
Molecular and Evolutionary Bases of Within-Patient Genotypic and Phenotypic Diversity in Extraintestinal Infections
Although polymicrobial infections, caused by combinations of viruses, bacteria, fungi and parasites, are being recognised with increasing frequency, little is known about the occurrence of within-species diversity in bacterial infections and the molecular and evolutionary bases of this diversity. We used multiple approaches to study the genomic and phenotypic diversity among 226 Escherichia coli isolates from deep and closed visceral infections occurring in 19 patients. We observed genomic variability among isolates from the same site within 11 patients. This diversity was of two types, as patients were infected either by several distinct E. coli clones (4 patients) or by members of a single clone that exhibit micro-heterogeneity (11 patients); both types of diversity were present in 4 patients. A surprisingly wide continuum of antibiotic resistance, outer membrane permeability, growth rate, stress resistance, red dry and rough morphotype characteristics and virulence properties were present within the isolates of single clones in 8 of the 11 patients showing genomic micro-heterogeneity. Many of the observed phenotypic differences within clones affected the trade-off between self-preservation and nutritional competence (SPANC). We showed in 3 patients that this phenotypic variability was associated with distinct levels of RpoS in co-existing isolates. Genome mutational analysis and global proteomic comparisons in isolates from a patient revealed a star-like relationship of changes amongst clonally diverging isolates. A mathematical model demonstrated that multiple genotypes with distinct RpoS levels can co-exist as a result of the SPANC trade-off. In the cases involving infection by a single clone, we present several lines of evidence to suggest diversification during the infectious process rather than an infection by multiple isolates exhibiting a micro-heterogeneity. Our results suggest that bacteria are subject to trade-offs during an infectious process and that the observed diversity resembled results obtained in experimental evolution studies. Whatever the mechanisms leading to diversity, our results have strong medical implications in terms of the need for more extensive isolate testing before deciding on antibiotic therapies.
Published in the journal:
. PLoS Pathog 6(9): e32767. doi:10.1371/journal.ppat.1001125
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1001125
Summary
Although polymicrobial infections, caused by combinations of viruses, bacteria, fungi and parasites, are being recognised with increasing frequency, little is known about the occurrence of within-species diversity in bacterial infections and the molecular and evolutionary bases of this diversity. We used multiple approaches to study the genomic and phenotypic diversity among 226 Escherichia coli isolates from deep and closed visceral infections occurring in 19 patients. We observed genomic variability among isolates from the same site within 11 patients. This diversity was of two types, as patients were infected either by several distinct E. coli clones (4 patients) or by members of a single clone that exhibit micro-heterogeneity (11 patients); both types of diversity were present in 4 patients. A surprisingly wide continuum of antibiotic resistance, outer membrane permeability, growth rate, stress resistance, red dry and rough morphotype characteristics and virulence properties were present within the isolates of single clones in 8 of the 11 patients showing genomic micro-heterogeneity. Many of the observed phenotypic differences within clones affected the trade-off between self-preservation and nutritional competence (SPANC). We showed in 3 patients that this phenotypic variability was associated with distinct levels of RpoS in co-existing isolates. Genome mutational analysis and global proteomic comparisons in isolates from a patient revealed a star-like relationship of changes amongst clonally diverging isolates. A mathematical model demonstrated that multiple genotypes with distinct RpoS levels can co-exist as a result of the SPANC trade-off. In the cases involving infection by a single clone, we present several lines of evidence to suggest diversification during the infectious process rather than an infection by multiple isolates exhibiting a micro-heterogeneity. Our results suggest that bacteria are subject to trade-offs during an infectious process and that the observed diversity resembled results obtained in experimental evolution studies. Whatever the mechanisms leading to diversity, our results have strong medical implications in terms of the need for more extensive isolate testing before deciding on antibiotic therapies.
Introduction
Polymicrobial infections, caused by combinations of viruses, bacteria, fungi and parasites, are being recognised with increasing frequency [1]. Polymicrobial infections due to bacteria of the same species have been less studied, as they are clearly more difficult to identify. However, recent molecular epidemiological tools allow their systematic detection, as well as the study of the relatedness between strains. Two kinds of within-species diversity have been reported: polyclonal diversity with infection caused by phylogenetically divergent clones and monoclonal diversity with infection by members of a single clone that exhibit genetic micro-heterogeneity. Such within-species polymicrobial infection have been reported mainly in chronic and/or open infections (Pseudomonas aeruginosa infections of lung in cystic fibrosis patients [2], [3] and of burn wounds [4], Helicobacter pylori gastric infection [5], Staphylococcus epidermidis joint prosthesis infection [6] and endocarditis [7], lung tuberculosis [8]) but also in septicemia [9], [10].
Beside these clinical observations, in vitro experimental evolution has provided some clues on the diversification process in a single clonal population. It has been shown that stable polymorphisms can emerge due to ecological interactions [11], [12], [13], [14]. More detailed analysis of variation with larger numbers of sampled co-evolving bacteria showed that a very complex polymorphism with considerable phenotypic diversity could emerge in a matter of days [15], revealing a kind of adaptive radiation. In this case, selection under nutrient limitation altered the SPANC balance, or the trade-off between self-preservation and nutritional competence [16]. Polymorphisms affecting the SPANC balance often result in altered cellular levels of the sigma factor RpoS, which results in modification of several phenotypes, such as nutritional abilities, general stress resistance, starvation survival, cell morphology [17]. RpoS is a central regulator of stress resistance. Strains with higher RpoS level are more resistant to external stress but metabolised fewer substrates whereas strains with lower RpoS level have broader nutritional capabilities but lower resistance to external stress. Experimental evolution has also studied cooperation and competition among single species bacteria according to relatedness [18]. Overall, many different ecological sources of polymorphism have been identified in more than 60 years of bacterial experimental evolution. Yet, few studies have connected the molecular bases of such polymorphism to the one observed in the course of an infection. Currently, it is impossible to distinguish whether bacterial adaptation in the course of an infection is the action of the immune system [19] or the kind of process observed in in vitro experimental systems. Are the selective pressures identified as a source of diversification in in vitro experimental systems actually relevant during an infection?
Escherichia coli is a commensal of the intestinal tract of vertebrates, including humans [20], that can cause intestinal (diarrhoea) and extraintestinal [urinary tract infection (UTI), pneumonia, neonatal meningitis, septicaemia] infections [21]. Human extraintestinal infections have high incidence and associated morbidity, mortality (500,000 estimated deaths a year worldwide), and costs [22]. E. coli species can be considered as having mainly a clonal genetic structure [23], [24] with four main phylogenetic groups (A, B1, B2 and D) [25], [26]. Numerous epidemiological and animal model studies have documented the association of specific “virulence” determinants and/or phylogenetic groups with the different clinical syndromes [21], [27], [28]. It is classically admitted that E. coli extraintestinal infections are caused by identical isolates originating from single clones. From this assumption, the identification and antibiotic testing are mainly based on the characterisation of a single colony from the pathological specimen isolation. However, as for other bacterial species [2], [3], [4], [5], [6], [7], [8], [9], [10], this assumption is questionable.
In this context, we studied the level of genomic and phenotypic polymorphism of E. coli in human severe and closed extraintestinal infections. The aim of our work was (i) to get a better appreciation of the occurrence of within-species diversity in closed infections, (ii) to decipher the molecular and evolutionary bases of this diversity and (iii) to test, using a mathematical model, whether SPANC balancing can be a contributing factor to the observed pathogen diversification.
Results
Two kinds of intra-patient E. coli genetic diversity were observed: polyclonal and monoclonal with micro-heterogeneity
To have a global distribution of the genetic diversity of the 226 E. coli isolates from 19 patients (Table 1), we used a combination of phylogenetic grouping, multi locus sequence typing (MLST), Enterobacterial Repetitive Intergenic Consensus (ERIC)-PCR and pulsed field gel electrophoresis (PFGE) analyses to characterise the two kinds of diversity, i.e. polyclonal and monoclonal with micro-heterogeneity. Polyclonal infections were identified by distinct sequence type and/or phylogenetic group/subgroup and unrelated ERIC-PCR and PFGE patterns whereas monoclonal infection with micro-heterogeneity were characterised by identical sequence type and phylogenetic group/subgroup and distinct but related patterns of ERIC-PCR and PFGE [29].

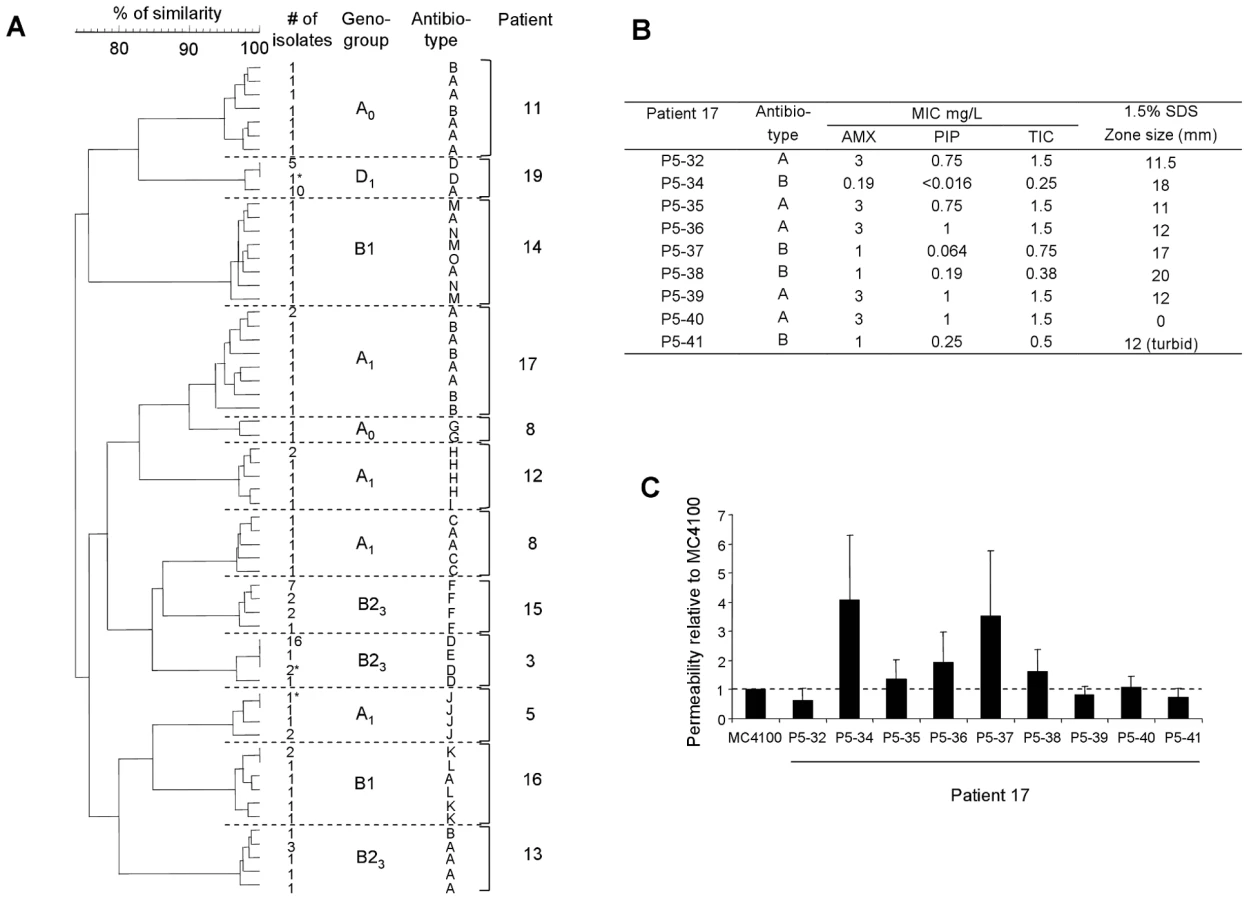
In 8 cases (42%) corresponding to 3 UTI, 1 meningitis, 1 pleural infection, 1 ascites, 1 placenta infection and 1 hepatic abscess (patients 1, 2, 4, 6, 7, 9, 10 and 18), no genetic diversity was observed (Table 1). In patients 5, 8, 15 and 16 (21%) (Table 1), corresponding to intra-abdominal infections, polyclonal infections were observed. In patient 16, three different clones corresponding to 3 distinct phylogenetic groups were observed among the isolates. In patients 8 and 15, the isolates belong to 2 different phylogenetic group/subgroups (Table 1). In patient 5 where the two isolates belong to the A1 subgroup (Table 1), MLST analysis clearly showed two distinct sequence types corresponding to a large divergence time. Among these infections, isolates exhibited different ERIC-PCR patterns with each of the three primers (see Fig. 1 for an example). Moreover, these 4 patients with polyclonal infections have at least one of the clone exhibiting micro-heterogeneity (Table 1), characterised by closely related but different patterns observed by ERIC-PCR with only one or two primers and by PFGE (see Fig. 1 and 2 for an example). In the remaining 7 patients (3, 11, 12, 13, 14, 17 and 19), monoclonal infections with micro-heterogeneity were detected by ERIC-PCR and PFGE (Table 1). In these cases of micro-heterogeneity, the isolates belong to the same phylogenetic group/subgroup and sequence type using 7 essential genes as well as the sequence of the gnd gene, one of the most variable genes in E. coli [30]. Altogether, presence of at least a clone with micro-heterogeneity was observed in 11 patients (58%). A dendrogram constructed from the PFGE patterns of E. coli isolates from these patients is shown in Fig. 3A. Within a patient, the level of similarity between isolates is above 94% (Fig. 3A). In patients 3 and 19 from which blood and an additional sample were available, the micro-heterogeneity was observed in both samples. Lastly, all the 19 patients were infected by unrelated clones (Table 1 and Fig. 3A).


The presence of 20 virulence factors (VFs) known to be associated to extra intestinal virulence [31] and representative of the main classes of VFs (protectin, adhesin, toxin, iron uptake system) were studied. A wide range of VF patterns was observed from the absence of detected gene in A group strains to the presence of 12 studied genes in the B2 group strains, in agreement with the well known link between virulence and phylogeny [27] (data not shown). Among the 4 patients (5, 8, 15 and 16) infected by several distinct clones, the VF patterns of each clone within a patient were always different. In 3 patients infected by a clone exhibiting a micro-heterogeneity (14, 17 and 19), the iucC and iutA genes, associated to traT gene, were either present or absent, indicating the variable presence of a plasmid bearing the aerobactin operon (Tables 1 and S1).
In summary, a high level of genetic diversity was frequently observed in the isolates originating from a single patient. Two kinds of genetic diversity were observed: (i) polyclonal infection and (ii) monoclonal infection with micro-heterogeneity. These two kinds of genetic diversity can be combined in a single patient.
Isolates from a single clone with genetic micro-heterogeneity exhibit variable antibiotic susceptibility and growth patterns
We then wanted to know if the genetic micro-heterogeneity observed within isolates of a single clone was associated with phenotypic variation. To this purpose, we studied in all the isolates two phenotypes having critical impact on strain fitness in an infection, i.e. antibiotic sensitivity and growth. Among the 226 E.coli isolates, 19 distinct antibiotypes were distinguished. We found 9 clones in 9 patients (3, 8, 11, 12, 13, 14, 16, 17 and 19) exhibiting heterogeneity in the antibiotic resistance pattern, all of which having a genetic micro heterogeneity (Tables 1 and S2, Fig. 3A and 4A). The antibiotics concerned were the β-lactams, tetracycline, streptomycin and kanamycin, the sulfonamides, trimethoprim and chloramphenicol. Within these single clones, 2 to 4 distinct antibiotypes were evidenced, the most sensitive phenotype being always observed in the minority of the isolates (Table S2). Several causes of variation in antibiotic susceptibility were likely. Firstly, loss of genetically mobile resistance genes was confirmed by a perfect correlation between the β-lactam resistance phenotype and the positive TEM (β-lactamase) PCR results in the isolates from 5 patients (3, 8, 12, 16 and 19). This mechanism is also likely involved in the loss of resistance to tetracycline, streptomycin, kanamycin, sulfonamides, trimethoprim and chloramphenicol, as the genes encoding for these resistances in clinical isolates are located on mobile elements such as transposons and integrons [32]. Patients 14 and 16 are particularly illustrative of this phenomenon as multiple combinations of antibiotic resistance losses were observed within a single patient (Table S2). Secondly, hyper-susceptibility to β-lactams was observed in 3 patients (11, 13 and 17). We have further studied the isolates of patient 17. Susceptibility to antibiotics (amoxicillin, piperacillin and ticarcillin) and detergents (SDS) was not constant and outer membrane permeability assays indicated heterogeneity in permeation rates due to variations in porin-mediated diffusion (Fig. 3B). Thirdly, the discrepancy between outer membrane permeability (Fig. 3C) and antibiotic/SDS susceptibility (Fig. 3B) points to yet other differences in envelope structure or efflux between isolates [32], suggesting an unexpectedly extensive range of surface variations inside a patient.
![Illustration of the phenotypic diversity observed in the <i>E. coli</i> isolates of patient 3 infected by a unique clone [isolates from blood (ID 42–50) and liver abscess (ID 51–58)].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/ba74b63c54247fb774880ec5aaf5ebdf.png)
Even more strikingly, heritable variation in growth characteristics was found amongst clonal isolates from 8 patients, all infected by at least one clone with genetic micro-heterogeneity (Tables 1 and 2). Impaired growth was characterised by longer lag time and/or generation time and/or reduced biomass in stationary phase (Table 2). Within a patient, impaired growth was found in 6 to 22% of isolates (mean 13%), the remaining isolates having identical unimpaired growth kinetics (Fig. 4B and Tables 1 and 2).

Commensal clonal populations are phenotypically less diverse than pathogenic ones
A crucial question is whether micro-heterogeneity is due to multiple infections of micro-evolved genotypes of the same clone from a common route and/or source of infection or to evolution during the infectious process. We thus compared within-clone phenotypic diversity in the 19 populations isolated from infections and in 15 clonal populations isolated from the faeces of 15 healthy subjects matched for geographic origin and sex with the infected patients (10 colonies studied per sample). We found in healthy subjects one change of antibiotic profile within a subject, affecting the resistance to sulfonamides in 7 isolates, and one change in growth ability within another subject, affecting one isolate from 10. This is a significantly different pattern as compared to the infected patients (Wilcoxon two sided test, p = 0.015), consistent with increased genetic differentiation during infection.
Mutators are present within monoclonal isolates of a unique patient
A way to increase the genetic and phenotypic diversity is to increase the mutation rate. We thus measured the frequencies of mutations conferring resistance to rifampin in the isolates of 9 patients where a strain with genetic micro-heterogeneity was identified. In patient 14 the isolates were all resistant to rifampin. The median value of mutagenesis for the 91 isolates from the remaining 8 patients was 3.33×10−9, a value not different from the previously reported E. coli collections [33]. Considering a threshold of 10-fold the median value for defining mutators, 3 isolates were mutators: one in patient 3, one in patient 12 and one among the A1 isolates in patient 17 (Table S3). Among these three isolates, one from patient 17 displayed a >50-fold increase in mutagenesis and was considered as a strong mutator. Such strong mutators have been shown to be essentially mismatch repair deficient in the wild [34].
Isolates exhibiting a micro-heterogeneity did not have a higher mutation rate than commonly found in the species E. coli. Hence, increased mutation rate should not be the primary cause of the observed phenotypic diversity, yet the presence of some mutator isolates at quite high frequency compared to their expected production by mutation, suggests that some strong adaptation might be under way in those populations [35].
The level of RpoS is variable within isolates of a single clone
In environments as in the host where stress and nutritional competition are both important, selection may well affect SPANC-related phenotypes [15], [36], [37].
We studied the variability of the SPANC-related phenotypes in the 8 patients showing differences in growth kinetics. Using an assay that tests the metabolic capacity of a strain to use a wide array of carbon sources (Phenotype MicroArrays) [38], we found that isolates from a single clone within a single patient have distinct patterns of substrate use (Fig. 4C and Tables 2 and S4). Interestingly, 9 of these substrates differentially used within isolates from a single patient (Table S4) are among the 13 substrates whose metabolism is stimulated by an rpoS disruption [39]. Motility and sensitivity to H2O2 and acid were also highly variable among isolates from a single patient (Figs 4D and E and Table 2). Thus, heterogeneity in motility pattern was evidenced in 7 of the 8 patient isolates, with the majority of isolates within a patient being motile and the minority being non motile whereas polymorphism in the H2O2 and acid sensitivity was observed in all and 5 patients, respectively. Furthermore, a significant relation was observed between growth, capacity to use the substrates, sensitivity to H2O2 and acid and motility. Strains showing impaired growth use fewer substrates, are more resistant to H2O2 and acid but less motile (Fig. S1), in accordance with the proposed SPANC trade-off [39]. We also studied the development of the red dry and rough (rdar) morphotype, which is depending of RpoS [40]. This morphotype is a multicellular behavior characterized by expression of the adhesive extracellular matrix components curli fimbriae and cellulose [41]. The morphotypes within isolates of single patients were highly variable (see Fig. 5A for an example). To confirm the role of RpoS in the observed polymorphism, we studied in more detail the 8 isolates of patient 3, the 7 isolates of the patient 13 and the 9 isolates of patient 17. The level of RpoS in the cell was assessed qualitatively by staining for presence of glycogen (RpoS regulates positively glgA, the glycogen synthase gene) and quantitatively by RpoS immunoblotting using specific antibodies. Both assays were consistent and showed isolate-specific endogenous levels of RpoS (Figs 5 and S2 and data not shown). Although the variations were not drastic except for few isolates, the fact that they were highly correlated between both techniques as well as to the nutritional and stress phenotypes and the rdar morphotypes, and the differentially expressed proteins for the patient 3 (see below and Fig. 6), is an argument for their physiologic relevance. Hence a major determinant of the polymorphism observed appear to be linked to the SPANC balance as observed in some experimental evolution settings.


Twenty-seven proteins are differentially expressed within the isolates of a single clone in relation to the SPANC-related phenotypes
To decipher the molecular bases of the RpoS phenotype, we first sequenced the rpoS promoter and gene but surprisingly did not detect any mutations amongst the 8 representative isolates of patient 3. We then performed differential proteomics on these 8 isolates by bidimensional electrophoresis (2-DE) and mass spectrometry. Twenty-seven proteins were significantly differentially expressed between the isolates, with a 1.8 to 24 fold range (Fig. S3 and Table 3). Among them, 16 were involved in central metabolism, 9 were membrane proteins and 2 were stress proteins. Furthermore, 14 out of the 27 differentially expressed proteins were known to be RpoS regulated (Table 3). Within the 8 isolates, the levels of the 27 proteins, except one putative outer membrane protein, were significantly related, as well as to the level of RpoS, the sensitivity to H2O2 and acid, the capacity to use substrates, the motility and the growth, with one group of proteins closely linked to the level of RpoS and the other with growth (Fig. 6).

Few mutations are associated with the phenotypic changes observed within the isolates of a single clone
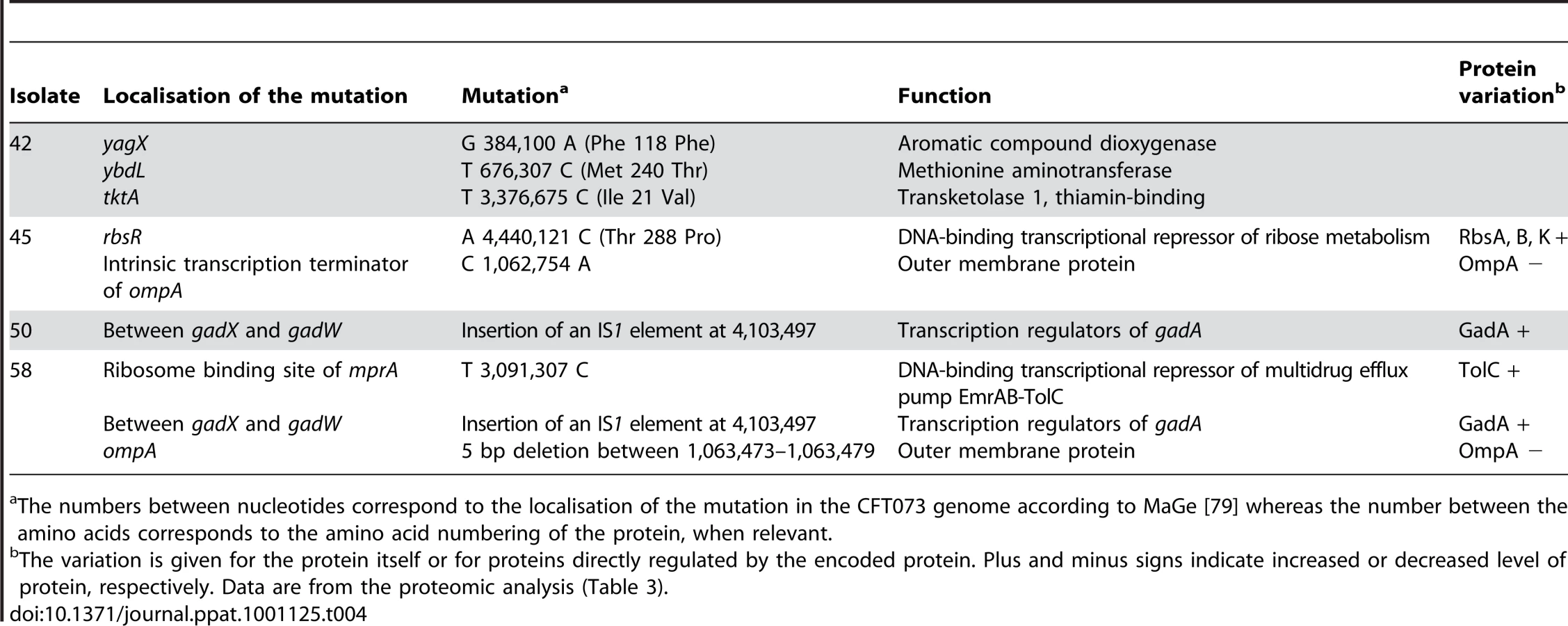
To identify the mutations responsible for these phenotypic changes, we performed whole-genome sequencing of 4 (42, 45, 50 and 58) of these 8 isolates of the patient 3 and searched by PCR for the identified mutations in the 4 (47, 51, 52 and 54) remaining isolates. Six point mutations, one small deletion and one IS insertion were identified, generating a star like phylogeny with each of the four sequenced isolates having its own combination of mutations (Fig. 7 and Table 4). The point mutations in 42, 45 and 58 isolates were absent from the remaining isolates in Fig. 7. The IS insertion was only identified in two of four sequenced isolates, but was shown by PCR to be in 47, 52 and 54 isolates as well. The genes affected were encoding for metabolic functions, including the acid resistance system dependent upon glutamate, and for membrane proteins. Of note, ompA was the target of 2 different molecular defects in isolates 45 and 58 (Table 4), which is a very strong sign of selection. A mutation in the repressor of the rbs operon involved in the D-ribose catabolic function, was observed. This operon was also shown to be the target of inactivating mutations selected during experimental evolution in glucose minimal medium [42]. Most of the mutations were corroborated by the proteomic changes in the corresponding isolate (Table 4).

Multiple genotypes with distinct levels of RpoS can co-exist as a result of the trade-off
Can SPANC balancing provide an explanation for E. coli diversity observed in the clonal isolates of a single patient described above? To test this hypothesis we consider a mathematical model that is built on a series of simple assumptions regarding bacterial metabolism and the environment in which bacteria reside, but that allows us examine in isolation the role of SPANC balancing in the creation and maintenance of diversity.
In our model we assume that E. coli isolates differ in their RpoS expression, x, normalized to 0≤x≤1 so that an rpoS - mutant has x = 0 and the rpoS+ with a maximal level of resistance has x = 1. Evolutionary changes in x are constrained by the SPANC balance trade-off in the following way: an increase in x leads to a decrease in the maximal resource uptake rate denoted by a decreasing function f(x); an increase in x also leads to an increase in the stress protection denoted by an increasing function c(x). Next we consider an E. coli population with n competing isolates each with a different value of the RpoS expression x, and define to be the density of an isolate with phenotype xi where i = 1…n and 0 = x1≤x2≤…≤xn = 1. We assume that mutations altering x occur at a rate ε taking into account both mutations in known regulators of RpoS levels as well as mutations that pleiotropically affect the levels of RpoS. A schematic representation of the model can be found in Fig. S4.
Solving numerically the system of equations (1) we find that starting with an isogenic rpoS+ population, the equilibrium population can support two or more partial rpoS- mutants depending on the shape of the SPANC balance trade-off namely on the shape of f(x) and c(x) (Fig. 8). For example, if mutations, that further decrease the levels of RpoS in a type that already has low levels of RpoS, do not lead to a marked decrease in stress protection (Fig. 8a), the long-term population structure supports two genotypes with distinct RpoS levels (Fig. 8d). On the other hand if mutations, that further decrease the levels of RpoS in a type that already has low levels of RpoS, lead to a clear decrease in stress protection (Fig. 8c), the long-term population structure supports three genotypes with distinct levels of RpoS (Fig. 8e).

Note that in our model the coexistence of multiple genotypes requires the assumption that changing RpoS levels from 1 to 0 induces three distinct levels of stress protection (Fig. 8a and c). While to our knowledge the shapes of the SPANC balance trade-off have not been determined in detail these forms are not unrealistic because stress responses to most environmental stresses involve not just RpoS, but also additional stress-specific responses (e.g. the heat shock or low pH responses) whose expression is directly or indirectly affected by the RpoS level. For example the acid stress resistance is influenced by numerous components including AR2 system whose expression can be initiated with either of two sigma factors RpoS and RpoD [43]. Similarly, DsrA, a small RNA, influences multiple acid resistance genes of E. coli as well as RpoS levels [44]. Therefore changing levels of DsrA, AR2 repressor H-NS and RpoS could give rise to non-linear relationships in the SPANC balance trade-offs as illustrated in Fig 8.
This simple model demonstrates that the SPANC balance is sufficient to create diversity in RpoS expressions within an E. coli population, and does not require any specific effect of the immune system.
The intrinsic virulence of isolates from a single clone is highly variable
Even if models predict that the mutants with variable levels of RpoS may emerge in the absence of immune selective pressure, we wanted to test the impact of RpoS phenotype on virulence. We therefore investigated the intrinsic extraintestinal virulence of each of the 8 representative isolates of patient 3, in a mouse model of septicemia [27]. Statistically significant differences were obtained between isolates, defining 4 groups (1 to 4), with the most virulent killing 100% of the mice in less than 15 hours (group 4) and the least virulent killing only 40% of the mice (group 1), the 2 other groups being intermediate (Fig. 4F). These groups are significantly correlated to growth properties; better growth gives greater virulence (Fig. 6). In confirmation, competition experiments in mice inoculated with a 1/1 ratio of isolate 58 (having an impaired growth) and reduced mouse killing (group 2) and isolate 42 (having a normal growth) and relatively high mouse killing (group 3) (Fig. 4F), showed a survival curve identical to the one observed in mice infected with isolate 42. Bacterial counts in the spleens of the killed mice showed a culture consisting > 99% of the 42 isolate. Identical results between these two strains were obtained after 24 h of in vitro competition in Luria Bertani broth (data not shown).
Hence the RpoS-associated impaired growth is not beneficial in the systemic phase of infection mimicked by the mouse model of septicemia, and the polymorphism observed may be driven mostly by the SPANC balance.
Discussion
Infections of a single patient by several distinct isolates is common in extraintestinal E. coli infections and result from different phenomena
To our knowledge, this work is the first one to investigate thoroughly, using molecular tools, numerous isolates from single patients in a large series of deep and severe extraintestinal E. coli infections. E. coli responsible for extraintestinal infections can be considered as opportunistic pathogens with the commensal reservoir being the intestinal tract [20], [21]. We observed an unexpectedly high level of within patient bacterial polymorphism as 11 of the 19 (58%) patients were infected by genotypically and/or phenotypically diverse bacteria. This diversity, as previously reported in other species [2], [3], [4], [5], [6], [7], [8], [9], [10], is of two types. Patients are infected either by several distinct E. coli clones or by a single clone with micro-heterogeneity. Both types of diversity can be observed in some patients. Patients with distinct E. coli clones were clearly infected from the intestinal commensal niche by different clones. Of note, these multiple infections are physically closely related to the commensal reservoir (Table 1). For the patients infected by a single clone that presents some micro-heterogeneity, two scenarios, not mutually exclusive, can be envisaged: the observed diversity could have pre-existed to the infection process in which a subset of the digestive tract diversity would have been sampled or the diversity could have emerged in the course of the infection that was initiated by a single isolate or few identical isolates. Several arguments favour the second hypothesis. First, in the cases with micro-heterogeneity, the infected organs were more diverse (kidney in 3 cases, digestive apparatus in 2 cases, pleural fluid and cerebrospinal fluid in one case each, associated in 2 cases to bacteremias) than in the patients infected by distinct clones, suggesting a less evident route for digestive tract bacteria to the infection zone. Second, we have studied the level of phenotypic polymorphism in E. coli isolates from stools of healthy subjects harbouring unique clones and found a significantly lower level of polymorphism in antibiotic resistance and growth patterns than in infections. Third, the tree reconstructed from the phenotypic and proteomic data (Fig. 7) of the 8 representative isolates of the patient 3 appears as a star like phylogeny and indicates a rapid diversification from a unique ancestor. Fourth, such scenario of diversification during the infectious process has been experimentally confirmed in a rat model of S. epidermidis foreign body-associated infection [7]. Similarly, genomic changes were observed in H. pylori during gastric experimental infection in rhesus monkey [45]. Lastly, it has been demonstrated in immunocompetent rodent models that bacteremias resulting from experimental colonisations are the products of very few bacteria [46], [47], [48], or even a single one [49].
The molecular bases of the within-clone phenotypic diversity
Some of the observed phenotypic diversity (antibiotic resistance, iron capture system) was easily explained by the loss of plasmid borne determinants. It has been shown that in stressful conditions, the induction of the SOS system mobilises transposons [50], integrons [51], as well as virulence genes [52]. The remaining phenotypic diversity was mainly RpoS related but we did not identified mutation in the rpoS gene or in the more than 20 diverse regulators that have been shown to influence RpoS levels [17]. Instead, we observed few mutations in metabolic and membrane related genes (Table 4). The convergence in ompA, the mutation in the rbs operon that has been shown to be under selection in experimental evolution system [42] and the presence of mutators (Table S3) are strong arguments for the fact that these mutations have been selected. Of note, ompA is among the 23 genes that show evidence of positive selection in E. coli [53]. The genomic mutations we identified suggest that SPANC balance can be affected by an even larger set of pleiotropic mutations, many of them yet unknown. For example, the overproduction of the EmrAB-TolC multi-drug efflux transport system in the mprA mutant, and the nutrient starvation in the ompA mutants could explain the altered growth and the RpoS response in isolates 45 and 58 (Table 4). In these cases, the RpoS phenotype would be secondary to the phenotype generated by the mutations. A limitation of our approach is that, (i) we could have missed IS based chromosomal reorganisations and gene amplification(s) and (ii) unlike in experimental evolution experiments, we do not have the ancestor clone that could allow us to perform reconstruction experiments with the mutated alleles and competitions [54], confirming the direct implications of the mutations in the observed phenotypes.
The evolutionary forces that drive the single clone diversity
A wide continuum of growth rate, stress resistance, outer membrane permeability, rdar morphotype and virulence properties were observed within isolates of single clones, directly linked to the SPANC trade-off and associated with distinct levels of the sigma factor RpoS in isolates (Figs 3, 4, 5, 6 and S1). rpoS mutations have been reported in E. coli laboratory and natural isolates as they are probably selected for in nutrient-limited environments [16]. Indeed, an important variation in the level of RpoS was observed in an E. coli clonal population grown in constant environment in a chemostat. This population radiated in more than five phenotypic clusters, two including rpoS mutant isolates [15]. In some other experimental evolution systems, duplications of rpoS have been reported [37], suggesting that higher or lower level could be achieved depending on the exact setting of the experiment. Our data in infected patients hence parallel these observations made in experimental evolution systems in the absence of immune selective pressure, and were explained by the simple mathematical model. This fully supports the idea that the SPANC balance is sufficient to promote a strong phenotypic diversification at intermediate stress levels (Fig. 8). Of course there are numerous other mechanisms [55] that could generate the diversity patterns observed in this study, such as environmental heterogeneities or immune system driven negative-frequency dependence. But the model provided here is a test of an alternative coexistence mechanism. The model assumptions have been deliberately kept simple in order to illustrate a minimal set of conditions under which the SPANC balance trade-off could lead to coexistence of E. coli genotypes with differing levels of RpoS. In particular the SPANC balance trade-off is defined through a single evolving phenotype, namely the RpoS expression and we conclude that complex but nonetheless realistic shapes of the trade-off are required for the diversity to be maintained. However, increasing the number of traits through which the trade-off exists will reduce the complexity of the shapes required to observe diversity [56].
The lack of mutations in rpoS itself that we observed suggests RpoS loss is detrimental under host infection conditions. Indeed, the role of RpoS in vivo is not clear as rpoS mutants are selected within the gut in commensal E. coli [57] but are avirulent in a mouse model of septicemia with Salmonella [58]. To test further if RpoS polymorphism is a virulence-linked effect, we tested the various isolates of patient 3 in a mouse model of infection. Yet we could not identify a virulence-linked benefit of high level of RpoS, as lower virulence was associated to lower growth rate (Fig. 4F and 6).
Another source of variation supported by theoretical and experimental approach is the one concerning iron uptake. Iron is essential for the growth of bacteria and they produce and release iron-chelating small molecules known as siderophores to scavenge iron from their hosts. Siderophore production is considered as an altruistic trait that is costly for the individual but provides a group benefit because other individuals can take up the siderophore-iron complex [18]. P. aeruginosa mutants that do not produce siderophore (cheaters) have been seen to evolve both in vitro [59] and in the lungs of cystic fibrosis patients [60]. Similarly, we observed within clone variability due to the coexistence in a same sample of isolates with different siderophore encoding gene content due to plasmid loss (Table S1). A frequent observed polymorphism in the monoclonal infection cases is also the presence or absence of penicillinase production (Fig. 3A and Table S2), due to a bla plasmid borne gene. As in the case of siderophore production, penicillinase production could be seen as an altruistic trait in the presence of antibiotics. The effect could also be reinforced by the fact that plasmids may carry other secreted (altruistic) proteins [61].
One emerging question related to the presence of some strong diversity in phenotypes as important as growth, resistance to stress or antibiotic resistance, is what are the consequences in term of the infection outcome. In other words, how such a polymorphism affects the virulence of the bacterial population. The emergence of cheaters lacking siderophore or eventually antibiotic resistance genes (and the other genes carried on the plasmid) clearly illustrates that selection may not always favour the most virulent genotypes, and that within-population selection may on the opposite select for genotypes that reduce the infectious power of the population as a whole [62]. Along the same line, our mathematical modelling suggests that the SPANC trade-off allows the emergence of some variable bacteria independently of the host immune system, and our animal model of infection revealed that the most stress resistant ones are not the more virulent. Yet, the existence of some strong variability along the SPANC balance may sometimes be beneficial as many different strategies are present within the population and some may be more efficient than other in the different stages of the infection.
Concluding remarks
Our results suggest that experimental evolution and the evolution of diversity in the course of an infection may have many parallels. Whether or not this diversity promotes increased virulence remains an open question, that will require further work to link the observed mutations to the various phenotypes and to some potential virulence linked effects.
Whatever the mechanisms leading to diversity (infections by several distinct isolates or diversification during the infectious process), our results have strong medical implications in term of antibiotic therapies. Indeed, although an individual can harbour several independent commensal clones [63], the classical medical assumption about extraintestinal E. coli infections is that they are strictly monoclonal. This is typified by the antimicrobial susceptibility testing performed on few (2 to 5) colonies obtained from the pathological sample to establish antibiotic treatment [64], [65]. This may fail to detect the presence of other, more resistant isolates, and lead to therapeutic failure [66]. Our work showing an impressive level of antibiotic resistance phenotype variability within the isolates of a single patient argues for the utilisation of a mix of numerous isolates to perform the antibiogram, or, alternatively for the realisation of simplified antibiograms with essential molecules on numerous isolates in addition to the classical antibiogram on a single isolate.
Materials and Methods
Bacterial strains
A total of 226 E. coli isolates responsible for severe infections in 19 epidemiologically unrelated adult patients from 3 university hospitals in France [Brest, Paris suburbs (Colombes and Clichy)], were studied (Table 1). To avoid contamination, only deep and closed visceral infections were selected (pyelonephritis, meningitis, pleurisy, cholecystitis, intra-abdominal deep abscess, ascitic fluid, peritoneal fluid and placental infections and bacteraemia). For 5 patients (1, 2, 3, 4 and 19), two samples were analysed (blood plus another site of infection), whereas only one sample was studied for the remaining patients. Except for urinary tract infection, samples were obtained during surgery or through sterile punctures. In all these infections, E. coli alone was recovered. For each sample, 5 to 20 colonies isolated on blood agar or CLED (cystine lactose electrolyte deficient) plates were randomly selected and stored with glycerol at −80°C before use. In addition, 150 commensal isolates belonging to 15 subjects (10 isolates per subject) matched for geographic origin (Brest, Paris suburbs) and sex with the patients [63] were studied as controls. Each subject exhibits identical isolates based on the phylogenetic group/subgroup and the virulence gene content, but the isolates of the different subjects belong to various phylogenetic group/sub groups. Isolates were stored with glycerol at −80°C before use.
Human ethics statement
All the sampling procedures of the infected patients were performed in the course of the clinical diagnosis. No additional procedure was performed in the patients for the present study. The study was approved by the institutional ethics committee (Comité de Protection des Personnes, Hôpital Saint-Louis, Paris; #2004-06). For the healthy subjects, the study was approved by the ethics committee of INSERM (IRB0000388, FWA00005831), with the opinion #01-014. In both studies, the participants were informed of their role in the study and written informed consent was provided by study participants.
PCR phylotyping and virulence factor pattern
All pathogenic isolates were assigned to the 7 E. coli phylogenetic group and subgroups using triplex PCR based on the presence or absence of three DNA fragments (chuA, yjaA and TSPE4.C2) [63]. The presence of 20 extraintestinal virulence factors (VFs) (neuC, sfa, iroN, iucC, iutA, iha, papC, papG, hlyC, cnf1, hra, sat, ire, usp, ompT, ibeA, malX, fyuA, irp2 and traT) was tested by PCR on all isolates, as previously described [31].
Enterobacterial Repetitive Intergenic Consensus (ERIC)-PCR
ERIC-PCR was performed on all pathogenic isolates, as previously described [67] using separately three different primers to increase the discriminatory power of the technique: ERIC1 (5′-GTGAATCCCCAGGAGCTTACAT-3′), ERIC1R (5′-ATGTAAGCTCCTGGGGATTCAC-3′) and ERIC2 (5′-AAGTAAGTGACTGGGGTGAGCG-3′).
Pulsed Field Gel Electrophoresis (PFGE) and analysis of strain relatedness
One hundred and ten pathogenic isolates from 11 patients (3, 5, 8, 11, 12, 13, 14, 15, 16, 17 and 19) in whom strain heterogeneity was observed in the antibiotypes and/or phylogenetic group/subgroups and/or virulence gene patterns and/or ERIC patterns, were further studied by PFGE. As previously described [68], PFGE was performed after digestion of chromosomal DNA with XbaI, which gave a convenient number of fragments. These fragments were separated in 0.5 X TBE buffer, pH 8.0, at 14°C and 6 V/cm with pulse times of 5–50 s for 21 h. DNA patterns obtained were analysed with Gel Compar software (Applied Maths, Kortrijk, Belgium). A similarity matrix was created by using the band-based Dice similarity coefficient, and Ward's algorithm was used to cluster the strains.
Multi Locus Sequence Typing (MLST)
MLST analysis based on the concatenated sequences of 6 essential genes (trpA, trpB, pabB, putP, icd and polB) [26] was performed in some cases to determine the relatedness between the isolates. In addition, the sequence of gnd, one of the most variable gene in E. coli due to a high level of recombination [30], was occasionally performed to further discriminate isolates.
Antibiotic susceptibility
Antimicrobial susceptibilities were determined on all the pathogenic and commensal isolates by the method of disc diffusion, according to the guidelines of European Committee for Antimicrobial Susceptibility Testing (www.eucast.org). The tested resistance markers were as follow: amoxicillin (AMX), amoxicillin-clavulanate, ticarcillin (TIC), piperacillin (PIP), piperacillin-tazobactam, imipenem, cefalotin (Cf), cefamandole, cefuroxime, cefoxitin, cefotaxime, ceftazidime, cefepime, aztreonam, mecillinam, streptomycin (St), kanamycin (Ka), gentamicin, tobramycin, netilmicin, amikacin, tetracycline (Te), minocycline (Mn), nalidixic acid (Na), pefloxacin, ciprofloxacin, chloramphenicol (Ch), fosfomycin, sulfonamides (Sul), trimethoprim (TMP). MICs were determined by the E-test method (AB Biodisk, Solna, Sweden) as recommended by the manufacturer. The production of penicillinase (Pase) was deduced from the resistance profile (resistance to amoxicillin, ticarcillin, piperacillin and intermediate susceptibility to amoxicillin-clavulanate, piperacillin-tazobactam and cefalotin) and confirmed by the detection of blaTEM by specific PCR amplification as previously described [69].
Outer membrane permeability
The isolates of patient 17 were electroporated with the plasmid pBR322 to introduce high levels of β-lactamase into all strains. Outer membrane permeability was measured by comparing β-lactamase activity in whole cells (with intact outer membrane) to that in disrupted cells with no barrier, using the permeable colorimetric substrate nitrocefin [70]. Bacterial strains were grown to exponential phase at 37°C in Luria-Bertani (LB) broth without salt containing either 100 µg/ml ampicillin or 15 µg/ml tetracycline. The optical density of the culture was measured and two 100 µl samples were taken. One aliquot (whole cells) was washed once, resuspended in the same volume of 0.1× M9 and the second aliquot was centrifuged and resuspended in 30% sucrose, 33 mM Tris. 5 µl 2 mg/ml lysozyme and 5 µl 0.2M EDTA and the mixture placed on ice for 15 min. Lysed bacteria were centrifuged for 5 min at 4°C, the pellet discarded and 3 µl 1M MgCl2 added to the supernatant. The broken cells and whole cells were assayed in 96 well micro titre plates with each well containing 160 µl 0.1×M9, 20 µl 50 µg/ml nitrocefin and 20 µl of the sample. The 96 well plate reader was configured for kinetics at 492nm for 20 min with readings taken every 2 min. The activity of the broken cells was very similar and quadruple assays were used to generate the values plotted in Fig. 3C. The activity is presented as the rate of optical density change/min/cell in the assay.
Bacterial growth
Cells grown overnight in LB broth were washed and transferred into a fresh LB medium at a dilution of 1∶250. The growth rates were then measured for all the pathogenic and commensal isolates at 37°C by monitoring the optical density at 600 nm (Tecan microplate reader) in LB medium. Experiments were repeated 3 times.
Carbon source utilisation
Biolog GN2 (Biolog, Inc., Hayward, CA) microplates were used to detect carbon utilization of 95 substrates in isolates of patients where a micro-heterogeneity was observed. Utilization of various C sources is coupled to reduction of a tetrazolium dye and production of purple colour [38]. Each strain was grown in LB medium, washed and resuspended to an optical density at 600 nm of 0.01 in mineral medium. Mineral medium was prepared as described elsewhere [38]. Plates were incubated at 37°C and colour changes were measured by optical density measurement (Tecan microplate reader) at a wavelength of 600 nm. The cut off point between negative results and positive results was an optical density of 0.2. Experiments were repeated 2 times. For the statistical analysis, the data considered were, for each isolate, the number of used substrates.
Hydrogen peroxide sensitivity
Bacterial isolates from patients showing differences in the growth kinetic were grown overnight in LB, washed, and resuspended in 0.9% NaCl to a concentration of about 1.108 cells ml-1. H2O2 was added to a final concentration of 50 mM. Aliquots of bacteria were removed at timed intervals, and numbers of viable cells were determined on LB plates. For the statistical analysis, only two categories were considered (resistant or sensitive). To delimit in categories the 8 isolates of patient 3, area between curves was used as a distance in order to compare curves. A k-means approach (two seeds, i.e. two clusters) on such distance matrix allows an automatic separation of clusters. As a control for the choice of the number of clusters, we represented these curves (according to the described distances) as points in a two dimensional Euclidean space thanks to an effective non-linear multidimensional algorithm named Data Driven High Dimensional Scaling (DD-HDS) [71]. Two clusters (categories) can be considered as fair.
Acid sensitivity
To assess the acid sensitivity phenotype of the isolates from patients showing differences in the growth kinetic, we used a protocol previously described [72] with some minor modifications. An inoculum containing about 108 cells from an overnight LB culture was introduced into LB acidified with HCl (35%) to pH 2.5. After two hours of exposure to pH 2.5, surviving cells were counted and the percentage of survivor was calculated. Strains which exhibited greater than 10% survival were designed acid resistant (low sensitivity), strains with 0.1% to 10% were designed intermediate in acid sensitivity, and those with less than 0.1% survival were considered acid sensitive (high sensitivity).
Motility
Cells of isolates from patients showing differences in the growth kinetic were grown overnight in LB, washed, and resuspended in minimum media (PO4 acetate) to a concentration of 1.102 cells ml−1. About 10 cells were grown in standard Petri dishes containing 20 ml minimum (PO4 acetate) media with 10mM glucose, solidified with 0.35% agar. Swim plates were incubated 48 hours at 37°C in humid atmosphere. Experiments were repeated 2 times. For the statistical analysis, only two categories were considered (motile or non motile).
Rdar morphotype
Cells of isolates from patients showing differences in the growth kinetic were grown on LB agar plates without NaCl (LB0) at 28°C. We determined colony morphology and colour using LB0 agar supplemented with Congo red (40µg.ml-1) and Coomassie brilliant blue (20 µg.ml-1), as described previously [41].
Iodine staining
Level of RpoS was obtained in 8 selected isolates of patient 3 by staining colonies on LB agar plates. Plates were incubated for 24 hours at 37°C and then left at 4°C for 20 hours before being flooded with Lugol (I2 concentration = 10g.L−1). Iodine staining is dependent on glycogen, whose synthesis is affected by RpoS [73]. Dark-brown colonies correspond to a high level of RpoS. A control corresponding to E. coli MG1655 ΔrpoS strain was used. Experiments were repeated 2 times. For the statistical analysis, a scale from 1 to 5 depending on the darkness was used.
RpoS immunoblot
Cells were harvested in stationary phase after 18 hours of culture in LB at 37°C, and proteins were extracted in SDS 1% at 100°C for 10 min. Protein concentration was measured by the method of Bradford (DC protein assay, Bio-Rad). SDS-PAGE was carried out in 10% polyacrylamide minigels (Mini Protean II; Bio-Rad). 10 µg of total proteins was loaded into each lane. The proteins were transferred to Amersham Hybond-P membrane (GE Healthcare) with a Mini TransBlot cell (Bio-Rad) in transfer buffer (25 mM Tris [pH 8.2], 192 mM glycine, 20% [vol/vol] ethanol). Western blot analysis was performed by serially incubating the membrane with polyclonal rabbit antibodies against the σS protein [74] overnight (1∶150000), and Amersham ECL anti-rabbit IgG (GE Healthcare), for one hour (1/20000). The RpoS protein was visualized using the Amersham ECL Plus Western Blotting Detection System (GE Healthcare). Quantifications were performed using a PhosphorImager (Molecular Dynamics). Data correspond to the ratio of the RpoS signal to a constant cross-reactive band. A control corresponding to E. coli MG1655 ΔrpoS strain was used. Experiments were repeated 2 times, values are the mean of the two experiments.
rpoS gene and promoter sequencing
DNA was prepared from 8 selected isolates of patient 3 using the Wizard Genomic DNA purification Kit (Promega). Two PCR products (product 1: primer forward ACAAGGGGAAATCCGTAAACC; primer reverse AGGATTTCGCGCAAACG, length: 1059 bp, and product 2: primer forward CCGTACTATTCGTTTCGGCCGA; primer reverse GAGACTGGCCTTTCTGACAG, length: 536 bp) were used to obtain the sequence of the rpoS gene and its promoter. Sequencing of PCR products was performed on both strands by classical Sanger technology.
Mouse extraintestinal virulence assay
A mouse model of systemic infection was used to assess the intrinsic virulence of 8 selected isolates of patient 3. For each isolate, 10 outbred female Swiss OF1 mice (3–4 weeks old, 14–18 g) received a 200 µL subcutaneous abdominal injection of 109 CFU/mL of stationary-phase bacteria. Mortality was monitored for 7 days [27]. Kaplan-Meier curves were drawn from the data. Isolates were classified in 4 significantly different virulence groups, from group 1 for the less virulent to group 4 for the more virulent group (p of log rank test < 0.05). This classification was used for the statistical analysis. Competition experiments were performed by inoculating 10 mice by a 1/1 ratio of strains 42 and 58. Cells were numbered in the spleens of animals after grinding and sprawl on LB agar plates. Distinction between isolates 42 and 58 was done using the iodine staining assay that stains glycogen (RpoS regulates positively glgA, the glycogen synthase gene and colonies from isolate 58 were darker than colonies from isolate 42).
Animal ethics statement
Animal experiments were performed in compliance with the recommendations of the French Ministry of Agriculture and approved by the French Veterinary Services (accreditation A 75-18-05).
Mutation frequency
Mutation frequencies of the isolates from 8 patients (91 isolates) showing a micro-heterogeneity were estimated by monitoring their capacity to generate mutation conferring resistance to rifampin in at least three independent cultures for each isolate, as described previously [33].
Bidimensional electrophoresis (2-DE) and mass spectrometry analysis
For each of the 8 representative isolates of patient 3, three independent protein extractions followed by 2-DE analysis were realised. Cells were harvested exactly at the same time at the beginning of stationary phase in LB and, after centrifugation washed three times in physiological serum. Proteins were extracted from cells by TCA-acetone precipitation according to [75], solubilized in R2D2 buffer [76] and quantified using the PlusOne 2-D Quant kit (Amersham Biosciences, Arlington Heights, IL). Isoelectrofocusing was carried out using 24-cm long, pH 4–7 Immobiline DryStrips (Amersham Biosciences) rehydrated in R2D2 solubilization buffer to which 300µg of protein extract was added. Full focusing was achieved after application of 84,000 V/hr at 20°C in a Protean IEF Cell (Bio Rad, Hercules, CA). Strips were equilibrated according to [77]. Second dimension electrophoresis was performed at 14°C (16 hr, 30 mA/gel) on a 24×24-cm gel (11% acrylamide, 2.9% of PDA crosslinker) in a Protean Plus Dodeca cell (Bio-Rad). Gels were stained by colloidal Coomassie blue and scanned using the PowerLook III scanner (Umax) and LabScan software (Amersham Biosciences). 2-DE gel image analysis was performed using Progenesis SameSpots (Nonlinear Dynamics). After optical density calibration, spot volumes were normalized according to total spot volume in each gel. Relative quantification of the detected spots was made in percentage of total spot volume (integrating optical density and spot area) for each gel, which allowed normalization of the values. Spots showing a significant variation between isolates (ANOVA test: p-value < 0.001) were excised with an EXQuest Spot Cutter (BioRad) and digested using a standard trypsin protocol as described by [78]. After digestion the supernatants were harvested and frozen at −20°C. Mass spectrometry identification of proteins was performed using a MALDI-TOF mass spectrometer Voyager DE super DTR system (Applied Biosystems, Foster City, CA, USA) equipped with a nitrogen laser emitting at 337 nm. Spectra were obtained in the mass range between 1000 and 2500 Da and were calibrated using internal calibration with autolytic trypsin fragments. Proteins were identified using the protein sequence database search program MS-FIT (http://prospector.ucsf.edu/prospector/4.0.7/html/msfit.htm) and the database MaGe (Genoscope, Evry France) [79]. Database search results were only accepted if the score reported by MS-FIT was higher than 1e4.
Whole genome sequencing
Four representative of the 8 isolates of patient 3 were fully sequenced. Genomic DNA was prepared using the Wizard genomic DNA purification kit (Promega) and sequenced with Solexa (Illumina) technology (Genoscope, Evry, France). The fastq files were then analysed though the Sanger pipeline SSAH2 [80], using CFT073 genome [79] as a reference genome. Preliminary analysis showed that it was the closest E. coli sequenced genome. We first confirmed this observation as the isolates are less than 0.1% diverged from CFT073 and share more than 95% of their genome with CFT073. For each isolate, using the mapping of reads on the reference genome, we could identify positions at which the sequence differed from this reference sequence. At all these sites, the number of reads supporting each of the four bases was recorded in all the isolates. For each site, an exact Fisher test was then performed among all pairs of isolates to detect any difference in the distribution of the counts supporting each base. Two kinds of significant deviation occurred. The ones with the highest p-values were due to change of the base presenting the highest counts between at least two isolates. These were all confirmed to be mutations through targeted sequencing. The other significant differences in distribution were due to change in the ratio of frequency between the dominant and the second most encountered base at the site. It only occurred in high coverage regions and the dominant base remained the same. None of the positions having such a profile we tested presented a different base across lines. As we do not have the ancestral genome, we used another approach in which instead of using the CFT073 genome, we used a concatenation of contigs resulting from a de novo assembly performed with Velvet software [81]. The same mutations events were detected. Through the SSAHA2 package we also identified small indels (<3bp long) compared to CFT073 genome, but none of them appeared to be specific to a subset of strains, they were all shared by all strains.
Finally we wanted to detect IS transpositions. For that purpose, we used the contigs assembled by Velvet software and looked for IS tails in the border of the produced contigs. A library of E. coli IS was recovered from ISFinder library (http://www-is.biotoul.fr/is.html). All IS tails were blasted against the assembled contigs. When a significant match was found, the 100 base pairs flanking the targeted sequence in the contig were blasted against the genome of CFT073. This provided a position of IS insertion element in the genome. A few IS insertions were identified as potentially different among the lines. To further comfort the specificity of IS elements integration, in all strains, we looked for the reads overhanging the suspected site of integration and see if any reads were mosaic (having one part of the chromosome and one part of the IS element border). After such a control a single site appeared as an IS integration specific to some strains. It was confirmed by targeted PCR analysis.
As deletion or other genomic rearrangement should also result in mosaic reads (matching two parts of the genome), we used the previous approach to scan all possible positions in the genome that were enriched in such mosaic reads in a specific subset of strains. In addition to the previous IS element, we found a 5 bp deletion in ompA uniquely in isolate 58 which was confirmed by targeted sequencing.
While with these different approaches we have scanned close to 90% of the genome for punctual mutations, small insertion or deletions, or IS integration, our approach is much more limited in repeated regions the genome (although the use of de novo assembled contigs suggest that in low copy regions no change were observed). Moreover our approaches, based on the use technology, fail to detect any IS based chromosomal reorganisation such as inversion and gene amplification.
All mutations detected by whole genome sequencing (Solexa) were verified by targeted sequencing using Sanger technology on PCR products or by PCR and agarose gel electrophoresis (for the IS detection) and searched on the other isolates of patient 3. The PCR primers were as follow: mprA, forward CATTATCATCCCAACACTGC, reverse ACGCCATAAACAACGTCTCG; rbsR, forward AATTTAACGGCGGGTTTGAC, reverse TTCAGCAGCAATGTTCATGC; ompA, forward ATCGTCTGGCAACGTCTGGC, reverse GACAAAATCTCCGCACGTGG; yagX, forward ACTGGTTGTTATAGACGCCG, reverse CGTCCCGCTCTATATTCATC; ybdL, forward AACTGTCTGGCAGCAGACTG, reverse TCACCAGAATATCGCGCTTC; tktA, forward GGCTGTAGATCAGCATGGAG, reverse GCAACATGCGAGCATGATCC; IS between gadA and gadW, forward GCCAGACTGTTTCTGTGTGC, reverse CACGGGAAACTTTGTGCTCT; deletion in ompA, forward GGGTTACCCAATCACTGACG, reverse CAGCGAAGACCGGAGAAAC.
Statistical analysis
In order to determinate if a link between various phenotypes (including RpoS level and 27 protein levels obtained by 2-DE analysis) exists we used a non-parametric test in the line of the Spearman's rank correlation test. There is no reason to a priori suspect that links between phenotypes are necessary linear. Values have been sorted (equal ranks are given for equal values, as usual in case of nonparametric analysis). However, according to the special characteristics of the dataset, the classical Pearson's rank correlation test cannot be applied here. Indeed, because the obtained values for each patient are not comparable, a unique sorting of data cannot be reached. We thus sort values for each patient. For a couple of phenotypes, the sum of absolute differences between the whole resulting ranks is related to the possible link between phenotypes (the strongest is the link, the lowest is the sum). A permutation test gives access to a p-value: ranks are sorted (by patient) in order to evaluate the distribution of such ranks in case of independence between phenotypes (10,000 permutations in our implementation). A similar analysis while ranks are reversed for one phenotype allows accounting of negative links between phenotypes. For each couple of phenotypes, we are thus able to evaluate the probability that a link is existing. Links detected as significant (at the 5% level) are drawn in Figs 6 and S1.
A distance tree of the 8 isolates of patient 3 has been constructed from the phenotype and proteomic data. Because measures of variables cannot be compared, we have rather considered ranks (just as it has been done for the statistical analysis). The tree is performed by neighbour-joining from Euclidean distances on ranks.
Mathematical modelling
The evolutionary model tracking the rate of change of different phenotypes within the E. coli population can be schematically represented as in Fig. S4 and mathematically written as:(1)For simplicity we consider a homogeneous environment containing a single limiting resource S and assume that bacterial growth on S is proportional to the rate of ATP production with r denoting the number of cells created per unit of resource and k representing the measure of affinity for the resource (for details see [82]). We also make a simplifying assumption that the resource (S0) diffuses into the system at the same constant rate D as the rate at which both resources and bacteria are depleted from the system. As mentioned above these simplifying assumptions regarding the environment and cell metabolism enable us to isolate the effects of SPANC balance trade-off on evolution of diversity of RpoS levels within an E. coli population.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. BrogdenKA
GuthmillerJM
TaylorCE
2005 Human polymicrobial infections. Lancet 365 253 255
2. DenamurE
PicardB
GoulletP
BingenE
LambertN
1991 Complexity of Pseudomonas aeruginosa infection in cystic fibrosis: combined results from esterase electrophoresis and rDNA restriction fragment length polymorphism analysis. Epidemiol Infect 106 531 539
3. RomlingU
FiedlerB
BosshammerJ
GrothuesD
GreipelJ
1994 Epidemiology of chronic Pseudomonas aeruginosa infections in cystic fibrosis. J Infect Dis 170 1616 1621
4. PirnayJP
De VosD
CochezC
BilocqF
PirsonJ
2003 Molecular epidemiology of Pseudomonas aeruginosa colonization in a burn unit: persistence of a multidrug-resistant clone and a silver sulfadiazine-resistant clone. J Clin Microbiol 41 1192 1202
5. IsraelDA
SalamaN
KrishnaU
RiegerUM
AthertonJC
2001 Helicobacter pylori genetic diversity within the gastric niche of a single human host. Proc Natl Acad Sci U S A 98 14625 14630
6. GaldbartJO
MorvanA
DesplacesN
el SolhN
1999 Phenotypic and genomic variation among Staphylococcus epidermidis strains infecting joint prostheses. J Clin Microbiol 37 1306 1312
7. Van EldereJ
PeetermansWE
StruelensM
DeplanoA
BobbaersH
2000 Polyclonal Staphylococcal endocarditis caused by genetic variability. Clin Infect Dis 31 24 30
8. WarrenRM
VictorTC
StreicherEM
RichardsonM
BeyersN
2004 Patients with active tuberculosis often have different strains in the same sputum specimen. Am J Respir Crit Care Med 169 610 614
9. ArbeitRD
SlutskyA
BarberTW
MaslowJN
NiemczykS
1993 Genetic diversity among strains of Mycobacterium avium causing monoclonal and polyclonal bacteremia in patients with AIDS. J Infect Dis 167 1384 1390
10. SharmaM
RiedererK
JohnsonLB
KhatibR
2001 Molecular analysis of coagulase-negative Staphylococcus isolates from blood cultures: prevalence of genotypic variation and polyclonal bacteremia. Clin Infect Dis 33 1317 1323
11. FriesenML
SaxerG
TravisanoM
DoebeliM
2004 Experimental evidence for sympatric ecological diversification due to frequency-dependent competition in Escherichia coli. Evolution 58 245 260
12. TrevesDS
ManningS
AdamsJ
1998 Repeated evolution of an acetate-crossfeeding polymorphism in long-term populations of Escherichia coli. Mol Biol Evol 15 789 797
13. RaineyPB
RaineyK
2003 Evolution of cooperation and conflict in experimental bacterial populations. Nature 425 72 74
14. RozenDE
LenskiRE
2000 Long-Term Experimental Evolution in Escherichia coli. VIII. Dynamics of a Balanced Polymorphism. Am Nat 155 24 35
15. MaharjanR
SeetoS
Notley-McRobbL
FerenciT
2006 Clonal adaptive radiation in a constant environment. Science 313 514 517
16. FerenciT
2005 Maintaining a healthy SPANC balance through regulatory and mutational adaptation. Mol Microbiol 57 1 8
17. Hengge-AronisR
2002 Signal transduction and regulatory mechanisms involved in control of the sigma(S) (RpoS) subunit of RNA polymerase. Microbiol Mol Biol Rev 66 373 395, table of contents
18. GriffinAS
WestSA
BucklingA
2004 Cooperation and competition in pathogenic bacteria. Nature 430 1024 1027
19. DeitschKW
MoxonER
WellemsTE
1997 Shared themes of antigenic variation and virulence in bacterial, protozoal, and fungal infections. Microbiol Mol Biol Rev 61 281 293
20. TenaillonO
SkurnikD
PicardB
DenamurE
2010 The population genetics of commensal Escherichia coli. Nat Rev Microbiol 8 207 217
21. KaperJB
NataroJP
MobleyHL
2004 Pathogenic Escherichia coli. Nat Rev Microbiol 2 123 140
22. RussoTA
JohnsonJR
2003 Medical and economic impact of extraintestinal infections due to Escherichia coli: focus on an increasingly important endemic problem. Microbes Infect 5 449 456
23. SelanderRK
LevinBR
1980 Genetic diversity and structure in Escherichia coli populations. Science 210 545 547
24. DesjardinsP
PicardB
KaltenbockB
ElionJ
DenamurE
1995 Sex in Escherichia coli does not disrupt the clonal structure of the population: evidence from random amplified polymorphic DNA and restriction-fragment-length polymorphism. J Mol Evol 41 440 448
25. HerzerPJ
InouyeS
InouyeM
WhittamTS
1990 Phylogenetic distribution of branched RNA-linked multicopy single-stranded DNA among natural isolates of Escherichia coli. J Bacteriol 172 6175 6181
26. Escobar-ParamoP
SabbaghA
DarluP
PradillonO
VauryC
2004 Decreasing the effects of horizontal gene transfer on bacterial phylogeny: the Escherichia coli case study. Mol Phylogenet Evol 30 243 250
27. PicardB
GarciaJS
GouriouS
DuriezP
BrahimiN
1999 The link between phylogeny and virulence in Escherichia coli extraintestinal infection. Infect Immun 67 546 553
28. Escobar-ParamoP
ClermontO
Blanc-PotardAB
BuiH
Le BouguenecC
2004 A specific genetic background is required for acquisition and expression of virulence factors in Escherichia coli. Mol Biol Evol 21 1085 1094
29. TenoverFC
ArbeitRD
GoeringRV
MickelsenPA
MurrayBE
1995 Interpreting chromosomal DNA restriction patterns produced by pulsed-field gel electrophoresis: criteria for bacterial strain typing. J Clin Microbiol 33 2233 2239
30. LecointreG
RachdiL
DarluP
DenamurE
1998 Escherichia coli molecular phylogeny using the incongruence length difference test. Mol Biol Evol 15 1685 1695
31. JohnsonJR
ClermontO
MenardM
KuskowskiMA
PicardB
2006 Experimental mouse lethality of Escherichia coli isolates, in relation to accessory traits, phylogenetic group, and ecological source. J Infect Dis 194 1141 1150
32. AlekshunMN
LevySB
2007 Molecular mechanisms of antibacterial multidrug resistance. Cell 128 1037 1050
33. DenamurE
BonacorsiS
GiraudA
DuriezP
HilaliF
2002 High frequency of mutator strains among human uropathogenic Escherichia coli isolates. J Bacteriol 184 605 609
34. DenamurE
MaticI
2006 Evolution of mutation rates in bacteria. Mol Microbiol 60 820 827
35. TenaillonO
ToupanceB
Le NagardH
TaddeiF
GodelleB
1999 Mutators, population size, adaptive landscape and the adaptation of asexual populations of bacteria. Genetics 152 485 493
36. ZambranoMM
SiegeleDA
AlmironM
TormoA
KolterR
1993 Microbial competition: Escherichia coli mutants that take over stationary phase cultures. Science 259 1757 1760
37. RiehleMM
BennettAF
LongAD
2001 Genetic architecture of thermal adaptation in Escherichia coli. Proc Natl Acad Sci U S A 98 525 530
38. BochnerBR
GadzinskiP
PanomitrosE
2001 Phenotype microarrays for high-throughput phenotypic testing and assay of gene function. Genome Res 11 1246 1255
39. KingT
IshihamaA
KoriA
FerenciT
2004 A regulatory trade-off as a source of strain variation in the species Escherichia coli. J Bacteriol 186 5614 5620
40. Robbe-SauleV
JaumouilleV
PrevostMC
GuadagniniS
TalhouarneC
2006 Crl activates transcription initiation of RpoS-regulated genes involved in the multicellular behavior of Salmonella enterica serovar Typhimurium. J Bacteriol 188 3983 3994
41. RomlingU
2005 Characterization of the rdar morphotype, a multicellular behaviour in Enterobacteriaceae. Cell Mol Life Sci 62 1234 1246
42. CooperVS
SchneiderD
BlotM
LenskiRE
2001 Mechanisms causing rapid and parallel losses of ribose catabolism in evolving populations of Escherichia coli B. J Bacteriol 183 2834 2841
43. WatermanSR
SmallPL
2003 Transcriptional expression of Escherichia coli glutamate-dependent acid resistance genes gadA and gadBC in an hns rpoS mutant. J Bacteriol 185 4644 4647
44. LeaseRA
SmithD
McDonoughK
BelfortM
2004 The small noncoding DsrA RNA is an acid resistance regulator in Escherichia coli. J Bacteriol 186 6179 6185
45. SolnickJV
HansenLM
SalamaNR
BoonjakuakulJK
SyvanenM
2004 Modification of Helicobacter pylori outer membrane protein expression during experimental infection of rhesus macaques. Proc Natl Acad Sci U S A 101 2106 2111
46. PluschkeG
MercerA
KusecekB
PohlA
AchtmanM
1983 Induction of bacteremia in newborn rats by Escherichia coli K1 is correlated with only certain O (lipopolysaccharide) antigen types. Infect Immun 39 599 608
47. MoxonER
MurphyPA
1978 Haemophilus influenzae bacteremia and meningitis resulting from survival of a single organism. Proc Natl Acad Sci U S A 75 1534 1536
48. RubinLG
1987 Bacterial colonization and infection resulting from multiplication of a single organism. Rev Infect Dis 9 488 493
49. MargolisE
LevinBR
2007 Within-host evolution for the invasiveness of commensal bacteria: an experimental study of bacteremias resulting from Haemophilus influenzae nasal carriage. J Infect Dis 196 1068 1075
50. AleshkinGI
KadzhaevKV
MarkovAP
1998 High and low UV-dose responses in SOS-induction of the precise excision of transposons tn1, Tn5 and Tn10 in Escherichia coli. Mutat Res 401 179 191
51. GuerinE
CambrayG
Sanchez-AlberolaN
CampoyS
ErillI
2009 The SOS response controls integron recombination. Science 324 1034
52. SotoSM
Jimenez de AntaMT
VilaJ
2006 Quinolones induce partial or total loss of pathogenicity islands in uropathogenic Escherichia coli by SOS-dependent or -independent pathways, respectively. Antimicrob Agents Chemother 50 649 653
53. PetersenL
BollbackJP
DimmicM
HubiszM
NielsenR
2007 Genes under positive selection in Escherichia coli. Genome Res 17 1336 1343
54. SleightSC
OrlicC
SchneiderD
LenskiRE
2008 Genetic basis of evolutionary adaptation by Escherichia coli to stressful cycles of freezing, thawing and growth. Genetics 180 431 443
55. ChessonP
2000 Mechanisms of maintenance of species diversity. Annual Review of Ecology and Systematics 31 343 366
56. DoebeliM
IspolatovI
2010 Complexity and diversity. Science 328 494 497
57. KrogfeltKA
HjulgaardM
SorensenK
CohenPS
GivskovM
2000 rpoS gene function is a disadvantage for Escherichia coli BJ4 during competitive colonization of the mouse large intestine. Infect Immun 68 2518 2524
58. FangFC
LibbySJ
BuchmeierNA
LoewenPC
SwitalaJ
1992 The alternative sigma factor katF (rpoS) regulates Salmonella virulence. Proc Natl Acad Sci U S A 89 11978 11982
59. WestSA
BucklingA
2003 Cooperation, virulence and siderophore production in bacterial parasites. Proc Biol Sci 270 37 44
60. De VosD
De ChialM
CochezC
JansenS
TummlerB
2001 Study of pyoverdine type and production by Pseudomonas aeruginosa isolated from cystic fibrosis patients: prevalence of type II pyoverdine isolates and accumulation of pyoverdine-negative mutations. Arch Microbiol 175 384 388
61. NogueiraT
RankinDJ
TouchonM
TaddeiF
BrownSP
2009 Horizontal gene transfer of the secretome drives the evolution of bacterial cooperation and virulence. Curr Biol 19 1683 1691
62. TurnerPE
ChaoL
1999 Prisoner's dilemma in an RNA virus. Nature 398 441 443
63. Escobar-ParamoP
GrenetK
Le Menac'hA
RodeL
SalgadoE
2004 Large-scale population structure of human commensal Escherichia coli isolates. Appl Environ Microbiol 70 5698 5700
64. EUCAST 2003 Determination of minimum inhibitory concentrations (MICs) of antibacterial agents by broth dilution. Clinical Microbiology and Infection 9 ix xx
65. TurnidgeJD
BellJM
2005 Antimicrobial susceptibility on solid media.
WilkinsLWa
Antibiotics in laboratory medicine 8 60
66. ProctorRA
2000 Editorial response: coagulase-negative staphylococcal infections: A diagnostic and therapeutic challenge. Clin Infect Dis 31 31 33
67. VersalovicJ
KoeuthT
LupskiJR
1991 Distribution of repetitive DNA sequences in eubacteria and application to fingerprinting of bacterial genomes. Nucleic Acids Res 19 6823 6831
68. BrangerC
BruneauB
LesimpleAL
BouvetPJ
BerryP
1997 Epidemiological typing of extended-spectrum beta-lactamase-producing Klebsiella pneumoniae isolates responsible for five outbreaks in a university hospital. J Hosp Infect 36 23 36
69. RasheedJK
JayC
MetchockB
BerkowitzF
WeigelL
1997 Evolution of extended-spectrum beta-lactam resistance (SHV-8) in a strain of Escherichia coli during multiple episodes of bacteremia. Antimicrob Agents Chemother 41 647 653
70. AngusBL
CareyAM
CaronDA
KropinskiAM
HancockRE
1982 Outer membrane permeability in Pseudomonas aeruginosa: comparison of a wild-type with an antibiotic-supersusceptible mutant. Antimicrob Agents Chemother 21 299 309
71. LespinatsS
VerleysenM
GironA
FertilB
2007 DD-HDS: A method for visualization and exploration of high-dimensional data. IEEE Trans Neural Netw 18 1265 1279
72. WatermanSR
SmallPL
1996 Characterization of the acid resistance phenotype and rpoS alleles of shiga-like toxin-producing Escherichia coli. Infect Immun 64 2808 2811
73. Hengge-AronisR
FischerD
1992 Identification and molecular analysis of glgS, a novel growth-phase-regulated and rpoS-dependent gene involved in glycogen synthesis in Escherichia coli. Mol Microbiol 6 1877 1886
74. CoynaultC
Robbe-SauleV
NorelF
1996 Virulence and vaccine potential of Salmonella typhimurium mutants deficient in the expression of the RpoS (sigma S) regulon. Mol Microbiol 22 149 160
75. DamervalC
De VienneD
ZivyM
ThiellementH
1986 Technical improvements in two-dimensional electrophoresis increase the level of genetic variation detected in wheat-seedling proteins. Electrophoresis 7 52 54
76. MechinV
ConsoliL
Le GuillouxM
DamervalC
2003 An efficient solubilization buffer for plant proteins focused in immobilized pH gradients. Proteomics 3 1299 1302
77. GörgA
PostelW
WeserJ
GüntherS
StrahlerJR
1987 Elimination of point streaking on silver stained two-dimensional gels by addition of iodoacetamide to the equilibration buffer. Electrophoresis 8 122 124
78. MechinV
BalliauT
Chateau-JoubertS
DavantureM
LangellaO
2004 A two-dimensional proteome map of maize endosperm. Phytochemistry 65 1609 1618
79. VallenetD
LabarreL
RouyZ
BarbeV
BocsS
2006 MaGe: a microbial genome annotation system supported by synteny results. Nucleic Acids Res 34 53 65
80. NingZ
CoxAJ
MullikinJC
2001 SSAHA: a fast search method for large DNA databases. Genome Res 11 1725 1729
81. ZerbinoDR
BirneyE
2008 Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18 821 829
82. GudeljI
BeardmoreRE
ArkinSS
MacLeanRC
2007 Constraints on microbial metabolism drive evolutionary diversification in homogeneous environments. J Evol Biol 20 1882 1889
83. ColletA
CosetteP
BeloinC
GhigoJM
RihoueyC
2008 Impact of rpoS deletion on the proteome of Escherichia coli grown planktonically and as biofilm. J Proteome Res 7 4659 4669
84. DongT
SchellhornHE
2009 Control of RpoS in global gene expression of Escherichia coli in minimal media. Mol Genet Genomics 281 19 33
85. RahmanM
HasanMR
ObaT
ShimizuK
2006 Effect of rpoS gene knockout on the metabolism of Escherichia coli during exponential growth phase and early stationary phase based on gene expressions, enzyme activities and intracellular metabolite concentrations. Biotechnol Bioeng 94 585 595
86. LacourS
LandiniP
2004 SigmaS-dependent gene expression at the onset of stationary phase in Escherichia coli: function of sigmaS-dependent genes and identification of their promoter sequences. J Bacteriol 186 7186 7195
87. IshihamaA
2000 Functional modulation of Escherichia coli RNA polymerase. Annu Rev Microbiol 54 499 518
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 9
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Structure of the Extracellular Portion of CD46 Provides Insights into Its Interactions with Complement Proteins and Pathogens
- The Length of Vesicular Stomatitis Virus Particles Dictates a Need for Actin Assembly during Clathrin-Dependent Endocytosis
- Inhibition of TIR Domain Signaling by TcpC: MyD88-Dependent and Independent Effects on Virulence
- The Coevolution of Virulence: Tolerance in Perspective
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy