
TOPO3α Influences Antigenic Variation by Monitoring Expression-Site-Associated Switching in
Homologous recombination (HR) mediates one of the major mechanisms of trypanosome antigenic variation by placing a different variant surface glycoprotein (VSG) gene under the control of the active expression site (ES). It is believed that the majority of VSG switching events occur by duplicative gene conversion, but only a few DNA repair genes that are central to HR have been assigned a role in this process. Gene conversion events that are associated with crossover are rarely seen in VSG switching, similar to mitotic HR. In other organisms, TOPO3α (Top3 in yeasts), a type IA topoisomerase, is part of a complex that is involved in the suppression of crossovers. We therefore asked whether a related mechanism might suppress VSG recombination. Using a set of reliable recombination and switching assays that could score individual switching mechanisms, we discovered that TOPO3α function is conserved in Trypanosoma brucei and that TOPO3α plays a critical role in antigenic switching. Switching frequency increased 10–40-fold in the absence of TOPO3α and this hyper-switching phenotype required RAD51. Moreover, the preference of 70-bp repeats for VSG recombination was mitigated, while homology regions elsewhere in ES were highly favored, in the absence of TOPO3α. Our data suggest that TOPO3α may remove undesirable recombination intermediates constantly arising between active and silent ESs, thereby balancing ES integrity against VSG recombination.
Published in the journal:
. PLoS Pathog 6(7): e32767. doi:10.1371/journal.ppat.1000992
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1000992
Summary
Homologous recombination (HR) mediates one of the major mechanisms of trypanosome antigenic variation by placing a different variant surface glycoprotein (VSG) gene under the control of the active expression site (ES). It is believed that the majority of VSG switching events occur by duplicative gene conversion, but only a few DNA repair genes that are central to HR have been assigned a role in this process. Gene conversion events that are associated with crossover are rarely seen in VSG switching, similar to mitotic HR. In other organisms, TOPO3α (Top3 in yeasts), a type IA topoisomerase, is part of a complex that is involved in the suppression of crossovers. We therefore asked whether a related mechanism might suppress VSG recombination. Using a set of reliable recombination and switching assays that could score individual switching mechanisms, we discovered that TOPO3α function is conserved in Trypanosoma brucei and that TOPO3α plays a critical role in antigenic switching. Switching frequency increased 10–40-fold in the absence of TOPO3α and this hyper-switching phenotype required RAD51. Moreover, the preference of 70-bp repeats for VSG recombination was mitigated, while homology regions elsewhere in ES were highly favored, in the absence of TOPO3α. Our data suggest that TOPO3α may remove undesirable recombination intermediates constantly arising between active and silent ESs, thereby balancing ES integrity against VSG recombination.
Introduction
Trypanosoma brucei proliferates in the bloodstream of its mammalian host and periodically escapes the antibody-mediated immune response. A single species of variant surface glycoprotein (VSG) is expressed at a given time, from among >1,000 VSG genes and pseudogenes [1], [2], and ∼10 million VSG molecules homogenously coat the surface of a parasite. Switching the expressed VSG causes antigenic variation (reviewed in [3]–[5]).
VSG genes are found in 15 expression sites (ESs) — polycistronic transcription units that are transcribed by RNA Polymerase I [3], [6]–[8] — of the Lister 427 strain [9]. These VSGs are located 40–60 kb downstream of their ES promoters and are flanked by 70-bp and telomere repeat sequences. Several expression-site-associated genes (ESAGs) with mostly unknown functions, and ESAG and VSG pseudogenes, are located between the promoter and the 70-bp repeat region. Only one ES is transcriptionally active at any time and the rest are silent. Many VSGs are found upstream of telomere repeats in minichromosomes but most are thought to reside in ‘telomere-distal’ arrays. Minichromosomal and telomere-distal VSGs lack promoters, but small numbers of 70-bp repeats are present upstream of these VSGs.
By analyzing switched variants, two major pathways of antigenic switching have been identified in T. brucei: in situ ES transcription switching and recombination-mediated switching [4], [5], [10]. In situ switching occurs by silencing the active ES and activating a silent ES, without DNA rearrangement [11], [12]. Recombination-mediated switching occurs mainly by gene conversion (GC) and can involve just the VSG or larger regions of the ES. VSG GC can occur by recombination between the active VSG and a silent ES-associated VSG, a minichromosomal VSG, or a telomere-distal VSG [13]–[18]. Gene conversion between larger regions can result in the duplication of an entire ES, including its VSG [12]. Crossover switches, where two VSGs are exchanged, have also been observed infrequently [19]–[22].
Deficiency of RAD51 or RAD51-3 (RAD51-related gene), or BRCA2, a mediator for RAD51 filament formation, decreased switching frequency in T. brucei [23]–[25]. Mre11 is essential for DNA damage response, as a sensor of double strand breaks (DSBs) that can be repaired by homologous recombination (HR) or non-homologous end joining (NHEJ) [26]–[28]. As in yeast and mammals, T. brucei mre11 null mutants exhibited growth defects, hypersensitivity to a DNA damaging agent, and gross chromosomal rearrangements (GCR), but no detectable decrease in VSG switching [29], [30], indicating that, although antigenic variation shares core features with classic HR, specific roles for recombination factors in antigenic variation remain to be determined.
Mitotic crossover can be detrimental, leading to unequal exchanges. Sgs1, a RecQ family helicase in yeast, is one of the major factors that control spontaneous crossovers [31]. Sgs1 forms a complex with Top3 (type IA topoisomerase) and Rmi1 (RecQ-mediated genome instability), and plays major roles in the suppression of genome instability by influencing mitotic and meiotic recombination, replication fork stability, and telomere maintenance [32]–[38]. At least one mechanism of crossover-suppression appears to involve ‘dissolution’ of double Holliday Junction (dHJ) intermediates. Sgs1-Top3-Rmi1, also known as the RTR (RecQ-Top3-Rmi1) complex, is well conserved in humans as the BLM (Bloom mutated)-TOPO3α-BLAP75/18 (Bloom associated protein 75kDa/18kDa, or RMI1/2). Mutations in any member of the RTR complex increase recombination frequency and crossover [31], [32], [39]–[43]. Defects in the BLM pathway are associated with elevated levels of sister chromatid exchanges (SCEs), chromosomal breaks and translocations [40], [41], [44]–[46].
Crossover has rarely been observed in VSG switching. Suppression of crossover is intriguing because, in principle, the outcome of duplicative VSG conversion holds no apparent advantage over crossover events, as re-expressing a VSG, either exchanged or duplicated, will be lethal in vivo. Given the similarities between HR and VSG switching, we hypothesized that certain yeast hyper-recombination mutants could be hyper-switchers in trypanosomes. Using new recombination and VSG switching assays, we took advantage of a potential member of T. brucei Sgs1 pathway, TbTOPO3α (Tb11.01.1280), to get better insights on how trypanosomes employ recombination factors to control antigenic variation.
Results
Type 1A toposiomerase TOPO3α is conserved in Trypanosoma brucei
Type IA topoisomerases cleave DNA by covalent attachment of one of the DNA strands through a 5′phosphodiester bond to a tyrosine residue in their catalytic domains [47]. In many organisms, type IA topoisomerases function in cooperation with helicases, as a combination of Top3-Sgs1 in yeasts and TOPO3α-BLM in humans. T. brucei expresses a 102.5-kDa TOPO3α protein with 918 amino acids. Figure 1 shows an alignment of TbTOPO3α with human TOPO3α and S. cerevisiae and S. pombe Top3. The primary sequences are well aligned at the N-terminal catalytic domain including the active site tyrosine. Both E. coli Top1 and human TOPO3α contain Zn-binding motif(s) in their C-terminal regions. E. coli Top3 and two yeast Top3 lack a Zn-binding domain (reviewed in [47]). TbTOPO3α seems to have a Zn-binding motif in the C-terminus (four cysteine residues written in red), although this region does not align well with human TOPO3α. The sequences of TOPO3α are very well conserved in T. brucei, T. cruzi and Leishmania major (Supporting Figure S1). T. brucei also has a type IA TOPO3β (http://www.genedb.org/genedb/tryp), but its function has not been studied.

topo3α−/− exhibits a minor growth defect in T. brucei
To explore the role of TOPO3α, we sequentially deleted both alleles. We used deletion-cassettes containing hygromycin (HYG) or puromycin (PUR) resistance genes fused to Herpes simplex virus thymidine kinase (HSVTK or TK) and flanked by loxP sites, allowing the markers to be removed by transient expression of Cre-recombinase and reused [48]. Deletion of both alleles was confirmed by PCR analyses (Supporting Figure S2).
Loss of Top3 causes a severe growth defect in budding yeast and is lethal in fission yeast [43], [49]. The absence of TOPO3α or TOPO3β results in embryonic lethality or shortened life span in mice [50], [51]. In contrast, TOPO3α null mutants exhibited only a minor growth defect in T. brucei (Figure 2A).

Tbtopo3α mutants are sensitive to phleomycin and hydoxyurea
Yeast Top3 is important for the maintenance of genome integrity. top3 mutants are sensitive to DNA-damaging agents and show defects in the activation of the cell-cycle checkpoint kinase Rad53 (CHK2 in mammals), in response to genotoxic stresses [52]–[54]. We therefore asked whether T. brucei TOPO3α is required for the DNA damage response, by assessing sensitivity to the DSB-inducing agent phleomycin or the replication inhibitor hydroxyurea (HU). Cells were treated with phleomycin for 24 hours and single cells were distributed in 96-well plates. The color of the medium turns from red to yellow when the culture becomes saturated. Yellow wells were counted after 7–8 days and the percent viability was calculated by normalizing to the untreated samples. In the null mutants, viability was reduced by 3-fold at 0.3 µg/ml and 10-fold at 0.6 µg/ml phleomycin (Figure 2B). Viability of the HU-treated null mutants was reduced by 3-fold in 0.04 mM HU (Figure 2C). topo3α−/+ was comparable to the wild type in both experiments. We conclude that TOPO3α is required for the response to DNA damage and replication block, similar to the roles of yeast Top3.
Tbtopo3α−/− is a ‘hyper-rec’
top3 was isolated as a hyper-recombination (‘hyper-rec’) mutant in a genetic screen designed to identify mutations that increase recombination frequency at SUP4-o locus in budding yeast [43]. We therefore hypothesized that Tbtopo3α could be a ‘hyper-rec’ mutant and this phenotype could be reflected in the frequency of recombination-mediated antigenic switching.
To test whether TOPO3α deficiency increases recombination frequency, we established a new recombination assay. Thus far, transfection-based recombination assays have been predominantly used, in which trypanosomes are transfected with linear DNA containing a selection marker flanked by targeting sequences, and the recombination frequency is calculated from the number of drug-resistant clones that arise. Although this method can give reliable measurements, it requires a high rate of recombination at the target site and is subject to variations in transfection efficiency. To allow a more convenient, natural and reliable measure of recombination efficiency, we established an assay (Figure 3A) in which HYG-TK can replace one allele of what we will call TbURA3 (the bifunctional orotidine-5-phosphate decarboxylase/orotate phosphoribosyltransferase Tb927.5.3810) on chromosome V. The frequency of loss of either the HYG-TK or TbURA3 allele represents the rate of gene conversion at this locus. The frequency of HYG-TK loss can be measured with gancyclovir (GCV), a nucleoside analog, as only the cells that had lost the TK gene can grow in the presence of GCV. The loss of TbURA3 can be measured with 5-FOA (5-fluoroorotic acid), as only the ura3− cells can grow in the presence of 5-FOA.

To remove the HYG-TK and PUR-TK markers that were used for the deletion of TOPO3α, Cre-recombinase was transiently transfected into the topo3α−/− cells and GCVR HYGS PURS clones were selected (Supporting Figure S2). One allele of TbURA3 was then replaced with HYG-TK and the targeting was confirmed by PCR. Gene-conversion frequencies were determined by counting total GCRR and FOAR cells, in three wild-type and five topo3α−/− independent HYG-TK clones. As shown in Figure 3B, Tbtopo3α gave indeed a hyper-recombination phenotype. Total gene-conversion frequency was increased 6-fold in topo3α−/− (5.12±0.15×10−5) compared to the wild type (0.87±0.70×10−5).
Tbtopo3α−/− is a VSG ‘hyper-switcher’ and this phenotype requires RAD51
To investigate the roles for TOPO3α in VSG switching, we generated a VSG switching reporter strain in which we could easily measure switching frequency and score different switching mechanisms. As illustrated in Figure 4, the parental strain expresses VSG 427-2 (221) in ES1, which was doubly marked with a blasticidin-resistance gene (BSD) downstream of the promoter and PUR-TK at the 3′ end of the 70-bp repeat region, without disrupting the co-transposed region (CTR), disruption of which has been shown to induce rapid VSG switching [55]. The 5′ boundaries for recombination-mediated VSG switching have been mapped at regions upstream of CTRs that are located between the 70-bp repeats and the VSG. Therefore, the PUR-TK gene will either be lost or repressed in switched cells. This will allow switchers, but not the parental cells, to grow in the presence of GCV.

Doubly marked wild-type and topo3α−/− cells were maintained in media containing blasticidin and puromycin, to exclude switchers from the starting population. The cells were allowed to switch in the absence of drugs for 3–4 days. Un-switched VSG 427-2-expressing cells were depleted by magnetic-activated cell sorting (MACS) [56]. The column flow-through, highly enriched with switchers, was serially diluted in medium containing 4 µg/ml GCV and distributed into 96-well plates. Switching frequency was determined as the ratio of GCV-resistant cells to the total number of cells prepared for the MACS column experiments. We analyzed three independent wild-type cultures and four topo3α−/− cultures. As shown in Figure 5A, TOPO3α deficiency caused a 10–40-fold increase in switching frequency (26±16×10−5) compared to wild type (1.01±0.45×10−5). This is the only known example of an increase in VSG switching frequency when a repair factor is deleted. To confirm that the column-mediated depletion of VSG 427-2-expressing cells was not biasing our results, other batches of cells were directly diluted in GCV-containing media and distributed into 96-well plates. Switching frequency was 10–30-fold increased in the absence of TOPO3α (data not shown).

We have determined the switching frequency in a strain without the TK marker but with a PUR marker inserted downstream of VSG 427-2 and obtained similar frequencies, ∼1×10−5, in wild type. In two different but closely related cell lines, with the same genotype except that one line has PUR-TK inserted at the 70-bp repeat and the other just PUR, again similar switching frequencies, ∼1×10−5, were observed [56] (personal communication with Nina Papavasiliou).
Reintroduction of wild-type TOPO3α complemented the hyper-switching phenotype of topo3α−/− (−/−/+ in Figure 5B), confirming that this phenotype is associated with the TOPO3α deficiency. The results were obtained from three complemented clones (−/−/+) and two cultures each of wild type and topo3α mutant.
RAD51-dependent recombination intermediates accumulate in top3 mutants and the removal of persistent intermediates requires the cleavage activity of Top3 [57], [58]. We examined whether the hyper-switching phenotype of topo3α−/− is dependent on RAD51. Both RAD51 alleles were sequentially deleted in the wild-type and topo3α−/− strains. We analyzed four independent cultures of rad51−/− and two of topo3α−/− rad51−/−. RAD51 deletion reduced the switching frequency of the wild type by 2-fold and abolished the hyper-switching phenotype of topo3α−/− (Figure 5C). Collectively, we concluded that TOPO3α functions as an important regulatory factor for recombination-mediated VSG switching and that, in the absence of TOPO3α, recombinogenic structures may accumulate between the active ES and VSG donors, and could then be resolved to give rise to switched variants.
T. brucei TOPO3α suppresses VSG GC and crossover
In other organisms, Top3 defects are associated with elevated crossover as well as hyper-recombination [32]–[34], [45], [46]. To learn how individual switchers had undergone antigenic variation, we analyzed total 296 cloned switchers. The rationales for the double marking of parental cells are as follows (Figure 4). First, switchers can be effectively counter-selected using GCV (Figure 4 and 5). Second, transcription is initiated at silent ESs but elongation is prematurely terminated [59]: genes that are located closer to silent ES promoters are not completely silenced. Therefore, in-situ switchers can be distinguished from recombination-mediated switchers using different concentrations of blasticidin. Based on our titration for blasticidin concentration, in-situ switchers can grow in 5µg/ml blasticidin but not in 100 µg/ml, while ES gene conversion (ES GC) switchers cannot grow in either concentration. VSG gene conversion (VSG GC) and VSG-exchange (crossover) switchers will be resistant to 100 µg/ml blasticidin, and these alternatives can be distinguished by the absence or presence of VSG 427-2, respectively, which can be analyzed by PCR. The strategies to score individual switching mechanisms are summarized in Table 1 and examples of PCR analyses are shown in Figure 6A (right).


We analyzed cloned switchers isolated from six independent cultures and were able to discriminate among the alternative switching mechanisms. The results are summarized in Table 2 and Figure 6A. Switchers from cultures 1 and 2 were isolated by the column method and switchers from culture 3 by directly plating in GCV. Switching occurred largely by gene conversion (Figure 6A). In both wild type and topo3α mutants, 64∼77% of switching exploited VSG GC. Crossovers were rare in wild type (∼3%) but, on average, 20% of switchers exchanged their VSGs in topo3α−/−. These data suggest that, in the absence of TOPO3α, recombination intermediates may be accumulated and these could be repaired mostly by duplicative VSG GC and crossover.
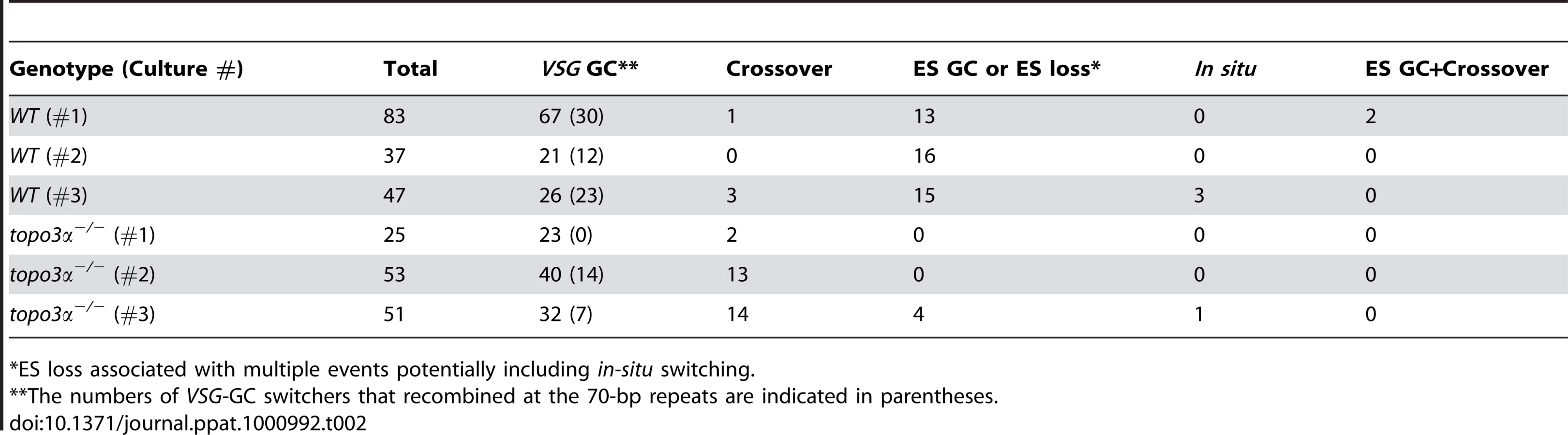
In a previous study designed to examine in-situ switching, using a cell line with TK marker inserted next to the active ES promoter, frequent loss of entire active ES was observed. This could be caused by duplicative transposition of a silent ES (ES GC) or by deletion of the active ES coupled with transcriptional activation of a silent ES [60]. In our experiments, ES GC and ES loss cannot be distinguished, as switchers that lost both BSD and VSG 427-2 genes could arise either by duplicative transposition of a silent ES or by ES breakage coupled with an ES transcriptional switch. The ‘ES GC or ES loss’ events were rather frequently detected in wild-type cells (average ∼30%), while they were either not detected (culture 1 and 2) or detected at a low frequency (4 our of 51 cloned switchers in culture 3) in the absence of TOPO3α (Figure 6A and Table 2). Interestingly, RAD51 deletion significantly decreased ‘ES GC or ES loss’ frequency (unpublished data), indicating that ‘ES GC or ES loss’ events are mainly under the control of RAD51-dependent recombination.
We noticed that some switched variants had growth disadvantages. Depending on how long it took to saturate the medium, wild-type switchers were categorized as ‘fast’, ‘medium’ or ‘slow’. ‘ES GC or ES loss’ switchers were prevalent in clones that grew up more slowly (data not shown). The functions of ESAGs are mostly unknown, but expressing different ESAGs might be advantageous when entering different hosts [61]. The slower-growth phenotype of some of these switchers may reflect impaired function of one or more ESAGs in the bovine serum-containing culture medium, which appears to favor stable transcription of the VSG 427-2-containing ES1.
In-situ switchers were rare in our assay. This phenotype is different from previous reports [24], [62], for reasons we do not understand. In our hands, in-situ switchers generally grew slower than VSG-GC switchers, so VSG-GC switchers would quickly take over the switched population if it was initially mixed, although this is unlikely because our switching population was initiated at 500–1000 cells/ml, while it was at 5,000–10,000 cell/ml in previous assays. Before this seeding, cells were grown in the presence of drugs that prevented switching.
T. brucei TOPO3α specifically regulates ES-associated VSG switching
The 70-bp repeat unit has been proposed to be a recombination hot spot, possibly as a potential target for a site-specific endonuclease playing a similar role to that of the HO-endonuclease in yeast. Such an endonuclease has not been identified in trypanosomes. The 70-bp repeats could serve as switching hot-spots because of their structural features [63], rather than require cleavage by a specific endonuclease. Early experiments suggested that the overall VSG switching-frequency was not reduced in the absence of 70-bp repeats or by inversion of the repeats although, when present in the correct orientation, the repeats were used more than 10% of the time [62]. More recently, however, it has been shown that the 70-bp repeats of the actively transcribed ES are prone to break, which could induce recombination-mediated switching, and that the switching frequency was greatly increased when breaks were experimentally induced at the 70-bp repeats, but not when induced elsewhere in the ES or in the absence of 70-bp repeats [56].
We mapped the region where the recombination occurred (or resolved) in the VSG-GC switchers from wild type and topo3α mutants, to learn whether the 70-bp repeat unit is the hot spot of duplicative VSG GC and whether TOPO3α can redirect this preference. ESAG1 genes are located immediately upstream of the 70-bp repeats, and their sequence polymorphisms allowed us to design ES1-specific-ESAG1 oligonucleotides for PCR analysis. PCR results from several VSG-GC switchers were shown in Figure 6A (right). The presence of ES1-specific ESAG1 in VSG-GC switchers indicates that gene conversion occurred at 70-bp repeat regions, and its absence indicates that recombination occurred upstream of ESAG1 (Figure 6B). Crossover and ‘ES GC or ES loss’ switchers were used to verify that the PCR primer set was amplifying only the ES1-specific ESAG1 gene. The ES1-specific ESAG1 was lost in all ‘ES GC or ES loss’ switchers but was detected in all crossover switchers examined, as expected. The ES1-specific ESAG1 gene was amplified in ∼63% of VSG-GC switchers in wild-type cells but ∼81% of VSG-GC switchers lost the ES1-specific ESAG1 gene in topo3α−/−, indicating that, in the absence of TOPO3α, the active ES recombined mostly with silent ESs upstream of ESAG1, rather than within the 70-bp repeats, but not with minichromosomal or telomere-distal VSGs. We concluded that the 70-bp repeat region is an important but not an essential element for recombination-mediated switching. Gene conversion upstream of 70-bp repeats, at ESAG2, has also been reported [64]. The primary function of TOPO3α may be to prevent accumulation of recombination intermediates constantly arising between the active and silent ESs, to maintain the integrity of ESs.
Recombination by a one-strand invasion event could replace VSGs by break-induced replication (BIR) [56]. Alternatively, a second strand invasion at homologous sequences within or downstream of the VSG could generate VSG-GC switchers. Duplication of a telomere-distal VSG into an active ES is a relatively rare event, at least in the modest extent to which switching events have been characterized experimentally, but it appears to serve as an important switching mechanism in later stage of infection and as a mechanism to further expand the expressed VSG repertoire [22], [65], [66]. The few telomere-distal VSG arrays so far characterized contain only short stretches of 70-bp repeats but lack telomeric repeats. To determine how VSG GC occurred, we analyzed the sequences downstream of the 3′ homology region of VSG 427-2 by PCR in all VSG-GC switchers (Supporting Figure S3). If the second strand invaded at this 3′ homology region, downstream sequences should be unchanged. We found, however, that the ES1-specific downstream sequences were lost in all the VSG-GC switchers obtained from wild-type and topo3α cells, indicating that VSG-GC switchers were most likely repaired by BIR, consistent with a recent report [56], and that internal-VSG duplication is extremely rare. PCR results from a selection of VSG-GC switchers were shown in Figure S3.
To confirm the duplicative translocation of newly expressed VSGs to the VSG 427-2 ES and to examine whether minichromosomal VSGs contribute to antigenic switching, 32 VSG-GC switchers from wild-type cells were further analyzed. Minichromosomes terminate with telomeres, VSGs and 70-bp repeats. Gene conversion with minichromosomal VSGs occurs frequently [56], but only when recombination is initiated at the 70-bp repeats. Therefore, we cloned and sequenced newly activated VSGs from VSG-GC switchers that utilized 70-bp repeats. From 32 switchers that had undergone at least one type of switching, VSG GC at the 70-bp repeats, we obtained eight different newly activated VSGs (Supporting Figure S5, left). It is possible that we have underestimated the number of independent switching events as these switchers may have used different sequences within or near the 70-bp repeats, which should be counted as independent. Some switchers might have arisen earlier than others, for examples VSG 427-32, as these were presented more often than others. Among these eight newly expressed VSGs, four were novel VSGs, 427-32, 33, 34 and 35, full or partial sequences of which can be found in the following website (http://tryps.rockefeller.edu). Switchers expressing VSGs 427-3, 11, 32, 33 and 35 were examined by rotating agarose gel electrophoresis (RAGE) and Southern blot [56]. As shown in Supporting Figure S5 (right panel), VSG 427-2 was lost in all the switchers and all newly expressed VSGs were duplicated and translocated to the 427-2 ES, except for 427-33, an intermediate chromosomal (IC) VSG. The original copy of 427-33 may be lost after recombination. VSGs 427-32 and 35 came from megabase chromosomes (MBC). We have not isolated any minichromosomal VSGs in these switchers, indicating that recombination between ES-associated VSGs was the major source for VSG switching.
Discussion
Repair by recombination serves to preserve genome integrity and can either homogenize or diversify genetic information, occasionally causing detrimental outcomes or benefiting certain organisms by providing adaptation systems to escape lethal situations. African trypanosomes escape the host immune response through a mechanism known as the antigenic variation. Here, we report that T. brucei TOPO3α, a member of a potential T. brucei RecQ-Top3-Rmi1 (RTR) complex, takes an important part in the regulation of recombination-mediated antigenic variation. Our results reveal a complex mechanism that has to balance ES integrity and VSG diversity to maximize the survival of a trypanosome population by suppressing crossovers on one hand and by promoting duplicative VSG gene conversions on the other.
Mechanism of recombination-mediated antigenic switching and roles for TOPO3α
As illustrated in Figure 7, ES structures seem to play a particular role in VSG switching. ES-associated VSG genes are located between the 70-bp and telomeric repeats. ESAGs and some pseudogenes are present upstream of the 70-bp repeats in all ESs, sometimes duplicated and sometimes missing [3], [9]. Strong sequence homologies are present throughout the ESs, with the exception of most of the VSG coding sequence and the immediately upstream ‘co-transposed region’ (CTR). VSG sequences are highly dissimilar except for ∼200-bp encoding the C-terminus and within the 3′ UTR [67]. The reason why every VSG cassette contains a unique CTR is unknown. The purpose of CTR could be to insulate the individuality of VSG cassettes, so that the VSG sequences can evolve separately from other regions in ESs, which maintain their sequences to serve for VSG recombination. When HR occurs, the CTR could block branch migration of HJ or dHJ downstream of the 70-bp repeats.

What roles does TOPO3α play in this scheme? Our study shows that TOPO3α deficiency increases VSG switching, especially VSG GC and crossover, and that the hyper-switching phenotype requires RAD51. The accumulation of toxic recombination intermediates accounts for the slow growth phenotype of yeast top3 mutants, which is suppressed by mutations in SGS1 or in the RAD51-pathway [43], [68], [69]. Recombination intermediates accumulate in cells over-expressing dominant-negative Top3-Y356F in response to methylmethane sulfonate in a RAD51-dependent manner [58]. The function of TOPO3α is not restricted to the 70-bp repeats in antigenic switching, as its absence appears to cause promiscuous recombination throughout the ESs. We therefore propose that TOPO3α removes recombinogenic structures constantly arising between ESs so as to maintain the albeit limited individuality of different ESs. In the absence of TOPO3α, recombination intermediates would accumulate during VSG switching and unresolved intermediates would have to be repaired either by GC associated with crossover or by placing a new duplicated VSG into the active ES by BIR (Figure 7).
Suppression of crossover in recombination-mediated VSG switching is an interesting result, considering that there are probably more than 200 potential VSG donors: ∼20 ESs with extensive sequence homology and ∼200 minichromosomal VSGs. Antigenic variation probably requires balancing preservation and variation of VSG information, but we cannot explain how suppression of crossover would be important for maintaining this balance. However, we think that by favoring duplicative GC over crossover, rather than crossover over GC, trypanosomes could slowly accumulate VSG diversity without abrupt loss of their functionalities, because duplicative GC requires VSG DNA synthesis, during which point mutations could be incorporated into newly synthesized VSGs, but VSG crossover does not require VSG DNA synthesis.
TOPO3α deficiency increased VSG GC far more than GC at the URA3 locus (Figures 3 and 6). GC at these two loci is probably mediated by different pathways. Recombination at URA3 locus would prefer flanking homologies, rather than BIR. In contrast, BIR would present a better option for VSG GC, as only one end homology appears to be involved (supporting Figure S3) [56]. It is possible that a second invasion could occur within the telomere repeats, but this is impossible to determine. The higher VSG GC rate could also be because the active ES is less stable than URA3 locus. Alternatively, TOPO3α may specifically suppress BIR-mediated VSG switching. The role of TOPO3α in BIR has not been extensively characterized elsewhere. Our results show a novel function of TOPO3α in VSG switching, which could be an excellent system to study BIR.
TbRTR complex and DSB-HR response in antigenic variation
DNA recombination involves many factors, of which only a few have been studied in the context of antigenic variation: RAD51, RAD51-related genes, BRCA2, KU70/80, MRE11, and MSH2/MLH1 [23]–[25], [29], [30], [70], [71]. Among these, only the deletion of RAD51, RAD51-3, and BRCA2 decreased VSG switching, in wild-type cells that already had a very low switching rate.
Our findings on TOPO3α in VSG switching suggest potential roles for numerous DSB-HR response factors in antigenic variation. Two RecQ family helicases are annotated in the T. brucei gene database (http://www.genedb.org/genedb/tryp). Rmi1 is required to load Top3 onto the substrates and stimulate its activity through the physical interaction [72]. We have identified a TbRMI1 homologue. All the phenotypes that we have examined in Tbrmi1 mutants were identical to those in topo3α mutants (unpublished data). Therefore, we believe that RecQ, TOPO3α and RMI1 are likely to function as a complex in antigenic variation in T. brucei.
Synthetic-lethality screens with sgs1 in budding yeast identified three pathways working in parallel with Sgs1 [73]; Mus81-Mms4, Slx1-Slx4, and Slx5-Slx8. Synthetic lethality of sgs1 mus81 or sgs1 mms4 requires HR factors [74]. Mus81-Mms4 is a structure-specific endonuclease that cleaves 3′ flap, replication fork, or HJ substrates [74]–[76]. Resolvase, an endonuclease, symmetrically cleaves HJs and the products can be resolved with crossover or non-crossover. Human and yeast resolvases have recently been characterized [77]. MUS81 appears to be present in T. brucei but a resolvase remains to be identified. Although we do not yet have functional data for these proteins, we propose, based on the studies from other organisms, that the regulation of antigenic variation is similar to that of mitotic HR. When present, TOPO3α could dissolve dHJs to prevent the ES instability, consequently generating non-crossover recombinants (no switching). In the absence of TOPO3α, resolvase (Figure 7a, grey box) or MUS81 may cleave the accumulated recombination intermediates arising between the ESs and generate crossover switchers. Alternatively, stalled replication forks can be cleaved by MUS81 and the broken leading strand can invade a silent ES to generate VSG-GC switchers (Figure 7b, grey box).
Although VSG switching has similarities with mitotic HR, it appears that specific elements are present for its regulation. A hyper-recombination phenotype does not always correlate with hyper-switching phenotype. The mismatch repair (MMR) pathway can abort recombination during strand exchange between non-identical substrates and mmr mutants can increase recombination frequency (reviewed in [78]). Consistent with their roles in repair and recombination, Tbmsh2 or Tbmlh1 mutants increased recombination frequency but did not change switching frequency [71]. Recombination is closely linked with DNA replication and checkpoint pathways as well [32], [57], [58], [79]. Therefore, we believe that roles for DNA replication, checkpoint, and recombination factors and their interactions need to be determined to fully understand the mechanisms of antigenic variation.
Measuring VSG switching has, until now, been time-consuming and not very reproducible. Our new switching assay circumvents previous technical difficulties and can effectively assign specific roles to individual proteins.
What triggers antigenic switching
It has recently been shown that a DSB introduced at the active 70-bp repeats by the I-SceI endonuclease causes a 250-fold increase in VSG switching and that the DSBs were repaired by BIR [56]. However, it is unknown whether the VSG switching is activated by targeted DSBs or by random chromosomal breaks, or whether recombinogenic ssDNA is a primary cause for the initiation of VSG switching. HR can be instigated by many different sources; random breaks, endonuclease cleavage at specific target sites, replication fork instability, unusual secondary DNA structure, or transcription.
The Mre11 complex, which consists of Mre11, Rad50, and Xrs2 (NBS1 in mammals), plays a central role in the DSB-HR response [26]–[28]. MRE11 deficiency, however, did not change the VSG switching frequency [29], [30], promoting the idea that ssDNA regions may generate recombinogenic structures for the initiation of switching. Uncoupling of leading and lagging strand DNA synthesis caused by DNA lesions can destabilize a replication fork, leaving ssDNA gaps behind the fork, which could be processed into recombinogenic structures. If an ssDNA gap is a major trigger for recombination-mediated switching, switching frequency should increase in cells suffering from replication challenge. To address this issue, we treated cells with aphidicolin, an inhibitor of lagging strand DNA synthesis, and HU, and measured the switching frequency in parallel (Supporting Figure S4). Cells were treated with the drugs at a sub-lethal dose to exclude a possibility of chromosome break-induced switching. No significant correlation was observed between these treatments and switching frequency. Therefore, an ssDNA gap may not be a major initiating factor for VSG switching. Rather, random breaks might be responsible for switching induction, consistent to a previous study [56]. However, it is still difficult to rule out the possibility that an ssDNA gap triggers switching, as ssDNA gaps might not be extensive enough to create recombinogenic structures at the low doses of aphidicolin or HU. The best way to test this hypothesis would be to use conditional mutants associated with replication defects. Unfortunately, we do not yet have such genetic tools, as nuclear DNA replication has not been studied in T. brucei.
A high transcription level can stimulate recombination, a mechanism known as transcription-associated recombination (TAR) (reviewed in [80]). Transcription has been shown to promote recombination in T. brucei [81], [82]. Interestingly, it was shown in budding yeast that transcription - and DSB-induced recombination events were similar, indicating that transcription affects only the initiation of recombination, not the mechanism of recombination [83]. ssDNA regions exposed in the active ES during transcription could be readily accessible by recombination factors. Alternatively, transcription-replication collision causes replication fork stalling, which could also induce switching. Studies of mammalian cells have shown that TAR is dependent on replication [84], and that transcription increases recombination frequency when a replication fork converges with transcription [85]. The active ES is more fragile than silent ESs [56]. The high level of transcription may explain why the active ES breaks more frequently, and this may induce VSG switching.
The 70-bp repeat has been proposed to be a potential endonuclease target site to induce switching, but such an enzyme has not been found. Instability of the 70-bp repeat [63] may play a role in the initiation of switching and could lead to template switching. However, according to our results and previous studies [62], [64], switching is not completely dependent on the 70-bp repeats. With the available data, it would be reasonable to conclude that random breaks may occur throughout the active ES but more frequently at 70-bp repeats, and these could initiate various switching events.
Gene conversion is used by several other pathogens, including Borrelia hermsii and Anaplasma marginale, as an evasion mechanism [10], [86]. Our study suggests that exploring how trypanosomes manipulate the HR machinery to gain advantage against their host's immunity, while successfully preserving their genomes, may reveal weaknesses that can be exploited to control infectivity and virulence.
Materials and Methods
Trypanosome strains and plasmids
Trypanosoma brucei bloodstream forms (strain Lister 427 antigenic type MITat1.2 clone 221a (VSG 427-2)) were cultured in HMI-9 at 37°C. The cell lines constructed for this study are listed in Supporting Table S1, and they are of ‘single marker’ (SM) background that expresses T7 RNA polymerase and Tet repressor (TETR) [87]. Stable clones were obtained and maintained in HMI-9 media containing necessary antibiotics at the following concentrations, unless otherwise stated: 2.5µg/ml, G418 (Sigma); 5µg/ml, blasticidin (Invivogen); 5µg/ml, hygromycin (Sigma); 0.1µg/ml, puromycin (Sigma); 1µg/ml, phleomycin (Invivogen). Plasmids used for this study are listed in Supporting Table S2.
Construction of topo3α−/− cell line and removal of markers using Cre recombinase-loxPs (Supporting Figure S2)
TOPO3α genes were sequentially deleted using deletion-cassettes containing either puromycin or hygromycin-resistance gene fused with HSVTK, Herpes simplex virus thymidine kinase (TK), PUR-TK and HYG-TK. These fusion genes are flanked by loxP sites so that the markers can be removed by transient expression of Cre recombinase (pLew100-Cre). The entire open reading frame (ORF) of TOPO3α was deleted by transfecting ‘single marker’ (SM) cells with a deletion-cassette that was amplified with primer 35 and 36 using pHJ18 (PUR-TK) as a template. Primer 35 and 36 contains 70 nt homologies to the target sites. This topo3α ‘single knock-out’ cells (sKO, HSTB-97) were used to PCR amplify a cassette containing a marker (PUR-TK) along with 453 nt upstream and 402 nt downstream sequences of TOPO3α gene. The PCR fragment was inserted into pGEM-easy-T vector by TA cloning to create pHJ63. pHJ64 was constructed by replacing a PUR-TK marker with a HYG-TK from pHJ17. topo3α ‘double knock-out’ (dKO) was generated by transfecting NotI-digested pHJ64 into topo3α sKO, HSTB-97. Deletion of both TOPO3α alleles was confirmed by PCR analyses.
To remove the selection markers, topo3α dKO cells were transfected with pLew100-Cre to transiently express Cre-recombinase, and the cells that lost both HYG-TK and PUR-TK were selected in 50µg/ml ganciclovir (GCV). Loss of markers was confirmed by resistance to puromycin and hygromycin, and by PCR analysis. The sequences of primers used here are available upon request.
Recombination assay
pLHTL-pyrFE [48]-linearized by PvuII digestion was transfected into wild-type (HSTB-188) and topo3α−/− (HSTB-328 and HSTB-330) cells, to replace one allele of TbURA3 with HYG-TK. The integration was confirmed by PCR analysis with primers 48 and 49. Three or five independent HYGR clones from wild-type or topo3−/− cells were analyzed. Cells were grown in the absence of hygromycin for 2 days to allow recombination to occur. Approximately 500,000 cells were diluted in HMI-9 media containing 30 µg/ml GCV or 6 µg/ml FOA, and distributed into 96-well plates. Yellow wells (phenol red indicating acidification due to growth) containing GCVR or FOAR cells were counted after 7–8 days of incubation and the GC frequency was determined. The sequences of primers used for genotyping are available upon request.
Switching assay and analyses of switchers
To create a doubly-marked switching reporter strain (Figure 4), pHJ23 was linearized by KpnI-NotI digestion and integrated downstream of the ES1 promoter, to confer resistance to blasticidin. These cells were then marked with PUR-TK at the 3′ end of 70-bp repeats by transfecting a PCR-amplified PUR-TK cassette. Ten µg/ml of puromycin, 100 times higher than normal usage, was added to select clones targeted specifically at the active ES. When determining switching frequency, the parental cells were maintained in the presence of blasticidin and puromycin to exclude switchers from the starting population. Cells were then allowed to switch in the absence of selection for 3–4 days. Switchers were enriched using a MACS [56]. Flow-through enriched with switchers was collected and serially diluted in media containing 4 µg/ml GCV, and distributed into 96-well plates. The switching frequency was determined by counting GCVR clones. Alternatively, switching frequency was determined without the column enrichment step. Cells were diluted in GCV-containing media and directly distributed into 96-well plates. Non-switchers that carry spontaneous mutation(s) in TK gene but not in PUR were ruled out by examining puromycin resistance. Non-switchers that carry mutations in PUR and TK were ruled out by western blot analysis using antibodies against VSG 427-2.
To determine switching mechanisms, cloned switchers were analyzed for blasticidin sensitivity at 5 µg/ml and 100 µg/ml concentrations. Genomic DNA was prepared from 296 switchers and PCR-analyses were performed at four regions: BSD, VSG 427-2, ESAG1, and VSG 427-2 downstream. The primer set designed for BSD-PCR can also amplify TETR (Tet Repressor) gene, which was used as a control for PCR analyses. The sequences of primers used here are available upon request.
Analysis of sensitivity to genotoxic agents
Wild type (SM), topo3α−/+ (HSTB-97), and topo3α−/− (HSTB-226 and HSTB-227) cells were incubated with indicated concentration of phleomycin for 24 hours. The same number of cells was distributed into 96-well plates. All the plating was duplicated. The wells that contain viable cells were counted after 7–8 days of incubation at 37°C and the viability was calculated by normalizing to untreated samples. Sensitivity to HU and aphidicolin was determined similarly. Cells were incubated with HU or aphidicolin for 2 or 3 days. The viability was calculated by normalizing to untreated samples.
Gene accession numbers
Database ID numbers (http://www.genedb.org and http://tritrypdb.org) for TOPO3α discussed in this paper are Tb11.01.1280, LmjF36.3200 and Tc00.1047053511589.120. What we refer to as TbURA3 is the bifunctional orotidine-5-phosphate decarboxylase/orotate phosphoribosyltransferase Tb927.5.3810.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. MarcelloL
BarryJD
2007 Analysis of the VSG gene silent archive in Trypanosoma brucei reveals that mosaic gene expression is prominent in antigenic variation and is favored by archive substructure. Genome Res 17 1344 1352
2. MarcelloL
MenonS
WardP
WilkesJM
JonesNG
2007 VSGdb: a database for trypanosome variant surface glycoproteins, a large and diverse family of coiled coil proteins. BMC Bioinformatics 8 143
3. HornD
BarryJD
2005 The central roles of telomeres and subtelomeres in antigenic variation in African trypanosomes. Chrom Res 13 525 533
4. BarryJD
McCullochR
2001 Antigenic variation in trypanosomes: enhanced phenotypic variation in a eukaryotic parasite.
BakerJR
MullerR
RollinsonD
Advances in Parasitology 49 London Academic Press Ltd 1 70
5. CrossGAM
2002 Antigenic variation in african trypanosomes and malaria.
MarrJ
KoumunieckiR
NilsenTW
Mol Med Parasitol Academic Press 89 110
6. RudenkoG
BishopD
GottesdienerK
van der PloegLHT
1989 Alpha-amanitin resistant transcription of protein coding genes in insect and bloodstream form Trypanosoma brucei. EMBO J 8 4259 4263
7. PalencharJ
BellofattoV
2006 Gene transcription in trypanosomes. Mol Biochem Parasitol 146 135 141
8. GunzlA
BrudererT
LauferG
SchimanskiB
TuLC
2003 RNA polymerase I transcribes procyclin genes and variant surface glycoprotein gene expression sites in Trypanosoma brucei. Eukaryot Cell 2 542 551
9. Hertz-FowlerC
FigueiredoLM
QuailMA
BeckerM
JacksonA
2008 Telomeric expression sites are highly conserved in Trypanosoma brucei. PLoS ONE 3 e3527
10. MachadoC
Augusto-PintoL
McCullochR
TeixeiraS
2006 DNA metabolism and genetic diversity in Trypanosomes. Mutat Res Rev-Mutat Res 612 40 57
11. ZomerdijkJCBM
OuelleteM
ten AsbroekALMA
KieftR
BommerAMM
1990 The promoter for a variant surface glycoprotein gene expression site in Trypanosoma brucei. EMBO J 9 2791 2801
12. JohnsonPJ
BorstP
1986 Mapping of VSG genes on large expression-site chromosomes of Trypanosoma brucei separated by pulse-field gradient electrophoresis. Gene 43 213 220
13. PaysE
van AsselS
LaurentM
DarvilleM
VervoortT
1983 Gene conversion as a mechanism for antigenic variation in trypanosomes. Cell 34 371 381
14. MylerP
NelsonRG
AgabianN
StuartK
1984 Two mechanisms of expression of a variant antigen gene of Trypanosoma brucei. Nature 309 282 284
15. LeeMG-S
van der PloegLHT
1987 Frequent independent duplicative transpositions activate a single VSG gene. Mol Cell Biol 7 357 364
16. de LangeT
KooterJM
MichelsPAM
BorstP
1983 Telomere conversion in trypanosomes. Nucl Acids Res 11 8149 8165
17. RobinsonNP
BurmanN
MelvilleSE
BarryJD
1999 Predominance of duplicative VSG gene conversion in antigenic variation in African trypanosomes. Mol Cell Biol 19 5839 5846
18. HoeijmakersJHJ
FraschACC
BernardsA
BorstP
CrossGAM
1980 Novel expression-linked copies of the genes for variant surface antigens in trypanosomes. Nature 284 78 80
19. PaysE
GuyauxM
AertsD
vanMeirvenneN
SteinertM
1985 Telomeric reciprocal recombination as a possible mechanism for antigenic variation in trypanosomes. Nature 316 562 564
20. AitchesonN
TalbotS
ShapiroJ
HughesK
AdkinC
2005 VSG switching in Trypanosoma brucei: antigenic variation analysed using RNAi in the absence of immune selection. Mol Microbiol 57 1608 1622
21. RudenkoG
McCullochR
DirksmulderA
BorstP
1996 Telomere exchange can be an important mechanism of variant surface glycoprotein gene switching in Trypanosoma brucei. Mol Biochem Parasitol 80 65 75
22. BernardsA
van der PloegLHT
GibsonWC
LeegwaterP
EijgenraamF
1986 Rapid change of the repertoire of variant surface glycoprotein genes in trypanosomes by gene duplication and deletion. J Mol Biol 190 1 10
23. ProudfootC
McCullochR
2005 Distinct roles for two RAD51-related genes in Trypanosoma brucei antigenic variation. Nucl Acids Res 33 6906 6919
24. McCullochR
BarryJD
1999 A role for RAD51 and homologous recombination in Trypanosoma brucei antigenic variation. Genes Dev 13 2875 2888
25. HartleyCL
McCullochR
2008 Trypanosoma brucei BRCA2 acts in antigenic variation and has undergone a recent expansion in BRC repeat number that is important during homologous recombination. Mol Microbiol 68 1237 1251
26. D'AmoursD
JacksonSP
2002 The Mre11 complex: at the crossroads of DNA repair and checkpoint signalling. Nat Rev Mol Cell Biol 3 317 327
27. WilliamsRS
MoncalianG
WilliamsJS
YamadaY
LimboO
2008 Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell 135 97 109
28. KanaarR
WymanC
2008 DNA repair by the MRN complex: break it to make it. Cell 135 14 16
29. TanKSW
LealSLG
CrossGAM
2002 Trypanosoma brucei MRE11 is non-essential but influences growth, homologous recombination and DNA double-strand break repair. Mol Biochem Parasitol 125 11 21
30. RobinsonNP
McCullochR
ConwayC
BrowittA
BarryJD
2002 Inactivation of Mre11 does not affect VSG gene duplication mediated by homologous recombination in Trypanosoma brucei. J Biol Chem 277 26185 26193
31. IraG
MalkovaA
LiberiG
FoianiM
HaberJE
2003 Srs2 and Sgs1-Top3 suppress crossovers during double-strand break repair in yeast. Cell 115 401 411
32. BachratiCZ
HicksonID
2008 RecQ helicases: guardian angels of the DNA replication fork. Chromosoma 117 219 233
33. SekiM
TadaS
EnomotoT
2006 Function of recQ family helicase in genome stability. Subcell Biochem 40 49 73
34. SungP
KleinH
2006 Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nat Rev Mol Cell Biol 7 739 750
35. ChangM
BellaouiM
ZhangC
DesaiR
MorozovP
2005 RMI1/NCE4, a suppressor of genome instability, encodes a member of the RecQ helicase/Topo III complex. EMBO J 24 2024 2033
36. MullenJR
NallasethFS
LanYQ
SlagleCE
BrillSJ
2005 Yeast Rmi1/Nce4 controls genome stability as a subunit of the Sgs1-Top3 complex. Mol Cell Biol 25 4476 4487
37. TsaiHJ
HuangWH
LiTK
TsaiYL
WuKJ
2006 Involvement of topoisomerase III in telomere-telomere recombination. J Biol Chem 281 13717 13723
38. HuangPH
PrydeFE
LesterD
MaddisonRL
BortsRH
2001 SGS1 is required for telomere elongation in the absence of telomerase. Curr Biol 11 125 129
39. NeffNF
EllisNA
YeTZ
NoonanJ
HuangK
1999 The DNA helicase activity of BLM is necessary for the correction of the genomic instability of bloom syndrome cells. Mol Biol Cell 10 665 676
40. SinghTR
AliAM
BusyginaV
RaynardS
FanQ
2008 BLAP18/RMI2, a novel OB-fold-containing protein, is an essential component of the Bloom helicase-double Holliday junction dissolvasome. Genes Dev 22 2856 2868
41. XuD
GuoR
SobeckA
BachratiCZ
YangJ
2008 RMI, a new OB-fold complex essential for Bloom syndrome protein to maintain genome stability. Genes Dev 22 2843 2855
42. WattPM
HicksonID
BortsRH
LouisEJ
1996 SGS1, a homologue of the Bloom's and Werner's syndrome genes, is required for maintenance of genome stability in Saccharomyces cerevisiae. Genetics 144 935 945
43. WallisJW
ChrebetG
BrodskyG
RolfeM
RothsteinR
1989 A hyper-recombination mutation in S. cerevisiae identifies a novel eukaryotic topoisomerase. Cell 58 409 419
44. WuL
HicksonID
2003 The Bloom's syndrome helicase suppresses crossing over during homologous recombination. Nature 426 870 874
45. HartungF
SuerS
KnollA
Wurz-WildersinnR
PuchtaH
2008 Topoisomerase 3alpha and RMI1 suppress somatic crossovers and are essential for resolution of meiotic recombination intermediates in Arabidopsis thaliana. PLoS Genet 4 e1000285
46. SekiM
NakagawaT
SekiT
KatoG
TadaS
2006 Bloom helicase and DNA topoisomerase IIIalpha are involved in the dissolution of sister chromatids. Mol Cell Biol 26 6299 6307
47. ChampouxJJ
2001 DNA topoisomerases: structure, function, and mechanism. Annu Rev Biochem 70 369 413
48. ScahillMD
PastarI
CrossGA
2008 CRE recombinase-based positive-negative selection systems for genetic manipulation in Trypanosoma brucei. Mol Biochem Parasitol 157 73 82
49. MaftahiM
HanCS
LangstonLD
HopeJC
ZigourasN
1999 The top3(+) gene is essential in Schizosaccharomyces pombe and the lethality associated with its loss is caused by Rad12 helicase activity. Nucl Acids Res 27 4715 4724
50. LiW
WangJC
1998 Mammalian DNA topoisomerase IIIalpha is essential in early embryogenesis. Proc Natl Acad Sci U S A 95 1010 1013
51. KwanKY
WangJC
2001 Mice lacking DNA topoisomerase IIIbeta develop to maturity but show a reduced mean lifespan. Proc Natl Acad Sci U S A 98 5717 5721
52. OakleyTJ
GoodwinA
ChakravertyRK
HicksonID
2002 Inactivation of homologous recombination suppresses defects in topoisomerase III-deficient mutants. DNA repair 1 463 482
53. OhM
ChoiIS
ParkSD
2002 Topoisomerase III is required for accurate DNA replication and chromosome segregation in Schizosaccharomyces pombe. Nucl Acids Res 30 4022 4031
54. ChakravertyRK
KearseyJM
OakleyTJ
GrenonM
de La Torre RuizMA
2001 Topoisomerase III acts upstream of Rad53p in the S-phase DNA damage checkpoint. Mol Cell Biol 21 7150 7162
55. DaviesKP
CarruthersVB
CrossGAM
1997 Manipulation of the VSG co-transposed region increases expression-site switching in Trypanosoma brucei. Mol Biochem Parasitol 86 163 177
56. BoothroydCE
DreesenO
LeonovaT
LyKI
FigueiredoLM
2009 A yeast-endonuclease-generated DNA break induces antigenic switching in Trypanosoma brucei. Nature 459 278 281
57. LiberiG
MaffiolettiG
LuccaC
ChioloI
BaryshnikovaA
2005 Rad51-dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Genes Dev 19 339 350
58. MankouriHW
HicksonID
2006 Top3 processes recombination intermediates and modulates checkpoint activity after DNA damage. Mol Biol Cell 17 4473 4483
59. VanhammeL
PoelvoordeP
PaysA
TebabiP
XongHV
2000 Differential RNA elongation controls the variant surface glycoprotein gene expression sites of Trypanosoma brucei. Mol Microbiol 36 328 340
60. CrossM
TaylorMC
BorstP
1998 Frequent loss of the active site during variant surface glycoprotein expression site switching in vitro in Trypanosoma brucei. Mol Cell Biol 18 198 205
61. BitterW
GerritsH
KieftR
BorstP
1998 The role of transferrin-receptor variation in the host range of Trypanosoma brucei. Nature 391 499 502
62. McCullochR
RudenkoG
BorstP
1997 Gene conversions mediating antigenic variation in Trypanosoma brucei can occur in variant surface glycoprotein expression sites lacking 70-base-pair repeat sequences. Mol Cell Biol 17 833 843
63. OhshimaK
KangS
LarsonJE
WellsRD
1996 TTA.TAA triplet repeats in plasmids form a non-H bonded structure. J Biol Chem 271 16784 16791
64. KooterJM
WinterAJ
de OliveiraC
WagterR
BorstP
1988 Boundaries of telomere conversion in Trypanosoma brucei. Gene 69 1 11
65. LiuAY
MichelsPA
BernardsA
BorstP
1985 Trypanosome variant surface glycoprotein genes expressed early in infection. J Mol Biol 175 383 386
66. TimmersHTM
de LangeT
KooterJM
BorstP
1987 Coincident multiple activations of the same surface antigen gene in Trypanosoma brucei. J Mol Biol 184 81 90
67. AlineRF
StuartKD
1989 Trypanosoma brucei: conserved sequence organisation 3′ to telomeric variant surface glycoprotein genes. Exp Parasitol 68 57 66
68. GangloffS
McDonaldJP
BendixenC
ArthurL
RothsteinR
1994 The yeast type I topoisomerase Top3 interacts with Sgs1, a DNA helicase homolog: a potential eukaryotic reverse gyrase. Mol Cell Biol 14 8391 8398
69. ShorE
GangloffS
WagnerM
WeinsteinJ
PriceG
2002 Mutations in homologous recombination genes rescue top3 slow growth in Saccharomyces cerevisiae. Genetics 162 647 662
70. ConwayC
McCullochR
GingerML
RobinsonNP
BrowittA
2002 Ku is important for telomere maintenance, but not for differential expression of telomeric VSG genes, in African trypanosomes. J Biol Chem 277 21269 21277
71. BellJS
McCullochR
2003 Mismatch repair regulates homologous recombination, but has little influence on antigenic variation, in Trypanosoma brucei. J Biol Chem 278 45182 45188
72. ChenCF
BrillSJ
2007 Binding and activation of DNA topoisomerase III by the Rmi1 subunit. J Biol Chem 282 28971 28979
73. MullenJR
KaliramanV
IbrahimSS
BrillSJ
2001 Requirement for three novel protein complexes in the absence of the Sgs1 DNA helicase in Saccharomyces cerevisiae. Genetics 157 103 118
74. Bastin-ShanowerSA
FrickeWM
MullenJR
BrillSJ
2003 The mechanism of Mus81-Mms4 cleavage site selection distinguishes it from the homologous endonuclease Rad1-Rad10. Mol Cell Biol 23 3487 3496
75. KaliramanV
MullenJR
FrickeWM
Bastin-ShanowerSA
BrillSJ
2001 Functional overlap between Sgs1-Top3 and the Mms4-Mus81 endonuclease. Genes Dev 15 2730 2740
76. CoteAG
LewisSM
2008 Mus81-dependent double-strand DNA breaks at in vivo-generated cruciform structures in S. cerevisiae. Mol Cell 31 800 812
77. IpSC
RassU
BlancoMG
FlynnHR
SkehelJM
2008 Identification of Holliday junction resolvases from humans and yeast. Nature 456 357 361
78. SurteesJA
ArguesoJL
AlaniE
2004 Mismatch repair proteins: key regulators of genetic recombination. Cytogenet Genome Res 107 146 159
79. CobbJA
SchlekerT
RojasV
BjergbaekL
TerceroJA
2005 Replisome instability, fork collapse, and gross chromosomal rearrangements arise synergistically from Mec1 kinase and RecQ helicase mutations. Genes Dev 19 3055 3069
80. GottipatiP
HelledayT
2009 Transcription-associated recombination in eukaryotes: link between transcription, replication and recombination. Mutagenesis 24 203 210
81. AlsfordS
HornD
2007 RNA polymerase I transcription stimulates homologous recombination in Trypanosoma brucei. Mol Biochem Parasitol 153 77 79
82. AlsfordS
HornD
2008 Single-locus targeting constructs for reliable regulated RNAi and transgene expression in Trypanosoma brucei. Mol Biochem Parasitol 161 76 79
83. Gonzalez-BarreraS
Garcia-RubioM
AguileraA
2002 Transcription and double-strand breaks induce similar mitotic recombination events in Saccharomyces cerevisiae. Genetics 162 603 614
84. GottipatiP
CasselTN
SavolainenL
HelledayT
2008 Transcription-associated recombination is dependent on replication in Mammalian cells. Mol Cell Biol 28 154 164
85. PradoF
AguileraA
2005 Impairment of replication fork progression mediates RNA polII transcription-associated recombination. EMBO J 24 1267 1276
86. PalmerGH
BraytonKA
2007 Gene conversion is a convergent strategy for pathogen antigenic variation. Trends Parasitol 23 408 413
87. WirtzE
LealS
OchattC
CrossGAM
1999 A tightly regulated inducible expression system for dominant negative approaches in Trypanosoma brucei. Mol Biochem Parasitol 99 89 101
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 7
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- RNA Virus Replication Complexes
- Functional Genetic Diversity among Complex Clinical Isolates: Delineation of Conserved Core and Lineage-Specific Transcriptomes during Intracellular Survival
- Extreme CD8 T Cell Requirements for Anti-Malarial Liver-Stage Immunity following Immunization with Radiation Attenuated Sporozoites
- A Systems Immunology Approach to Plasmacytoid Dendritic Cell Function in Cytopathic Virus Infections
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy