
Two Genes on A/J Chromosome 18 Are Associated with Susceptibility to Infection by Combined Microarray and QTL Analyses
Although it has recently been shown that A/J mice are highly susceptible to Staphylococcus aureus sepsis as compared to C57BL/6J, the specific genes responsible for this differential phenotype are unknown. Using chromosome substitution strains (CSS), we found that loci on chromosomes 8, 11, and 18 influence susceptibility to S. aureus sepsis in A/J mice. We then used two candidate gene selection strategies to identify genes on these three chromosomes associated with S. aureus susceptibility, and targeted genes identified by both gene selection strategies. First, we used whole genome transcription profiling to identify 191 (56 on chr. 8, 100 on chr. 11, and 35 on chr. 18) genes on our three chromosomes of interest that are differentially expressed between S. aureus-infected A/J and C57BL/6J. Second, we identified two significant quantitative trait loci (QTL) for survival post-infection on chr. 18 using N2 backcross mice (F1 [C18A]×C57BL/6J). Ten genes on chr. 18 (March3, Cep120, Chmp1b, Dcp2, Dtwd2, Isoc1, Lman1, Spire1, Tnfaip8, and Seh1l) mapped to the two significant QTL regions and were also identified by the expression array selection strategy. Using real-time PCR, 6 of these 10 genes (Chmp1b, Dtwd2, Isoc1, Lman1, Tnfaip8, and Seh1l) showed significantly different expression levels between S. aureus-infected A/J and C57BL/6J. For two (Tnfaip8 and Seh1l) of these 6 genes, siRNA-mediated knockdown of gene expression in S. aureus–challenged RAW264.7 macrophages induced significant changes in the cytokine response (IL-1 β and GM-CSF) compared to negative controls. These cytokine response changes were consistent with those seen in S. aureus-challenged peritoneal macrophages from CSS 18 mice (which contain A/J chromosome 18 but are otherwise C57BL/6J), but not C57BL/6J mice. These findings suggest that two genes, Tnfaip8 and Seh1l, may contribute to susceptibility to S. aureus in A/J mice, and represent promising candidates for human genetic susceptibility studies.
Published in the journal:
. PLoS Pathog 6(9): e32767. doi:10.1371/journal.ppat.1001088
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1001088
Summary
Although it has recently been shown that A/J mice are highly susceptible to Staphylococcus aureus sepsis as compared to C57BL/6J, the specific genes responsible for this differential phenotype are unknown. Using chromosome substitution strains (CSS), we found that loci on chromosomes 8, 11, and 18 influence susceptibility to S. aureus sepsis in A/J mice. We then used two candidate gene selection strategies to identify genes on these three chromosomes associated with S. aureus susceptibility, and targeted genes identified by both gene selection strategies. First, we used whole genome transcription profiling to identify 191 (56 on chr. 8, 100 on chr. 11, and 35 on chr. 18) genes on our three chromosomes of interest that are differentially expressed between S. aureus-infected A/J and C57BL/6J. Second, we identified two significant quantitative trait loci (QTL) for survival post-infection on chr. 18 using N2 backcross mice (F1 [C18A]×C57BL/6J). Ten genes on chr. 18 (March3, Cep120, Chmp1b, Dcp2, Dtwd2, Isoc1, Lman1, Spire1, Tnfaip8, and Seh1l) mapped to the two significant QTL regions and were also identified by the expression array selection strategy. Using real-time PCR, 6 of these 10 genes (Chmp1b, Dtwd2, Isoc1, Lman1, Tnfaip8, and Seh1l) showed significantly different expression levels between S. aureus-infected A/J and C57BL/6J. For two (Tnfaip8 and Seh1l) of these 6 genes, siRNA-mediated knockdown of gene expression in S. aureus–challenged RAW264.7 macrophages induced significant changes in the cytokine response (IL-1 β and GM-CSF) compared to negative controls. These cytokine response changes were consistent with those seen in S. aureus-challenged peritoneal macrophages from CSS 18 mice (which contain A/J chromosome 18 but are otherwise C57BL/6J), but not C57BL/6J mice. These findings suggest that two genes, Tnfaip8 and Seh1l, may contribute to susceptibility to S. aureus in A/J mice, and represent promising candidates for human genetic susceptibility studies.
Introduction
Staphylococcus aureus is an important human pathogen whose clinical spectrum ranges from asymptomatic colonization to endocarditis, shock, and death. Although the importance of genetic factors in determining host susceptibility to S. aureus colonization and infection [1]–[3] is generally accepted, the specific genes responsible for this susceptibility are largely unknown. As with most infectious diseases, the genetics of host susceptibility to S. aureus is complex, resulting from variation in multiple genes of small to moderate effect, and from the interaction of these genes with non-genetic factors [4].
Mouse models offer an attractive strategy for investigating complex diseases such as S. aureus infections. Abundant breeding, short gestation periods, and the availability of extensive sequence data for inbred strains (http://mouse.perlegen.com/mouse/index.html and http://www.informatics.jax.org/) facilitate genetic research in mice. In addition, inbred mouse strains demonstrate considerable strain variation in susceptibility to various pathogens [5]–[11], including S. aureus [3]. The determinants of this differential susceptibility to pathogens are probably multigenic, and largely unknown [12].
Among the inbred mouse strains, A/J was recently shown to be highly susceptible to S. aureus infection compared with C57BL/6J [3]. By chance, these same genetic backgrounds (A/J and C57BL/6J) were used to construct murine chromosome substitution strains (CSS). CSS were developed to speed the genetic mapping of heritable traits, and were created from a host C57BL/6J strain and a donor A/J inbred strain [13]. Each CSS mouse is homosomic for a specified A/J chromosome but otherwise has a C57BL/6J background. Breeding strategies using these CSS strains greatly enhance the detection of quantitative trait loci (QTLs). Using CSS mice, the impact of a single A/J chromosome can be effectively isolated by eliminating the contribution to variance in phenotype from the other A/J chromosomes. CSS strains have been used to identify QTL for several complex traits, including anxiety [14], diet-induced obesity [15] serum levels of sterols and amino acids [15], testicular cancer [16], [17], pubertal timing [18], airway hyperresponsiveness [19], and seizures [20], and provide an ideal means of identifying genetic determinants of murine susceptibility to S. aureus sepsis.
In this study, we sought to identify genetic factors associated with susceptibility to S. aureus using a multi-step selection process (Figure 1). First, we identified individual chromosomes governing susceptibility to S. aureus infection in A/J mice by phenotyping a complete panel of CSS mice. Next, we used peripheral blood mRNA expression to identify genes on these chromosomes that were differentially expressed between susceptible (A/J) and resistant (C57BL/6J) mouse strains in the setting of S. aureus infection. Following this, we identified two QTL regions on chromosome 18 that are significantly associated with susceptibility to infection with S. aureus. From these two QTL regions, we identified 10 candidate genes that were differentially expressed between A/J and C57BL/6J. We found evidence of biological relevance for two of these genes (Tnfaip8 and Seh1l) using siRNA mediated knockdown and Luminex-based cytokine profiles in S. aureus-challenged RAW264.7 mouse macrophages, as well as peritoneal macrophages from CSS18 mice.

Results
Differential susceptibility to S. aureus in A/J and C57BL/6J
When mice were injected (intraperitoneally [i.p.]) with the Sanger 476 strain of S. aureus, C57BL/6J mice demonstrated a resistant phenotype (median survival >120 h), whereas A/J mice demonstrated a susceptible phenotype (median survival: 22 h) (Figure 2A). This pattern of differential susceptibility to S. aureus between C57BL/6J and A/J mice persisted when the experiments were repeated using both a different S. aureus strain (MW2, a methicillin resistant S. aureus [MRSA] isolate) (Figure 2B), and when mice were infected by intravenous rather than i.p. route (Figure 2C). A/J mice are known to be deficient in complement factor C5, an important component in neutrophil and macrophage recruitment [21]–[23]. Thus, the potential impact of C5 deficiency on susceptibility to S. aureus infection is important to consider. To ensure that A/J's susceptibility to S. aureus is not primarily due to C5 deficiency, we challenged three additional C5 deficient mouse strains (B10.D2/oSnJ, B10.D2-Hc0 H2d H2-T18c/o2SnJ, and NOD/LtJ) with an intraperitoneal injection of S. aureus (107 CFU/g). All three additional C5-deficient mice were resistant to S. aureus infection (median survival >120 h, Figure 2D), suggesting that factors other than C5 deficiency were responsible for the susceptibility of A/J mice to S. aureus.

Genetic susceptibility to S. aureus in mice localizes to A/J chromosomes 8, 11, and 18
Since we identified C57BL/6J mice as resistant and A/J mice as susceptible to S. aureus infection, we used CSS from these backgrounds to further localize the chromosomal regions responsible for these divergent phenotypes. CSS mice with chromosome 8, 11 or 18 from the susceptible background (A/J) transferred individually into the otherwise resistant background (C57BL/6J) were significantly more susceptible to S. aureus infection compared to C57BL/6J mice (p<0.005, log rank test) (Table 1) or other chromosome substituted strains. This suggests that genetic loci mediating susceptibility to S. aureus are located on each of these 3 chromosomes.

Rapid mortality in CSS mice with A/J “susceptibility” chromosomes 8, 11, and 18 is associated with increased bacterial load in the peritoneal fluid and kidneys
To further understand the basis of the enhanced susceptibility to S. aureus infection, we measured the tissue burden of S. aureus in A/J and C57BL/6J mice and the CSS mice 24 hours after infection with S. aureus. The animals were euthanized and bacterial load in the kidney and peritoneal fluid was determined. The bacterial load in the peritoneal fluid of susceptible A/J mice was significantly higher than in the resistant C57BL/6J mice (178.9±16.3×106 CFU/ml vs. 3.40±1.55×106 CFU/ml; P = 0.0001, F-test; Figure 3A). The bacterial load of the peritoneal fluid was also significantly higher in the CSS mice with A/J chromosome 8 (119.3±47.8×106 CFU/ml, P = 0.0017, F-test), chromosome 11 (220±74.7×106 CFU/ml, P = 0.016, F-test) or chromosome 18 (132.7±25.7×106 CFU/ml, P = 0.019, F-test) compared to C57BL/6J mice and to the rest of the chromosome substituted strains (1.531±5.5×106). Similarly, the bacterial load in the kidney of the susceptible A/J mice was significantly higher than in C57BL/6J mice (111.9±99.8×106 CFU/g vs. 33.1±18.7 CFU/g; P = 0.003, F-test; Figure 3B). Finally, the bacterial count in the kidney of CSS mice with chromosome 8 (44.8±27.8×106 CFU/g, P<0.001, F-test), chromosome 11 (3.5693±0.1940×106 CFU/g, P<0.001, F-test) and chromosome 18 (1.3451±0.2491×106 CFU/g, P = 0.0013, F-test) was significantly higher than that in C57BL/6J mice and the remainder of the chromosome substituted strains (4.75±86.39×103 CFU/g, P = 0.016, F-test). Thus, increased mortality due to S. aureus sepsis is associated with a higher bacterial load in the kidneys and peritoneal fluid of susceptible A/J, CSS8, CSS11 and CSS18 mice as compared to the resistant C57BL/6J mice and the remainder of the consomic mouse strains (CSS1–CSS7, CSS9–10, CSS12–CSS17, CSS19, CSSX and CSSY). This correlation of susceptibility with bacterial burden provides important insight into the pathogenesis of early death in murine staphylococcal sepsis, as the genes involved in susceptibility of A/J to S. aureus sepsis appear to be associated with a relative inability of the host to control infection.

Genetic susceptibility to S. aureus in mice is not sex linked and has variable forms of inheritance
We next generated F1 progeny by crossing the resistant C57BL/6J and susceptible CSS 8, 11, and 18 mice. These crosses result in offspring that, for the relevant chromosome pair (8, 11, or 18), have one C57BL/6J chromosome and one A/J chromosome. Using the same model of infection as described above, we observed no significant difference in the median survival of each F1 cohort when the parents were of either sex, indicating that the alleles of the loci mediating susceptibility to S. aureus are not imprinted (Table 2). F1 mice from the C57BL/6J×CSS8 (C8A) and C57BL/6J×CSS18 (C18A) crosses retained susceptibility to S. aureus infection (median survival <48 h), whereas F1 mice from C57BL/6J×CSS11 cross (C11A) were resistant (median survival >120 h) to S. aureus infection. These results indicate that the individual loci mediating susceptibility to S. aureus in A/J mice have different modes of inheritance (i.e., autosomal dominant for loci on chromosomes 8 and 18 and autosomal recessive for loci on chromosome 11). Similar to the homozygous CSS mice, the bacterial load in the kidneys of the susceptible F1 mice (C8A and C18A) was significantly higher than in the resistant C57BL/6J or C11A mice (data not shown). Taken together, these results provide further evidence that genes governing murine susceptibility to S. aureus infection reside on chromosomes 8, 11 and 18.

Genes on chromosomes 8, 11, and 18 are differentially expressed between A/J and C57BL/6J mice
Next, to identify genes on A/J chromosomes 8, 11, and 18 that contribute to susceptibility, we compared whole blood genome transcription profiles of A/J and C57BL/6J mice in both an uninfected state and following S. aureus infection. A total of 675 genes on chromosomes 8, 11, and 18 were differentially expressed between uninfected A/J and C57BL/6J mice, and 751, 683, 1223 and 639 were differentially expressed at 2, 4, 6, and 12 hours after infection, respectively. However, only 191 of these genes were differentially expressed at all four post-infection time points (Table 3 and Table S1). Of the 191 genes, 37 genes were similarly expressed in uninfected A/J and C57BL/6J mice. The remaining 155 genes were differentially expressed between C57BL/6J and A/J mice in both uninfected and infected states.

QTL for susceptibility to S. aureus infection on chromosome 18
To further fine-map the locus (or loci) on chromosome 18 involved in determining susceptibility to S. aureus infection, we performed a QTL analysis, generating N2 backcross mice by mating F1 mice (C18A) to C57BL/6J mice (Figure S1). A total of 144 N2 backcross mice were generated and infected with S. aureus, and their survival times were measured. No sex-linked susceptibility in N2 backcross mice was identified (Figure S2). Using J/qtl software, two significant QTLs were identified as linked to survival time after S. aureus infection (Figure 4A), one located at 41 to 54 Mb, and one located at 57 to 67Mb. Approximately 317 genes are contained within these two significant intervals.
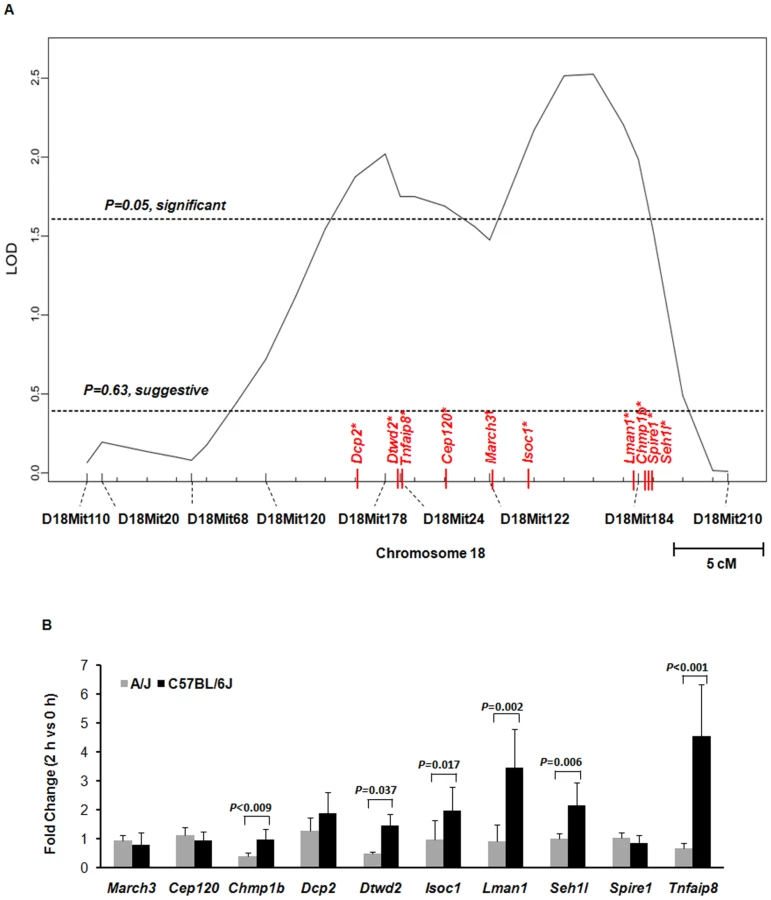
Next, we identified genes implicated by both our expression array-based and QTL-based selection strategies. A total of 10 genes (March3, Cep120, Chmp1b, Dcp2, Dtwd2, Isoc1, Lman1, Spire1, Tnfaip8, and Seh1l) mapping within our two significant QTLs were differentially expressed between S. aureus infected A/J and C57BL/6J mice. Four genes (Dcp2, Dtwd2, Tnfaip8, and Cep120) were located within the first significant QTL (41 to 54 Mb) and one gene (March3) was located in the suggestive interval for this QTL (56 Mb). The remaining 5 genes (Isoc1, Lman1, Chmp1b, Spire1, and Seh1l) fell within the second significant QTL (57 to 67 Mb). For 6 of these genes (Dtwd2, Tnfaip8, Isoc1, Lman1, Chmp1b, and Seh1l), real-time PCR confirmed significant differences in expression between S. aureus-infected A/J and C57BL/6J mice (P<0.05, paired two-tailed t-test) (Figure 4B).
Assessing biological relevance: Neutrophil and macrophage function in A/J and C57BL/6J
Because of the central role of neutrophils in the host response to S. aureus, we considered whether neutrophil dysfunction might be a primary cause of A/J susceptibility to S. aureus. To do this, we assessed ex vivo the bactericidal activity of neutrophils from A/J, C57BL/6J, or CSS18 mice by incubating them with S. aureus, and then comparing the number of viable bacteria in the extracellular (unphagocytosed) and intracellular (phagocytosed) fractions of neutrophils between mouse strains. As shown in Figure S3A, the percentage of surviving S. aureus was similar in both fractions across all three mouse strains. Thus, no significant strain-specific differences in the function or capacity of neutrophils to kill S. aureus were noted. We also found no significant strain-specific differences in bactericidal capacity when we repeated this experiment using peritoneal macrophages from A/J and C57BL/6J mice (Figure S3B). Based on these findings, we considered whether the observed susceptibility of A/J mice to S. aureus might be due in part to perturbations in the initial host inflammatory response.
Assessing biological relevance: Cytokine response in A/J and C57BL/6J
To further evaluate the potential importance of our 10 candidate genes in host immune responses to S. aureus, we treated mouse macrophage RAW264.7 cells with short interfering RNA (siRNA) oligonucleotides. Knockdown expression of each of the 10 target genes in cells was confirmed by real-time-PCR (Figure S4). The cells were then stimulated with S. aureus, and a Luminex-based multiplex cytokine assay was used to evaluate the impact of each genes' individual knockdown expression on the host inflammatory response to S. aureus. Cell-culture supernatants from pre - and post - S. aureus stimulation conditions were compared (Figure S5). Only TNFAIP8 and SEH1L knockdown cells significantly influenced cytokine levels in S. aureus-challenged siRNA-transfected RAW264.7 cells (Figure 5A). Compared to S. aureus-challenged negative controls, S. aureus-challenged cells with knockdown expression of Tnfaip8 produced a significantly lower level of IL-1β, while those with knockdown expression of Seh1l produced a significantly higher level of GM-CSF.

To evaluate whether our cytokine results from the RAW264.7 knockdown experiments mimicked what was encountered in vivo, we first demonstrated by real time PCR that expression levels of Tnfaip8 and Seh1l are significantly lower in whole blood from A/J mice than from C57BL/6J mice (Figure 4B). Next, we confirmed that the same reduced expression pattern was also encountered in peritoneal macrophages (PMΦ) from A/J mice (54±13% [Tnfaip8] and 70±10% [Seh1l] of C57BL/6J PMΦ mRNA expression, respectively) and CSS18 mice (42±3% [Tnfaip8] and 78±5% [Seh1l] of C57BL/6J PMΦ mRNA expression, respectively). We then compared cytokine production of PMΦ from C57BL/6J and CSS18 after stimulation with S. aureus (Figure S6). We used PMΦ from CSS18 (which contain Chr. 18 from A/J but are otherwise C57BL/6J) in order to eliminate the potential confounding effect on the results of additional unidentified susceptibility genes on Chr. 8 and 11 of A/J mice. As shown in Figure 5B, we encountered the same pattern of IL-1β and GM-CSF in S. aureus-stimulated PMΦs from CSS18 mice as we did in the TNFAIP8 and SEH1L knockdown in RAW264.7 cells. Specifically, IL-1β was significantly decreased and GM-CSF was significantly increased in PMΦ of CSS18 compared to that of C57BL/6J (P = 0.009 for IL-1β, P = 0.04 for GM-CSF; unpaired two tailed t-test).
Presence of SNPs in the QTL regions
All but one (Dtwd2) of the 10 differentially expressed genes in the two QTLs on chr. 18 contained SNPs in the region flanking the first and last exon. Such SNPS could potentially lie within transcriptional regulatory sites. A total of 22 genes within the two QTLs on chromosome 18 contained non-synonymous coding SNPs. These genes included Dcp2, Gm5505, Ccdc112, Pggt1b, Commd10, Gm5095, Gm3790, Ftmt, Zfp474, Fbn2, Slc27a6, Gm3957, Synpo, Tcof1, Csf1r, Ppargc1b, Il17b, Afap1l1, Sh3tc2, Wdr7, Alpk2, Malt1, and 5330437l02Rik.
Discussion
Host genetic determinants of susceptibility to S. aureus are poorly understood. In the current study, we localized the genetic determinants of S. aureus susceptibility in A/J inbred mice to chromosomes 8, 11, and 18. From the ∼4200 genes on these three chromosomes, we identified 191 which are differentially expressed between the susceptible A/J and the resistant C57BL/6J inbred mouse strains. We also pursued QTL analysis for chr. 18 to corroborate the findings of our array-based approach with a second strategy. We found two QTLs on chr. 18 over the significance threshold. This suggests that there are at least two genes, one (or more) of which are associated with each peak and which are involved in susceptibility to S. aureus infection in A/J mice. In the current report, we have identified two genes, one on each of the two QTL peaks, which are strongly associated with susceptibility to S. aureus infection.
We hypothesized that our candidate genes influenced susceptibility to S. aureus in A/J mice by regulating macrophage cytokine production. There are several lines of evidence in support of this hypothesis. Macrophages play an important role in initiating host defense to S. aureus by secreting cytokines and chemokines [24]–[26], and impairment of this cytokine production by macrophages results in impaired host inflammatory responses [27], [28]. In support of our hypothesis, we found that macrophages from the two mouse lineages did differ significantly in their cytokine response to S. aureus. In addition, we found no difference in the bactericidal capacity of either macrophages or neutrophils from A/J and C57BL/6J mice, suggesting that functional deficits within these cells are not the primary cause of the observed differences in susceptibility between the two murine strains. For these reasons, we pursued siRNA-mediated knockdown of our candidate genes in mouse macrophage cells and analyzed the cytokine profiles of knockdown cells exposed to S. aureus. As evidenced by real-time PCR, six genes were significantly differentially expressed. More notably, knockdown expression of two of the six genes, Tnfaip8 and Seh1l, significantly altered the specific patterns of cytokine production in S. aureus - stimulated RAW cells in a manner that was also observed in PMФ of CSS18, but not C57BL/6J mice. These two genes are prime candidates for influencing susceptibility to S. aureus.
Of the 191 genes on chromosomes 8, 11, or 18 that are also differentially expressed between A/J and C57BL/6J mice, 28 have previously been shown to play key roles in the response to various infectious diseases. For example, Cd209d has been known to be able to capture Gram-negative bacteria such as Escherichia coli and Salmonella typhimurium as well as Gram-positive bacteria such as Streptococcus pneumoniae [29], [30]. Tbkbp1 may play a role in antiviral innate immunity as part of the TNF/NFkB interaction pathway [31]. Map2k3 is induced by various stimuli, including cellular stress, inflammatory cytokines and cell surface receptors, and mediates the activation of p38 mitogen-activated protein kinase (p38MAPK) [32]. Grb2 [33], Prdx2 [34], and Ppp2cb [35] are also involved in the MAPKKK pathway. Activation of the MAPKKK cascade confers resistance to both Gram - negative and Gram-positive pathogens, indicating that signaling pathways initiated by various pathogens converge into a MAPKKK cascade[36], [37]. Interestingly, almost one-third of these known 28 genes were associated with apoptosis (Birc5, Cln8, Cyfip2, Psme3, Rffl, Rtn4, Trp53, Tnfaip8, and Xaf1). Although the precise reasons for the high frequency of apoptosis-related genes are not known, it suggests that this pathway may represent a particularly important area for further study. Finally, our strategy also identified a number of novel genes about which little is known. For example, 20 of the 191 differentially expressed genes on chromosomes 8, 11, and 18 were identified entirely by RIKEN cDNA information, and had no known biological function (Table S1).
The TNFAIP8 family of proteins consists of TNFAIP8, TIPE1 (TNFAIP8Like1), TIPE2 and TIPE3. TIPE2 is known as a negative regulator of innate and adaptive immune processes[38]. Woodward et al. showed that TNFAIP8 regulates glucocorticoid-mediated apoptosis of thymocytes [39]. Interestingly, RAW264.7 cells in which TNFAIP8 had been knocked down produced significantly more GM-CSF and significantly less IL-1β compared to control cells. GM-CSF is a critical Th1 cell-derived cytokine that mediates pulmonary inflammation in vivo, and controls neutrophil and macrophage numbers [40], [41]. Interestingly, overexpression of GM-CSF leads to severe inflammation [41]. IL-1β is a key inflammatory cytokine in orchestrating host defense against S. aureus, with mice deficient in this cytokine exhibiting markedly increased susceptibility to S. aureus [24], [42]. Taken together, Tnfaip8 is a strong candidate gene governing susceptibility to S. aureus through IL-1β and GM-CSF regulation in mice. Seh1l, the other candidate gene, is a part of nuclear pore complex Nup107–160 that was recently found to play a role in embryonic development. Although the infectious diseases-related function of this gene is unknown, its knockdown was also shown to increase GM-CSF production in S. aureus-stimulated macrophages. Thus, it may be that SEH1L contributes to susceptibility of A/J to S. aureus by increasing, along with TNFAIP8, macrophage production of GM-CSF.
The biological relevance of Tnfaip8 and Seh1l is strengthened by several key observations. First, Tnfaip8 and Seh1l exhibited decreased mRNA expression in both peripheral blood and PMΦ of A/J and CSS18 mice as compared with C57BL/6J mice. Second, of the 10 differentially expressed genes on the significant QTL regions of chromosome 18, only Tnfaip8 and Seh1l induced significant changes in cytokine production from siRNA-inhibited, S. aureus-challenged macrophages. Third, the cytokines altered by this siRNA-mediated knockdown of TNFAIP8 and SEH1L are critically important components of host response to S. aureus[24], [42]. Finally, the cytokine patterns elicited by TNFAIP8 and SEH1L knockdowns were also seen in the peritoneal macrophages of CSS18 mice, but not C57BL/6J mice. Taken together, these data support the importance of our two candidate genes, Tnfaip8 and Seh1l, to the susceptibility to S. aureus, both in vivo and in vitro.
Although regulatory variation can play an important role in various complex traits [43]–[46], its role in the current study is unresolved. Nine of the 10 differentially expressed candidate genes on our two QTLs contained SNPs in the region flanking the first and last exon. In addition, 22 other genes on our two QTLs contained non-synonymous coding SNPs. Such SNPs could also potentially contribute to strain-specific phenotypes via modification of protein function. Only 10 of these genes (Ftmt, Fbn2, Synpo, Tcof1, Csf1r, Ppargc1b, Il17b, Sh3tc2, Wdr7, and Malt1) were identified in the map of disease genes described in Online Mendelian Inheritance in Man (OMIM). Of these, three (Csf1r, Il17b, and Malt1) are known to be involved in immune response. For example, Csf1r encodes a tyrosine kinase growth factor receptor for colony-stimulating factor-1, a cytokine which controls the production, differentiation, and function of macrophages[47]. IL-17b plays an important role in the pathogenesis of inflammatory arthritis [48] in the animal model. MALT1 is essential for NF-kappa-B activation, cytokine production, and proliferation of primary T and B lymphocytes [49]. Future studies will investigate the role of these genes, if any, on the susceptibility of A/J mice to S. aureus infection.
The different susceptibilities of A/J and C57BL/6J mice to S. aureus infection were recently confirmed by other investigators [3]. von Kockritz-Blickwede et al. suggested that resistance to S. aureus in C57BL/6 mice is critically dependent on an effective and fast recruitment of neutrophils to the site of infection due to different kinetic profiles in expression of KC (Cxcl1) and MIP-2 (Cxcl2). These two chemokines play a central role in mediating neutrophil recruitment to sites of infection by binding to receptors on the surface of these cells. In support of this hypothesis, we found that Cxcl1 was significantly differentially expressed between A/J and C57BL/6J mice at 2, 6, and 12 hours after infection with S. aureus. Although the gene coding for Cxcl1 is located on mouse chromosome 5, CSS5 mice infected with S. aureus in the current study retained the resistant phenotype. This finding suggests that genes on mouse chromosome 5 are not directly responsible for A/J's susceptibility, but that the reduced chemokine production observed in S. aureus - infected A/J mice [3] may be regulated by one or more genes on chromosomes 8, 11, or 18.
Although C5 deficiency has been shown to contribute to susceptibility to a variety pathogens, including Pseudomonas aeruginosa, Mycobacterium tuberculosis, Listeria monocytogenes, and Candida albicans [22], [50]–[53], its contribution to susceptibility to S. aureus is unresolved. For example, Crequetti et al. (1983) showed that C5-deficient mice had impaired lung clearance of S. aureus [53], while Toews and Pierce (1984) demonstrated similar lung clearance of S. aureus and PMN recruitment in both C5-deficient and sufficient mice [54]. In addition, Easmon and Glynn reported decreased survival of C5-deficient mice compared with C5-sufficient controls after intraperitoneal injection with mucoid S. aureus strains [55], while Cunnion et al. observed similar survival in both C5-deficient and sufficient mice challenged via intravenous injection with S. aureus [56]. Finally, von Kockritz-Blickwede and colleagues recently demonstrated a protective role of complement C5a in an experimental model of S. aureus bacteremia [23]. In the present study, all three of the additional C5-deficient mouse strains we tested were resistant to S. aureus. Our consomic experiments also suggest that C5-deficiency is not the primary determinant of susceptibility to S. aureus in A/J mice. Although the gene for C5 (Hc) resides on chromosome 2, CSS2 mice (which contain A/J chromosome 2 and thus would be C5 deficient) remained resistant to S. aureus infection. Based on this body of evidence, we conclude that factors other than C5-deficiency are likely to be primarily responsible for the susceptibility to S. aureus seen in A/J mice.
The current study has limitations. First, linkage scores of the two QTLs on chr. 18 imply that there may be more than one gene on the larger significant peak that plays a role in susceptibility to S. aureus. One possible additional susceptibility gene is Lman1. In the Figure S5, knockdown of LMAN1 showed dramatically reduced IL-1α compared to control when stimulated with S. aureus. Although it was not statistically significant (P = 0.08), the quantity of IL-1α in LMAN1 knockdown cells was below the detection value of standard minimum. Thus, it is possible that testing of a much larger number of samples would reveal a statistical significance of additional candidate genes such as Lman1 in the larger peak. Second, there may be genes on other chromosomes that contribute to a lesser extent, or that have a strong effect only when acting in conjunction with genes on another chromosome. Third, our strategy will miss genes on chromosomes 8, 11, or 18 that contribute to phenotype at the protein level (e.g., coding SNPs) or by modulating the gene expression patterns of genes on other chromosomes. Fourth, our strategy would not allow detection of polymorphic regulatory sites more than 2kb away from genes, such as those within gene deserts. Finally, we have only completed QTL linkage analysis for chr. 18. Thus, additional work is required, including QTL mapping analysis for A/J chromosomes 8 and 11, defining the pathogenesis of Tnfaip8 and Seh1l using knockout mice, and evaluating the impact of Tnfaip8 and Seh1l on host susceptibility to different pathogen classes. Ultimately, the importance of the identified candidate genes, or their human homologs, will need to be evaluated in patients with S. aureus infections.
Despite these limitations, this study makes several key observations. First, using expression array-based strategies, we have identified 191 genes that are differentially expressed between susceptible and resistant mice, and are found on three different chromosomes where loci conferring susceptibility to S. aureus infection have been mapped. Second, we have identified two QTLs on chr. 18 that are linked to survival time after infection with S. aureus. Ten differentially expressed genes mapped to the significant - or suggestive threshold of these two QTLs. Of these 10 genes, two (Tnfaip8 and Seh1l) significantly affect the production of IL-1β and GM-CSF in S. aureus-stimulated mouse macrophages in a pattern also seen in peritoneal macrophages from mice containing A/J chromosome 18, thereby supporting a potential role for these two genes in host response to S. aureus. These genes in general, and Tnfaip8 and Seh1l in particular, represent promising candidates for the genetic basis of host susceptibility to this serious, common pathogen.
Materials and Methods
Ethics statement
All animal experiments were performed in accordance to NIH guidelines, the Animal Welfare Act, and US federal law. Such experiments were carried out following approval by the Duke University's Institutional Animal Care & Use Committee (IACUC) which has been accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) International. All animals were housed in a centralized and AAALAC-accredited research animal facility that is fully staffed with trained husbandry, technical, and veterinary personnel.
Mouse strains
C57BL/6J and A/J mice were purchased from The Jackson Laboratory (Bar Harbor, ME). Chromosome substitution strains (CSS) were generated by transferring a single, full-length chromosome from the donor strain (A/J) onto the genetic background of the host strain (C57BL/6J) by repeated backcrossing [15]. Male CSS mice (CSS1–19, CSSX and CSSY) were also obtained from the Jackson Laboratory (Bar Harbor, ME).
Preparation of bacterial cells
S. aureus clinical strains, Sanger 476 or MW2 were used in the mortality studies. For preparation of S. aureus for injection, an overnight bacterial culture of S. aureus was diluted with fresh tryptic soy broth (TSB) and incubated (37°C) with aeration to log-phase growth (optical density at wavelength 600 nm (O.D.600) of ∼1.0) [57]. S. aureus was harvested by centrifugation, rinsed, and resuspended in saline. To mimic the natural course of S. aureus infection in humans, which typically arises from a primary focus of infection and disseminates to other sites, we employed an intraperitoneal (i.p.) route of infection in our animal model [58].
Murine phenotype definitions, injection (S. aureus), and specimen procurement
Mouse phenotypes were defined as either resistant (survival for >120 hours following S. aureus infection) or susceptible (survival for <48 hours following S. aureus infection). Mice were injected via an intraperitoneal (i.p.) route with 1×107 CFU/g S. aureus depending on the experiment and observed every 8 h for morbidity. Mice were sacrificed using CO2 asphyxiation if they appeared moribund. Pain/distress was assessed using a numerical scale for the following characteristics: Appearance (0 = normal; 1 = lack of grooming; 2 = rough hair coat; 3 = abnormal posture); Behavior (0 = normal; 1 = moving slowly; 2 = moving slowly, irregular ambulation; 3 = immobile). A score of 3 indicated significant pain and distress and culminated in the early euthanasia of the animal. Peritoneal lavage was performed as described previously [2]. Kidneys were harvested and either frozen (−80°C) or fixed in 10% buffered formalin, as appropriate, for subsequent analysis. For experiments involving RNA analysis, blood was collected by intracardiac puncture and stored in RNAlater at −20°C.
Quantifying bacterial load in murine tissues and blood
Kidneys collected from euthanized animals were homogenized in phosphate buffered saline (PBS) and diluted 10 fold serially. Peritoneal lavage (50 µl) was serially diluted in PBS. The serial dilutions (50 µl) were plated in Tryptic Soy Agar (TSA) plates and incubated (37°C, overnight) to count the number of colony forming units (CFU) of S. aureus.
Generation of F1 and N2 progeny
For the current study, susceptible CSS mice with chromosome 8, 11 and 18 from A/J mice on a C57BL/6J background (CSS8, CSS11 and CSS18) were mated with resistant C57BL/6J strain in reciprocal crosses [C57BL/6J male×CSS8, 11 or 18 female) and (CSS8, 11 or 18 male×C57BL/6J female)] to generate an F1 population that was heterozygous for the chromosome of interest on an otherwise uniform C57BL/6J genomic background. To generate N2 backcross mice for QTL linkage analysis on chromosome 18, we bred F1 mice (C18A) to C57BL/6J (Figure S1).
Microarray
Total RNA was prepared from mouse blood using the Mouse RiboPure Blood RNA isolation kit (Ambion) following the manufacturer's instruction. Globin mRNA was removed from whole blood RNA samples using the Globinclear kit (Ambion). A total of 30 samples passed the quality criteria of the Agilent Bioanalyzer and were used for microarray analysis. Since the total RNA yield of many samples was low, 1 round of linear amplification was performed for all samples (Ambion MessageAmp Primier). Mouse Genome 430 2.0 Array-Chips were used (Affymetrix). The biotin-labeled cDNA was hybridized to the arrays for 16 hours at 45°C following the manufacturer's instruction. The arrays were then washed and labeled with streptavidinphycoerythrin (strep-PE), and the signal was amplified using biotinylated antistreptavidin followed by another round of staining with strep-PE. Washing and staining were performed on the Affymetrix fluidics station according to the recommended fluidics protocol. Labeled gene chips were scanned using an Affymetrix Genechip Scanner 7G (Affymetrix). This array contains 45,101 probe sets to analyze the expression level of over 39,000 transcripts and variants from over 34,000 well characterized mouse genes. The microarray data have been deposited in the NCBI GEO and are accessible through GEO series accession no. GSE19668.
Analysis of gene expression microarray data
To identify genes for which differential expression between A/J and C57BL/6J mice could contribute to host susceptibility to S. aureus infection, we compared the gene expression profiles between uninfected A/J and C57BL/6J mice and between infected A/J and C57BL/6J mice at 2, 4, 6, and 12 hours after infection. We used a multi-step approach that included i) preprocessing to determine probeset expression values, ii) filtering based on probeset annotations, iii) identification of genes with statistically significant differential expression between A/J and C57BL/6J mice after infection, and iv) ranking based on fold change (FC) in gene expression and Gene Ontology (GO) (http://www.geneontology.org) annotation.
Preprocessing was conducted using the Robust Multichip Analysis (RMA) [59] implementation in the Bioconductor “affy” package (http://www.bioconductor.org/), with an additional step to account for differences in probe hybridization resulting from single nucleotide polymorphisms (SNP) between A/J and C57BL/6J mice. The additional step is referred to as SNP masking [60] and is applied after background correction and quantile-quantile normalization but prior to the determination of probeset expression values. Genomic locations hybridized by each probe were obtained from the Ensembl database (http://www.ensembl.org/index.html), and these genomic locations were compared to the locations of SNPs for which A/J and C57BL/6J mice have different alleles. Probes that hybridize to such SNPs within their target transcripts were excluded from the determination of probeset expression values. There were 7714 probesets for which some of the probes were excluded. In addition, there were four probesets excluded from further analysis because all probes in the set hybridize to SNPs: 1437478_s_at, 1416030_a_at, 1452066_a_at, and 1449635_at. SNP data were obtained from the Perlegen SNP database (http://mouse.perlegen.com/mouse/index.html).
We next filtered the probesets based on probeset annotations. We eliminated from further analysis 6041 probesets with no gene symbol annotation, 2441 probesets with “_x_at” in the probeset name, and 2091 probesets with “_s_at” in the probset name (including 1437478_s_at mentioned above). Probesets with the “_x_at” designation are those that are known to cross-hybridize in an unpredictable manner. Probesets with the “_s_at” designation are those for which multiple different probesets have probes in common.
The remaining 34,706 probesets were analyzed using Analysis of Variance (ANOVA) to determine whether the difference in mean expression levels between A/J and C57BL/6J mice are statistically significant. The following generalized linear model was used:where B1 corresponds to IVT batch effects, B2 to hybridization batch effects, T to time main effects, and S*T to strain-time interaction effects, where the time factor levels correspond to the uninfected state and 2, 4, 6, and 12 hours post-infection). A list of candidate genes for further analysis was compiled by identifying those genes on chromosomes 8, 11, or 18 with p<0.05 for the S*T model coefficient for all post-infection times (i.e. 2, 4, 6, and 12 hours post-infection). There are 191 such genes (Table S1). We have not used a generalized family-wise error rate (gFWER), as stringent P-value thresholds have been shown to reduce the reproducibility of lists of differentially expressed genes [61]. We have chosen, therefore, to use non-adjusted p values and to further refine the candidate gene list based on biological rather than statistical criteria, namely differential expression between strains for multiple post-infection timepoints, as well as GO annotations and FC as discussed below. The estimated false discovery rate ranges from 0.09 (6 hours post-infection) to 0.3 (12 hours post-infection) [62].
SNPs allele differences between A/J and C57BL/6J
Single nucleotide polymorphism (SNP) data were obtained from the Mouse Genome Informatics database (MGI) (http://www.informatics.jax.org/) for the A/J and C57BL/6J mouse strains [63] for all genes on the candidate gene list. We identified those SNP loci within the gene boundaries or within 2 kilobases of the gene boundaries and which have allele differences between the two strains. We next identified the functional class of each SNP as assigned in the MGI database. MGI assigns functional classes according to those defined by dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/). The functional classes are: coding-nonsynonymous, coding-synonymous, intron, locus-region, splice-site, mRNA-UTR, contig-reference, coding, and coding-exception. SNPs in untranscribed regions but within 2 kilobases of the 5′ boundary of a gene or within 0.5 kilobases of the 3′ boundary of a gene are assigned the locus-region functional class. SNPs that lie within promoter or enhancer sites are therefore most likely assigned the locus-region functional class.
QTL linkage analysis
Polymorphic microsatellite markers for the cross between C57BL/6J and A/J were chosen from a database maintained by Mouse Genomic Informatics (http://www.informatics.jax.org/). To cover chromosome 18, nine microsatellite marker were selected with an average inter-marker distance of 5.3 cM. A total of 144 N2 backcross were generated, all of which were genotyped for each microsatellite marker by PCR amplification and gel electrophoresis. PCR genotyping reactions were used Platinum Taq polymerase (Invitrogen Co.). J/qtl software was used to analyze phenotype and genotype data for association between survival time after infection with S. aureus and marker location. Phenotypes were defined as either sensitive or resistant based on the dichotomization of survival data (Survival of less than 1day is “0” and survival of longer than 1.5 days is “1”, respectively). Because our analyses focused solely upon one chromosome, our significance threshold reflected a chromosome – level analysis [64]–[66], rather than a genome-wide approach [67]. All linkage analysis results were expressed as LOD scores. LOD score was considered “suggestive” if > = 0.41 (P = 0.63) and “Significant” if > = 1.64 (P = 0.05). Threshold values for linkage were determined by a 1,000 permutation test by using J/qtl.
Isolation of bone marrow-derived neutrophils
Bone marrow-derived neutrophils were isolated as described previously [2]. Briefly, bones from the femurs and tibias of 8-week-old male A/J and C57BL/6J mice were removed of all muscles. The bone marrow cells were flushed out with Hank's buffer (without calcium and magnesium) and filtered through a 70-µm nylon cell strainer to remove cell clumps. Neutrophils were separated from the remaining cells by centrifugation over discontinuous Percoll (Amersham Biosciences) gradients at 500×g for 30 min at 4°C, consisting of 52% (vol/vol), 69% (vol/vol), and 78% (vol/vol) Percoll in PBS. Neutrophils from the interface of the 65 and 75% fractions were washed with Hank's buffer (without calcium and magnesium) and cultured (37°C, 10% CO2) in RPMI 1640 supplemented with 10% fetal calf serum (both from the American Type Culture Collection).
Neutrophil and macrophage bactericidal assay
Killing of S. aureus was assayed as described [68] with modification. S. aureus cells were opsonized with 10% mouse serum at 37°C for 30 min and washed twice in PBS. In brief, 1 ml of the RPMI 1640 supplemented with 10% fetal calf serum containing neutrophils (2×106 per ml) and S. aureus (2×106/ ml) was placed in the sterile 1.5 ml tubes. The tubes were incubated at 37°C with end-over-end rotation (6 rpm). After 90 min of incubation, the neutrophils were centrifuged at 100×g for 5 min at 4°C and washed twice with 1 ml of cold PBS. Supernatants were used for evaluation of the count of extracellular bacteria. The neutrophil pellet was lysed in sterile water (pH 11) to release intracellular bacteria. The each resulting suspension was serially diluted in PBS and bacterial numbers were determined after plating onto TSB agar. Peritoneal macrophage bactericidal assay was performed in the same manner as neutrophil bactericidal assay except the final step for bacterial counting [69], [70]. After 90 min of incubation, the each sample was diluted in sterile water for 5 min to lyse the macrophage, and serially diluted in PBS. The dilutions were plated on TSB agar, and the plates were incubated overnight at 37oC. The amount of killing was determined by plate count of surviving bacteria (extracellular and intracellular bacteria).
Small interfering RNA (siRNA) experiments
To test the role of each candidate gene on cytokine production by host defense cells, we transfected siRNAs into the mouse macrophage cell line RAW264.7. All siRNAs were purchased from Ambion. Cells were transfected by using siPORT Amine Transfection Agent in 48-well plate (Ambion), according to the manufacturer's instructions. Transfections were carried out using 30 nM siRNA and 400,000 cells per well. Cells were maintained in DMEM supplemented with 10% FBS. Forty-eight hours after transfection, S. aureus Bioparticles (Invitrogen) was added to a final concentration of 10 µg/ml. After 5 h, supernatants (medium) and cells were collected, stored at −70°C for multiplex cytokine assay (described below) and real-time PCR analyses, respectively. RNA was isolated for detecting the level of mRNA expression. The target gene mRNA expression was analyzed by real-time PCR (TaqMan) using 1 µg total RNA from untreated cells or from cells treated with oligonucleotides. The reaction was performed using the High-Capacity cDNA Reverse Transcription kit (Applied Biosystems) and was followed by Taqman real-time PCR. A full list of gene names, siRNA ID numbers, sequences, and Taqman assay ID numbers are shown in Table S2.
Peritoneal macrophage (PMΦ) isolation and stimulation
Resident PMΦs were obtained by lavage of the peritoneal cavity of uninfected mice (CSS18 and C57BL/6J) as described elsewhere [69], [71]. Cells were washed once with PBS by centrifugation (300×g, 5 min at 4°C) and resuspended at a concentration of 1×106 cells/ml of RPMI1640 containing 10% FBS. Cells were incubated in a 24-well plate for 2 h and the remaining nonadherent cells were removed by washing with media. The macrophage monolayers were then stimulated to produce cytokines by incubation with 10 µg of S. aureus Bioparticles for 48 h at 37°C. Incubation time was determined based on the previous similar studies [72], [73]. Culture supernatants for Luminex assay were collected pre - and post - S. aureus stimulation.
Measurement of cytokine/chemokine production
Cytokine production was assayed from the collected supernatant of the S. aureus-challenged siRNA transfected macrophages and the S. aureus-challenged peritoneal macrophages from C57BL/6J and CSS18 using a multiplex cytokine assay kit and Luminex technology (Bio-Rad) available at Duke Human Vaccine Institute. Twenty cytokines were tested: FGF basic, TNFα, IL-1α, IL-1β, IL-2, IL-4, IL-5, IL-6, IL-10, IL-12p40/p70, IL-13, IL-17, IFNγ, IP-10, KC, MIG, MIP-1α, MCP-1, VEGF, and GM-CSF. Of the 20 tested cytokines, 9 (FGF basic, IFNγ, IL-2, IL-4, IL-5, IL-13, IL-17, MIG, and IP-10) were excluded from our analysis due to weak signal in both pre - and post - S. aureus stimulated supernatant (5 cytokines); lack of response to S. aureus (2 cytokines), and cross-contamination (2 cytokines) (Figure S5). The concentrations for cytokines and chemokines were compared between two groups (negative control vs target gene) with a two-tailed t-test for unpaired samples. Differences were considered to be statistically significant when the P-value was smaller than 0.05.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. FournierB
PhilpottDJ
2005 Recognition of Staphylococcus aureus by the innate immune system. Clin Microbiol Rev 18 521 540
2. DeshmukhHS
HamburgerJB
AhnSH
McCaffertyDG
YangSR
2009 Critical role of NOD2 in regulating the immune response to Staphylococcus aureus. Infect Immun 77 1376 1382
3. von Kockritz-BlickwedeM
RohdeM
OehmckeS
MillerLS
CheungAL
2008 Immunological mechanisms underlying the genetic predisposition to severe Staphylococcus aureus infection in the mouse model. Am J Pathol 173 1657 1668
4. HillPC
WongCG
VossLM
TaylorSL
PottumarthyS
2001 Prospective study of 125 cases of Staphylococcus aureus bacteremia in children in New Zealand. Pediatr Infect Dis J 20 868 873
5. BullockAE
SlobeBS
VazquezV
CollinsAC
1997 Inbred mouse strains differ in the regulation of startle and prepulse inhibition of the startle response. Behav Neurosci 111 1353 1360
6. LogueSF
PaylorR
WehnerJM
1997 Hippocampal lesions cause learning deficits in inbred mice in the Morris water maze and conditioned-fear task. Behav Neurosci 111 104 113
7. PaylorR
CrawleyJN
1997 Inbred strain differences in prepulse inhibition of the mouse startle response. Psychopharmacology (Berl) 132 169 180
8. RalphRJ
PaulusMP
GeyerMA
2001 Strain-specific effects of amphetamine on prepulse inhibition and patterns of locomotor behavior in mice. J Pharmacol Exp Ther 298 148 155
9. VartyGB
WaltersN
Cohen-WilliamsM
CareyGJ
2001 Comparison of apomorphine, amphetamine and dizocilpine disruptions of prepulse inhibition in inbred and outbred mice strains. Eur J Pharmacol 424 27 36
10. WillottJF
TannerL
O'SteenJ
JohnsonKR
BogueMA
2003 Acoustic startle and prepulse inhibition in 40 inbred strains of mice. Behav Neurosci 117 716 727
11. ZaasAK
LiaoG
ChienJW
WeinbergC
ShoreD
2008 Plasminogen alleles influence susceptibility to invasive aspergillosis. PLoS Genet 4 e1000101
12. MarquisJF
GrosP
2008 Genetic analysis of resistance to infections in mice: A/J meets C57BL/6J. Curr Top Microbiol Immunol 321 27 57
13. NadeauJH
SingerJB
MatinA
LanderES
2000 Analysing complex genetic traits with chromosome substitution strains. Nat Genet 24 221 225
14. SingerJB
HillAE
NadeauJH
LanderES
2005 Mapping quantitative trait loci for anxiety in chromosome substitution strains of mice. Genetics 169 855 862
15. SingerJB
HillAE
BurrageLC
OlszensKR
SongJ
2004 Genetic dissection of complex traits with chromosome substitution strains of mice. Science 304 445 448
16. MatinA
CollinGB
AsadaY
VarnumD
NadeauJH
1999 Susceptibility to testicular germ-cell tumours in a 129.MOLF-Chr 19 chromosome substitution strain. Nat Genet 23 237 240
17. YoungrenKK
NadeauJH
MatinA
2003 Testicular cancer susceptibility in the 129.MOLF-Chr19 mouse strain: additive effects, gene interactions and epigenetic modifications. Hum Mol Genet 12 389 398
18. KrewsonTD
SupelakPJ
HillAE
SingerJB
LanderES
2004 Chromosomes 6 and 13 harbor genes that regulate pubertal timing in mouse chromosome substitution strains. Endocrinology 145 4447 4451
19. AckermanKG
HuangH
GrasemannH
PumaC
SingerJB
2005 Interacting genetic loci cause airway hyperresponsiveness. Physiol Genomics 21 105 111
20. WinawerMR
KupermanR
NiethammerM
ShermanS
RabinowitzD
2007 Use of chromosome substitution strains to identify seizure susceptibility loci in mice. Mamm Genome 18 23 31
21. GerardC
GerardNP
1994 C5A anaphylatoxin and its seven transmembrane-segment receptor. Annu Rev Immunol 12 775 808
22. MullickA
LeonZ
Min-OoG
BerghoutJ
LoR
2006 Cardiac failure in C5-deficient A/J mice after Candida albicans infection. Infect Immun 74 4439 4451
23. von Kockritz-BlickwedeM
KonradS
FosterS
GessnerJE
MedinaE
2009 Protective role of complement C5a in an experimental model of Staphylococcus aureus bacteremia. J Innate Immun 2 87 92
24. ShimadaT
ParkBG
WolfAJ
BrikosC
GoodridgeHS
2010 Staphylococcus aureus evades lysozyme-based peptidoglycan digestion that links phagocytosis, inflammasome activation, and IL-1beta secretion. Cell Host Microbe 7 38 49
25. LowyFD
1998 Staphylococcus aureus infections. N Engl J Med 339 520 532
26. Bubeck WardenburgJ
WilliamsWA
MissiakasD
2006 Host defenses against Staphylococcus aureus infection require recognition of bacterial lipoproteins. Proc Natl Acad Sci U S A 103 13831 13836
27. SmithAM
RahmanFZ
HayeeB
GrahamSJ
MarksDJ
2009 Disordered macrophage cytokine secretion underlies impaired acute inflammation and bacterial clearance in Crohn's disease. J Exp Med 206 1883 1897
28. CailhierJF
SawatzkyDA
KipariT
HoulbergK
WalbaumD
2006 Resident pleural macrophages are key orchestrators of neutrophil recruitment in pleural inflammation. Am J Respir Crit Care Med 173 540 547
29. TakaharaK
YashimaY
OmatsuY
YoshidaH
KimuraY
2004 Functional comparison of the mouse DC-SIGN, SIGNR1, SIGNR3 and Langerin, C-type lectins. Int Immunol 16 819 829
30. KangYS
KimJY
BrueningSA
PackM
CharalambousA
2004 The C-type lectin SIGN-R1 mediates uptake of the capsular polysaccharide of Streptococcus pneumoniae in the marginal zone of mouse spleen. Proc Natl Acad Sci U S A 101 215 220
31. BouwmeesterT
BauchA
RuffnerH
AngrandPO
BergaminiG
2004 A physical and functional map of the human TNF-alpha/NF-kappa B signal transduction pathway. Nat Cell Biol 6 97 105
32. RaingeaudJ
WhitmarshAJ
BarrettT
DerijardB
DavisRJ
1996 MKK3 - and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol Cell Biol 16 1247 1255
33. ZhangS
WeinheimerC
CourtoisM
KovacsA
ZhangCE
2003 The role of the Grb2-p38 MAPK signaling pathway in cardiac hypertrophy and fibrosis. J Clin Invest 111 833 841
34. KamataH
HondaS
MaedaS
ChangL
HirataH
2005 Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 120 649 661
35. Arachchige DonAS
DallapiazzaRF
BenninDA
BrakeT
CowanCE
2006 Cyclin G2 is a centrosome-associated nucleocytoplasmic shuttling protein that influences microtubule stability and induces a p53-dependent cell cycle arrest. Exp Cell Res 312 4181 4204
36. AsaiT
TenaG
PlotnikovaJ
WillmannMR
ChiuWL
2002 MAP kinase signalling cascade in Arabidopsis innate immunity. Nature 415 977 983
37. LuHT
YangDD
WyskM
GattiE
MellmanI
1999 Defective IL-12 production in mitogen-activated protein (MAP) kinase kinase 3 (Mkk3)-deficient mice. Embo J 18 1845 1857
38. SunH
GongS
CarmodyRJ
HilliardA
LiL
2008 TIPE2, a negative regulator of innate and adaptive immunity that maintains immune homeostasis. Cell 133 415 426
39. WoodwardMJ
de BoerJ
HeidornS
HubankM
KioussisD
2010 Tnfaip8 is an essential gene for the regulation of glucocorticoid-mediated apoptosis of thymocytes. Cell Death Differ 17 316 323
40. HamiltonJA
StanleyER
BurgessAW
ShadduckRK
1980 Stimulation of macrophage plasminogen activator activity by colony-stimulating factors. J Cell Physiol 103 435 445
41. ShiY
LiuCH
RobertsAI
DasJ
XuG
2006 Granulocyte-macrophage colony-stimulating factor (GM-CSF) and T-cell responses: what we do and don't know. Cell Res 16 126 133
42. MillerLS
PietrasEM
UricchioLH
HiranoK
RaoS
2007 Inflammasome-mediated production of IL-1beta is required for neutrophil recruitment against Staphylococcus aureus in vivo. J Immunol 179 6933 6942
43. MackayTF
2001 The genetic architecture of quantitative traits. Annu Rev Genet 35 303 339
44. DoebleyJ
1992 Mapping the genes that made maize. Trends Genet 8 302 307
45. StamLF
LaurieCC
1996 Molecular dissection of a major gene effect on a quantitative trait: the level of alcohol dehydrogenase expression in Drosophila melanogaster. Genetics 144 1559 1564
46. LemonWJ
SwintonCH
WangM
BerbariN
WangY
2003 Single nucleotide polymorphism (SNP) analysis of mouse pulmonary adenoma susceptibility loci 1–4 for identification of candidate genes. J Med Genet 40 e36
47. GlennG
van der GeerP
2008 Toll-like receptors stimulate regulated intramembrane proteolysis of the CSF-1 receptor through Erk activation. FEBS Lett 582 911 915
48. YamaguchiY
FujioK
ShodaH
OkamotoA
TsunoNH
2007 IL-17B and IL-17C are associated with TNF-alpha production and contribute to the exacerbation of inflammatory arthritis. J Immunol 179 7128 7136
49. Ruefli-BrasseAA
FrenchDM
DixitVM
2003 Regulation of NF-kappaB-dependent lymphocyte activation and development by paracaspase. Science 302 1581 1584
50. JagannathC
HoffmannH
SepulvedaE
ActorJK
WetselRA
2000 Hypersusceptibility of A/J mice to tuberculosis is in part due to a deficiency of the fifth complement component (C5). Scand J Immunol 52 369 379
51. GervaisF
DesforgesC
SkameneE
1989 The C5-sufficient A/J congenic mouse strain. Inflammatory response and resistance to Listeria monocytogenes. J Immunol 142 2057 2060
52. CerquettiMC
SordelliDO
BellantiJA
HookeAM
1986 Lung defenses against Pseudomonas aeruginosa in C5-deficient mice with different genetic backgrounds. Infect Immun 52 853 857
53. CerquettiMC
SordelliDO
OrtegonRA
BellantiJA
1983 Impaired lung defenses against Staphylococcus aureus in mice with hereditary deficiency of the fifth component of complement. Infect Immun 41 1071 1076
54. ToewsGB
PierceAK
1984 The fifth component of complement is not required for the clearance of Staphylococcus aureus. Am Rev Respir Dis 129 597 601
55. EasmonCS
GlynnAA
1976 Comparison of subcutaneous and intraperitoneal staphylococcal infections in normal and complement-deficient mice. Infect Immun 13 399 406
56. CunnionKM
BenjaminDKJr
HesterCG
FrankMM
2004 Role of complement receptors 1 and 2 (CD35 and CD21), C3, C4, and C5 in survival by mice of Staphylococcus aureus bacteremia. J Lab Clin Med 143 358 365
57. RiceKC
FirekBA
NelsonJB
YangSJ
PattonTG
2003 The Staphylococcus aureus cidAB operon: evaluation of its role in regulation of murein hydrolase activity and penicillin tolerance. J Bacteriol 185 2635 2643
58. ThakkerM
ParkJS
CareyV
LeeJC
1998 Staphylococcus aureus serotype 5 capsular polysaccharide is antiphagocytic and enhances bacterial virulence in a murine bacteremia model. Infect Immun 66 5183 5189
59. IrizarryRA
HobbsB
CollinF
Beazer-BarclayYD
AntonellisKJ
2003 Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4 249 264
60. WalterNA
McWeeneySK
PetersST
BelknapJK
HitzemannR
2007 SNPs matter: impact on detection of differential expression. Nat Methods 4 679 680
61. ShiL
JonesWD
JensenRV
HarrisSC
PerkinsRG
2008 The balance of reproducibility, sensitivity, and specificity of lists of differentially expressed genes in microarray studies. BMC Bioinformatics 9 Suppl 9 S10
62. StoreyJD
TibshiraniR
2003 Statistical significance for genomewide studies. Proc Natl Acad Sci U S A 100 9440 9445
63. FrazerKA
EskinE
KangHM
BogueMA
HindsDA
2007 A sequence-based variation map of 8.27 million SNPs in inbred mouse strains. Nature 448 1050 1053
64. PetryshenTL
KirbyA
HammerRPJr
PurcellS
O'LearySB
2005 Two quantitative trait loci for prepulse inhibition of startle identified on mouse chromosome 16 using chromosome substitution strains. Genetics 171 1895 1904
65. ChurchillGA
DoergeRW
1994 Empirical threshold values for quantitative trait mapping. Genetics 138 963 971
66. ProwsDR
HafertepenAP
WinterbergAV
GibbonsWJJr
WesselkamperSC
2008 Reciprocal congenic lines of mice capture the aliq1 effect on acute lung injury survival time. Am J Respir Cell Mol Biol 38 68 77
67. LanderE
KruglyakL
1995 Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 11 241 247
68. ClarkeTB
DavisKM
LysenkoES
ZhouAY
YuY
2010 Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat Med 16 228 231
69. LeijhPC
van ZwetTL
ter KuileMN
van FurthR
1984 Effect of thioglycolate on phagocytic and microbicidal activities of peritoneal macrophages. Infect Immun 46 448 452
70. WilsonKR
NapperJM
DenvirJ
SollarsVE
YuHD
2007 Defect in early lung defence against Pseudomonas aeruginosa in DBA/2 mice is associated with acute inflammatory lung injury and reduced bactericidal activity in naive macrophages. Microbiology 153 968 979
71. van FurthR
Diesselhoff-den DulkMM
1980 Method to prove investigation of particles by macrophages with light microscopy. Scand J Immunol 12 265 269
72. ZellwegerR
ZhuXH
WichmannMW
AyalaA
DeMasoCM
1996 Prolactin administration following hemorrhagic shock improves macrophage cytokine release capacity and decreases mortality from subsequent sepsis. J Immunol 157 5748 5754
73. WhitakerSM
ColmenaresM
PestanaKG
McMahon-PrattD
2008 Leishmania pifanoi proteoglycolipid complex P8 induces macrophage cytokine production through Toll-like receptor 4. Infect Immun 76 2149 2156
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 9
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Structure of the Extracellular Portion of CD46 Provides Insights into Its Interactions with Complement Proteins and Pathogens
- The Length of Vesicular Stomatitis Virus Particles Dictates a Need for Actin Assembly during Clathrin-Dependent Endocytosis
- Inhibition of TIR Domain Signaling by TcpC: MyD88-Dependent and Independent Effects on Virulence
- The Coevolution of Virulence: Tolerance in Perspective
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy