against the PI3K catalytic subunit p110α (Figure 1G). Taken together, these findings indicate that T. gondii induces rapid Akt activation in non-hematopoietic cells in a manner that is dependent on PI3K.
Blockade of Akt induces accumulation of the autophagy protein LC3 around the parasite, vacuole-lysosome fusion and killing of T. gondii dependent on autophagy proteins
We performed studies to investigate whether blockade of Akt signaling promotes killing of T. gondii. HBMEC were incubated with or without Akt inhibitor IV followed by challenge with T. gondii. The percentage of infected cells at 2 hours and 24 hours post-challenge were determined. Akt inhibitor IV did not impair the percentage of infected cells at 2 h (Figure 2A). However, treatment with Akt inhibitor IV markedly reduced the percentage of infected cells at 24 h (p<0.01) (Figure 2A). Changes in the percentage of infected cells were not due to preferential cell loss in Akt inhibitor IV-treated cells since cell densities as determined with an eyepiece grid were similar in all experimental groups and inhibition of Akt did not induce a detectable increase in apoptosis of T. gondii-infected cells (not shown). Akt inhibitor IV not only induced a significant decrease in the numbers of parasites per 100 HBMEC at 24 h but it also caused a profound reduction in the numbers of T. gondii-containing vacuoles per 100 HBMEC (p<0.01) (Figure 2A, Figure S1A). Similar results were obtained with mouse endothelial cells (mHEVc; Figure 2A, Figure S1A) and human RPE cells (p<0.01) (Figure 2A, Figure S1A). Not only pharmacologic inhibition of Akt but also Akt knockdown in HBMEC reduced the parasite load and the number of T. gondii-containing vacuoles (p<0.01) (Figure 2B, Figure S1A). The vacuoles that persisted after Akt knockdown had similar numbers of parasites as those from control cells (Figure S1B). These results indicate that blockade of Akt caused parasite killing. T. gondii infection causes Akt activation in macrophages [31]. Similar to endothelial and epithelial cells, treatment with Akt inhibitor IV caused anti-T. gondii activity in the mouse macrophage line RAW 264.7 and in mouse microglia line BV-2 (p<0.01) (Figure 2C and not shown). These findings revealed an important role of Akt activation in promoting survival of T. gondii within host cells.

T. gondii survives within mammalian cells by avoiding delivery of the lysosomal contents into the parasitophorous vacuole [32]–[34]. Akt is a negative regulator of autophagy [35], a cellular mechanism that results in lysosomal degradation and killing of T. gondii [13]–[15]. First, we examined T. gondii-infected cells after Akt inhibition to determine the distribution of LC3, a protein associated with the autophagosome membrane. mHEVc-LC3-EGFP cells were treated with or without Akt inhibitor IV and challenged with T. gondii-RFP. Akt inhibitor IV led to significant accumulation of LC3 around the parasite (p<0.01) (Figure 2D). Electron microscopy studies were performed since a double membrane isolation membrane that encircles portions of cytoplasm or organelles is formed during autophagy [16]. Indeed, a double membrane structure was noted around the parasitophorous vacuole membrane in HBMEC treated with Akt inhibitor IV (Figure 2E). Next, we examined the effects of Akt inhibition on the distribution of the late endosomal/lysosomal molecule LAMP-1. Endothelial cells were incubated with or without Akt inhibitor IV, challenged with T. gondii-YFP followed by staining with anti-LAMP-1 mAb. Treatment with Akt inhibitor IV resulted in a remarkable increase in the percentage of parasites surrounded by LAMP-1 (p<0.01) (Figure 2F). To explore whether the killing of T. gondii during inhibition of Akt is dependent on the autophagy machinery, we examined the effects of knockdown of the autophagy proteins Beclin 1 or Atg7 on T. gondii survival. Transfection with Beclin 1 siRNA or Atg7 siRNA effectively diminished expression of Beclin 1 or Atg7 respectively (Figure 2G, H). Endothelial cells transfected with Beclin1 siRNA or Atg7 siRNA were incubated with or without Akt inhibitor and challenged with T. gondii. Cells transfected with Beclin1 siRNA (Figure 2G) or Atg7 siRNA (Figure 2H) were unable to control the parasite in the presence of the Akt inhibitor IV. Since autophagosomes deliver their contents to lysosomes for degradation, we examined the role of lysosomal degradation in killing of T. gondii utilizing the lysosomal protease inhibitors leupeptin and pepstatin. mHEVc and RPE cells were treated with or without Akt inhibitor IV and infected with T. gondii. 1 h post infection cells were treated with or without leupeptin plus pepstatin. Lysosomal protease inhibitors impaired the anti-T. gondii activity induced by Akt inhibition (p<0.05) (Figure 2I and not shown). Finally, the anti-T. gondii activity induced by Akt inhibitor IV in mouse microglia and human RPE cells was impaired by 3-methyl adenine, an inhibitor of autophagy (p<0.05) (Figure 2J and not shown). Taken together, these results indicate that T. gondii-induced Akt activation is critical to promote parasite survival because it prevents killing of T. gondii dependent on the autophagy pathway and lysosomal protease activity.
T. gondii infection induces EGFR activation in mammalian cells that prevents autophagy pathway dependent killing of the parasite
Akt activation classically occurs downstream of cell membrane receptors that include growth factor receptors, G protein-coupled receptor (GPCR) and TLR [36]. To examine the role of GPCR in Akt activation in non-hematopoietic cells, HBMEC were incubated with or without Pertussis toxin (PTx), an inhibitor of GPCR signaling, followed by challenge with T. gondii tachyzoites. PTx did not affect the initial percentage of infected cells (data not shown). Incubation with PTx decreased basal Akt phosphorylation. However, PTx did not prevent the increased Akt phosphorylation induced by T. gondii (Figure 3A) indicating that T. gondii can activate Akt independently of GPCR signaling. In contrast, PTx inhibited Akt activation induced by lysophosphatidic acid (LPA), a GPCR ligand [37] (Figure 3A). To examine the potential role of TLR signaling in Akt, MyD88 was knocked-down in HBMEC using siRNA. Knockdown of MyD88 did not affect T. gondii-induced Akt activation (Figure 3B). In contrast, as assessed by FACS, the ICAM-1 upregulation induced by LPS (1 µg/ml) in HBMEC was inhibited in cells transfected with MyD88 siRNA compared to those transfected with control siRNA (cMFI: Control siRNA = 10,682±1,053; MyD88 siRNA = 3,250±527; p<0.05). These studies indicate that GPCR and TLR are unlikely to play a major role in Akt phosphorylation induced by T. gondii in non-hematopoietic cells.

Relevant to the possibility of activation of growth factor receptors during T. gondii-host cell interaction is the fact that host cell invasion by T. gondii requires the secretion of parasite micronemal proteins (MICs) with the potential to activate such receptors [38]. MICs exist as multiprotein complexes, the most important being MIC1/4/6, MIC3/8, MIC2/M2AP, and a complex of the microneme protein TgAMA1 with rhoptry neck proteins RON2/RON4/RON6/RON8 [39]–[41]. MIC3, MIC6 and MIC8 have multiple domains with homology to EGF [42] and are therefore termed EGF-MICs. As an initial experiment, we examined whether T. gondii induces autophosphorylation at 2 major tyrosine residues of EGFR (1068 and 1148). HBMEC were incubated with RH T. gondii tachyzoites followed by determination of EGFR phosphorylation by immunoblot. T. gondii induced activation of EGFR, as indicated by phosphorylation of tyrosine residue 1068 (Figure 4A). Moreover, the parasite caused phosphorylation of tyrosine residue 1148, a site that appears to be phosphorylated only by ligand binding to EGFR [43] (Figure 4A). Similar results were found using the ME49 strain of T. gondii (not shown). Immunoblot analysis revealed that EGFR activation occurred in HBMEC upon challenge with viable but not killed parasites (Figure 4B). EGFR autophosphorylation was not only observed in endothelial cells but also in human RPE cells and mouse microglia incubated with T. gondii (Figure 4C, 4D). Thus, T. gondii causes EGFR activation in various mammalian cells.

Next, we examined whether EGFR signaling is involved in activation of Akt triggered by T. gondii. Endothelial cells were transiently transfected with a plasmid that encodes either control siRNA or EGFR siRNA followed by challenge with T. gondii. The efficiency of EGFR knockdown was confirmed by immunoblot (Figure 5A). EGFR knockdown ablated the ability of T. gondii to induce activation of Akt at all time points tested (Figure 5A). Next, we explored the role of EGFR signaling on Akt activation in professional phagocytes. Mouse microglia were treated with vehicle or AG1478, a pharmacological inhibitor of EGFR kinase activity, followed by challenge with T. gondii. Inhibition of EGFR kinase activity ablated parasite-induced Akt activation in mouse microglia (Figure 5B).

We assessed whether EGFR activation affects T. gondii survival within host cells. HBMEC were treated with vehicle or AG1478 followed by challenge with T. gondii. While AG1478 did not affect the percentage of infected cells at 2 h, AG1478 caused a marked reduction in the percentage of infected cells 24 h post-challenge (p<0.05) (Figure 6A). In addition, there was a significant reduction in the numbers of parasites per 100 endothelial cells (p<0.01) (Figure 6A). Similar results were obtained whether HBMEC or human retinal endothelial cells were infected with RH or ME49 strains of T. gondii (not shown). The role of EGFR in affecting parasite survival was confirmed with a genetic approach since knockdown of EGFR in human RPE cells resulted in enhanced killing of T. gondii (p<0.01) (Figure 6B). Similar to the studies of blockade of Akt, inhibition of EGFR signaling not only reduced the percentages of infected cells but also caused a reduction in the numbers of vacuoles per 100 cells without affecting the numbers of parasites in the vacuoles that persisted after EGFR blockade (not shown). The effects of EGFR signaling inhibition were not restricted to non-hematopoietic cells since mouse bone marrow-derived macrophages also acquired anti-T. gondii activity when treated with AG1478 (p<0.05) (Figure 6C). To further explore the role of EGFR in the survival of T. gondii, we took a reverse approach and infected parental CHO cells, known to be EGFR null [44], and CHO cells expressing human EGFR (CHO-EGFR). A reduction in the percentage of infected cells and a reduction in parasite load at 24 h were observed in parental CHO cells compared to CHO-EGFR cells (p<0.05) (Figure 6D). These findings revealed an important role of EGFR in promoting Akt activation and T. gondii survival within host cells.

We investigated whether T. gondii killing induced by inhibition of EGFR is dependent on autophagy proteins. Knockdown of EGFR in mHEVc cells or treatment of these cells with AG1478 resulted in an enhanced accumulation of LC3 and LAMP-1 around the parasite (p<0.05) (Figure 6E and 6F). Moreover, silencing of Beclin 1 or Atg7 prevented induction of anti-T. gondii activity in endothelial cells subjected to EGFR knock-down or treated with AG1478 (p<0.01) (Figure 6G and 6H). Taken together, activation of EGFR signaling promoted survival of T. gondii within host cells by inhibiting autophagy protein-dependent killing of the parasite.
T. gondii MICs can induce phosphorylation of EGFR and Akt in host cells
EGFR ligands exist as precursors transmembrane proteins that are shed from the plasma membrane by members of the ADAM (a disintegrin and metalloprotease) family of zinc-dependent metalloproteases [45]. This results in an autocrine or paracrine EGFR activation, a phenomenon that explains how proteins such GPCR activate EGFR [45]. We explored whether EGFR activation triggered by T. gondii could be due to this mechanism of autocrine/paracrine signaling. HBMEC were treated with GM6001, a broad spectrum ADAM inhibitor, followed by challenge with T. gondii. GM6001 did not affect the percentage of infected cells (data not shown) and did not prevent the ability of T. gondii to induce EGFR activation (Figure 7A). Moreover, EGFR phosphorylation after T. gondii infection took place despite incubation with PTx (Figure 7B). These findings suggest that ADAM - and GPCR-dependent EGFR activation do not play a major role in EGFR phosphorylation induced by T. gondii.

As stated above, MIC3, MIC6, MIC8 have multiple domains with homology to EGF [42]. MIC7 and MIC9 also express EGF-like domains but these MICs have poor or no expression in tachyzoites [42]. We examined the effect of deficiency of MICs on the ability to induce activation of EGFR and Akt. HBMEC were infected with wild type (WT), MIC1 ko (lacks MIC1, resulting in deficient secretion of MIC6 [46]), MIC3 ko (lacks MIC3), MIC1-3 ko (lacks MIC6 secretion and MIC3) parasites followed by determination of EGFR and Akt activation. These MIC ko parasites still express MIC8 (MIC8 deficiency results in parasites that are unable to infect mammalian cells). The multiplicity of infection was adjusted so that the initial percentages of infected HBMEC were similar for all strains of the parasite (Figure 8A). Compared to WT T. gondii, MIC1 ko and MIC3 ko parasites caused a partial reduction in EGFR and Akt phosphorylation (p<0.05) (Figure 8B, 8C). MIC1-3 ko parasites caused further decrease in EGFR and Akt phosphorylation compared to MIC1 ko and MIC3 ko parasites (p<0.05) (Figure 8B, 8C). However, even in cells infected with MIC1-3 ko parasites the reduction in EGFR and Akt phosphorylation was not complete. MIC1-3 ko parasites still express MIC8, a molecule that has EGF-like domains. We used conditional MIC8 knockout T. gondii previously generated using a tetracycline-inducible system to explore the potential role of MIC8 in signal activation [47]. Incubation of these parasites with anhydrotetracycline (ATc) results in almost complete ablation of MIC8 [47]. Parasites previously grown in the absence or presence of ATc were incubated with HBMEC. We could not detect an appreciable decrease in Akt phosphorylation in cells exposed to MIC8 deficient parasites (Figure S2). To further explore the role of MICs in the activation of EGFR and Akt, HBMEC were incubated with Pichia pastoris-derived MIC3. Although the EGF-like domains alone do not appear to promote the adhesion of MIC3 to mammalian cells [48], it was still possible that MIC3 could cause EGFR and Akt activation. Indeed, compared to recombinant MIC4 (a control that does not express EGF-like domains) incubation with recombinant MIC3 caused enhanced phosphorylation of EGFR and Akt in HBMEC (Figure 8D, 8E). Moreover, incubation with E. coli-derived MIC6 but not M2AP caused EGFR-Akt phosphorylation (Figure 8D, 8E). This response was unlikely to be mediated by LPS since M2AP and MIC6 preparations had similar concentrations of LPS (12 ng/ml and 12.4 ng/ml respectively). In addition, LPS at concentrations between 10–1,000 ng/ml failed to induce EGFR phosphorylation in HBMEC (not shown). Taken together, EGF-MICs (MIC3 and MIC6) can induce EGFR-Akt activation and parasites deficient on these MICs have diminished capacity to activate EGFR and Akt.

EGFR signaling and MICs impair autophagic killing of T. gondii
Cells stimulated with CD154 (CD40 ligand) exhibit accumulation of LC3 around T. gondii and killing that is dependent on autophagy proteins [13]–[15]. We examined whether targeting of the parasite by LC3+ structures in CD154-treated cells can be affected by EGFR signaling. Endothelial cells were treated with or without CD154 followed by challenge with T. gondii in the presence or absence of EGF. EGF did not affect the initial percentage of infected cells (not shown). As previously reported [15], CD154 caused accumulation of LC3 around T. gondii (Figure 9A). Targeting of parasites by LC3+ structures was inhibited in cells that were exposed to EGF (p<0.05) (Figure 9A), The effect of EGF was specific since addition of AG1478 to cells treated with EGF restored LC3 accumulation around T. gondii (Figure 9A). Similar results were obtained using rapamycin, a well-described stimulator of autophagy (Figure 9B). Next, we explored the role of MICs on the distribution of LC3+ structures in endothelial cells treated with CD154. Endothelial cells were treated with or without CD154 followed by challenge with WT, MIC1 ko, MIC3 ko, MIC1-3 ko and their respective complemented parasites. Infection with MIC1 ko, MIC3 ko or MIC1-3 ko parasites induces a partial decrease in EGFR-Akt activation (see Figures 8B, 8C). Indeed, in control endothelial cells (no CD154 treatment) there were no differences in the low level LC3 accumulation around the parasites (Figure 9C). After treatment with CD154, enhanced accumulation of LC3 around the parasites was similar in endothelial cells infected with WT, MIC1 ko or MIC3 ko parasites (Figure 9C). In contrast, cells infected with MIC1-3 ko parasites (the strain that was the weakest inducer of EGFR-Akt activation) exhibited a significant further increase in LC3 accumulation (p<0.05) (Figure 9C). These results were specific because the phenotype was lost in the complemented strain of T. gondii (MIC1-3 ko+MIC1-3) (Figure 9C). Examination of the parasite load revealed that the loads of MIC1 ko, MIC3 ko and MIC1-3 ko parasites were not significantly different from those of WT parasites in control endothelial cells (no CD154 treatment) (Figure 9D). When cells were treated with CD154, MIC1-3 ko T. gondii displayed increased susceptibility to CD154-induced anti-T. gondii activity (p<0.05) (Figure 9D). Similar to the studies of LC3 expression, the phenotype of MIC1-3 ko parasites was lost in the complemented strain (MIC1-3 ko+MIC1-3) (Figure 9D). Next, we examined whether increased killing of MIC1-3 ko parasites was observed in cells treated with another autophagy inducer (rapamycin) or in cells treated with IFN-γ, a cytokine that triggers anti-T. gondii activity independently of autophagic degradation [13]–[15]. Similar to CD154-stimulated cells, MIC1-3 ko parasites were more susceptible to rapamycin-induced killing (p<0.05) (Figure 9E). Moreover, in contrast to the results obtained with CD154-stimualtion, anti-T. gondii activity induced by IFN-γ/TNF-α was similar in all parasite strains tested including MIC1-3 ko T. gondii (Figure 9F). Finally, we explored the effects of recombinant MICs on CD154-induced killing of MIC1-3 ko T. gondii. In initial experiments, recombinant MICs did not affect the load of T. gondii in non-activated (control) endothelial cells or cells treated with IFN-γ/TNF-α (not shown). Next, control or CD154-activated endothelial cells were challenged with WT or MIC1-3 ko parasites in the presence of absence of recombinant MICs. Whereas treatment of endothelial cells with MIC4 and M2AP did not affect the load of WT or MIC1-3 ko parasites in CD154-activated cells, treatment with MIC3 or MIC6 inhibited CD154-induced T. gondii activity (p<0.05) (Figure 9G). Moreover, the phenotype of MIC1-3 ko parasites of increased susceptibility to CD154-mediated anti-T. gondii activity was lost in the presence of either MIC3 or MIC6 since the loads of WT and MIC1-3 ko parasites were no longer different in cells treated with these EGF-MICs (Figure 9G). Taken together, our findings indicate that EGFR, MIC3 and MIC6 negatively regulate autophagic killing of T. gondii.

Discussion
Avoidance of lysosomal degradation is pivotal for the survival of numerous intracellular pathogens including T. gondii. Our studies indicate that, in addition to exclusion of type I transmembrane proteins from the PVM, T. gondii also activates EGFR-Akt signaling in the host cell to prevent targeting of the parasite by LC3+ structures and pathogen killing that is dependent on autophagy proteins and lysosomal protease activity. Thus, these studies identified EGFR-Akt signaling as a pathway critical for pathogen survival. In addition, they suggest that EGF-MICs may be involved in pathogen virulence not only by allowing parasite invasion of host cells but also by activating host cell signaling that counter-regulates autophagy.
Various bacteria and viruses encode virulence factors that impair the function of autophagy proteins and as a result, avoid their degradation via the autophagy pathway [18]–[24]. It has been suggested that HIV-1 and M. tuberculosis may prevent autophagic degradation by affecting signaling cascades that regulate the autophagy pathway [25], [26]. Our studies indicate that indeed a pathogen can act at the level of a regulatory pathway to avoid its degradation by the autophagy machinery. Relevant to our findings is the report that HIV-1 tat impairs autophagy by stimulating counter-regulatory cascades (Akt and STAT3), although these studies did not examine whether these pathways would prevent lysosomal degradation of the virions [49].
Our studies indicate that T. gondii-induced EGFR activation is a major event upstream of Akt phosphorylation in endothelial and RPE cells, a finding consistent with the important role of EGFR and other growth factor receptors as activators of Akt signaling [36], [50]. PI3K is a classical link between growth factor receptors and Akt activation. However, in contrast inhibition of EGFR signaling, the effect of PI3K inhibition on Akt activation appeared to be more transient. These findings may be explained by the fact that, besides PI3K, there are additional activators of Akt that might be engaged by growth factor receptors [51]. T. gondii has been reported to activate Akt in macrophages, a phenomenon that was inhibited by PTx [31]. Our studies indicate that EGFR also contributes to Akt activation in macrophages/microglia since the parasite caused EGFR autophosphorylation and inhibition of EGFR signaling impaired parasite-induced Akt activation. Moreover, not only activation of Akt but also activation of EGFR in endothelial cells, RPE cells and macrophages/microglia prevented killing of T. gondii dependent on autophagy proteins and lysosomal enzymes. The fact that Akt activation has been linked to inhibition of apoptosis of T. gondii-infected cells [31] raises the possibility that parasite-induced EGFR - Akt signaling may not only promote parasite survival by preserving the non-fusogenic nature of the PV but also by avoiding death of infected cells subjected to pro-apoptotic signals. While EGFR is a central mediator of Akt activation in the early stages after T. gondii, Akt phosphorylation has recently been reported at 24 h post-infection with the parasite [52]. This raises the possibility that T. gondii may also activate Akt through additional mechanisms besides parasite engagement of EGFR.
Although T. gondii causes EGFR - Akt activation and these signaling molecules have been shown to inhibit autophagy [35], [53], [54], T. gondii does not appear to prevent autophagosome formation in infected cells. Indeed, large LC3+ structures were readily detected within infected cells during early stages post-infection (see Figure 2D), a finding previously reported in host cells at 24 h post-infection [55]. Moreover, there is no decrease in the levels of LC3 II (the lipidated form of LC3 that associates with the autophagosome membrane) during the early stages of infection (Muniz-Feliciano and Subauste, unpublished observations). In fact, T. gondii has been reported to increase LC3 II levels and autophagosome formation in host cells at 24 h post-infection, presumably as an attempt to gain access to nutrients [55]. Our studies indicate that while global autophagy did not appear to be inhibited by T. gondii, engagement of EGFR impaired targeting of the PV by LC3+ structures. Future studies that identify how autophagosomes target the PV will likely shed light on the molecular mechanism by which EGFR - Akt diminish autophagic targeting of the parasite.
Various pathogens can target EGFR. Pseudomonas aeruginosa and Helicobacter pylori can cause EGFR phosphorylation that is mediated by the release of membrane-bound EGF ligands and transactivation of EGFR [56], [57]. Klebsiella pneumonia causes EGFR activation that appears to be dependent on bacterial capsule polysaccharide engagement of TLR4 and subsequent Src-dependent EGFR activation [58]. In addition, proteins from oncogenic viruses activate EGFR to mediate transformation [59]. Much less is known on whether microbial products can directly engage and activate EGFR. It has been suggested that H. influenza may activate EGFR through the presence of bacterial-derived molecules with EGF-like properties [60]. Uptake of Influenza A virus causes EGFR activation, a process that may be dependent on multivalent binding of hemagglutinin to sialic acids present on EGFR or ganglioside GM1 leading to aggregation of rafts, clustering of EGFR and its activation [61]. Our studies suggest that EGF-MICs play a role in mediating EGFR-Akt activation of host cells and prevention of parasite killing since: recombinant EGF-MICs (MIC3 and MIC6) induce EGFR-Akt activation while MICs that lack EGF domains do not cause appreciable phosphorylation of EGFR and Akt; EGFR signaling inhibits LC3 accumulation around T. gondii; parasites deficient in 2 EGF-MICs (MIC3 and MIC6: MIC1-3 ko parasites) cause markedly impaired EGFR-Akt activation and exhibit increased encasing by LC3+ structures as well as killing in cells treated with autophagy stimulators; MIC3 and MIC6 impair parasite killing mediated by the autophagy pathway.
It was interesting to note that MIC1-3 ko parasites are not targeted by LC3+ structures and are not more likely to be killed in unstimulated cells despite the markedly weakened EGFR-Akt signaling. MIC1-3 ko parasites only display increased susceptibility to autophagic targeting and killing when autophagy is stimulated by CD154 or rapamycin. Of relevance to our findings, other studies support the existence of signaling thresholds that need to be achieved in order for autophagy to take place [62], [63]. For example, in Drosophila both the Ret-like receptor tyrosine kinase Stitcher (Stit) and insulin receptor (InR) are required for cell growth and proliferation through the PI3K-I/TORC1 pathway in the wing disc [63]. A decrease in either Stit or InR signaling diminishes TORC1 activity and suppresses growth [63]. However, this decrease in TORC1 activity is not sufficient to trigger autophagy in the wing [63]. Autophagy only takes place when both Stit and InR are impaired [63]. It was proposed that the simultaneous inactivation of Stit and InR reduces PI3K-I activity and TORC1 signaling below a critically low level at which autophagy in the wing can no longer be prevented [63]. Given that the EGFR-Akt pathway inhibits autophagy by regulating TORC1 activity, a similar phenomenon could be at play in the case of T. gondii infection. The reduction in EGFR-Akt observed in cells infected with MIC1 ko or MIC3 ko parasites does not translate in increased autophagic killing of these parasites either in unstimulated cells or in cells treated with stimulators of autophagy. The further reduction in EGFR-Akt signaling observed in cells infected with MIC1-3 ko may still be sufficient to prevent autophagic killing in unstimulated cells but results in enhanced killing in cells treated with autophagy stimulators. Finally, further inhibition of EGFR-Akt signaling (by genetic or pharmacological approaches) triggers autophagic targeting of T. gondii even in unstimulated cells. Thus, our studies suggest that the effects of MIC deficiency on the levels of EGFR-Akt activation likely explain the differences in outcome observed after infection. Taken together, in addition to being key for invasion of host cells, EGF-MICs (MIC3 and MIC6) contribute to the induction of a signaling cascade within these cells that is required to avoid lysosomal degradation of the parasite.
While MIC1-3 ko parasite exhibited a marked defect in EGFR-Akt activation in host cells, phosphorylation of these molecules still took place. Although we cannot rule out a role of MIC8 in activation of this cascade, it appears that the residual ability of MIC1-3 ko parasites to activate EGFR-Akt may not be explained by their expression of MIC8 (an EGF-MIC). Conditional MIC8 ko parasites did not exhibit a noticeable defect in signal activation in host cells. These findings are likely explained by the fact that MIC8 ko parasites do not exhibit defects in attachment to host cells and they secrete MICs [47]. The presence of an additional mechanism of EGFR-Akt activation that normally cooperates with MIC-dependent EGFR signaling may explain why MIC1-3 ko T. gondii have residual capacity to activate the EGFR-Akt pathways.
T. gondii is very successful as a pathogen and utilizes various strategies to manipulate host cell signaling to ensure its survival [64]–[67]. Here we report that the parasite activates EGFR - Akt to maintain the non-fusogenic nature of PV a process that appears to be dependent at least in part on EGF-MICs. These findings may be of therapeutic relevance since various inhibitors of EGFR are being used for treatment of cancer. The fact that EGFR inhibition induced parasite killing in cells not treated with immune activators, raises the possibility that this approach may be effective even in immunocompromised hosts.
Materials and Methods
Mammalian cells
Primary human brain microvascular endothelial cells (HBMEC) were obtained from ScienCell Research Laboratories (Carlsbad, CA) and cultured in fibronectin-coated tissue culture flasks and basal medium supplemented with Endothelial Cell Growth Supplement (ECGS) and 5% fetal bovine serum (FBS) all from ScienCell. The mouse high endothelial venule cell line (mHEVc) (gift from Joan Cook-Mills, Northwestern University, Chicago, IL) and mHEVc cells stably expressing LC3-EGFP construct (mHEVc-LC3-EGFP) or hmCD40 plus LC3-EGFP (hmCD40 mHEVc-LC3-EGFP) [15] were cultured in DMEM plus 10% FBS (HyClone; Logan, UT). A human RPE cell line (ARPE-19; American Type Culture Collection, Manassas, VA), a mouse macrophage line (RAW 264.7) and mouse microglia line (BV-2) were cultured in DMEM plus 10% FBS. Mouse bone marrow-derived macrophages were obtained as described and cultured in DMEM plus 30% L929-conditioned medium, 10% FBS and 5% horse serum [68]. Parental Chinese Hamster Ovary (CHO) cells and CHO cells expressing human EGFR (CHO-EGFR) were cultured in MEM plus 10% FBS.
T. gondii and infection
Experiments were conducted using tachyzoites of the RH strain of T. gondii (Type I strain), RH that express cytoplasmic YFP [69] or cytoplasmic DsRed (RFP) [69], tachyzoites of the ME49 (type II strain), transgenic parasites deficient in micronemal proteins MIC1 ko, MIC3 ko, MIC1-3 ko and the complemented strains (MIC1ko+MIC1, MIC3 ko+MIC3 and MIC1-3 ko+MIC1-3; gift from Maryse Lebrun, Universite de Montpellier 2, France) [39], as well as conditional MIC8 ko parasites (gift from Markus Meissner, University of Glasgow). Parasites were maintained in human foreskin fibroblasts following standard procedures [70]. In order to deplete MIC8, conditional MIC8 ko parasites were cultured in HFF in the presence of anhydrotetracycline (1 µg/ml) for 48 h. T. gondii tachyzoites were killed by incubation in 1% paraformaldehyde in PBS.
A potassium buffer shift was used to synchronize T. gondii invasion of serum-starved (0.1% FBS) mammalian cells as described [71]. Briefly, freshly egressed tachyzoites were resuspended in Endo buffer and incubated with cells for 20 minutes at 37°C. The Endo buffer was replaced for a low-potassium permissive medium to allow parasite invasion. In certain experiments, mammalian cells were incubated with Akt inhibitor IV (1.25 µM; EMD Millipore, Billerica, MA), PI3K inhibitor (LY294002; 20 µM; Sigma-Aldrich; St. Louis, MO), EGFR inhibitor (AG1478; 1 µM; EMD Millipore), a broad spectrum ADAM inhibitor (GM6001; 10 µM; EMD Millipore) (all 1 h prior to challenge with T. gondii), Pertussis Toxin (PTx; 100 ng/ml; EMD Millipore; 4 h prior to challenge), leupeptin (10 µM; EMD Millipore) and pepstatin (10 µM; EMD Millipore; both 1 h after challenge with T. gondii), 3-methyl adenine (3MA; 10 mM; Sigma Chemical) and rapamycin (1 µM; EMD Milipore; both 2 h after challenge with T. gondii) or vehicle. To induce CD40 signaling, mHEVc cells were treated with cell-free supernatants containing either multimeric human CD154 or a non-functional CD154 mutant [72] (T147N; both obtained from Dr. Richard Kornbluth, Univ. of California San Diego, current address Multimeric Biotherapeutics Inc., La Jolla, CA) for 18 h at 37°C as previously described [73] prior to challenge with parasites. Monolayers were fixed at indicated time points and stained with Diff-Quick (Dade Diagnostics, Aguada, Puerto Rico). The percentage of infected cells, the numbers of tachyzoites and vacuoles per 100 cells as well as the numbers of parasites per vacuole were determined by light microscopy by counting at least 200 cells or 200 vacuoles per monolayer as previously described [15].
T. gondii proteins
For expression of MIC3 and MIC4 in P. pastoris, amplified DNA fragments were cloned into a pPICZα A vector (Invitrogen; Carlsbad, CA). The pPICZα A vector contains the S. cervisiae α-factor secretion signal that allows for the secretion of folded proteins from P. pastoris. Cells were grown in BMGY media, washed and resuspended in BMMY media for an initial OD600 of 20–40. The culture was then incubated in a 28°C incubator with vigorous shaking. The culture was then grown for 1–5 days depending on the optimal period of expression. Inhibition of glycosylation during culture required the addition of 20 µg/ml of tunicamycin. The supernatant is then passed through a HiTrap Q HF Column (GE Healthcare; Little Chalfont, UK). The eluted fraction was buffer exchanged into nickel-column binding buffer (50 mM Tric-HCl, pH 8.0, 50 mM NaCl). If needed further protein purification was achieved by a further nickel affinity step and gel filtration.
M2AP and MIC6 encompassing residues 87 to 197 (including EGF2 and EGF3 motifs) were generated using a pET32 Xa/LIC plasmid (Novagen, EMD Millipore) in the Origami (D3) Escherichia coli strain (Stratagene) [74], [75]. Expressions of the fusion protein was induced by adding 1 mM IPTG and harvested after overnight culture at 18°C. The cells were collected by centrifugation and lysed by French Press. The fusion protein incorporating a hexahistidine tag was purified by bench top chromatography using a nickel-nitrilotriacetic acid resin (QIAGEN). The fusion partner of protein was cleaved by factor Xa and removed by an additional chromatography step and the factor Xa was removed by agarose resins (Novagen). Protein samples were concentrated to 0.5 mM in 50 mM NaCl, 50 mM potassium phosphate and 5% D2O at pH 5.8. Endotoxin concentrations were similar in MIC3 and MIC4 as well as in M2AP and MIC6.
Transfection
Cells were transiently transfected with a plasmid that encodes Akt-PH-GFP [76], human PI3K p110α siRNA [77], human Akt siRNA [78], mouse Beclin1 siRNA [79], mouse Atg7 siRNA [79], human MyD88 siRNA [80], human EGFR siRNA [81] or control siRNA (Dharmacon) using Lipofectamine 2000 (Invitrogen) or an Amaxa nucleofector as described [13], [15]. siRNA against mouse EGFR was synthesized using siRNA construction kit (Ambion) [82] following manufacturer's recommendation and used for mouse EGFR knock-down after transfection using Lipofectamine 2000.
Fluorescent microscopy
To assess for LC3 accumulation around the parasite, mHEVc-LC3-EGFP cells were cultured with or without Akt inhibitor IV or transfected with either control siRNA or EGFR siRNA or treated with or without EGF (50 ng/ml; PeproTech). Monolayers were challenged with RH T. gondii that express cytoplasmic RFP (T. gondii-RFP). Five hours post-challenge, monolayers were fixed with 4% paraformaldehyde, slides were mounted using Fluoromount G and assessed for LC3-EGFP accumulation around T. gondii as described [13], [15]. In certain experiments, hmCD40 mHEVc-LC3-EGFP cells treated with or without CD154 were infected with WT, MIC1 ko, MIC1 ko+MIC1, MIC3 ko, MIC3 ko+MIC3, MIC1-3 ko or MIC1-3 ko+MIC1-3 tachyzoites. Monolayers were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 and incubated with rabbit anti-T. gondii Ab (BioGenex; San Ramon, CA) for 30 minutes. Monolayers were then washed with PBS and incubated for 1 h at room temperature with goat anti-rabbit Alexa 568-conjugated secondary antibody (Jackson ImmunoResearch Laboratories Inc., West Grove, PA). HBMEC transfected with a plasmid encoding PH-Akt-GFP were cultured with or without LY294002 followed by challenge with T. gondii-RFP. Distribution of PH-Akt-GFP was examined 5 min. post-challenge. In certain experiments, endothelial cells were treated with either Akt inhibitor IV or AG1478 and challenged with T. gondii-YFP were fixed with 4% paraformaldehyde at 8 h post-infection, permeabilized with ice-cold methanol. Monolayers were incubated overnight at 4°C with either mouse anti-human LAMP-1 or rat anti-mouse LAMP-1 (all from Developmental Studies Hybridoma Bank; Iowa City, IA). Monolayers were washed with PBS plus 1% BSA, then incubated for 1 h at room temperature with Alexa 568-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories Inc.). Specificity of staining was determined by incubating monolayers with secondary antibody alone. Slides were analyzed using a Leica DMI 6000 B automated microscope equipped for epifluorescence microscopy. Experimental groups had triplicate samples and at least 100 cells per sample were counted.
Electron microscopy
Endothelial cells were seeded onto a sterilized Aclar Embedding Film (Electron Microscopy Sciences, PA) and incubated with or without T. gondii tachyzoites in the presence of Akt inhibitor IV or vehicle. At 5 h post-challenge, the Aclar sheets with their attached cells were fixed as described [83]. After a soak in acidified uranyl acetate, the specimen was dehydrated in ethanol, passed through propylene oxide, and embedded in Poly/Bed (Polysciences, PA). Sections were cut in a horizontal plane parallel to that of the Aclar film to provide panoramic views of the endothelial cells. Thin sections were stained with acidified uranyl acetate in 50% methanol followed by triple lead stain of Sato. These sections were examined in a JEOL 1200 EX electron microscope (Tokyo, Japan).
Immunoblot
Cells were lysed in buffer supplemented with protease and phosphatase inhibitors (Cell Signaling). Equal amounts of protein were subjected to either 7.5% or 10% SDS-PAGE (Bio-Rad) and transferred to PVDF membranes. Membranes were probed with either antibody to total Akt (Cell Signaling), phospho-Ser473 Akt (Cell Signaling), total EGFR (Santa Cruz Biotechnology), phospho-tyrosine 1068 EGFR (Invitrogen), phospho-tyrosine 1148 EGFR (Cell Signaling), Atg7 (Cell Signaling), Beclin 1 (BD Biosciences), PI3K p110α (Cell Signaling) or MyD88 (Cell Signaling) followed by incubation with secondary antibody conjugated to horseradish peroxidase (Santa Cruz Biotechnology). Bands were visualized by using enhanced chemiluminescence kit (Pierce Bioscience). Intensities of phospho-Akt and phospho-EGFR were calculated using ImageJ (NIH) and normalized against total Akt and total EGFR respectively.
Statistics
Results from pooled experiments were analyzed for statistical significance using 2-tailed Student's t test and ANOVA (InStat version 3.0, GraphPad; La Jolla, CA). Differences were considered statistically significant when P was<0.05.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. CarruthersVB, BoothroydJC (2007) Pulling together: an integrated model of Toxoplasma cell invasion. Curr Opin Microbiol 10 : 83–89.
2. CarruthersVB, TomleyFM (2008) Receptor-ligand interaction and invasion: micronemeproteins in apicomplexans. Subcell Biochem 47 : 33–45.
3. AnantharamanV, IyerLM, BalajiS, AravindL (2007) Adhesion molecules and other secreted host-interaction determinants in Apicomplexa: insights from comparative geneomics. Int Rev Cytol 262 : 1–74.
4. TomleyFM, SoldatiDS (2001) Mix and match modules: structure and function of microneme proteins in apicomplexan parasites. Trends Parasitol 17 : 81–88.
5. SibleyLD (2011) Invasion and intracellular survival by protozoan parasites. Immunol Rev 240 : 72–91.
6. Soldati-FavreD (2008) Molecular dissection of host cell invasion by the apicomplexans: the glideosome. Parasite 15 : 197–205.
7. BoothroydJC, DubremetzJF (2008) Kiss and spit: the dual roles of Toxoplasma rhoptries. Nat Rev Microbiol 6 : 79–88.
8. BesteiroS, DubremetzJF, LebrunM (2011) The moving junction of apicomplexan parasites: a key structure for invasion. Cell Microbiol 13 : 797–805.
9. MordueDG, DesaiN, DustinM, SibleyLD (1999) Invasion by Toxoplasma gondii establishes a moving junction that selectively excludes host cell plasma membrane proteins on the basis of their membrane anchoring. J Exp Med 190 : 1783–1792.
10. MordueDG, HakanssonS, NiesmanI, SibleyLD (1999) Toxoplasma gondii resides in a vacuole that avoids fusion with host cell endocytic and exocytic vescicular pathways. Exp Parasitol 92 : 87–99.
11. MartensS, ParvanovaI, ZerrahnJ, GriffithsG, SchellG, et al. (2005) Disruption of Toxoplasma gondii parasitophorous vacuoles by the mouse p47-resistance GTPases. PLoS Pathogens 1 : 187–201.
12. ZhaoZ, FuxB, GoodwinM, DunayIR, StrongD, et al. (2008) Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe 4 : 458–469.
13. AndradeRM, WessendarpM, GubbelsMJ, StriepenB, SubausteCS (2006) CD40 induces macrophage anti-Toxoplasma gondii activity by triggering autophagy-dependent fusion of pathogen-containing vacuoles and lysosomes. J Clin Invest 116 : 2366–2377.
14. PortilloJ-AC, OkenkaG, ReedE, SubausteA, Van GrolJ, et al. (2010) The CD40-autophagy pathway is needed for host protection despite IFN-γ-dependent immunity and CD40 induces autophagy via control of p21 levels. Plos One 5: e14472.
15. Van GrolJ, Muniz-FelicianoL, PortilloJ-AC, BonilhaVL, SubausteCS (2013) CD40 induces anti-Toxoplasma gondii activity in non-hematopoietic cells dependent on autophagy proteins. Infect Immun 81 : 2002–2011.
16. MizushimaN, YoshimoriT, OhsumiY (2010) The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 27 : 107–132.
17. LevineB, MizushimaN, VirginHW (2011) Autophagy in immunity and inflammation. Nature 469 : 323–335.
18. OgawaM, YoshimoriT, SuzukiT, SagaraH, MizushimaN, et al. (2004) Escape of intracellular Shigella from autophagy. Science 307 : 727–731.
19. TalloczyZ, VirginHW, LevineB (2006) PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy 2 : 24–29.
20. OrvedahlA, AlexanderD, TalloczyZ, SunQ, WeiY, et al. (2007) HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host & Microbe 1 : 23–35.
21. YoshikawaY, OgawaM, HainT, YoshidaM, FukumatsuM, et al. (2009) Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat Cell Biol 11 : 1233–1240.
22. EX, HwangS, OhS, LeeJ-S, JeongJH, et al. (2009) Viral Bcl-2-mediated evasion of autophagy aids chronic infection of gHerpesvirus 68. PLoS Pathog 5: e10000609.
23. KyeiGB, DinkinsC, DavisAS, RobertsE, SinghSB, et al. (2009) Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J Cell Biol 186 : 255–268.
24. ChoyA, DancourtJ, MugoB, O'ConnorTJ, IsbergRR, et al. (2012) The Legionella effector RavZ inhibits host autophagy through irreversible Atg8 deconjugation. Science 338 : 1072–1076.
25. BlanchetFP, MorisA, NikolicDS, LehmannM, CardinaudS, et al. (2010) Human immunodeficiency virus-1 inhibition of immunoamphisomes in dendritic cells impairs early innate and adaptive immune responses. Immunity 32 : 654–669.
26. KumarDM, NathL, KamalMA, VarshneyA, JainA, et al. (2010) Genome-wide analysis of the host intracellular network that regulates survival of Mycobacterium tuberculosis. Cell 140 : 731–743.
27. AlessiDR, AndjelkovicM, CaudwellB, CronP, MorriceN, et al. (1996) Mechanisms of activation of protein kinase B by insulin and IGF-1. EMBO J 15 : 6541–6551.
28. ChanTO, RittenhouseSE, TsichlisPN (1999) AKT/PKB and other D3 phosphoinositide-regulated kinases: kinase actvation by phosphoinositide-dependent phosphorylation. Annu Rev Biochem 68 : 965–1014.
29. StokoeD, StephensLR, CopelandT, GaffneyPRJ, ReeseCB, et al. (1997) Dual role of phosphatidylinositol-3,4,5-triphosphate in the activation of protein kinase B. Science 277 : 567–570.
30. ServantG, WeinerOD, HerzmarkP, BallaT, SedatJW, et al. (2000) Polarization of chemoattractant receptor signaling during neutrophil chemtoaxis. Science 287 : 1037–1040.
31. KimL, DenkersEY (2006) Toxoplasma gondii triggers Gi-dependent PI3-kinase signaling required for inhibition of host cell apoptosis. J Cell Sci 119 : 2119–2126.
32. JoinerKA, FuhrmanSA, MietinnenH, KasperLH, MellmanI (1990) Toxoplasma gondii: fusion competence of parasitophorous vacuoles in Fc receptor transfected fibroblasts. Science 249 : 641–646.
33. MordueDG, SibleyLD (1997) Intracellular fate of vacuoles containing Toxoplasma gondii is determined at the time of formation and depends on the mechanisms of entry. J Immunol 159 : 4452–4459.
34. CoppensI, DunnJD, RomanoJD, PypaertM, ZhangH, et al. (2006) Toxoplasma gondii sequesters lysosomes from mammalian hosts in the vacuolar space. Cell 125 : 261–274.
35. ScarlattiF, BauvyC, VentrutiA, SalaG, CluzeaudF, et al. (2004) Ceramide-mediated macroautophagy involves inhibition of protein kinase B and up-regulation of Beclin 1. J Biol Chem 279 : 18384–18391.
36. HemmingsBA, RestucciaDF (2012) PI3K-PKB/Akt pathway. Cold Spring Harb Perspect Biol 4: a011189.
37. TigyiG, ParrillA (2003) Molecular mechanisms of lysophosphatidic acid action. Prog Lipid Res 42 : 498–526.
38. CarruthersVB (2002) Host cell invasion by the opportunistic pathogen Toxoplasma gondii. Acta Tropica 81 : 111–122.
39. CeredeO, DubremetzJF, SoeteM, DesleeD, VialH, et al. (2005) Synergistic role of micronemal proteins in Toxoplasma gondii virulence. J Exp Med 201 : 453–463.
40. HuynhM-H, CarruthersVB (2006) Toxoplasma MIC2 is a major determinant of invasion and virulence. PLoS Pathogens 2: e84.
41. SheinerL, SantosJM, KlagesN, ParussiniF, JemmelyN, et al. (2010) Toxoplasma gondii transmembrane microneme proteins and their modular design. Mol Microbiol 77 : 912–929.
42. MeissnerM, ReissM, ViebigN, CarruthersVB, TourselC, et al. (2002) A family of transmembrane microneme proteins of Toxoplasma gondii contain EGF-like domains and function as escorters. J Cell Sci 115 : 563–574.
43. MoroL, DolceL, CabodiS, EB, Boeri ErbaE, et al. (2002) Integrin-induces epidermal growth factor (EGF) receptor activation requires c-Src and p130Cas and leads to phosphorylation of specific EGF receptor tyrosines. J Biol Chem 277 : 9405–9414.
44. KrugAW, SchusterC, GassnerB, FreudingerR, MildenbergerS, et al. (2002) Human epidermal growth factor receptor-1 expression renders chinese hamster ovary cells sensitive to alternative aldosterone signaling. J Biol Chem 277.
45. YardenY, SliwkowskiMX (2001) Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2 : 127–137.
46. ReissM, ViebigN, BrechtS, FourmauxMN, SoeteM, et al. (2001) Identification and characterization of an escorter for two secretory adhesins in Toxoplasma gondii. J Cell Biol 152 : 563–578.
47. KesslerH, Hern-GotzA, HeggeS, RauchM, Soldati-FavreD, et al. (2008) Micronem protein 8 - a new essential invasion factor in Toxoplasma gondii. J Cell Sci 121 : 947–956.
48. CeredeO, DubremetzJF, BoutD, LebrunM (2002) The Toxoplasma gondii protein MIC3 requires pro-peptide cleavage and dimerization to function as adhesin. EMBO J 21 : 2526–2536.
49. Van Grol J, Subauste MC, Andrade RM, Fujinaga K, Nelson JA, et al.. (2010) HIV-1 inhibits autophagy in bystander macrophages/monocytic cells through Src-Akt and STAT3. PLoS One: e11733.
50. WangX, McCulloughKD, FrankeTF, HolbrookNJ (2000) Epidermal growth factor receptor-dependent Akt activation by oxidative stress enhances cell survival. J Biol Chem 275 : 14624–14631.
51. MahajanK, MahajanNP (2012) PI3K-independent AKT activation in cancers: a treasure trove for novel therapeutics. J Cell Physiol 227 : 3178–3184.
52. ZhouW, QuanJ-H, LeeY-H, ShinD-W, ChaG-H (2013) Toxoplasma gondii proliferation require down-regulation of host Nox4 expression via activation of PI3 kinase/Akt signaling pathway. Plos One 8: e66306.
53. SobolewskaA, GajewskaM, ZarzynskaJ, GajkowskaB, MotylT (2009) IGF-I, EGF, and sex steroids regulate autophagy in bovine mammary epithelial cells via the mTOR pathway. Eur J Cell Biol 88 : 117–130.
54. MaynardAA, DvorakK, KhailovaL, DobrenenH, ArganbrightKM, et al. (2010) Epidermal growth factor reduces autophagy in intestinal epithelium and in the rat model of necrotizing enterocolitis. Am J Physiol Gastrointest Liver Physiol 299: G614–G622.
55. WangY, WeissLM, OrlofskyA (2009) Host cell autophagy is induced by Toxoplasma gondii and contributes to parasite growth. J Biol Chem 284 : 1694–1701.
56. KoffJL, ShaoMXG, KimS, UekiIF, NadelJA (2006) Pseudomonas lipopolysaccharide accelerates wound repair via activation of a novel epithelial cell signaling cascade. J Immunol 177 : 8693–8700.
57. KeatesS, SougioultzisS, KeatesAC, ZhaoD, PeekRMJ, et al. (2001) cag+ Helicobacter pylori induce transactivation of the epidermal growth factor receptor in AGS epithelial cells. J Biol Chem 276 : 48127–48134.
58. FrankCG, ReguerloV, RotherM, MorantaD, MaeurerAP, et al. (2013) Klebsiella pneumoniae targets an EGF receptor-dependent pathway to subvert inflammation. Cell Microbiol 15 : 1212–33.
59. KungC-P, MeckesDG, Raab-TraubN (2011) Epstein-Barr virus LMP1 activates EGFR, STAT3, and ERK through effects on PKCd. J Virol 85 : 4399–4408.
60. MikamiF, GuH, JonoH, AndalabiA, KaiH, et al. (2005) Epidermal growth factor receptor acts as a negative regulator for bacterium nontypable Haemophilus influenzae-induced Toll-like receptor 2 expression via an Src-dependent p38 mitogen-activated protein kinase signaling pathway. J Biol Chem 280 : 36185–36194.
61. EierhoffT, HrinciusER, RescherU, LudwigS, EhrhardtC (2010) The epidermal growth factor receptor (EGFR) promotes uptake of Influenza A viruses (IAV) into host cells. PLoS Pathog 6: e1001099.
62. ChhipaRR, WuY, IpC (2011) AMPK-mediated autophagy is a survival mechanism in androgen-dependent prostate cancer cells subjected to androgen deprivation and hypoxia. Cell Signal 23 : 1466–1472.
63. O'FarrellF, WangS, KathederN, RustenTE, SamkovlisC (2013) Two-tiered control of epithelial growth and autophagy by the insulin receptor and the Ret-like receptor, Stitcher. PLoS Biol 11: e1001612.
64. BoothroydJC, DubremetzJF (2008) Kiss and spit: the dual roles of Toxoplasma rhoptries. Nat Rev Microbiol 6 : 79–88.
65. LaliberteJ, CarruthersVB (2008) Host cell manipulation by the human pathogen Toxoplasma gondii. Cell Mol Life Sci 65 : 1900–1915.
66. LengJ, ButcherBA, DenkersEY (2009) Dysregulation of macrophage signal transduction by Toxoplasma gondii: past progress and recent advances. Parasite Immunol 31 : 717–728.
67. PollardAM, KnollLJ, MordueDG (2009) The role of specific Toxoplasma gondii molecules in manipulation of innate immunity. Trends Parasitol 25 : 491–494.
68. AndradeRM, PortilloJ-AC, WessendarpM, SubausteCS (2005) CD40 signaling in macrophages induces anti-microbial activity against an intracellular pathogen independently of IFN-γ and reactive nitrogen intermediates. Infect Immun 73 : 3115–3123.
69. GubbelsMJ, LiC, StriepenB (2003) High-throughput growth assay for Toxoplasma gondii using yellow fluorescent protein. Antimicrob Agents Chemother 47 : 309–316.
70. RoosDS, DonaldRG, MorrissetteNS, MoultonAL (1994) Molecular tools for genetic dissection of the protozoan parasite Toxoplasma gondii. Methods Cell Biol 45 : 27–63.
71. KafsackBFC, BeckersCJ, CarruthersVB (2004) Synchronous invasion of host cells by Toxoplasma gondii. Mol Biochem Parasitol 136 : 309–311.
72. BajorathJ, SeyamaK, NonoyamaS, OchsHD, AruffoA (1996) Classification of mutations in the human CD40 ligand, gp39, that are associated with X-linked hyper IgM syndrome. Protein Sci 5 : 531–534.
73. PortilloJ-AC, Van GrolJ, ZhengL, OkenkaG, GentilK, et al. (2008) CD40 mediates retinal inflammation and neuro-vascular degeneration. J Immunol 181 : 8719–8726.
74. LiuB, SawmynadenK, MarchantJ, SimpsonP, MatthewsS (2009) Complete resonance assigments for the MIC2 associated protein from Toxoplasma gondii. Biomol NMR Assign 3 : 81–83.
75. SaourosS, Edwards-JonesB, ReissM, SawmynadenK, CotaE, et al. (2005) A novel galectin-like domain from Toxoplasma gondii micronemal protein 1 assists the folding, assembly, and transport of a cell adhesion complex. J Biol Chem 280 : 38583–38591.
76. KwonY, HofmannT, MontellC (2007) Integration of phosphoinositide - and calmodulin-mediated regulation of TRPC6. Mol Cell 25 : 491–503.
77. MengQ, XiaC, FangJ, RojanasakulY, JiangB-H (2006) Role of PI3K and AKT specific isoforms in ovarian cancer cell migration, invasion and proliferation through the p70S6K1 pathway. Cell Signal 18 : 2262–2271.
78. KuoPL, HsuYL, ChoCY (2006) Plumbagin induces G2-M arrest and autophagy by inhibiting the Akt/mammalian target of rapamycin pathway in breast cancer cells. Mol Cancer Ther 5 : 3209–3221.
79. YuL, AlvaA, SuH, DuttP, FreundtE, et al. (2004) Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 304 : 1500–1502.
80. HirataY, OhmaeT, ShibataW, MaedaS, OguraK, et al. (2006) MyD88 and TNF Receptor-Assocaited Factor 6 are critical signal transducers in Helicobacter pylori-infected human epithelial cells. J Immunol 176 : 3796–3803.
81. KangC-S, ZhangZ-Y, JiaZ-F, WangG-X, QiuM-Z, et al. (2006) Suppression of EGFR expression by antisense or small interference RNA inhibits U251 glioma cell growth in vitro and in vivo. Cancer Gene Ther 13 : 530–538.
82. KimS-E, ChoiK-Y (2007) EGF receptor is involved in WNT3a-mediated proliferation and motility of NIH3T3 cells via ERK pathway activation. Cellular Signaling 19 : 1554–1564.
83. FujiokaH, TandlerB, ConsoloMC, KarnikP (2013) Division of mitochondria in cultured human fibroblasts. Microsc Res Tech [Epub ahead of print].
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2013 Číslo 12
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Influence of Mast Cells on Dengue Protective Immunity and Immune Pathology
- Host Defense via Symbiosis in
- Coronaviruses as DNA Wannabes: A New Model for the Regulation of RNA Virus Replication Fidelity
- Myeloid Dendritic Cells Induce HIV-1 Latency in Non-proliferating CD4 T Cells
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy