
NK Cells and γδ T Cells Mediate Resistance to Polyomavirus–Induced Tumors
NK and γδ T cells can eliminate tumor cells in many experimental models, but their effect on the development of tumors caused by virus infections in vivo is not known. Polyomavirus (PyV) induces tumors in neonatally infected mice of susceptible strains and in adult mice with certain immune deficiencies, and CD8+ αβ T cells are regarded as the main effectors in anti-tumor immunity. Here we report that adult TCRβ knockout (KO) mice that lack αβ but have γδ T cells remain tumor-free after PyV infection, whereas TCRβ×δ KO mice that lack all T cells develop tumors. In addition, E26 mice, which lack NK and T cells, develop the tumors earlier than TCRβ×δ KO mice. These observations implicate γδ T and NK cells in the resistance to PyV-induced tumors. Cell lines established from PyV-induced tumors activate NK and γδ T cells both in culture and in vivo and express Rae-1, an NKG2D ligand. Moreover, these PyV tumor cells are killed by NK cells in vitro, and this cytotoxicity is prevented by treatment with NKG2D-blocking antibodies. Our findings demonstrate a protective role for NK and γδ T cells against naturally occurring virus-induced tumors and suggest the involvement of NKG2D-mediated mechanisms.
Published in the journal:
. PLoS Pathog 6(5): e32767. doi:10.1371/journal.ppat.1000924
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1000924
Summary
NK and γδ T cells can eliminate tumor cells in many experimental models, but their effect on the development of tumors caused by virus infections in vivo is not known. Polyomavirus (PyV) induces tumors in neonatally infected mice of susceptible strains and in adult mice with certain immune deficiencies, and CD8+ αβ T cells are regarded as the main effectors in anti-tumor immunity. Here we report that adult TCRβ knockout (KO) mice that lack αβ but have γδ T cells remain tumor-free after PyV infection, whereas TCRβ×δ KO mice that lack all T cells develop tumors. In addition, E26 mice, which lack NK and T cells, develop the tumors earlier than TCRβ×δ KO mice. These observations implicate γδ T and NK cells in the resistance to PyV-induced tumors. Cell lines established from PyV-induced tumors activate NK and γδ T cells both in culture and in vivo and express Rae-1, an NKG2D ligand. Moreover, these PyV tumor cells are killed by NK cells in vitro, and this cytotoxicity is prevented by treatment with NKG2D-blocking antibodies. Our findings demonstrate a protective role for NK and γδ T cells against naturally occurring virus-induced tumors and suggest the involvement of NKG2D-mediated mechanisms.
Introduction
Virus-induced tumors mostly develop in immune-compromised hosts, suggesting that the immune system provides protection against the induction and/or progression of these tumors. T cells expressing α and β TCR and recognizing viral peptide epitopes are thought to be important for this protection. However, other cell types of the immune system, including NK cells and γδ T cells, are also endowed with effector functions similar to those of αβ T cells, but their role in the control of virus-induced tumors is largely unexplored.
A growing body of experimental evidence suggests that tumor cells can be recognized and eliminated by NK cells and γδ T cells. In a variety of human cancers such as lung, colon and renal cell carcinomas NK cells and γδ T cells can be found among tumor infiltrating lymphocytes (TIL) [1], [2], [3], [4]. Moreover, NK cell infiltration of tumors was noted to be associated with improved prognosis in some human cancers [4], [5], [6]. Implanted syngeneic tumors, including those induced by tumor viruses, grow more aggressively in mice if no functional NK cells are present [7]. γδ T cells can also protect mice against transplanted hematopoietic tumors [8], and mice deficient in γδ T cells have an increased susceptibility to chemically induced cutaneous tumor formation [9]. Acute virus infections, as well as other NK cell activating agents, can augment the rejection of implanted tumor cells [7]. Nevertheless, evidence that NK and γδ T cells can control the formation and progression of naturally occurring virus-induced tumors is lacking.
Polyomavirus (PyV), a small DNA tumor virus that carries potent oncogenes, can transform a variety of cells in culture readily, but infection of adult immune competent mice (the natural host for PyV) does not lead to tumor formation. However, PyV infection causes a wide variety of tumors affecting multiple tissues and cell types when neonatal mice of some “susceptible” mouse strains are infected, and it also causes tumors in adult mice with certain immune-deficiencies [10], [11]. Neonatal mice of the tumor susceptible mouse strains rapidly gain resistance after birth, and become refractory to tumor induction by the virus within a few days. The importance of the immune system in tumor resistance is indicated by observations that mouse strains highly resistant to PyV-induced tumor formation could be rendered tumor susceptible with immune suppressive treatments such as neonatal thymectomy, irradiation, and administration of anti-lymphocyte serum [12], [13], [14].
A high level of virus replication and spread seems to be a prerequisite for PyV-induced tumor development. Therefore, antiviral immune responses which decrease the virus load and reduce the levels of virus persistence may also decrease the chances of tumor formation. This does not mean, however, that antiviral resistance is always coupled with resistance against tumors and vice versa. For example, antibody responses to PyV reduce the virus load, but they do not prevent tumor formation [15]. CD8 T cells specific for PyV viral peptides, on the other hand, reduce virus load and also have a role in preventing the formation of virus-induced tumors [16], [17], [18]. Endogenous super-antigens encoded by a mouse mammary tumor provirus Mtv-7 have been shown to increase susceptibility of neonatal mice to PyV-induced tumors by eliminating Vβ6+ T cells from their T cell repertoire; Vβ6T cells make up the majority of the CTLs reactive to the dominant middle T peptide epitope in H2k mice [19]. Of note is that in the rejection of PyV-induced tumor cells transferred into PyV-immune mice both CD4 and CD8 αβ T cells were shown to play a role [20]. Consistent with the role of αβ T cells mediating tumor resistance, athymic nude mice infected as adults developed tumors, as did mice lacking β2m, an essential component of MHC class I molecules [17]. However, it is possible that there is a redundancy in antitumor responses and that CD8 T cells specific for viral peptides do not act only by themselves against the PyV-transformed tumor cells. The contribution of various other cell types capable of cytotoxic activity, such as γδ T cells or NK cells to PyV tumor resistance in vivo has not been investigated so far.
Here we report that γδ T and NK cells can prevent tumor formation in PyV-infected αβ T cell-deficient mice. PyV-induced tumor cell lines activate γδ T cells and NK cells in vitro and in vivo. Moreover, these tumor cell lines express Rae-1 ligands recognized by NKG2D, an activating receptor expressed on all NK cells and some γδ T cells, they are efficiently killed by NK cells in vitro and this killing is NKG2D-mediated. These data taken together show a major role for γδ T cells and NK cells in tumor resistance in a naturally occurring virus-induced tumor model, implicate NKG2D-NKG2D ligand interactions in these antitumor responses, and suggest that αβ T cells, γδ T cells and NK cells may work together to prevent tumor formation during life-long virus persistence.
Results
PyV infection induces a high incidence of tumor development in TCRβ×δ KO and E26 (T and NK cell-deficient), but not in TCRβ KO mice
In order to understand which components of the immune system are required to control PyV -induced tumor formation in adult mice we tested the outcome of long-term PyV infection in TCRβ KO mice that lack αβ T cells, TCRβ×δ KO mice that lack all αβ and γδ T cells, and E26 mice that lack all T cells and NK cells [21]. Following intranasal infection with 2×106 p.f.u. of PyV strain A2 (“high tumor” strain) ∼80% of TCRβ KO mice survived for ten months and all of them were free of tumors, whereas only 1 out of 20 (5%) TCRβ×δ KO mouse was alive at this time point and ∼60% of the TCRβ×δ KO mice had developed large tumors (Fig. 1A and B). The tumors were detected by visual observation and palpation, usually when their size exceeded ∼0.5 cm in diameter. The TCRβ×δ KO mice which died but are not counted as tumor bearing had hind leg paralysis, a condition seen in many previous studies [22], [23] and likely due to bone tumors of the spine or possibly other PyV-induced neurological conditions, or died of unknown causes. The tumors found by gross examination were almost all salivary gland tumors, often occurring bilaterally. The lack of a wide spectrum of tumors in these mice is similar to findings previously reported for B6/β2m KO mice infected as adults [22]. Similar results were obtained with mice infected intraperitoneally (i.p.). For example, in a more limited study, 0/8 TCRβ×δ KO mice, 4/5 TCRβ KO mice and 10/10 B6 mice were alive and tumor-free at 5 months post infection when PyV was given i.p. There was an earlier appearance of tumors in E26 mice than in TCRβ×δ KO mice (Fig. 1C), suggesting that NK cells may also contribute to tumor resistance and delay the onset of tumor formation.

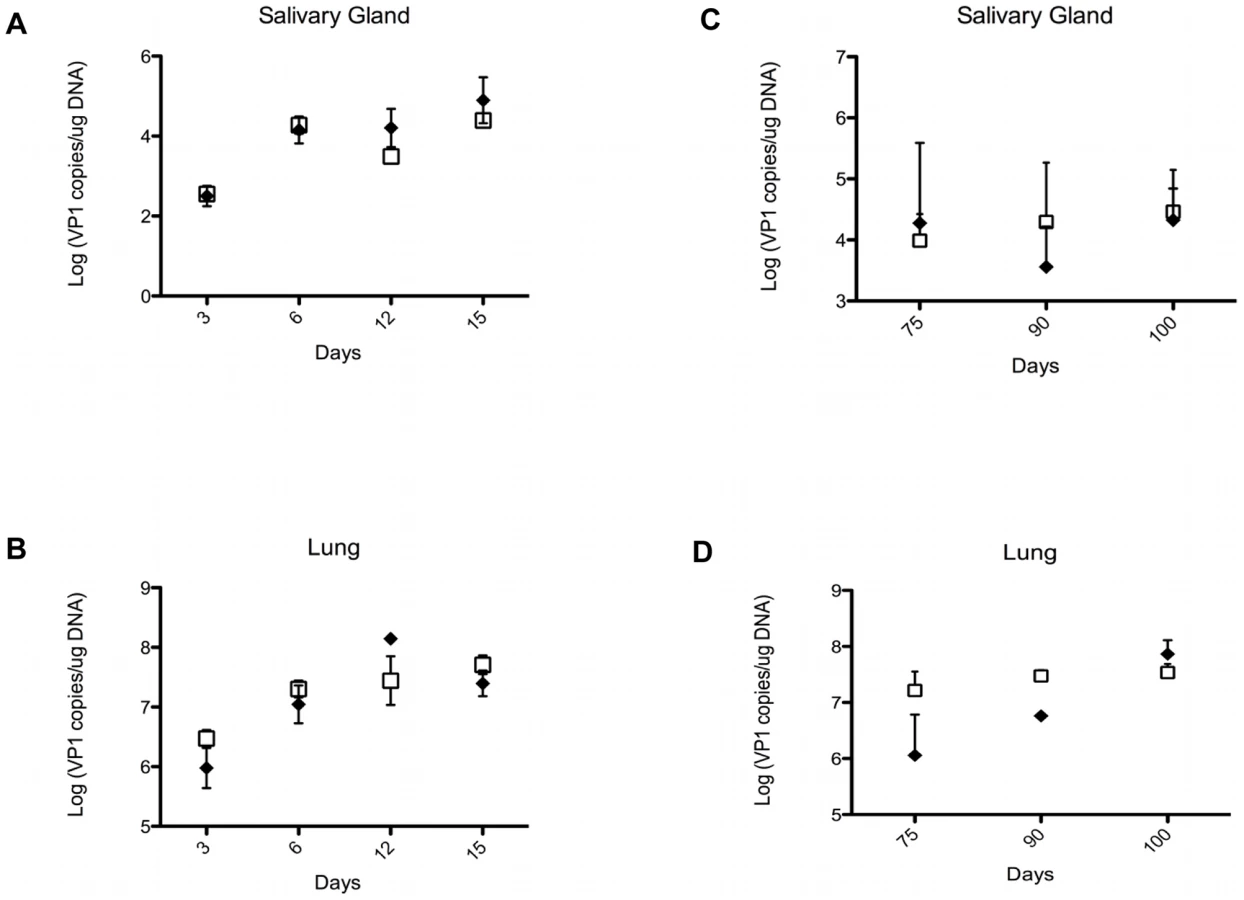
PyV load is not different in organs of TCRβ KO and TCRβ×δ KO mice
NK cells and γδ T cells may provide protection against PyV-induced tumors by directly eliminating the emerging tumor cells, or indirectly, by lowering the PyV titer in various organs and thereby decreasing the chances of cell transformation and tumor development. To test the effect of γδ T cells on viral load we compared the number of PyV genome copies in the salivary glands and lungs of TCRβ KO and TCRβ×δ KO mice at various time points after intranasal PyV infection, but before tumors were detectable in TCRβ×δ KO mice. The number of viral genome copies on days 3, 6, 12 and 15 (Fig. 2A and B; short-term infection) and on days 75, 90 and 100 (Fig. 2C and D; long-term infection), were not significantly different in the lungs or salivary glands between TCRβ KO and TCRβ×δ KO mice. This observation suggests that the γδ T cells provide protection against PyV-induced tumors by controlling tumor development, rather than by reducing viral load in the target organs prior to PyV-induced tumor formation. Previous studies in our lab showed that in vivo NK cell depletion did not increase PyV titers in the kidneys and spleens of SCID mice one week after PyV infection i.p., suggesting that NK cells did not have a direct role in the control of PyV levels in those organs at the acute phase of the infection [24].
PyV-induced salivary gland tumor cell lines express the NKG2D ligand Rae-1
NK cells and γδ T cells may recognize PyV-induced tumor cells by detecting tumor cell antigens on their surface via activating receptors, such as NKG2D. We established several cell lines from salivary gland tumors isolated from PyV-infected TCRβ×δ KO mice and tested them for the expression of the NKG2D ligands Rae-1, H60 and MULT1. These cell lines, PyVTu1, PyVTu2 and PyVTu3, were all positive for Rae-1, H60 and MULT1 transcripts by reverse transcriptase (RT) PCR (Fig. 3A). The Rae-1 specific primers could amplify cDNA reverse transcribed from mRNA of all Rae-1 family members, α, β, γ, δ and ε, and gave the same size PCR product. We also tested the expression of NKG2D ligand surface proteins on PyVTu cells by staining them with monoclonal antibodies against Rae-1, H60 and MULT1. All of the cell lines tested, PyVTu1, PyVTu2 and PyVTu3, had a high expression of Rae-1 but H60 or MULT1 surface protein expression was not detectable on these cell lines despite the abundant H60 and MULT1 transcripts (Fig. 3B). YAC-1 T cell lymphoma cells expressed Rae-1, H60 and MULT1 messages and surface proteins, whereas RMA T cell lymphoma cells did not express Rae-1, H60, only low levels of MULT1 mRNA, and none of these surface proteins, and served as negative controls. NK cells can also detect the expression of class I MHC molecules on their targets and preferentially kill cells displaying low levels of class I MHC molecules. However, we found that PyVTu cells express high levels of class I MHC molecules, unlike YAC-1 cells which have low levels of class I expression (Fig. 3B).

PyVTu cells activate NK cells and γδ T cells in-vitro and in-vivo
To test the ability of PyVTu cells to activate NK cells and γδ T cells in vitro, spleen cells of uninfected TCRβ KO mice were incubated with PyVTu cells with or without PMA and ionomycin treatment, followed by intracellular cytokine staining for IFNγ and granzyme-B. PMA and ionomycin are commonly used for non-specific stimulation of cytokine production in various cell types and they were also reported to increase the IFNγ mRNA half-life in activated NK cells [25]. The PyVTu cells were incubated with spleen leukocytes at a ratio of 10∶1. Spleen cells from TCRβ KO mice were used in these experiments, because they contain a higher percentage of γδ T cells than do spleens of B6 mice. The addition of PyVTu cells and PMA and ionomycin treatment together resulted in a significant increase in the percentage of IFNγ producing NK cells and γδ T cells and also a major increase in the mean fluorescent intensity (MFI) of intracellular IFNγ staining in these cells compared to NK and γδ T cells only treated with PMA and ionomycin (Fig. 4A, B and C), suggesting that PyVTu cells played an important role in NK and γδ T cell activation. Incubation of NK cells and γδ T cells with PyVTu cells also led to increased granzyme-B production in these cell types compared to cultures without PyVTu cells, even in the absence additional PMA and ionomycin stimulation (Fig. 4D, E and F). The increase in granzyme-B production by γδ T cells following their co-culture with PyVTu cells was small, but reproducible, a consistent observation in multiple experiments.

We also tested whether PyVTu cells activate NK cells and γδ T cells in vivo. PyVTu cells were injected i.p. into TCRβ KO mice (5×106 cells/ mouse), and 3 days later peritoneal exudate cells (PEC) were harvested and incubated for 4 hours with or without PMA and ionomycin stimulation, and tested for IFNγ and granzyme-B production by intracellular cytokine staining. There was an increase in the percentage of NK cells in the PEC population after injection of tumor cell lines. In one representative experiment 3 days after PyVTu cell injection there were 22.3% NK1.1+/ CD3 − cells in the peritoneum, whereas control mice that did not receive PyVTu cells had approximately 3.8% NK cells (Fig. 5A). The total number of PEC recovered from mice three days after PyVTu cell injection was 7–10 times higher than that from untreated mice, and the number of NK cells in PyVTu-injected mice was ∼46 times higher than in untreated mice (Fig. 5A). The percentage of γδ T cells did not change after PyVTu cell injection, but the number of γδ T cells in the peritoneum of PyVTu–injected mice was ∼8 times higher than in the untreated controls (Fig. 5A).

NK cells from the PEC of PyVTu cell-injected mice had increased IFNγ production compared to untreated mice, a higher percentage of cells was IFNγ+, and the IFNγ staining had a higher MFI. This difference was also consistent, but smaller in magnitude in the absence of ex vivo PMA and ionomycin stimulation. However, γδ T cells in the PEC did not have similar increases in IFNγ production after PyVTu cell injection (Fig. 5B, C and D). In the same experiments both NK cells and γδ T cells from PEC had increased granzyme-B production after PyVTu cell injection i.p. compared to untreated mice, and this was manifested by higher percentages of granzyme-B positive NK and γδ T cells and higher MFI of granzyme-B staining for NK cells (Fig. 5E, F and G). The IFNγ production was greatly stimulated by a brief in vitro PMA and ionomycin treatment, but granzyme-B production did not require any stimulation in vitro. I.p. injection of RMA cells into mice did not result in an increase in peritoneal NK cells and NK or γδ T cell activation, as judged by intracellular IFNγ and granzyme-B staining (see Fig. S1A and B). This result indicates that the ability to activate NK cells and γδ T cells in the peritoneal cavity is somewhat selective for PyVTu cells, and it does not occur in response to injection of any murine tumor cell line.
CD107a and CD107b (LAMP1 and LAMP2) is expressed on the surface of NK cells and CD8 T cells as they undergo activation-induced degranulation, and these markers can be used as a direct measure of the cytotoxic potential of these cells [26]. There was a significant increase in CD107a and CD107b expression on NK cells from PEC of mice injected i.p. with PyVTu cells in comparison to the NK cells from untreated mice (Fig. 6). This result is consistent with our other findings and gives additional support for the notion that PyVTu cells induce cytolytically active NK cells in vivo. Similar experiments performed with γδ T cells did not indicate CD107a or CD107b up-regulation.

PyVTu cell lines harbor a high number of PyV DNA genomes (5×108 copies/µg of cell DNA) and also shed some infectious virus particles. This raises the question whether the activation of NK cells and γδ T cells by PyVTu cells in vivo could be merely due to the released infectious virions. Several lines of experimental evidence suggest that this was not the case. First, i.p. infection of mice with 2×106 p.f.u. of PyV (a virus dose several orders of magnitude higher than the amount of virus which would be shed by injected PyVTu cells) did not result in increases of PEC, NK or γδ T cell percentages or numbers comparable to the ones observed after injection of PyVTu cells (Fig. S2A). Second, PyV infection i.p. activated NK and γδ T cells to produce IFNγ as did PyVTu injection (Fig. S2B), but in contrast to PyVTu cells, the viral infection did not lead to high levels of granzyme-B production (Fig. S2C). Based on these findings we reason that the activation and expansion of NK and γδ T cells by PyVTu cells could not be merely due to infectious virus release by PyVTu cells.
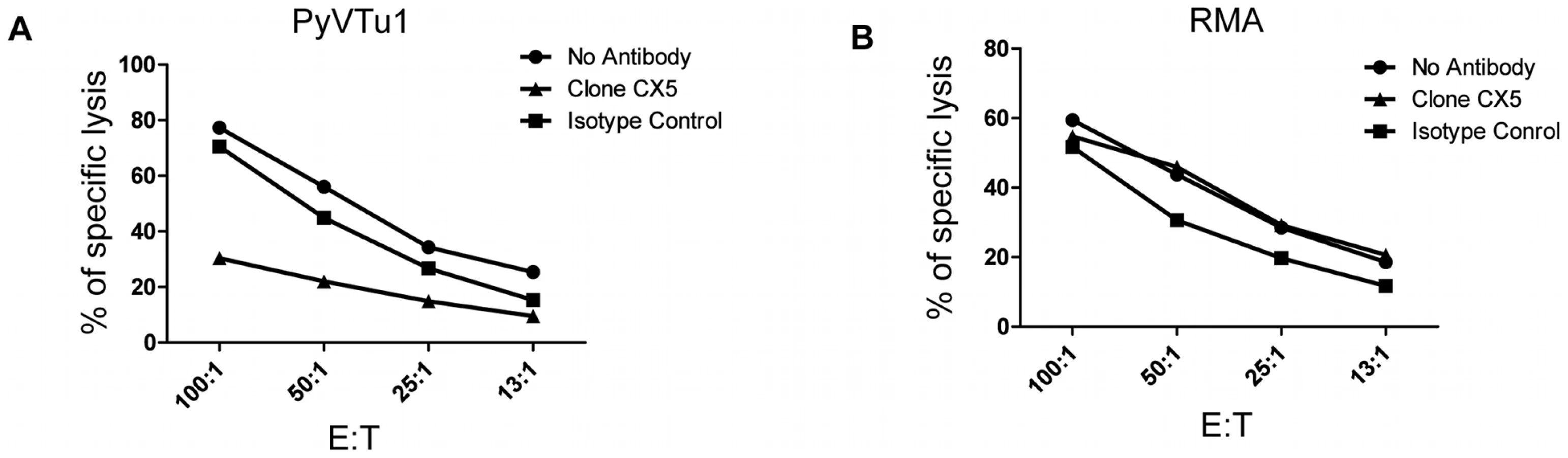
NK cells kill PyVTu cells by a NKG2D-dependent mechanism
NK cells are thought to exert a potent antitumor activity by directly killing tumor cells. We tested the ability of in vivo activated NK cells to kill PyVTu cells in vitro in chromium release cytotoxicity assays. PyVTu and YAC-1 cell targets were efficiently killed by PEC effectors taken from TCRβ KO mice injected with PyVTu cells three days prior to the PEC harvest. This killing was completely abolished, however, when the PEC was taken from PyVTu-injected, NK cell-depleted mice (Fig. 7A). These results demonstrated that PyVTu cells are sensitive to activated NK cell-mediated killing. NK cells enriched from spleens of untreated SCID mice by MACS separation likewise killed PyVTu targets, and importantly, this killing was completely prevented by treatment with an NKG2D blocking monoclonal antibody CX5, but not by treatment with an isotype control antibody (Fig. 7B and C). Experiments with another NKG2D-specific blocking monoclonal antibody, MI6, gave similar results (data not shown). NK cell-mediated killing of RMA cell targets, on the other hand, was not prevented by CX5 treatment. RMA cells do not express known NKG2D ligands, and therefore they may be recognized and killed by activated NK cells by NKG2D-NKG2D ligand-independent mechanisms (Fig. 8A and B). From these studies we conclude that the interaction of the activating receptor NKG2D on NK cells with NKG2D ligands, such as Rae-1 expressed on PyVTu cells was essential for the NK cell-mediated killing of these virus-induced tumor cells.

Discussion
In this report for the first time we provide evidence for the critical role of γδ T cells and NK cells in mice in the resistance to naturally occurring virus-induced tumors. Following infection with PyV, a natural mouse pathogen with strong oncogenic potential, mice that lacked αβ T cells, but had γδ T cells, remained tumor free, but mice lacking both αβ and γδ T cells developed tumors by six months post infection. An additional role for NK cells was suggested by an earlier onset of tumor formation in mice lacking both NK and T cells in comparison to T cell-deficient mice that have NK cells.
An increasing body of data obtained in both mice and humans suggests that γδ T cells may constitute an important component of the immunological resistance to tumors, although direct evidence showing that they control tumors induced by viral infections in vivo has been lacking so far. γδ T cells were found among tumor infiltrating lymphocytes (TIL) in human tumors such as lung cancer, colon carcinomas and renal cell carcinomas [1], [2], [3], [27], [28]. Moreover, γδ T cell clones established from TIL or PBMC of cancer patients could kill autologous tumors in vitro in cytotoxicity assays [27], [29], [30], [31], and the growth of human melanoma cells engrafted on SCID mice was inhibited by transfusion of Vδ1 γδ T cells and NK cells from the same patient [32]. Mice deficient in γδ T cells have been shown to have increased susceptibility to chemically induced cutaneous tumors [9], but their role in resistance to natural virus-induced tumor formation had never been demonstrated.
NK cells preferentially kill cells with low MHC class I expression, and tumor cells often down-regulate class I expression to escape from CD8+ T cell responses [33]. Therefore, tumor cell killing was postulated decades ago to be a major function for NK cells [7]. Indeed, low NK cell-mediated cytotoxicity of PBMC correlates with increased risk of tumor development in people [34]. Moreover, NK cell infiltration of tumors is associated with a better prognosis in a variety of carcinomas [4], [5], [6], and transfer of alloreactive NK cells increases the survival of patients with leukemia [35]. Implanted syngeneic tumors grow more aggressively in mice if no functional NK cells are present. Administration of cytokines known to enhance NK cell function can lead to accelerated elimination of implanted tumors [7]. The role of NK and γδ T cells in the control of naturally occurring or spontaneous tumors, however, has been less understood. Here we show data indicating a major role for both γδ T cells and NK cells in tumor immunity in a natural model of tumors induced by virus infection.
NKG2D is a molecule known to be involved in target recognition of both γδ T cells and NK cells [36], it is a type II trans-membrane glycoprotein with a C-type lectin binding domain, expressed as a disulfide-linked homodimer. Its ligands, Rae-1 (α–ε), H60 and Mult-1 in mice, are regarded as markers of cell stress [37], [38]. Here we demonstrate that cell lines established from PyV-induced tumors express Rae-1. These cells activate NK and γδ T cells in vitro and in vivo, and NK cells kill these tumor cells in vitro in an NKG2D-dependent fashion. These findings suggest that γδ T cells and NK cells may recognize emerging PyV-transformed cells expressing Rae-1 stress molecules by their NKG2D receptors. NKG2D is a major activating receptor on NK cells, although NK cell activation is influenced by a variety of other receptors as well. The signals received from NKG2D are transmitted via DAP10, and in mice also the DAP12 signaling molecules [36], [39], [40], [41]. γδ TCR and NKG2D are both major sources of activating signals on γδ T cells. Whether the γδ TCRs get any stimulation from from PyV-induced tumors is an open question.
Freshly isolated γδ T cells lack in vitro cytolytic activity in most studies, and there are only a few reports on in vitro cytotoxic activity using target cells infected by pathogens [42]. In contrast to the easily demonstratable cytotoxic activity mediated by NK cells, we could not demonstrate in vitro killing of PyVTu cells by γδ T cells in this study. Nevertheless, the tumor resistance of TCRβ KO and susceptibility of TCRβ×δ KO mice strongly argues for the involvement of some γδ T cell effector mechanisms in the antitumor responses in vivo.
A variety of virus-infected cells also express Rae-1 and other stress molecules recognized by NKG2D. Examples for the viruses which induce these stress molecules are HCMV, MCMV, influenza A and Epstein Barr Virus [43]. Acutely PyV-infected TCRβ KO mice on day 7 post infection, however, did not show an increase in Rae-1 transcripts in spleen and salivary gland tissues (Fig. S3A). Moreover, in SCID mice on day 6 post infection, when spleens have a high PyV load, macrophages and DC, cell types known to be infected with PyV, did not up-regulate Rae-1 expression (data not shown). Therefore PyV infection of these cells in vivo does not seem to induce Rae-1 expression on their surface. In vitro infection of NIH 3T3, UC1b and MC57G cells with PyV does not change their expression of Rae-1 protein, and only in primary mouse embryonic fibroblast (MEF) cultures was an increase in Rae-1 expression seen after PyV infection (Fig. S3B).
Killing of PyVTu cells by activated NK cells in vitro was prevented by NKG2D blocking antibodies, showing that NKG2D-dependent mechanisms play an essential role in NK cell-mediated cytotoxic responses to PyVTu cells. These findings suggest that γδ T cells and NK cells may control the outgrowth of PyV-induced tumors via NKG2D but would not eliminate permissively infected cells by the same mechanisms. This scenario is supported by our findings that TCRβ KO and TCRβ×δ KO mice did not differ in viral load at various time points post PyV infection. Thus, γδ T cells do not have a significant effect on the control of PyV levels in vivo. Of note is that the titers of T cell-independent antiviral IgG responses are not significantly different in PyV-infected TCRβ KO and TCRβ×δ KO mice, therefore γδ T cells do not contribute as helper cells enhancing antiviral humoral immunity either [44]. Previously we have also reported that NK depletion did not lead to increased viral titers in various organs of PyV-infected SCID mice [24], suggesting that NK cells have no direct antiviral role in these animals. Thus, we conclude that γδ and NK cells seem to mount an antitumor, but not an antiviral response in PyV-infected mice.
PyV can infect a variety of cell types and replicate in a broad range of tissues in mice, and it also has a strong ability to transform cells and induce tumors. Nevertheless, PyV-induced tumors are rare in nature or in wild type adult mice. This suggests that the immune system applies a very successful, multi-pronged strategy against PyV-induced tumor formation. Persisting high virus levels seem to be a prerequisite for tumor formation. Therefore the control of viral spread and replication by B cells and αβ T lymphocytes decreases the likelihood of cell transformation and the generation of tumors. Humoral immunity reduces the viral levels even in the absence of T cell help [15], but B cell responses by themselves do not prevent tumor formation, as TCRβ×δ KO mice containing B cells develop tumors. CD8+ αβ T cells act against virus-infected cells and also against tumor cells expressing viral epitopes [11]. A reduced population of middle T epitope-specific Vβ6+ CD8 αβ T cells in H2k mice carrying Mtv-7 superantigens seems to correlate with increased tumor-susceptibility of these mice when infected as neonates [19]. Our study now shows for the first time, that when αβ T cells are not functional, NK cells and γδ T cells can provide an additional potent line of defense against virus-induced tumor development, by responses that are not specific for viral-coded proteins, but instead probably directed against tumor cell-expressed stress molecules.
The high tumor incidence observed in PyV-infected TCRβ×δ KO mice that have NK cells brings up the question of why NK cells cannot overcome tumor development in the absence of γδ T cells, and why NK cells only delay but don't prevent tumor formation. We speculate that this finding can be explained by the concept of immune editing, which describes the interaction of emerging tumors with the host immune system in three phases, elimination, equilibrium and escape [45]. NK cell responses may represent the first phase of immune editing, eliminating only a fraction of the emerging tumors. The remaining tumor cells eventually may overwhelm the NK cells, perhaps by escaping recognition and/ or loss of NK functionality. This hypothetical scenario is supported by our preliminary findings that large tumors freshly isolated from TCRβ×δ KO mice although express Rae-1 mRNA, lack Rae-1 protein expression on the cell surface (R. Mishra unpublished). Mice which have γδ T cells in addition to NK cells, however, have a numerical advantage of effector cells against the emerging tumors. As a consequence, either all tumor cells may be eradicated at the elimination phase, or if residual tumor cells are left, they may be handled by a second wave of effectors, which may sense and attack the tumor cells, perhaps by another mechanism.
The findings of this study have implications to human cancer. It has been known for decades that most people harbor polyomaviruses, such as BK and JC virus, which persist at low levels, but are harmless in healthy individuals, similar to PyV in normal mice. Patients with impaired immunity, however, suffer from severe pathology associated with the reactivation of these viruses. New human polyomaviruses have recently been identified [46], and one of them is associated with the malignancy Merkel cell carcinoma. This neuroepithelial cancer is rare, and develops mostly in immune-compromised individuals. A large majority of the population is seropositive for the Merkel cell carcinoma-associated polyomavirus and may have a low level persistent infection. New insights obtained in the mouse PyV model may, in addition to elucidating general mechanisms of tumor control, also help to understand and treat polyomavirus –associated human malignancies.
Materials and Methods
Mice and infections
All the mice used in the studies were on the C57BL/6 (B6) background. TCRβ KO, TCRβ×δ KO and SCID mice were obtained from the Jackson Laboratory (Bar Harbor, Maine), and colonies of these mice were maintained in the Department of Animal Medicine of the University of Massachusetts under specific pathogen free conditions. E26 mice which express the human CD3E transgene in high copy numbers and are defective in T cells and NK cells [21] were originally obtained on the CBA/J×C57BL/6 mixed background from the Jackson Laboratory, and were fully back crossed onto the C57BL/6 background and bred at the University of Massachusetts Medical School. Mice were used between 8 and 12 wk of age, virus infections were done i.n. or i.p. with 2×106 PFU of PyV strain A2. All the procedures using animals were done according to the protocols “Immunology of virus infections” approved by the University of Massachusetts Medical School Animal Care and Use Committee.
Quantitative PCR (qPCR) to measure viral DNA genome copy number
DNA was prepared from organ homogenates by digestion with proteinase K (Sigma) at 55°C overnight, followed by phenol extraction and RNase-A treatment (10u/µl, Promega). The PCR amplification was performed as described previously [47]. 50 µl reaction mix containing 50 mM Tris pH 8.0, 0.5 µg/ml BSA, 3 mM MgCl2, 0.25 mM of each deoxynucleotide triphosphate, 0.5 U of Taq polymerase (Promega), 0.66 U SYBR-Green (Molecular Probes), 0.1mM each of forward and reverse primer (Invitrogen), 5nM Fluorescein (Bio-Rad) and 1 µg of the DNA sample tested was used. The following primers were used: β actin forward CGA GGC CCA GAG CAA GAG AG; β actin reverse CGG TTG GCC TTA GGG TTC AG; PyV VP1 forward CCC CCG GTA CAG GTT CAG TCC CAT CAT; VP1 reverse GGC ACA ACA GCT CCA CCC GTC CTG CAG. The amplification for VP1 started with one cycle at 95°C for 3 min, 37 cycles of 95°C for 30 sec, 65°C for 20 sec, 72°C for 45 sec. PCR amplification with the β actin primers started with one cycle at 95°C for 150 sec, then 40 cycles of 95°C for 30 sec, 62°C for 25 sec, and 72°C for 25 sec. Negative controls included a sample with no DNA substrate, and DNA from uninfected mouse organs. Three-fold serial dilutions of DNA prepared from uninfected mouse organs (spanning 1µg - 31 ng) were used to generate a standard curve for β actin PCR. For PyV we used a recombinant plasmid containing the VP1 coding sequences and made dilution series from 2×108 copies to 20 copies and mixed these plasmids with 1 µg of DNA from uninfected mouse organs. All the reactions were run in duplicates. The obtained PyV copy numbers were normalized for β actin which reflected the amount of mouse genomic DNA present, and the results were expressed as PyV genome copies/ µg organ DNA.
Generation of PyV tumor cell lines
Salivary gland tumors were aseptically excised from euthanized PyV-infected TCRβ×δ KO mice. The tumor tissues were rinsed with sterile DMEM containing antibiotics and cut into small pieces and digested with type I collagenase (100U/ml) in 10ml of DMEM containing 10% FCS for 1 hr. The cells were then harvested and plated in DMEM containing 10% FCS. Subsequently the adherent cells were trypsinized and propagated through multiple passages to obtain the transformed cell lines. The three salivary gland tumor cell lines used in these studies, PyVTu1, PyVTu2 and PyVTu3, were independently derived from three tumor bearing TCRβ×δ KO mice. These cell lines have been propagated for over 30 to 40 passages so far without showing major changes in growth or survival.
Reverse transcriptase (RT) PCR and qRT PCR to detect NKG2D ligand messages
RNA samples from various organs or cell lines were isolated by Trizol (Invitrogen) or using the RNeasy mini kit (Qiagen) following the manufacturer's protocol. Two µg of total RNA from each samples was used to synthesize 1st strand cDNA using 0.5µg of oligo dT (Invitrogen) and superscript II RT (Invitrogen) following the manufacturer's protocol. The PCR amplification was carried out in a total volume of 50µl containing 0.2mM of each dNTP, 0.5 U of Taq polymerase (Invitrogen) in 1× PCR buffer supplied by the manufacturer and 20 pM each of forward and reverse primer (Invitrogen). The following primers were used: β-actin forward primer 5′ CGA GGC CCA GAG CAA GAG AG and β-actin reverse primer 5′ CGG TTG GCC TTA GGG TTC AG; Rae-1 forward primer 5′TGA GCT GGA GAT CAG CTA ATG A and Rae-1 reverse primer 5′ GAA GCG GGG AAG TTG ATG TA; H60 forward primer 5′ CAT GGA GCA GTG GAA GAA CA and H60 reverse primer 5′ CAC TCA GAC CCT GGT TGT CA; Mult1 forward primer 5′ CAA TGT CTC TGT CCT CGG AA and Mult1 reverse primer 5′CTG AAC ACG TCT CAG GCA CT. For qPCR SYBR green master mix (Applied Biosystem) was used. PCR amplification with the β-actin primers started with one cycle at 95°C for 10 minutes, then 37 cycles of 95°C for 30 sec, 62°C for 25 sec, and 72°C for 25 sec. Negative controls included a sample with no DNA substrate. For Rae-1, H60 and Mult1 primers PCR cycles started with 95°C for 10 minutes, then 32 cycles of 95°C for 30 sec, 55°C for 30 sec, and 72°C for 30 sec. For determining relative Rae-1 expression, Rae-1 copy numbers were normalized for β-actin obtained from standard curves.
Detection of NKG2D L expression by flow cytometry
To stain for NKG2D ligands 2.5×105 to 5×105 PyVTu cells, YAC-1 and RMA cells were treated with anti-CD16/32 (Fc block; clone 2.4G2; BD Pharmingen) and then stained with the following antibodies: PE-anti-mouse Rae-1 (pan-specific, clone 186107), PE-anti-mouse H60 (clone 205326) and Rat IgG2A isotype control-PE (clone 54447) from R&D, PE-anti-mouse MULT-1 (clone 5D10) and Armenian hamster IgG isotype control(clone ebio299Arm) from eBioscience, and PE-anti-mouse MHC class I H-2Kb (clone AF6-88.5; BD Bioscience) or class I H-2Kk (clone 36-7-5; BD Bioscience).
NK cell and γδ T cell activation assays, intracellular IFNγ, granzyme-B and CD107a/b staining
For in-vitro assays single cell suspensions were prepared from spleens. Two times 106 spleen leukocytes were incubated with 105 PyVTu cells for 6 hours in 0.2 ml RPMI containing 10% FCS. For stimulation 50 ng/ml of PMA (Sigma) and 500 ng/ml of ionomycin (Sigma) were added after 2 hr of incubation, and parallel cultures were left unstimulated. For the final 3 hours of the incubation time 0.2µl of golgi plug (BD Bioscience) and 0.13 µl of golgi stop (BD Bioscience) was added to allow accumulation of intracellular proteins.
For in vivo assays 5×106 PyVTu cells in HBSS were injected i.p. into TCR β KO mice, control mice were injected i.p. with HBSS with no cells. Three days after injection PEC were harvested from the mice, RBC were removed by lysis and 2×106 of the PEC were incubated for 4 hours in RPMI/ 10% FCS with or without PMA and ionomycin stimulation. Similarly to the in vitro assays, for the final 3 hours of the incubation time 0.2µl of golgi plug (BD Bioscience) and 0.13 µl of golgi stop (BD Bioscience) was added to allow accumulation of intracellular proteins. The cells tested for in vitro or in vivo activation were then treated with anti-CD16/32 (Fc block; clone 2.4G2; BD Pharmingen) and surface stained with FITC-anti-mouse-CD3 (clone - 145-2C11; BD Pharmingen), PerCPCy5.5-anti-mouse NK.1.1 (clone - PK136; BD Pharmingen), and PE-anti-mouse-γδ TCR (clone-GL3; BD Pharmingen) antibodies for 25 minutes at room temperature. Cells were then washed and permeabilized with Cytofix/Cytoperm buffer (BD Biosciences), followed by staining with PE Cy7-anti-mouse-IFNγ (clone XMG1.2; BD Pharmingen) and APC-anti-human-granzyme-B (clone GB11; Invitrogen) for 20–25 minutes at room temperature. For CD107 staining both FITC-anti-mouse-CD107a (clone-1D4B; BD Pharmingen) and FITC-anti-mouse-CD107b (clone-ABL-93; BD Pharmingen) antibodies were added along with golgi plug and golgi stop for four hours and then treated with anti-CD16/32 (Fc block; clone 2.4G2; BD Pharmingen) and surface stained with PerCPCy5.5-anti-mouse NK.1.1 (clone PK-136; BD Pharmingen), PE-anti-mouse-γδ TCR (clone GL3; ) for 25 minutes. The cells were finally analyzed by flow cytometry.
In vitro cytotoxicity assays
Standard 4 h 51Cr release microcytotoxicity assays were used to determine NK cell activity [48]. Activated PECs or spleen cells of SCID mice were used as effector cells, in some experiments the NK cells were enriched by using an NK cell isolation kit (MACS, Miltenyi Biotech) following the manufacturer's protocol. The PECs were activated in vivo by an injection of 4×106 to 5×106 PyVTu cells i.p. two days prior to their harvest. 51Cr-labelled YAC-1 cells, PyVTu cells or RMA cells were used as targets, and 104 target cells were plated into wells of microtiter plates with varying numbers of effectors to achieve the planned effector to target (E∶T) ratios. After 4 hours of incubation 51Cr release into the supernatants was measured. The percentage of specific 51Cr release was calculated as described before [42]. For NKG2D blocking, anti-NKG2D blocking antibodies (clone CX5; eBioscience or clone MI-6; eBioscience ) or corresponding isotype controls (Rat IgG1 isotype HRPN; BioXcell and Rat IgG2a isotype eBioscience) were used at 5µg/ml. For depletion of NK cells anti-NK1.1 antibody (clonePK136; BioXcell ) was injected at the dose of 40 µg/mouse i.p. one day prior to injection of PyVTu cells.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. ZocchiMR
FerrariniM
MigoneN
CasoratiG
1994 T-cell receptor V delta gene usage by tumour reactive gamma delta T lymphocytes infiltrating human lung cancer. Immunology 81 234 239
2. CorvaisierM
Moreau-AubryA
DiezE
BennounaJ
MosnierJF
2005 V gamma 9V delta 2 T cell response to colon carcinoma cells. J Immunol 175 5481 5488
3. TodaroM
D'AsaroM
CaccamoN
IovinoF
FrancipaneMG
2009 Efficient killing of human colon cancer stem cells by gammadelta T lymphocytes. J Immunol 182 7287 7296
4. TakanamiI
TakeuchiK
GigaM
2001 The prognostic value of natural killer cell infiltration in resected pulmonary adenocarcinoma. J Thorac Cardiovasc Surg 121 1058 1063
5. CocaS
Perez-PiquerasJ
MartinezD
ColmenarejoA
SaezMA
1997 The prognostic significance of intratumoral natural killer cells in patients with colorectal carcinoma. Cancer 79 2320 2328
6. IshigamiS
NatsugoeS
TokudaK
NakajoA
CheX
2000 Prognostic value of intratumoral natural killer cells in gastric carcinoma. Cancer 88 577 583
7. WelshRM
1984 Natural killer cells and interferon. Crit Rev Immunol 5 55 93
8. PenningerJM
WenT
TimmsE
PotterJ
WallaceVA
1995 Spontaneous resistance to acute T-cell leukaemias in TCRV gamma 1.1J gamma 4C gamma 4 transgenic mice. Nature 375 241 244
9. GirardiM
OppenheimDE
SteeleCR
LewisJM
GlusacE
2001 Regulation of cutaneous malignancy by gammadelta T cells. Science 294 605 609
10. BenjaminTL
2001 Polyoma virus: old findings and new challenges. Virology 289 167 173
11. SwansonPA2nd
LukacherAE
Szomolanyi-TsudaE
2009 Immunity to polyomavirus infection: the polyomavirus-mouse model. Semin Cancer Biol 19 244 251
12. AllisonAC
LawLW
1968 Effects of antilymphocyte serum on virus oncogenesis. Proc Soc Exp Biol Med 127 207 212
13. LawLW
TingRC
LeckbandE
1967 Prevention of virus-induced neoplasms in mice through passive transfer of immunity by sensitized syngeneic lymphoid cells. Proc Natl Acad Sci U S A 57 1068 1075
14. AllisonAC
MongaJN
HammondV
1974 Increased susceptibility to virus oncogenesis of congenitally thymus-deprived nude mice. Nature 252 746 747
15. Szomolanyi-TsudaE
WelshRM
1996 T cell-independent antibody-mediated clearance of polyoma virus in T cell-deficient mice. J Exp Med 183 403 411
16. LukacherAE
WilsonCS
1998 Resistance to polyoma virus-induced tumors correlates with CTL recognition of an immunodominant H-2Dk-restricted epitope in the middle T protein. J Immunol 160 1724 1734
17. LukacherAE
MoserJM
HadleyA
AltmanJD
1999 Visualization of polyoma virus-specific CD8+ T cells in vivo during infection and tumor rejection. J Immunol 163 3369 3378
18. BerkeZ
PalmerS
BergmanT
WesterD
SvedmyrJ
1996 A short peptide eluted from the H-2Kb molecule of a polyomavirus-positive tumor corresponds to polyomavirus large T antigen peptide at amino acids 578 to 585 and induces polyomavirus-specific immunity. J Virol 70 3093 3097
19. LukacherAE
MaY
CarrollJP
Abromson-LeemanSR
LaningJC
1995 Susceptibility to tumors induced by polyoma virus is conferred by an endogenous mouse mammary tumor virus superantigen. J Exp Med 181 1683 1692
20. LiunggrenG
LiunggrenHG
DalianisT
1994 T cell subsets involved in immunity against polyoma virus-induced tumors. Virology 198 714 716
21. WangB
BironC
SheJ
HigginsK
SunshineMJ
1994 A block in both early T lymphocyte and natural killer cell development in transgenic mice with high-copy numbers of the human CD3E gene. Proc Natl Acad Sci U S A 91 9402 9406
22. DrakeDR3rd
LukacherAE
1998 Beta 2-microglobulin knockout mice are highly susceptible to polyoma virus tumorigenesis. Virology 252 275 284
23. McCanceDJ
SebestenyA
GriffinBE
BalkwillF
TillyR
1983 A paralytic disease in nude mice associated with polyoma virus infection. J Gen Virol 64(Pt 1) 57 67
24. Szomolanyi-TsudaE
DundonPL
JorisI
ShultzLD
WodaBA
1994 Acute, lethal, natural killer cell-resistant myeloproliferative disease induced by polyomavirus in severe combined immunodeficient mice. Am J Pathol 144 359 371
25. WilderJA
YuanD
1995 Regulation of IFN-gamma mRNA production in murine natural killer cells. Int Immunol 7 575 582
26. BettsMR
BrenchleyJM
PriceDA
De RosaSC
DouekDC
2003 Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods 281 65 78
27. WatanabeN
HizutaA
TanakaN
OritaK
1995 Localization of T cell receptor (TCR)-gamma delta+T cells into human colorectal cancer: flow cytometric analysis of TCR-gamma delta expression in tumour-infiltrating lymphocytes. Clin Exp Immunol 102 167 173
28. KowalczykD
SkorupskiW
KwiasZ
NowakJ
1996 Activated gamma/delta T lymphocytes infiltrating renal cell carcinoma. Immunol Lett 53 15 18
29. VieyE
FromontG
EscudierB
MorelY
Da RochaS
2005 Phosphostim-activated gamma delta T cells kill autologous metastatic renal cell carcinoma. J Immunol 174 1338 1347
30. BensussanA
LagabrielleJF
DegosL
1989 TCR gamma delta bearing lymphocyte clones with lymphokine-activated killer activity against autologous leukemic cells. Blood 73 2077 2080
31. ChoudharyA
DavodeauF
MoreauA
PeyratMA
BonnevilleM
1995 Selective lysis of autologous tumor cells by recurrent gamma delta tumor-infiltrating lymphocytes from renal carcinoma. J Immunol 154 3932 3940
32. LozuponeF
PendeD
BurgioVL
CastelliC
SpadaM
2004 Effect of human natural killer and gammadelta T cells on the growth of human autologous melanoma xenografts in SCID mice. Cancer Res 64 378 385
33. KarreK
LjunggrenHG
PiontekG
KiesslingR
1986 Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature 319 675 678
34. ImaiK
MatsuyamaS
MiyakeS
SugaK
NakachiK
2000 Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: an 11-year follow-up study of a general population. Lancet 356 1795 1799
35. RuggeriL
MancusiA
BurchielliE
AversaF
MartelliMF
2007 Natural killer cell alloreactivity in allogeneic hematopoietic transplantation. Curr Opin Oncol 19 142 147
36. BauerS
GrohV
WuJ
SteinleA
PhillipsJH
1999 Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 285 727 729
37. CerwenkaA
BakkerAB
McClanahanT
WagnerJ
WuJ
2000 Retinoic acid early inducible genes define a ligand family for the activating NKG2D receptor in mice. Immunity 12 721 727
38. CarayannopoulosLN
NaidenkoOV
FremontDH
YokoyamaWM
2002 Cutting edge: murine UL16-binding protein-like transcript 1: a newly described transcript encoding a high-affinity ligand for murine NKG2D. J Immunol 169 4079 4083
39. LanierLL
CorlissBC
WuJ
LeongC
PhillipsJH
1998 Immunoreceptor DAP12 bearing a tyrosine-based activation motif is involved in activating NK cells. Nature 391 703 707
40. WuJ
CherwinskiH
SpiesT
PhillipsJH
LanierLL
2000 DAP10 and DAP12 form distinct, but functionally cooperative, receptor complexes in natural killer cells. J Exp Med 192 1059 1068
41. RosenDB
ArakiM
HamermanJA
ChenT
YamamuraT
2004 A Structural basis for the association of DAP12 with mouse, but not human, NKG2D. J Immunol 173 2470 2478
42. SelinLK
SantolucitoPA
PintoAK
Szomolanyi-TsudaE
WelshRM
2001 Innate immunity to viruses: control of vaccinia virus infection by gamma delta T cells. J Immunol 166 6784 6794
43. EagleRA
TrowsdaleJ
2007 Promiscuity and the single receptor: NKG2D. Nat Rev Immunol 7 737 744
44. Szomolanyi-TsudaE
LeQP
GarceaRL
WelshRM
1998 T-Cell-independent immunoglobulin G responses in vivo are elicited by live-virus infection but not by immunization with viral proteins or virus-like particles. J Virol 72 6665 6670
45. DunnGP
OldLJ
SchreiberRD
2004 The three Es of cancer immunoediting. Annu Rev Immunol 22 329 360
46. zur HausenH
2008 Novel human polyomaviruses–re-emergence of a well known virus family as possible human carcinogens. Int J Cancer 123 247 250
47. Szomolanyi-TsudaE
SeedhomMO
CarrollMC
GarceaRL
2006 T cell-independent and T cell-dependent immunoglobulin G responses to polyomavirus infection are impaired in complement receptor 2-deficient mice. Virology 352 52 60
48. WelshRMJr
1978 Cytotoxic cells induced during lymphocytic choriomeningitis virus infection of mice. I. Characterization of natural killer cell induction. J Exp Med 148 163 181
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 5
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Quorum Sensing Inhibition Selects for Virulence and Cooperation in
- The Role of Intestinal Microbiota in the Development and Severity of Chemotherapy-Induced Mucositis
- Crystal Structure of HIV-1 gp41 Including Both Fusion Peptide and Membrane Proximal External Regions
- Susceptibility to Anthrax Lethal Toxin-Induced Rat Death Is Controlled by a Single Chromosome 10 Locus That Includes
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy