
Genome-Wide Mutagenesis Reveals That ORF7 Is a Novel VZV Skin-Tropic Factor
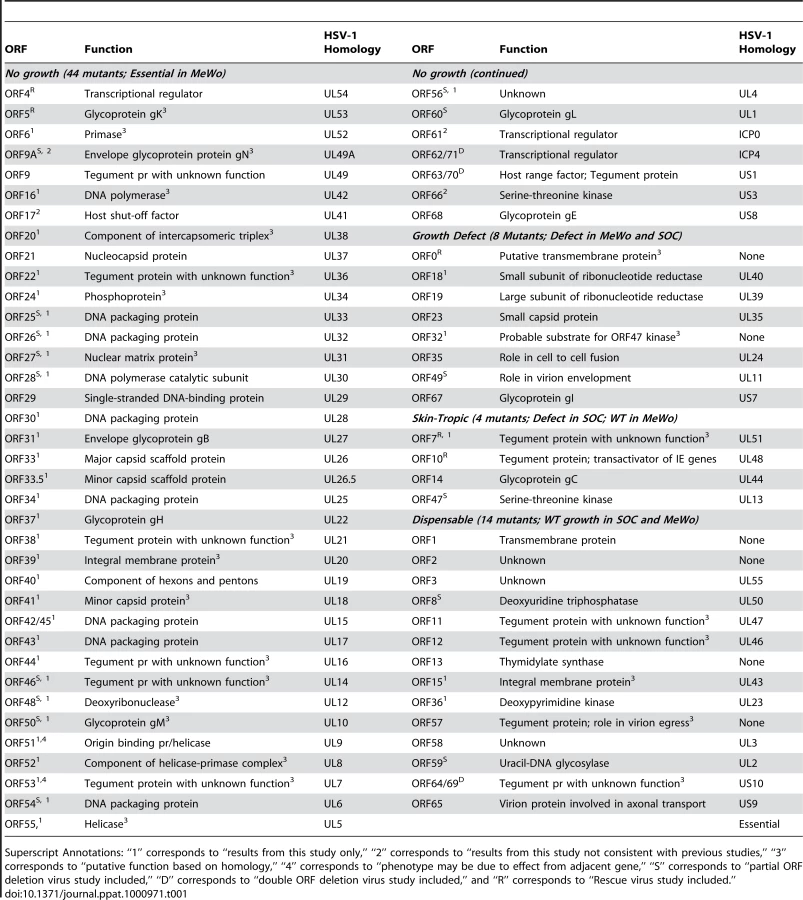
The Varicella Zoster Virus (VZV) is a ubiquitous human alpha-herpesvirus that is the causative agent of chicken pox and shingles. Although an attenuated VZV vaccine (v-Oka) has been widely used in children in the United States, chicken pox outbreaks are still seen, and the shingles vaccine only reduces the risk of shingles by 50%. Therefore, VZV still remains an important public health concern. Knowledge of VZV replication and pathogenesis remains limited due to its highly cell-associated nature in cultured cells, the difficulty of generating recombinant viruses, and VZV's almost exclusive tropism for human cells and tissues. In order to circumvent these hurdles, we cloned the entire VZV (p-Oka) genome into a bacterial artificial chromosome that included a dual-reporter system (GFP and luciferase reporter genes). We used PCR-based mutagenesis and the homologous recombination system in the E. coli to individually delete each of the genome's 70 unique ORFs. The collection of viral mutants obtained was systematically examined both in MeWo cells and in cultured human fetal skin organ samples. We use our genome-wide deletion library to provide novel functional annotations to 51% of the VZV proteome. We found 44 out of 70 VZV ORFs to be essential for viral replication. Among the 26 non-essential ORF deletion mutants, eight have discernable growth defects in MeWo. Interestingly, four ORFs were found to be required for viral replication in skin organ cultures, but not in MeWo cells, suggesting their potential roles as skin tropism factors. One of the genes (ORF7) has never been described as a skin tropic factor. The global profiling of the VZV genome gives further insights into the replication and pathogenesis of this virus, which can lead to improved prevention and therapy of chicken pox and shingles.
Published in the journal:
. PLoS Pathog 6(7): e32767. doi:10.1371/journal.ppat.1000971
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1000971
Summary
The Varicella Zoster Virus (VZV) is a ubiquitous human alpha-herpesvirus that is the causative agent of chicken pox and shingles. Although an attenuated VZV vaccine (v-Oka) has been widely used in children in the United States, chicken pox outbreaks are still seen, and the shingles vaccine only reduces the risk of shingles by 50%. Therefore, VZV still remains an important public health concern. Knowledge of VZV replication and pathogenesis remains limited due to its highly cell-associated nature in cultured cells, the difficulty of generating recombinant viruses, and VZV's almost exclusive tropism for human cells and tissues. In order to circumvent these hurdles, we cloned the entire VZV (p-Oka) genome into a bacterial artificial chromosome that included a dual-reporter system (GFP and luciferase reporter genes). We used PCR-based mutagenesis and the homologous recombination system in the E. coli to individually delete each of the genome's 70 unique ORFs. The collection of viral mutants obtained was systematically examined both in MeWo cells and in cultured human fetal skin organ samples. We use our genome-wide deletion library to provide novel functional annotations to 51% of the VZV proteome. We found 44 out of 70 VZV ORFs to be essential for viral replication. Among the 26 non-essential ORF deletion mutants, eight have discernable growth defects in MeWo. Interestingly, four ORFs were found to be required for viral replication in skin organ cultures, but not in MeWo cells, suggesting their potential roles as skin tropism factors. One of the genes (ORF7) has never been described as a skin tropic factor. The global profiling of the VZV genome gives further insights into the replication and pathogenesis of this virus, which can lead to improved prevention and therapy of chicken pox and shingles.
Introduction
Human varicella-zoster virus (VZV) is a widespread human alpha-herpesvirus, and the majority of the US population has been previously exposed [1]. VZV is the causative agent of chicken pox and shingles, the latter of which is associated with a significant incidence of post-herpetic neuralgia [2], [3]. A universal chicken pox vaccine (v-Oka strain) was first introduced to the United States in 1995, and this immunization program has dramatically reduced chicken pox incidence [4]–[10]. However, outbreaks of chicken pox are still seen [11]–[13], and shingles remains an important concern because the current shingles vaccine only reduces the risk of infection by about 50% [14]. Therefore, VZV is still an important pathogen and remains a public health concern in the U.S. [7], [15]. A better understanding of the biology and pathogenesis of VZV is essential to improve the medical prevention and the treatment of VZV infections.
VZV is the smallest member of the human herpesvirus family, with a linear double-stranded DNA genome (125 kb) that encodes 70 unique ORFs. As a result of the recent development of a VZV cosmid system and of the severe combined immunodeficient mouse model with xenografts of human tissue (SCID-hu), many viral ORFs have been investigated in both biochemical and functional studies, shedding light upon several VZV gene functions [16]–[18]. However, the majority of VZV's 70 unique ORFs have not been studied, and their roles in viral replication and cell-/tissue-specific pathogenesis remain unclear. This is partly due to the absence of an efficient genetic tool to quickly isolate a large number of mutants and a true animal model to screen for in vivo virulence factors on a large scale [2]. Though the functions of many ORFs can only be predicted based on their homologies to other herpesviruses, such as herpes simplex virus 1, our direct manipulation of VZV's ORFs has enabled us to provide functional annotations for the entire VZV genome.
The knowledge of VZV replication and pathogenesis is limited, in part because of its highly cell-associated nature in cultured cells and the difficulty of generating recombinant viruses. In order to circumvent some of these problems, we cloned the VZV (p-Oka strain) genome as a bacterial artificial chromosome (BAC) carrying both green fluorescent protein (GFP) and luciferase reporter genes [19]. We then systematically deleted every open reading frame in the VZV genome. An overview of our method for genome-wide mutagenesis is shown in Figure 1. With a highly efficient homologous recombination system and the dual-reporter system, the recombinant viruses were isolated and analyzed.

Human fetal skin organ culture (SOC) has been previously established to mimic VZV skin infection, which allows for the study of VZV replication and pathogenesis [20]. We further combined SOC with the luciferase assay-based viral detection method to facilitate screening of skin tropism determinants. Although many investigators utilize SCID-hu models (grafts of human tissue in severe combined immunodeficient mice) to study VZV pathogenesis in vivo [21], SOC is a more suitable and cost efficient approach for genome-wide screening the VZV mutant phenotypes. Nevertheless, any interesting findings can be further verified by further in-depth SCID-hu model studies.
The luciferase VZV BAC (VZVLuc) was used to individually delete and/or mutate each of the 70 unique ORFs by employing the E. coli DY380 strain recombination system [22]. As a result, a library of whole-ORF deletion mutants was created. Each mutant DNA obtained from E. coli was transfected into human melanoma (MeWo) cells, and the results provide direct evidence of that 44 ORFs are essential for viral replication in cultured MeWo cells and 26 are non-essential. Moreover, among the non-essential gene group, 8 ORF deletion mutants showed significant growth defects compared to the wild-type strain (p-value <6.07×10−21; see “Statistical Analysis of Mutant Growth Kinetics” section in Materials and Methods), while 18 ORFs were dispensable. All 26 non-essential ORF deletion mutant VZV variants obtained have been tested in SOC. Interestingly, four ORFs were found to be required for optimal viral replication in cultured skin tissue samples, but not in MeWo cells, suggesting their potential roles as skin tropism factors. The results obtained from this study are in agreement with most of those regarding these particular ORFs that have been published to date, and we have provided explanations of all possible discrepancies in the literature. Overall, we provide 51% novel functional annotations to the VZV proteome (36 ORFs).
Results
Generation of VZV ORF deletion and rescue mutants
All VZV ORF deletion mutants were constructed from BAC mutants with a luciferase reporter (VZVLuc) using a PCR-based approach [19], [22] (also see Supplementary Text S1). Construction of ORF rescued BAC mutants was carried out by adapting a two-step homologous recombination approach in E. coli [19], [22] (also see Supplementary Text S1). The generation of a rescue virus is important in order to prove that the deleted fragment was responsible for any growth defect observed in analyses of the mutants. The rescue virus should be able to fully restore the wild-type phenotypes. Because of the large number of ORFs, we chose a small subset of VZV open reading frames to rescure and we have shown these rescue mutants behave as the wild –type strain. A detailed description of these protocols is provided in [19], [22] and an overview is shown in Figure 1. Previous studies in our laboratory have shown that the BAC mutant has an identical growth curve to the wild-type virus [19] and that addition of the luciferase reporter to the BAC virus does not change its growth properties [22].
Identification of essential VZV genes
All of VZV's 70 unique ORFs were deleted and analyzed based on a bioluminescence detection method, as described previously [19]. For 14 ORFs that overlap with adjacent ORFs (ORF8, ORF9A, ORF25, ORF26, ORF27, ORF28, ORF46, ORF47, ORF48, ORF49, ORF50, ORF54, ORF59 and ORF60), respective partial ORF deletions have been constructed and analyzed. A detailed description of these partial ORFs is included in Supplementary Table S2. The results suggest that 44 ORFs are essential for viral replication in cultured MeWo cells (Table 1 and Figure 2). We have confirmed that ORF4 and ORF5 are essential by making genetic rescue viruses. For the essential group, we provide novel functional annotations for 31 of 44 ORFs. All of these VZV essential genes have HSV-1 homologies (Table 1), and the majority of them are conserved among other herpesviruses. These ORFs encode important viral structural proteins, enzymes involved in DNA replication, and transcriptional regulatory proteins.

Among VZV's 44 essential ORFs, the majority encodes proteins with vital functions throughout the viral life cycle. Most VZV proteins that regulate transcription (ORF4, ORF62/71, ORF63/70, and ORF 61) were found to be essential in this study. ORF4 and ORF62/71 are incorporated into the viral tegument, and both encode immediate-early (IE) proteins with transcriptional regulatory activity [23]–[26]. ORF4 and ORF62/71 have been extensively studied, and their essential natures have been suggested previously [27], [28]. Both ORF63/70 and ORF61 encode phosphoproteins primarily localized to the nuclei of infected cells [25]. Although it has been suggested that ORF 63/70 is not essential for viral replication in vitro [29], we could not generate a viable virus from a 63/70 double deletion; this result is in agreement with several other studies [30], [31].
Most of the VZV ORFs that encode glycoproteins are essential. Glycoprotein K (gK) (encoded by ORF5) [32], gB (ORF31), gH (ORF37), gM (ORF50) [33], gL (ORF60) [34], [35], and gE (ORF68) [32], [36] are required for viral replication, and many of them had previously been investigated and reported. Only glycoprotein C (ORF14) [37], [38] and gI (ORF67) [36], [39], [40] deletion mutants produced viable viral progenies, and both of these mutants appeared to suffer a severe growth defect. The results regarding the essentiality of VZV glycoprotein genes in this study are in agreement with the published data.
Essential VZV genes have significantly different enrichment for functional categories than do non-essential genes (Figure 3A). In order to make this calculation, we first listed every gene in a functional category, such as “DNA replication” for a DNA polymerase gene. Then, we compared the proportion of essential (and then of non-essential) genes in each functional category to the background rate expected by chance (e.g. the proportion of genes in that functional category for the entire VZV genome). This calculation was performed using a hypergeometric test. For example, essential genes are significantly enriched for DNA replication (Bonferroni corrected p-value <10−4) and for DNA packaging (Bonferroni corrected p-value <10−4); ORF28 encodes the catalytic subunit of VZV DNA polymerase and ORF16 encodes the subunit of the viral DNA polymerase processivity factor [2]. DNA binding proteins include proteins encoded by ORF6 (primase), ORF29, ORF33 (capsid protein), ORF41 (capsid protein), ORF51 (helicase), ORF52 (component of helicase/primase complex), and ORF55 (component of helicase/primase complex) [41], [42]. Not surprisingly, almost all of the ORFs that encode DNA packaging proteins—including ORF25, ORF26, ORF30, ORF34, ORF42/45, ORF43, ORF54, and nucleocapsid proteins including ORF21, ORF33.5, and ORF40—also fall into the essential gene category. In contrast, non-essential genes were significantly enriched for other (Bonferroni corrected p-value <10−3) and unknown (Bonferroni corrected p-value <0.01) functional categories (Figure 3B).

Identification of non-essential genes
In this study, we found that 26 ORFs are non-essential genes and 6 of these lack HSV-1 homologies (ORF0, ORF1, ORF2, ORF13, ORF32, and ORF57) (Table 1). According to the growth kinetics (in cultured MeWo cells), 8 ORF mutants had significant growth defects (p-value <6.07×10−21), and the peak signals of the viral detection assay were at least 5-fold less than were those of the wild-type parental strain (Figure 4A). Two of these VZV ORF deletion growth phenotypes, ORF18 and ORF32, have not been previously reported, and two others (ORF23 and ORF35 deletions) have been confirmed to have growth defects in vitro, which is in accordance with previously published data [43], [44]. ORF0 deletion's growth defect has been confirmed by making its genetic revertant [19]. ORF18 and ORF19 respectively encode the small and large subunits of ribonucleotide reductase, and both of them diminished viral growth when deleted in this study. The result on ORF19 is in accordance with previous publications [45]. ORF32 encodes a phosphoprotein that is post-translationally modified by ORF47 protein kinase [46]. Among these 8 viral mutants showing severe growth defects, atypical morphology of virally infected cells was frequently observed, including reduced plaque sizes and altered syncytia formation.
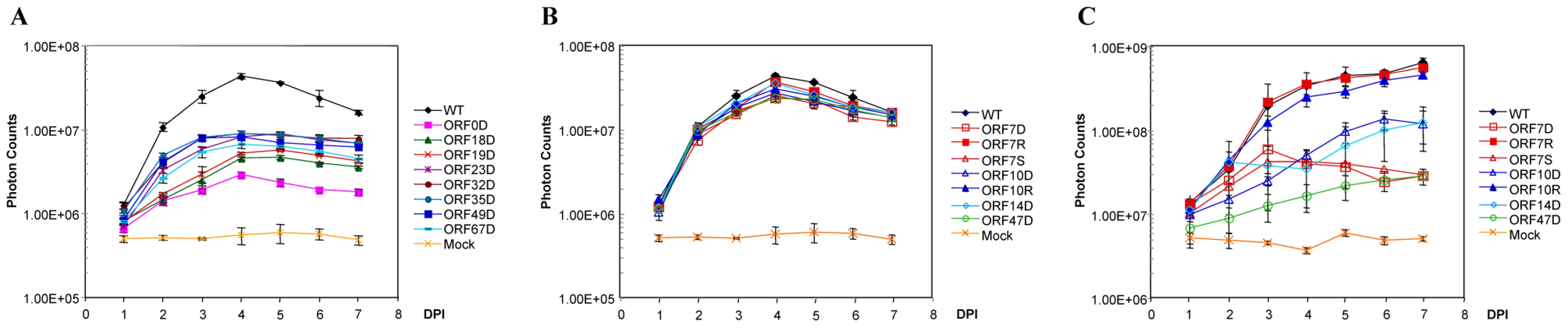
The remaining 18 VZV ORFs had wild type growth curves for viral replication in cultured MeWo cells. In vitro growth curve analysis showed that these ORF deletion mutants had the same growth kinetics as their wild-type parental strain, VZVLuc (Table 1). Previous studies have reported that 15 of these genes (ORF1, ORF2, ORF3, ORF8, ORF10, ORF11, ORF12, ORF13, ORF14, ORF47, ORF57, ORF59, ORF58, ORF64/69, and ORF65) are non-essential [17], [31], [38], [46]–[54]. In this study, three of these ORF mutants (ORF7, ORF15, and ORF36) have been shown to be dispensable for in vitro viral replication for the first time.
The human fetal skin organ culture (SOC) model has been proven to be a simple and convenient alternative to the SCID-hu mouse model in the study of VZV pathogenesis [20], especially in the case of an initial genome-wide screening for skin tropism determinants. Although 26 VZV ORFs were found to be non-essential for viral replication in cultured MeWo cells, it was possible that some of these viral genes encode proteins critical for optimal viral infection in skin tissue. To test this hypothesis, all 26 non-essential ORF deletion viruses were further analyzed in cultured skin-tissue samples.
Every deletion mutant that showed severe growth defects in cultured MeWo cells also demonstrated significantly slow growth kinetics in human skin samples when compared to wild-type VZV (p-value <9.05×10−19). Only two of these genes (ORF35 and ORF67) have been previously reported to be required for viral growth in SCID-hu skin mouse models [40], [44]. Therefore, we have been able to provide novel functional annotations for the other 6 deletion mutants with severe growth defects.
Many non-essential genes appear to cluster together, particularly between ORF0 to ORF15 (Figure 2). More than 70% of ORFs (12 out of 17) in this region are non-essential, compared to 37% of the entire genome. Four out of six VZV ORFs without HSV-1 homologues are also located in this region, so this region may be more evolutionarily variable compared to other highly conserved regions
Among the 18 VZV ORFs dispensable for viral replication in cultured MeWo cells, 14 were also dispensable for viral replication in skin tissue (Table 1). Among the above 14 deletion mutants, we have been able to provide novel ex vivo functional annotations for all but one of these 18 genes (ORF64/69) [31].
Identification of skin-tropic genes
Interestingly, among non-essential VZV ORFs, four ORFs (ORF7, ORF10, ORF14, and ORF47) appear to have selective impacts on viral replication in skin tissue. The growth of each virus in SOC was compared with its growth in cultured MeWo cells. These ORF deletion mutants grew like the wild-type strain in vitro (Figure 4B). In contrast, they showed significant growth defects in skin organ cultures (p-value <9.51×10−19; Figure 4C). For instance, the ORF10 deletion mutant had a growth defect in SOC. The bioluminescence signal kept increasing during the entire 7-day experiment period; the total photon count values consistently remained approximately 10-fold less than those of the wild-type strain. The ORF7 deletion mutant virus quickly reached its growth peak around 3 days after inoculation, and then bioluminescence steadily declined. To prove that the VZV ORF7 and ORF10 growth defects observed in cultured skin-tissue samples were due to the functions of the deleted genes rather than to undesirable mutations in other regions of the genome, rescue viruses ORF7R and ORF10R were generated. The growth curve analysis indicated that ORF7R and ORF10R viruses grew in MeWo cells indistinguishably from wild-type VZV, as expected (Figure 4B). In skin organ cultures, they were also able to fully recover the growth defects of the corresponding deletion mutant viruses and grew as well as the wild-type strain (Figure 4C). In contrast, ORF47 deletion virus had a more severe growth defect, approximately 80–100 fold (2 log) less than wild-type VZV. Our results suggest these three ORFs are important for viral replication in human skin tissue but not in cultured MeWo cells.
Three skin-tropic virulence factors (ORF10, ORF14, and ORF47) have been previously identified. VZV ORF10 encodes a tegument protein that enhances transactivation of VZV genes, and it was shown to be dispensable for VZV replication in vitro [47]. Recent studies showed that ORF10 protein is required for efficient VZV virion assembly and is a specific determinant of VZV virulence in SCID-hu skin xenografts but not in human T cells in vivo [55], [56]. ORF14 (gC) has been reported to have reduced infectivity in an SCID-hu skin model [38]. VZV ORF47 encodes a serine/threonine protein kinase and was shown to be dispensable for viral replication in cultured MeWo cells [48]. It has been designated as a virulence factor for both skin tissue and T cells in SCID-hu models [38]. The findings of these three skin-tropic ORFs not only confirmed previous studies but also further verified the similarity between SCID-skin and SOC systems.
In the current study, ORF7 has been identified as a novel skin-tropic virulence factor. In order to confirm the accuracy of our results, we also produced a premature stop-codon mutant (ORF7S) by mutating the 5th codon from TGT to the TGA stop codon (see Table S1). Just like ORF7D, ORF7S showed wild-type growth in MeWo (Figure 4B) but had a growth defect in SOC (Figure 4C).
Discussion
In this study, a global functional analysis of the entire VZV genome was performed that emphasized the identification of viral ORFs important for viral replication both in cultured MeWo cells and human fetal skin organs. We took full advantage of the highly efficient luciferase VZV BAC system and obtained a library of single ORF deletion mutants. Advanced live culture bioluminescence imaging technology allowed us to systematically test a large number of mutant viruses for comparing viral growth kinetics in different systems.
VZV has a 125-kb DNA genome encoding 70 unique open reading frames (Table 1, Figure 2). In this study, all of the predicted 70 ORFs were individually deleted. Our results directly showed that 44 ORFs encode essential genes and 26 ORFs encode non-essential genes. Among the non-essential group, 8 ORF deletion mutants suffered severe growth defects in MeWo cells. Fourteen ORFs were shown to be dispensable for viral replication, both in MeWo cells and in SOC. We also found 4 tissue tropic factors (ORF7, ORF10, ORF14, and ORF47) that showed a growth defect in SOC but normal growth in MeWo. Three of these tissue-tropic factors (ORF10, ORF14, and ORF47) have been previously identified, but ORF7 has never been previously studied.
In the current study, we have reported ORF7 as a novel VZV skin-tropic factor. ORF7 encodes a 29-kDa tegument protein, and its function remains unknown. The homolog of the VZV ORF7 protein in the herpes simplex virus is the UL51 protein. Recent studies showed that deletion of HSV-1 UL51 causes reduced size plaque formation and low infectivity [56]. Similarly, the function of the UL51 gene product of the pseudorabies virus (PrV) has been investigated by generating a deletion mutant, and the result suggested that the UL51 protein is involved in viral egress, but is not essential for viral replication [57]. Our result suggests that VZV ORF7 might serve as a skin-specific virulence factor. However, the role of ORF7 in pathogenesis needs further investigation.
Despite the large differences between herpesvirus genomes (ranging from 125 kb to >230 kb), all the herpes viruses studied thus far have a similar number of essential genes. For example, HSV-1 encodes 37 essential genes and 48 non-essential genes [58]; human cytomegalovirus (HCMV), which is one of the largest human DNA viruses, encodes 45 essential genes and 117 non-essential genes [59]. Our data suggest that VZV, which contains the smallest genome, encodes 44 essential genes and 26 non-essential genes. A comparison between the essentiality of HSV-1 and HCMV homologues to essential VZV genes is provided in Supplementary Table S3. Of the 44 essential genes, 26 have essential homologues in HSV, and all essential gene homologues conserved in CMV (18 of 44 essential VZV genes) are essential. Therefore, we believe that several of these essential genes perform core functions for all of these herpesviruses.
Unlike the other functional profiling studies performed on HCMV [59], our results did not reveal any VZV-encoded factors that repress viral replication in cultured MeWo cells or in human fetal skin tissue. If such VZV temperance genes existed, enhanced growth kinetics should have been observed by making the corresponding ORF deletion mutants.
There is also an apparent size difference between essential and non-essential ORFs. Essential ORFs are significantly larger in size compared to non-essential ones (μ = 1250 bp vs. μ = 970 bp, respectively, p = 6×10−4 by t-test). The 10 largest VZV ORFs are all essential, while 8 out of 11 VZV ORFs less than 600 bp are non-essential.
All of our results are in agreement with previous VZV functional annotations, except for those on ORFs 9A, 17, 61 and 66, for which we could not generate viral deletion mutant progenies with sufficient titers for growth studies. For example, previous studies indicated that was ORF9A not essential viral growth in cell culture (due to insertion of a premature stop codon) yet they also showed that failure to express either of these genes resulted in growth defects [60]. Therefore, we believe that our findings are at least in partial agreement with previous studies because this previous study utilized a premature stop codon (thus allowing expression of a partial protein), whereas we completely removed ORF9A from the VZV genome.
Although some studies have shown ORF17 to be dispensable for viral replication [61], other studies have shown the gene to be essential for growth under certain conditions [62]. Therefore, we believe this discrepancy can probably be explained by subtle differences in experimental design (such as the temperature of the growth culture, as described in [62], and we believe that our analysis for ORF17 deletion best reflects conditions in vivo.
ORF61 has also been suggested to be a non-essential gene for viral replication in vitro in a previous study [63], [64]. However, we could not retrieve enough infectious viral progeny from the ORF61 deletion clone, even after repeated transfection and extensive incubation. Large deletion mutants of ORF61 [63] and promoter bashing experiments [64] have shown ORF61 to be important for viral replication (albeit non-essential) due to a considerable growth defect shown in the deletion. However, no complete deletion virus has ever been created, so it is possible that the large deletions may have only been sufficient to cause a growth defect, whereas our complete deletion results in a complete loss of VZV replication.
ORF66 has been previously cited as dispensable for viral replication, but we have found it to be essential [65]–[67]. In previous studies, a premature stop codon mutant of ORF66 resulted in a decrease in viral titer, but not in a complete loss of viral replication [65], [66]. Premature stop codons were inserted such that more than 50% of the original coding sequence remained and was able to be expressed, so we believe this discrepancy can be explained by the possible attenuated activity of the partial protein (which did have a substantial growth defect), while our ORF66 deletion removed the entire sequence. For the cosmid-based studies [67], [68], a premature stop codon mutant (with a 21-amino acid partial protein expressed) had to be used to assess the impact of ORF66 on viral replication. However, the authors [68] were also unable to produce infectious virus with a complete ORF66 deletion mutant (which is identical to our results).
In this study, we have presented novel functional annotations for 36 VZV genes. Due to the global nature of our study and the lack of well-defined upstream and downstream regulatory regions for most VZV genes, some of our annotations may have to be redefined by more detailed studies (genes most likely to be affected by adjacent genes are specifically noted in Table 1). Moreover, the current profiling study has provided the first global view of VZV genomic functions in viral replication, which is likely to serve as the basis for further investigative studies on many VZV genes.
Materials and Methods
Cells, virus and PCR primers
Human melanoma (MeWo) cells were grown in DMEM supplemented with 10% fetal calf serum, 100U of penicillin-streptomycin/ml, and 2.5ug of amphotericin B/ml, as previously described, and used to propagate VZV in vitro [18], [69]. VZVLuc containing the entire p-Oka VZV genome was constructed as previously described [19]. Recombinant VZVLuc virus was derived by transfection methods [19], [22] (also see Supplementary Text S1). All primer sequences are listed in Supplementary Table S1. Primer sequences were designed based upon the Dumas VZV strain (Accession Number: NC_001348).
Growth analysis of viral mutants in vitro
VZVLuc DNAs were transfected into MeWo cells using the FuGene 6 transfection kit (Roche, Indianapolis, IN) [19], [22] (also see Supplementary Text S1). Recombinant viruses were titrated by infectious focus assay. MeWo cells were seeded in 6-well tissue culture plates and inoculated with serial dilutions of VZV-infected MeWo cell suspensions. Plaques were counted by fluorescence microscopy at 3 days after inoculation. All transfections were performed a minimum of 3 times. Since VZV is highly cell-associated under tissue culture conditions, mutant VZV-infected MeWo cells were harvested, titrated and stored in liquid nitrogen. Wild-type infections served as positive controls and mock infections served as negative controls.
In vitro growth curve analyses were carried out by live-cell bioluminescence detection assay. MeWo cells were infected with 100 PFU of infected MeWo cell suspensions on 6-well tissue culture plates. Every 24 h, the cell culture medium was replaced with medium containing 150 ug/ml D-luciferin (Xenogen, Alameda, CA). After incubation at 37°C for 10 min, the bioluminescent signals were quantified and recorded using an IVIS Imaging System (Xenogen), following the manufacturer's instructions. After each measurement, the luciferin-containing medium was replaced with fresh cell culture medium. Measurements were taken daily from the same plate for 7 days. Bioluminescence signal data from each sample were quantified by manually demarcating regions of interest and analyzed using LivingImage analysis software (Xenogen). It has been demonstrated previously that both the infectious center assay and the luciferase assay correlate well [19], [22].
Growth analysis of viral mutants in SOC
Human fetal skin-tissue samples (∼20 weeks gestational age) were acquired from Advance Biosciences Resources (Alameda, CA). Skin organ-culture techniques were as previously described [20]. Ex vivo growth curve analyses were carried out by live-tissue bioluminescence assay. Infected MeWo cells were titrated and then re-suspended in skin organ culture media (SOCM). After 24 h of incubation, each skin-tissue section was injected five times with 10 ul of the virus-infected cell suspension (total inoculation was 5×103 PFU per tissue) by a 1-ml syringe fitted with a 27-gauge needle attached to a volumetric stepper (Tridak, Brookfield, CT). After inoculation, the sections were placed individually on 500 um mesh NetWell inserts (Corning, Corning, NY) that rested above 1ml of SOCM in each well of 12-well plates and followed by a 24 h incubation in a tissue culture incubator, 37°C, 5%CO2. Each 24 h, SOCM was replaced with media containing 150ug/ml of D-luciferin. Following 10 min incubation at 37°C, the bioluminescence being emitted from individual cultured skin-tissue samples was recorded using the IVIS Imaging System. After the measurements, each sample (still on a NetWell insert) was transferred onto new 12-well plates with fresh SOCM. The measuring process was repeated every 24 h for 7 days. Bioluminescence signals from manually defined regions of interest were quantified and analyzed. All experiments were performed in triplicate. Wild-type infections served as positive controls and mock infections served as negative controls.
Statistical analysis of mutant growth kinetics
Wild type and mutant growth curves (7 time points, 3 replicates each) were compared using the “timecourse” Bioconductor package [70], [71]. The difference in growth rate for wild type and mutant growth curves was estimated by the mb.long function was used to estimate a Hotelling T2 test statistic using the mb.long function. P-values for the T2 test statistic were calculated using an F-distribution. The T2 test statistic did an excellent job of quantifying the difference in growth curves, but a very strict p-value cutoff was required in order to define statistically significant growth defects (implying that the test statistic may be too sensitive). Therefore, we used a Mann-Whitney U test in order to determine which individual time points significantly differed between wild type and deletion mutant strains. All strains with reported growth defects have at least 6 significantly reduced time points (p<0.05).
Supporting Information
Zdroje
1. AbendrothA
ArvinA
1999 Varicella-zoster virus immune evasion. Immunol Rev 168 143 156
2. CohenJI
StrausSE
ArvinAM
2007 Varicella-Zoster Virus Replication, Pathogenesis, and Management.
KnipeDM
HowleyPM
Fields Virology. 5th ed Philadelphia, PA Lippincott Williams & Wilkins 2773 2818
3. GildenDH
Kleinschmidt-DeMastersBK
LaGuardiaJJ
MahalingamR
CohrsRJ
2000 Neurologic complications of the reactivation of varicella-zoster virus. N Engl J Med 342 635 645
4. Centers for Disease Control and Prevention 2003 Decline in annual incidence of varicella—selected states, 1990–2001. MMWR Morb Mortal Wkly Rep 52 884 885
5. GershonAA
ArvinAM
ShapiroE
2007 Varicella vaccine. New England Journal of Medicine 356 2648 2649
6. LopezAS
KolasaMS
SewardJF
2008 Status of school entry requirements for varicella vaccination and vaccination coverage 11 years after implementation of the varicella vaccination program. J Infect Dis 197 Suppl 2 S76 81
7. MarinM
MeissnerHC
SewardJF
2008 Varicella prevention in the United States: a review of successes and challenges. Pediatrics 122 e744 751
8. SewardJF
WatsonBM
PetersonCL
MascolaL
PelosoJW
2002 Varicella disease after introduction of varicella vaccine in the United States, 1995–2000. Jama-Journal of the American Medical Association 287 606 611
9. VazquezM
LaRussaPS
GershonAA
NiccolaiLM
MuehlenbeinCE
2004 Effectiveness over time of varicella vaccine. Jama-Journal of the American Medical Association 291 851 855
10. VazquezM
LaRussaPS
GershonAA
SteinbergSP
FreudigmanK
2001 The effectiveness of the varicella vaccine in clinical practice. New England Journal of Medicine 344 955 960
11. GalilK
LeeB
StrineT
CarraherC
BaughmanAL
2002 Outbreak of varicella at a day-care center despite vaccination. New England Journal of Medicine 347 1909 1915
12. GershonAA
SteinbergSP
GelbL
GalassoG
BorkowskyW
1984 Live Attenuated Varicella Vaccine - Efficacy for Children with Leukemia in Remission. Jama-Journal of the American Medical Association 252 355 362
13. IzurietaHS
StrebelPM
BlakePA
1997 Postlicensure effectiveness of varicella vaccine during an outbreak in a child care center. Jama-Journal of the American Medical Association 278 1495 1499
14. OxmanMN
LevinMJ
JohnsonGR
SchmaderKE
StrausSE
2005 A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. New England Journal of Medicine 352 2271 2284
15. ArvinAM
2001 Varicella-Zoster Virus.
KnipeDM
HowleyPM
Fields Virology Philadelphia, PA Lippincott Williams & Wilkins 2731 2767
16. NiizumaT
ZerboniL
SommerMH
ItoH
HinchliffeS
2003 Construction of varicella-zoster virus recombinants from parent Oka cosmids and demonstration that ORF65 protein is dispensable for infection of human skin and T cells in the SCID-hu mouse model. J Virol 77 6062 6065
17. CohenJI
SeidelKE
1993 Generation of varicella-zoster virus (VZV) and viral mutants from cosmid DNAs: VZV thymidylate synthetase is not essential for replication in vitro. Proc Natl Acad Sci U S A 90 7376 7380
18. MoffatJF
SteinMD
KaneshimaH
ArvinAM
1995 Tropism of varicella-zoster virus for human CD4+ and CD8+ T lymphocytes and epidermal cells in SCID-hu mice. J Virol 69 5236 5242
19. ZhangZ
RoweJ
WangW
SommerM
ArvinA
2007 Genetic analysis of varicella-zoster virus ORF0 to ORF4 by use of a novel luciferase bacterial artificial chromosome system. J Virol 81 9024 9033
20. TaylorSL
MoffatJF
2005 Replication of varicella-zoster virus in human skin organ culture. J Virol 79 11501 11506
21. ArvinAM
2006 Investigations of the pathogenesis of Varicella zoster virus infection in the SCIDhu mouse model. Herpes 13 75 80
22. ZhangZ
HuangY
ZhuH
2008 A highly efficient protocol of generating and analyzing VZV ORF deletion mutants based on a newly developed luciferase VZV BAC system. J Virol Methods 148 197 204
23. DefechereuxP
MelenL
BaudouxL
Merville-LouisMP
RentierB
1993 Characterization of the regulatory functions of varicella-zoster virus open reading frame 4 gene product. J Virol 67 4379 4385
24. MoriuchiH
MoriuchiM
SmithHA
CohenJI
1994 Varicella-zoster virus open reading frame 4 protein is functionally distinct from and does not complement its herpes simplex virus type 1 homolog, ICP27. J Virol 68 1987 1992
25. MoriuchiH
MoriuchiM
StrausSE
CohenJI
1993 Varicella-zoster virus (VZV) open reading frame 61 protein transactivates VZV gene promoters and enhances the infectivity of VZV DNA. J Virol 67 4290 4295
26. PereraLP
MoscaJD
Sadeghi-ZadehM
RuyechanWT
HayJ
1992 The varicella-zoster virus immediate early protein, IE62, can positively regulate its cognate promoter. Virology 191 346 354
27. SatoB
ItoH
HinchliffeS
SommerMH
ZerboniL
2003 Mutational analysis of open reading frames 62 and 71, encoding the varicella-zoster virus immediate-early transactivating protein, IE62, and effects on replication in vitro and in skin xenografts in the SCID-hu mouse in vivo. J Virol 77 5607 5620
28. SatoB
SommerM
ItoH
ArvinAM
2003 Requirement of varicella-zoster virus immediate-early 4 protein for viral replication. J Virol 77 12369 12372
29. CohenJI
CoxE
PesnicakL
SrinivasS
KrogmannT
2004 The varicella-zoster virus open reading frame 63 latency-associated protein is critical for establishment of latency. J Virol 78 11833 11840
30. BaikerA
BagowskiC
ItoH
SommerM
ZerboniL
2004 The immediate-early 63 protein of Varicella-Zoster virus: analysis of functional domains required for replication in vitro and for T-cell and skin tropism in the SCIDhu model in vivo. J Virol 78 1181 1194
31. SommerMH
ZaghaE
SerranoOK
KuCC
ZerboniL
2001 Mutational analysis of the repeated open reading frames, ORFs 63 and 70 and ORFs 64 and 69, of varicella-zoster virus. J Virol 75 8224 8239
32. MoC
SuenJ
SommerM
ArvinA
1999 Characterization of Varicella-Zoster virus glycoprotein K (open reading frame 5) and its role in virus growth. J Virol 73 4197 4207
33. YamagishiY
SadaokaT
YoshiiH
SomboonthumP
ImazawaT
2008 Varicella-zoster virus glycoprotein M homolog is glycosylated, is expressed on the viral envelope, and functions in virus cell-to-cell spread. J Virol 82 795 804
34. DuusKM
GroseC
1996 Multiple regulatory effects of varicella-zoster virus (VZV) gL on trafficking patterns and fusogenic properties of VZV gH. J Virol 70 8961 8971
35. MaresovaL
KutinovaL
LudvikovaV
ZakR
MaresM
2000 Characterization of interaction of gH and gL glycoproteins of varicella-zoster virus: their processing and trafficking. Journal of General Virology 81 1545 1552
36. MalloryS
SommerM
ArvinAM
1998 Analysis of the glycoproteins I and E of varicella-zoster virus (VZV) using deletional mutations of VZV cosmids. Journal of Infectious Diseases 178 S22 S26
37. GharaviS
SadeghizadehM
HosseinkhaniS
SabahiF
2007 A study of varicella zoster virus glycoprotein C regulatory region response to viral activators in vitro. Pak J Biol Sci 10 2140 2145
38. MoffatJF
ZerboniL
KinchingtonPR
GroseC
KaneshimaH
1998 Attenuation of the vaccine Oka strain of varicella-zoster virus and role of glycoprotein C in alphaherpesvirus virulence demonstrated in the SCID-hu mouse. J Virol 72 965 974
39. MalloryS
SommerM
ArvinAM
1997 Mutational analysis of the role of glycoprotein I in Varicella-Zoster virus replication and its effects on glycoprotein E conformation and tracking. Journal of Virology 71 8279 8288
40. MoffatJ
ItoH
SommerM
TaylorS
ArvinAM
2002 Glycoprotein I of varicella-zoster virus is required for viral replication in skin and T cells. J Virol 76 8468 8471
41. StallingsCL
SilversteinS
2005 Dissection of a novel nuclear localization signal in open reading frame 29 of varicella-zoster virus. J Virol 79 13070 13081
42. RobertsCR
WeirAC
HayJ
StrausSE
RuyechanWT
1985 DNA-binding proteins present in varicella-zoster virus-infected cells. J Virol 55 45 53
43. ChaudhuriV
SommerM
RajamaniJ
ZerboniL
ArvinAM
2008 Functions of Varicella-zoster virus ORF23 capsid protein in viral replication and the pathogenesis of skin infection. J Virol 82 10231 10246
44. ItoH
SommerMH
ZerboniL
BaikerA
SatoB
2005 Role of the varicella-zoster virus gene product encoded by open reading frame 35 in viral replication in vitro and in differentiated human skin and T cells in vivo. J Virol 79 4819 4827
45. HeinemanTC
CohenJI
1994 Deletion of the varicella-zoster virus large subunit of ribonucleotide reductase impairs growth of virus in vitro. J Virol 68 3317 3323
46. ReddySM
CoxE
IofinI
SoongW
CohenJI
1998 Varicella-zoster virus (VZV) ORF32 encodes a phosphoprotein that is posttranslationally modified by the VZV ORF47 protein kinase. J Virol 72 8083 8088
47. CohenJI
SeidelK
1994 Varicella-zoster virus (VZV) open reading frame 10 protein, the homolog of the essential herpes simplex virus protein VP16, is dispensable for VZV replication in vitro. J Virol 68 7850 7858
48. HeinemanTC
CohenJI
1995 The varicella-zoster virus (VZV) open reading frame 47 (ORF47) protein kinase is dispensable for viral replication and is not required for phosphorylation of ORF63 protein, the VZV homolog of herpes simplex virus ICP22. J Virol 69 7367 7370
49. SatoH
PesnicakL
CohenJI
2002 Varicella-zoster virus open reading frame 2 encodes a membrane phosphoprotein that is dispensable for viral replication and for establishment of latency. J Virol 76 3575 3578
50. CohenJI
SeidelKE
1995 Varicella-zoster virus open reading frame 1 encodes a membrane protein that is dispensable for growth of VZV in vitro. Virology 206 835 842
51. CohenJI
SeidelKE
1994 Absence of varicella-zoster virus (VZV) glycoprotein V does not alter growth of VZV in vitro or sensitivity to heparin. J Gen Virol 75(Pt 11) 3087 3093
52. CoxE
ReddyS
IofinI
CohenJI
1998 Varicella-zoster virus ORF57, unlike its pseudorabies virus UL3.5 homolog, is dispensable for viral replication in cell culture. Virology 250 205 209
53. YoshiiH
SadaokaK
MatsuuraM
NagaikeK
TakahashiM
2008 Varicella-zoster virus ORF 58 gene is dispensable for viral replication in cell culture. Virol J 5 54
54. CohenJI
SatoH
SrinivasS
LekstromK
2001 Varicella-zoster virus (VZV) ORF65 virion protein is dispensable for replication in cell culture and is phosphorylated by casein kinase II, but not by the VZV protein kinases. Virology 280 62 71
55. CheX
ZerboniL
SommerMH
ArvinAM
2006 Varicella-zoster virus open reading frame 10 is a virulence determinant in skin cells but not in T cells in vivo. J Virol 80 3238 3248
56. CheXB
BerarducciB
SommerM
RuyechanWT
ArvinAM
2007 The ubiquitous cellular transcriptional factor USF targets the varicella-zoster virus open reading frame 10 promoter and determines virulence in human skin xenografts in SCIDhu mice in vivo. Journal of Virology 81 3229 3239
57. KluppBG
GranzowH
KlopfleischR
FuchsW
KoppM
2005 Functional analysis of the pseudorabies virus UL51 protein. Journal of Virology 79 3831 3840
58. RoizmanB
KnipeDM
WhitleyRJ
2007 Herpes simplex viruses.
KnipeDM
HowleyPM
Fields Virology. 5th ed Philadelphia, PA Lippincott Williams & Wilkins 2399 2460
59. DunnW
ChouC
LiH
HaiR
PattersonD
2003 Functional profiling of a human cytomegalovirus genome. Proc Natl Acad Sci U S A 100 14223 14228
60. RossJ
WilliamsM
CohenJI
1997 Disruption of the varicella-zoster virus dUTPase and the adjacent ORF9A gene results in impaired growth and reduced syncytia formation in vitro. Virology 234 186 195
61. DeslogesN
RahausM
WolffMH
2005 The varicella-zoster virus-mediated delayed host shutoff: open reading frame 17 has no major function, whereas immediate-early 63 protein represses heterologous gene expression. Microbes and Infection 7 1519 1529
62. SatoH
CallananLD
PesnicakL
KrogmannT
CohenJI
2002 Varicella-zoster virus (VZV) ORF17 protein induces RNA cleavage and is critical for replication of VZV at 37 degrees C but not 33 degrees C. Journal of Virology 76 11012 11023
63. CohenJI
NguyenH
1998 Varicella-zoster virus ORF61 deletion mutants replicate in cell culture, but a mutant with stop codons in ORF61 reverts to wild-type virus. Virology 246 306 316
64. WangL
SommerM
RajamaniJ
ArvinAM
2009 Regulation of the ORF61 Promoter and ORF61 Functions in Varicella-Zoster Virus Replication and Pathogenesis. Journal of Virology 83 7560 7572
65. HeinemanTC
SeidelK
CohenJI
1996 The varicella-zoster virus ORF66 protein induces kinase activity and is dispensable for viral replication. Journal of Virology 70 7312 7317
66. KinchingtonPR
FiteK
SemanA
TurseSE
2001 Virion association of IE62, the varicella-zoster virus (VZV) major transcriptional regulatory protein, requires expression of the VZV open reading frame 66 protein kinase. Journal of Virology 75 9106 9113
67. ErazoA
YeeMB
OsterriederN
KinchingtonPR
2008 Varicella-zoster virus open reading frame 66 protein kinase is required for efficient viral growth in primary human corneal stromal fibroblast cells. Journal of Virology 82 7653 7665
68. SchaapA
FortinJF
SommerM
ZerboniL
StamatisS
2005 T-Cell tropism and the role of ORF66 protein in pathogenesis of varicella-zoster virus infection. Journal of Virology 79 12921 12933
69. MarchiniA
LiuH
ZhuH
2001 Human cytomegalovirus with IE-2 (UL122) deleted fails to express early lytic genes. J Virol 75 1870 1878
70. GentlemanRC
CareyVJ
BatesDM
BolstadB
DettlingM
2004 Bioconductor: open software development for computational biology and bioinformatics. Genome Biology 5
71. TaiYC
SpeedTP
2006 A multivariate empirical Bayes statistic for replicated microarray time course data. Annals of Statistics 34 2387 2412
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 7
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- RNA Virus Replication Complexes
- Functional Genetic Diversity among Complex Clinical Isolates: Delineation of Conserved Core and Lineage-Specific Transcriptomes during Intracellular Survival
- Extreme CD8 T Cell Requirements for Anti-Malarial Liver-Stage Immunity following Immunization with Radiation Attenuated Sporozoites
- A Systems Immunology Approach to Plasmacytoid Dendritic Cell Function in Cytopathic Virus Infections
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy