
Host-Detrimental Role of Esx-1-Mediated Inflammasome Activation in Mycobacterial Infection
The Esx-1 (type VII) secretion system is a major virulence determinant of pathogenic mycobacteria, including Mycobacterium marinum. However, the molecular events and host-pathogen interactions underlying Esx-1-mediated virulence in vivo remain unclear. Here we address this problem in a non-lethal mouse model of M. marinum infection that allows detailed quantitative analysis of disease progression. M. marinum established local infection in mouse tails, with Esx-1-dependent formation of caseating granulomas similar to those formed in human tuberculosis, and bone deterioration reminiscent of skeletal tuberculosis. Analysis of tails infected with wild type or Esx-1-deficient bacteria showed that Esx-1 enhanced generation of proinflammatory cytokines, including the secreted form of IL-1β, suggesting that Esx-1 promotes inflammasome activation in vivo. In vitro experiments indicated that Esx-1-dependent inflammasome activation required the host NLRP3 and ASC proteins. Infection of wild type and ASC-deficient mice demonstrated that Esx-1-dependent inflammasome activation exacerbated disease without restricting bacterial growth, indicating a host-detrimental role of this inflammatory pathway in mycobacterial infection. These findings define an immunoregulatory role for Esx-1 in a specific host-pathogen interaction in vivo, and indicate that the Esx-1 secretion system promotes disease and inflammation through its ability to activate the inflammasome.
Published in the journal:
. PLoS Pathog 6(5): e32767. doi:10.1371/journal.ppat.1000895
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1000895
Summary
The Esx-1 (type VII) secretion system is a major virulence determinant of pathogenic mycobacteria, including Mycobacterium marinum. However, the molecular events and host-pathogen interactions underlying Esx-1-mediated virulence in vivo remain unclear. Here we address this problem in a non-lethal mouse model of M. marinum infection that allows detailed quantitative analysis of disease progression. M. marinum established local infection in mouse tails, with Esx-1-dependent formation of caseating granulomas similar to those formed in human tuberculosis, and bone deterioration reminiscent of skeletal tuberculosis. Analysis of tails infected with wild type or Esx-1-deficient bacteria showed that Esx-1 enhanced generation of proinflammatory cytokines, including the secreted form of IL-1β, suggesting that Esx-1 promotes inflammasome activation in vivo. In vitro experiments indicated that Esx-1-dependent inflammasome activation required the host NLRP3 and ASC proteins. Infection of wild type and ASC-deficient mice demonstrated that Esx-1-dependent inflammasome activation exacerbated disease without restricting bacterial growth, indicating a host-detrimental role of this inflammatory pathway in mycobacterial infection. These findings define an immunoregulatory role for Esx-1 in a specific host-pathogen interaction in vivo, and indicate that the Esx-1 secretion system promotes disease and inflammation through its ability to activate the inflammasome.
Introduction
One third of the world's population is infected with Mycobacterium tuberculosis, a human specific pathogen responsible for nearly 2 million deaths annually [1]. To facilitate fundamental studies of M. tuberculosis infection, safer and experimentally more amenable species are often used as models. Among these, the closely related M. marinum is used increasingly to study pathogenesis [2], [3]. M. marinum is a pathogen of fish and amphibians causing disease with many features of tuberculosis [2], and is also able to infect immunocompetent humans where it induces formation of dermal granulomas pathologically similar to those formed in tuberculosis [2], [4]. Importantly, the Esx-1 (Early secreted antigen 6 kilodaltons [Esat-6] secretion system 1) secretion system is highly conserved between M. tuberculosis and M. marinum, and required for virulence of both species [5]–[8]. Esx-1 is encoded primarily by genes within the chromosomal region of difference 1 (RD1) [9]; indeed, attenuation of the Mycobacterium bovis BCG vaccine strain is in large part due to a deletion of RD1, emphasizing the general significance of this secretory apparatus in mycobacterial virulence [10]. However, the biological function of Esx-1 during infection remains incompletely understood.
Macrophages infected with mycobacteria secrete proinflammatory cytokines, including IL-1β and IL-18 [11]–[15]. Both M. tuberculosis and M. marinum induce secretion of IL-1β in an Esx-1-dependent manner in vitro [13], [14]. The cysteine protease caspase-1 is a critical component of inflammasomes and is required for proteolytic activation and release of IL-1β and IL-18. Analysis of M. marinum infection demonstrates that Esx-1 is required to activate an inflammasome containing NLRP3 (Nalp3) and ASC [13]. In agreement with these findings, M. bovis BCG, which lacks Esx-1, is unable to activate caspase-1 efficiently [16]. However, nothing is known about the relevance of inflammasome activation to the progression of mycobacterial infection in vivo.
We examined the role of the inflammasome in M. marinum infection of mice by a ‘genetics squared’ approach, in which host and pathogen genetic strategies are combined in a single experimental system [17]. By this approach we are able to attribute a pathogenic role for Esx-1 in a defined host-pathogen interaction in vivo.
Results
Quantification of disease and inflammation demonstrates a requirement for Esx-1 in M. marinum virulence in mice
Mice were infected via tail vein injection with wild type or Esx-1 deficient (ΔRD1) M. marinum and observed for development of disease (Figure 1). In wild type M. marinum infection, visible lesions appeared in tails ∼1 week post infection, and over time these lesions increased in size and became more numerous (Figure 1A). Determination of the accumulated length of all visible lesions in individual tails allowed quantitative kinetic analysis of disease progression (Figure 1B). Wild type M. marinum caused severe tail disease, whereas ΔRD1 infected mice developed very few, or no, lesions (Figure 1A, B). Moreover, lesions in ΔRD1 infected tails were small, and did not significantly increase in size over time. Complementation of ΔRD1 bacteria with the M. tuberculosis-derived RD1-locus (ΔRD1::RD1) restored ability to cause disease (Figure S1), confirming a specific role for Esx-1 in pathogenesis and, importantly, demonstrating functional conservation of this secretory pathway between M. tuberculosis and M. marinum in vivo.

Infected mice did not lose weight over the course of the experiments (Figure S2A), suggesting that M. marinum did not cause significant systemic effects. Tail infection was not simply the result of inoculation and growth at the site of injection because infection by intracardiac injection caused similar tail disease (Figure S2B), showing that M. marinum can spread via the blood to cause disease in this tissue. Thus, M. marinum cause disease confined to the tail, which is likely due the low optimal growth temperature (∼32°C) of the bacteria and the cooler environment provided in the tail [18].
Initial histopathological studies indicated significant bone erosion of vertebrae in tails of mice infected with wild type M. marinum, suggesting that direct and quantitative analysis of erosion could be used as a separate readout of inflammation caused by infection. To this end, we measured the bone volume of tail vertebrae over time by micro-computed tomography (micro-CT), a technique capable of quantifying inflammatory bone damage and subsequent repair in mice (Figure 1C, D) [19]. During the first 3 weeks of infection, wild type M. marinum induced considerably more bone loss than did ΔRD1, showing that wild type infection caused more bone erosion—and by inference, a stronger inflammatory response (Figure 1D). However, by 28 days post infection vertebrae from both ΔRD1 and wild type infections showed similarly reduced bone volume (Figure 1D), suggesting that infection with Esx-1-deficient bacteria does induce bone erosion, but with delayed kinetics. In addition, at this late time point, there was significant bone regeneration in tails of mice infected with wild type (Figure 1C; note massive sprouting of new bone in wild type infected tails at 28 days post infection), probably due to normal osteoblastic response to bone destruction [19] as well as to resolution of the acute phase of infection. Thus, two separate quantitative traits, visible lesions and bone volume, indicated that wild type M. marinum cause significantly more disease and inflammation than Esx-1-deficient bacteria.
M. marinum cause formation of granulomas similar to those formed in tuberculosis
Hematoxylin and eosin (H&E) staining showed PMN infiltration at sites of infection in both wild type and ΔRD1 infected tails one day post infection, and immunohistochemistry revealed few macrophages and T cells at this time (not shown). At 14 days post infection, lesions in wild type M. marinum infections demonstrated a peripheral ring containing macrophages and T cells, with granulomatous architecture (Figure 2A; Figure S3); the cellularity of this peripheral lining increased over time (Figure 2B, C; Figures S4 and S5), and developed into a solid border of macrophages and epitheloid macrophages with juxtaposed T cells by 21 days post infection. Granulomas in wild type infections exhibited central acellular necrosis from 14 days post infection, and the amount of central necrosis increased over time (Figure 2D; Figure S6). Thus, wild type M. marinum induced formation of granulomas with central caseous necrosis, histologically very similar to those formed in human tuberculosis, but distinct from those in murine infection with M. tuberculosis, which generally lack central necrosis. In contrast, the lesions present in infections by ΔRD1 M. marinum were smaller, did not develop into well-delineated granulomas during the timeframe of the infection (Figure 2A–C), and did not exhibit central necrosis until 28 days post infection (not shown), indicating that Esx-1 is required for a normal granulomatous response. During the first 21 days of infection ΔRD1 lesions also contained more T cells (Figure S7), which localized throughout the entire structure rather than organized to the periphery as in wild type infection (Figure 2A–C), implying that Esx-1 affects T cell functions in vivo via unappreciated mechanisms. After 28 days, however, few T cells were observed in both in wild type and ΔRD1 induced lesions (Figure S7; Figure 2C).

M. marinum grows specifically in tails and escapes phagosomes in an Esx-1-dependent manner
To address the ability of wild type and ΔRD1 M. marinum to grow during infection, mice were analyzed for colony forming units (CFUs) in blood, lung, liver, and tail (Figure 3A, B). Similar numbers of wild type and ΔRD1 bacteria were retrieved from blood and the three tissues analyzed one day after infection. Subsequently, both strains were similarly cleared from blood, lung and liver, suggesting that M. marinum is seeded systemically upon injection, but is unable to colonize internal organs productively (Figure 3A, B). In contrast, both wild type and ΔRD1 bacteria maintained colonization in the tails, where wild type showed modest growth (Figure 3B). The number of wild type bacteria in infected tails dropped to the level of ΔRD1 between 21 and 28 days post infection, a feature that might be explained by the onset of an adaptive immune response, which is typically initiated ∼20 days post infection in M. tuberculosis infected mice [20], [21].

Analysis ∼2.5 and 4 months after infection demonstrated similar bacterial numbers (∼1×106 CFU/g and ∼3×105 CFU/g, respectively) in the tails of wild type and ΔRD1 infected mice (Figure S8A, B). Concomitant analysis of visible tail lesions demonstrated that disease induced by wild type M. marinum decreased to a level comparable to that of ΔRD1 over time (Figure S8C), suggesting that both strains are able to similarly persist in the tails with minimal pathology for extended periods of time. Thus, Esx-1 may exert its major pathogenic role during the acute phase of infection.
Histochemical analysis suggested that both wild type and ΔRD1 bacilli were scattered throughout the lesions in infected tails, with a preference for peripheral regions (not shown). Analysis of this region by transmission electron microscopy (TEM) in both wild type M. marinum and ΔRD1 lesions 21 days post infection indicated that bacteria resided preferentially in host cells with morphology consistent with macrophages (Figure 3C). Furthermore, 76.8% of wild type bacteria were observed without an apparent surrounding host membrane, whereas 93.6% of ΔRD1 bacteria were found within membranous vesicles (Figure 3C, D; Figure S9), suggesting that intracellular M. marinum escapes from phagosomes in an Esx-1-dependent manner in vivo.
Esx-1 promotes secretion of proinflammatory IL-1β in vivo
Tail specimens allowed for detailed analysis of proteins in the diseased tissue (Figure 4). Wild type M. marinum induced more TNFα and less IFNγ as compared to ΔRD1 (Figure 4A), suggesting a more proinflammatory response during wild type M. marinum infection. The amount of IL-12p40 was high but unaffected by Esx-1 (Figure 4A). Similarly, total IL-1β protein also was greatly increased in tails infected with both wild type and Esx-1-deficient bacteria (Figure 4A).

IL-1β is synthesized as a ∼31 kDa inactive proprotein, which is secreted to the extracellular environment after proteolytic processing into its biologically active mature form (∼17 kDa) by caspase-1. To examine the amount of mature IL-1β specifically, we analyzed tail proteins by Western blot, which separates mature from pro-IL-1β by molecular weight. Such analysis demonstrated a 2.6-fold increase of mature IL-1β in the tails of mice infected with wild type M. marinum compared to ΔRD1 infection (Figure 4B), suggesting that Esx-1 promotes caspase-1 activation in vivo. Because IL-1β has significant pro-inflammatory effects, this feature may contribute to the dramatic difference in inflammation between wild type and ΔRD1 infections.
Esx-1 is required for M. marinum activation of the NLRP3/ASC-inflammasome in vitro
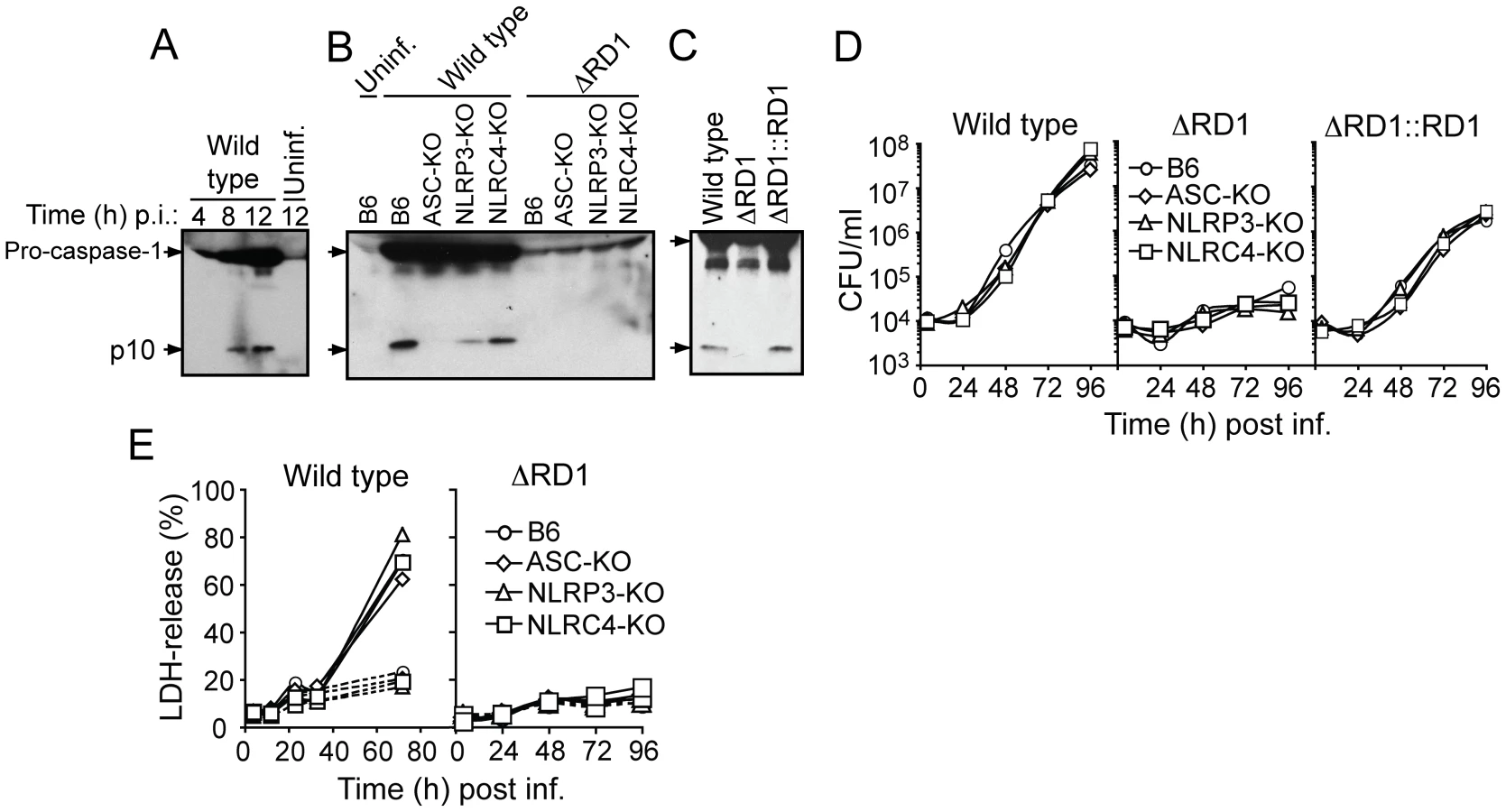
Caspase-1 is autoprocessed into 20 kDa (p20) and 10 kDa (p10) subunits upon assembly of an inflammasome. These subunits become part of the active inflammasome, and can also be used as markers for caspase-1 activation in Western blot analysis. Kinetic analysis in B6 macrophages infected with wild type M. marinum showed that caspase-1 p10 appeared 8 hrs post infection, suggesting that the bacteria interacted with the host cytoplasm at this time to activate an inflammasome (Figure 5A). Detailed analysis of bacterial and host genetic requirements demonstrated that wild type but not ΔRD1 activated caspase-1 in a process involving the host proteins ASC and NLRP3 but not NLRC4 (Ipaf), indicating that M. marinum activates the NLRP3/ASC-inflammasome in an Esx-1-dependent manner (Figure 5B). Of note, infection with wild type M. marinum induced higher levels of pro-caspase-1 than ΔRD1, independent of the inflammasome (Figure 5B), which might be explained by secretion of Esat-6, a major Esx-1 substrate that has been proposed to induce caspase-1 gene expression in macrophages [22]. Like wild type, ΔRD1::RD1 bacteria caused caspase-1 activation in infected macrophages, indicating a specific role for Esx-1 in this process and further emphasizing the functional conservation of Esx-1 between M. tuberculosis and M. marinum (Figure 5C). In agreement with these findings, and with a previous analysis of cytokine secretion from mycobacteria infected macrophages [13], Esx-1-proficient bacteria induced ASC - and NLRP3-dependent secretion of IL-1β and IL-18 (Figure S10).
Analysis of TNFα and IL-6 demonstrated Esx-1-dependent secretion (Figure S11), implying that Esx-1 promotes NFκB activation in macrophages in vitro, which could account for the increased TNFα seen in infections by wild type M. marinum in vivo. However, in agreement with previous reports [5], [23], IL-12p40 secretion was repressed in macrophages infected with wild type M. marinum (Figure S11), implying that Esx-1 down-regulates secretion of this cytokine via NFκB-independent mechanisms. The finding that wild type and ΔRD1 M. marinum caused similar levels of IL-12p40 in infected tails (Figure 4A) might be explained by additional stimulation provided in the complex in vivo environment, or by contributions from other cell types [24].
Activation of inflammasomes is normally accompanied by pyroptosis, whereby infected host cells succumb to a necrotic-like cell death commonly believed to represent an altruistic mechanism to restrict bacterial growth [25]. As expected, wild type but not Esx-1-deficient bacteria were able to grow in infected macrophages (Figure 5D). However, bacterial growth was unaffected by the inflammasome, as wild type M. marinum grew similarly well in B6, ASC-KO and NLRP3-KO macrophages (Figure 5D). Moreover, while M. marinum caused cytotoxicity to infected macrophages in an Esx-1-dependent manner, this feature was similarly independent of the inflammasome (Figure 5E). Thus, M. marinum-induced activation of the inflammasome is separable from its effect on macrophage viability, and intracellular bacterial growth is unaffected by inflammasome activation in vitro.
Esx-1-dependent activation of the inflammasome exacerbates inflammation without restricting bacterial growth in vivo
Because these in vitro experiments left uncertain how inflammasome activation affected the course of mycobacterial infection in vivo, B6 and ASC-KO mice were infected with wild type and ΔRD1 bacteria, respectively (Figure 6). Analysis of visible tail lesions and bone volume of tail vertebrae showed that development of disease was dependent on Esx-1 in both mouse strains (Figure 6A, B). However, infection with wild type M. marinum caused less visible pathology in ASC-KO than in B6 mice (Figure 6A), indicating that deficient inflammasome activation results in less disease. Consistent with this finding, micro-CT-analysis 21 days post infection demonstrated reduced loss of bone volume in tail vertebrae in ASC-KO compared to B6 mice upon challenge with wild type M. marinum (Figure 6B), confirming that lack of inflammasome activation leads to a milder inflammatory response. Thus, Esx-1-dependent activation of the inflammasome causes increased disease and inflammation in infected mice.

Histological analysis of granulomas formed in tails of B6 and ASC-KO mice infected with wild type M. marinum was performed 21 days post infection, and demonstrated similar overall architecture and cellularity of granulomas formed in the two mouse strains (Figure 6C). Notably, granulomas in wild type M. marinum infections in both mouse strains showed caseous necrosis, consistent with the ASC-independence of M. marinum-induced macrophage death in vitro. However, granulomas in ASC-KO mice contained increased numbers of T cells as compared to B6 mice (Figure 6C), suggesting that Esx-1's effect on T cells (Figure 2; Figure S7) may in part be mediated via the inflammasome.
The decreased inflammation observed in ASC-KO mice cannot be explained by decreased bacterial growth, because in agreement with our in vitro findings (Figure 5D), CFU analysis indicated a similar bacterial burden in tails of wild type infected ASC-KO and B6 mice (Figure 6D). Taken together, these findings demonstrate that Esx-1-dependent activation of the inflammasome in vivo exacerbates disease and inflammation without significantly limiting bacterial growth, suggesting that inflammasome activation is detrimental to the host in mycobacterial infection.
Discussion
Experimental infections of laboratory animals are increasingly important to our understanding of microbial pathogenesis, as these may elucidate mechanisms by which pathogens exploit the host that might not be appreciated using reductionist in vitro models. In addition, the ability to manipulate both host and pathogen genetically has become increasingly important for understanding the molecular basis of virulence [17]. The importance of this ‘genetic squared’ approach is well illustrated by the study of M. marinum infection, in which host-pathogen interactions have been dissected extensively in vitro and in vivo using infections of both Drosophila and zebrafish embryos [26]–[29]. However, the fruit fly and zebrafish embryos lack aspects of a mammalian immune system, including functional T cells, which are generally believed to play an important role in the host response to mycobacteria. Interestingly, early work in the 1960s through 1980s demonstrated that M. marinum is able to infect primarily cooler anatomical regions in mice [18], [30]. However, neither mycobacterial genetics nor mouse immunology was sufficiently developed to take full advantage of this model, and it has largely been abandoned for the past 30 years. In light of recent advances in these areas, we re-examined this model and found that its unique features allow a detailed analysis of infection leading to new insights into the biological role of Esx-1, a major virulence determinant generally involved in mycobacterial pathogenesis [2], [9], [31].
M. marinum injection into mice caused local infection in the tail. Measurement of disease by two separate analyses, the visible area of diseased tissue and the extent of bone destruction, demonstrated a major role in pathogenesis for the Esx-1 secretion system. The difference in pathology is likely not due to the difference in bacterial growth between the two strains, because this difference was small and resolved completely by 28 days, a time point at which the difference in disease was still significant. Furthermore, after the initial acute phase of infection (≥28 days), both wild type and ΔRD1 bacilli persisted equally well, with little pathology in tails of infected animals, suggesting Esx-1-independent establishment of latent disease. Taken together, these findings suggest that a major role for Esx-1 in vivo is to manipulate the inflammatory response during the early events of infection. This is consistent with the recent discovery of an important role for Esx-1 in M. marinum infected zebrafish embryos, which, unlike adult zebrafish, lack a functional adaptive immune response [32]. That study found a role for Esx-1 in promoting bacterial spread and expansion of granulomatous lesions, in part by influencing macrophage chemotaxis [32].
Because Esx-1-dependent pathology appeared to relate to perturbation of the host immune response, we analyzed cytokines in tails infected with wild type and Esx-1-deficient bacteria. Quantification of TNFα and IFNγ suggested that Esx-1 alters the immune response by decreasing the T cell response and enhancing inflammatory cytokine production. IL-12p40 was unaffected by Esx-1 in vivo, which differs from the markedly decreased levels of IL-12p40 secretion observed in wild type infected macrophages in vitro. This apparent paradox might be explained by in vivo contribution from dendritic cells, whose secretion of IL-12p40 is not repressed by infection with Esx-1-proficient M. tuberculosis [11]. Previous analyses have suggested that secretion of TNFα is unaffected or even decreased during macrophage infection with wild type compared to Esx-1-deficient mycobacteria [5], [13]. In contrast, our findings indicated that secretion of this cytokine is enhanced by infection with Esx-1-proficient bacteria both in vivo and in non-primed macrophages in vitro. While the reasons for these differences remain unknown, these anomalies stress the importance of translating in vitro findings into the more complex in vivo environment.
IL-1β is upregulated in the lungs of tuberculosis patients [33], and both IL-1β and IL-18 are secreted from M. tuberculosis and M. marinum infected macrophages in vitro [11], [13]. While IL-18 might have a minor role in experimental M. tuberculosis mouse infections [34], [35], IL-1β is commonly believed to play a role in the host response elicited by mycobacteria. IL-1β was highly upregulated in M. marinum infected mouse tails, and Esx-1 promoted processing of this cytokine into its biologically active form in vivo. Studies in primary macrophages indicated that M. marinum activates the NLRP3/ASC-inflammasome in an Esx-1-dependent manner. However, while M. marinum caused Esx-1-dependent cell death to infected macrophages, this was not dependent on ASC or NLRP3, suggesting that the macrophage death so apparent in vitro and possibly underlying caseous necrosis in vivo is distinct from caspase-1 activation. Indeed, ASC was dispensable also for development of caseation in vivo.
A role for inflammasomes in determining the fate of cellular macrophage infections has been analyzed in vitro for several bacterial pathogens [25], [36]. However, the biological role of inflammasomes in vivo remains elusive. For M. marinum, activation of the inflammasome exacerbated disease and inflammation without significantly limiting bacterial growth, indicating that inflammasome activation is detrimental to the host in mycobacterial infection and that disease, at least in part, is a function of the inflammatory response rather than direct bacterial mechanisms. Possibly Esx-1 has evolved to increase the inflammatory response in order to promote bacterial spread to new hosts, as when granulomas rupture into bronchi during tuberculosis, or into the skin during piscine infection by M. marinum. Interestingly, however, ASC-deficiency does not completely abolish the ability of wild type bacteria to cause disease and inflammation, suggesting that inflammasome activation is part of a broader repertoire of Esx-1-mediated virulence mechanisms; it is likely that Esx-1-mediated regulation of TNFα and IFNγ, as well as Esx-1-mediated activation of the host metalloprotease MMP-9 [37], also contributes to inflammation and disease progression.
In infected host cells in vitro, a fraction of M. marinum bacilli escapes the phagosome in an Esx-1-dependent manner [8], [38], which may promote bacterial spread to uninfected neighboring cells [8], [38]. While the ability of M. tuberculosis to escape the phagosome remains highly controversial, Esx-1-dependent communication with host cell cytoplasm might play a similar role also in M. tuberculosis infection, and might also contribute to MHC class I presentation of mycobacterial antigens [6], [39]–[42]. Our study suggests that phagosome escape occurs in vivo as well as in vitro, a point previously uninvestigated. Thus, this may be a pathogenic role for the Esx-1 secretion system during infection, and might explain the requirement for Esx-1 in activation of the inflammasome, which generally responds to cytoplasmic signals.
Infection with wild type M. marinum caused formation of granulomas with a cellularity and architecture similar to those formed in tuberculosis [43], [44]. The granulomas also developed central caseating necrosis, an important feature in M. marinum infected fish and in human tuberculosis not replicated in the M. tuberculosis mouse model [7], [43], [45]. Thus, the mouse model of M. marinum infection might provide unique opportunities to study the development of caseous necrosis in the context of an experimentally amenable mammalian immune system. In contrast, Esx-1-deficient bacteria were unable to attract significant numbers of macrophages or to induce formation of proper granulomas, implying that Esx-1 has evolved to actively influence the genesis of granulomas. A similar requirement for Esx-1 has been observed in zebrafish embryos, where Esx-1-deficient bacteria are able to grow within macrophages but unable to recruit new macrophages to sites of M. marinum infection and induce their aggregation into granulomatous structures [7]. Importantly, our analysis extends this work and suggests that Esx-1's role in granuloma formation is significantly more complex, since compared to wild type, lesions formed in mouse tails in response to ΔRD1 M. marinum exhibited earlier T cell recruitment, aberrant T cell distribution, and a significant delay in developing central necrosis. Intriguingly, granulomas formed in response to wild type M. marinum in ASC-KO mice also exhibited increased numbers of T cells compared to similarly infected B6 mice, implying that at least part of Esx-1's effect on T cells is mediated via the inflammasome; this might be explained by IL-1β, which has been shown to functionally impair antigen presenting dendritic cells [46]. Although this hypothesis is consistent with data indicating that virulent M. tuberculosis decreases T cell activation by dendritic cells in vivo [47], future studies will be required to elucidate a possible role for Esx-1 and the inflammasome in influencing the adaptive host response during mycobacterial infection.
Our findings in the M. marinum-mouse model confirm and extend knowledge gained from other more established model systems, including the M. marinum-zebrafish and M. tuberculosis-mouse models. One advantage of the model we describe over zebrafish infection is the greater ability to probe the host immune response, particularly adaptive immunity. At the same time, this model is infection in a non-natural host, a defect that is shared by M. tuberculosis infections of rabbits, guinea pigs, and mice. As a result, there are almost certainly important adaptations M. marinum has made to its piscine and amphibian hosts that will not be discovered in murine infection, and it is equally likely that many adaptations M. tuberculosis has made to its human host will not be reflected in our model. Future studies are needed to establish how closely the M. marinum-mouse model mimics events in human tuberculosis. For example, it remains to be confirmed that granuloma formation in the tail progresses through the same mechanisms as in the lung of tuberculosis patients; initial insight into this important question may come from experimental aerosol infection of guinea pig or rabbit with wild type and Esx-1-deficient M. tuberculosis.
In summary, the mouse model of M. marinum infection has unique features that open up new avenues to analyze fundamental aspects of mycobacterial pathogenesis. Here we demonstrate that Esx-1-dependent activation of the inflammasome is host-detrimental, identifying an immunoregulatory function for Esx-1 in a defined host-pathogen interaction in vivo and suggesting that activation of caspase-1 during mycobacterial infection is a manifestation of bacterial virulence rather than a manifestation of host response.
Methods
Ethics statement
All animal studies followed the ethical guidelines of the M. marinum mouse infection protocol and the mouse bone marrow-derived macrophage protocol, which were created by FC, CD, JK and EJB and received ethical approvals by the Institutional Animal Care and Use Committee (IACUC) at Genentech.
Bacterial strains
Wild type M. marinum M-strain and an isogenic deletion mutant lacking RD1 (ΔRD1) have been described previously [48]. ΔRD was complemented with RD1-2F9 by integration of this cosmid into the chromosomal attB-site [13].
Macrophage infections
Bone marrow derived macrophages (BMDM) were obtained and cultured from C57BL/6 wild type, ASC-KO, NLRP3-KO and NLRC4-KO mice as described previously [13]. For analysis of caspase-1 activation and cytokine secretion, 3×106 BMDMs/well were infected at an MOI of 5, essentially as described [13]. For analysis of bacterial intracellular growth and LDH-release, 5x104 BMDMs/well were infected at an MOI of 5 or 0.1, as indicated in figure legends. All infections were performed at 32°C.
For analysis of caspase-1 activation and cytokine secretion upon M. marinum infection of bone marrow-derived macrophages, supernatants from infected cells were collected at indicated time points and immediately supplemented with complete, EDTA-free, protease inhibitor cocktail (Roche). Suspensions were centrifuged (5.500 rpm, 10 min, 4°C) to pellet remaining bacteria and cells, and subsequently concentrated 3-fold using Vivaspin 15R (2.000 MWCO; Sartorius Biolab). For Western blot analysis of caspase-1 activation, equal amounts were separated by SDS-PAGE. Caspase-1 p10 was detected with polyclonal rabbit anti-mouse caspase-1 p10 (M-20) Abs (Santa Cruz Biotechnology) followed by donkey anti-rabbit HRP-conjugated secondary Abs, and membranes were developed with West Pico (Pierce). Cytokines were measured by Luminex analysis (see below). For analysis of intracellular bacterial growth, infected macrophages were lysed with 0.1% (final concentration) Triton-X for 10 min at indicated time points, and serial dilutions were plated on 7H10 plates for CFU analysis. Cytotoxicity was assessed by analysis of LDH-release using cytotox 96 non-radioactive cytotoxicity assay (Promega), as described by the manufacturer. As control, uninfected macrophages were lysed with Triton-X, which causes complete lysis, as described above.
Mouse infections
Female C57BL/6 (B6) mice and ASC-KO mice were infected with 1x107 bacteria in 200 µl phosphate buffered saline (PBS) via tail vein or intracardiac injection, as indicated, at 12 weeks of age. Matched control mice were similarly injected with PBS. All ASC-KO mice used were backcrossed to B6 ≥18 times.
For mouse infections, bacteria were grown to logarithmic growth phase (OD600 = 0.7±0.2) in 7H9-broth, and collected by centrifugation (3500 rpm, 10 min). Cells were washed twice in PBS, and needled three times through a 26G1/2 needle (Becton Dickinson) to disrupt bacterial aggregates. Aggregates were pelleted by two separate centrifugation steps (2000 rpm, 1 min), where the supernatants, enriched for single cell bacteria, were transferred to new tubes. Bacterial suspensions were subsequently analyzed by light microscopy to confirm the absence of aggregates. Finally, the bacterial concentration was determined using a hemacytometer, and suspensions were diluted to 5×107 bacteria/ml (final concentration).
Analysis of visible tail lesions and bone erosion
The length (broadest width) of individual visible lesions was measured at indicated time points, and the accumulated length of all lesions in individual tails was calculated, and presented in centimeters.
Micro-computed tomography (micro-CT) imaging was performed on an ex-vivo micro-CT scanner (microCT 40, SCANCO Medical, Switzerland) at 12 µm isotropic voxel size, 1000 projections/rotation, 300 ms integration time, 70 keV photon energy, and 114 µA current. For each mouse, three corresponding tail vertebrae at or near the site of infection were scanned (except for the 14 days post infection time point in Fig 1C and D, where two vertebrae per mouse were scanned). The bone was segmented by applying a lower threshold (0.738 gHA/cc) to the 3D image data sets. Mean bone volume within the segmented bone was measured for each vertebra, and the average bone volume was calculated for each animal. Image analysis was performed using Analyze software package (AnalyzeDirect, Inc., Lenexa, KS, USA).
Analysis of bacterial growth in vivo
Blood collected via cardiac puncture was subjected to serial dilutions and plated on 7H10 plates and the amount of bacteria presented as CFUs per ml. Tails were cut into ∼5 mm pieces and homogenized in 3 ml DMEM supplemented with 0.1% Triton-X, using an AHS200 homogenizer (VWR) with saw tooth adaptors (10×105 mm, Troemner). Livers and lungs were similarly homogenized. Organ and tail suspensions were serially diluted and plated on 7H10 plates. Bacterial load is presented as CFUs/gram tissue.
Preparation of tail suspensions and cytokine analysis
Tails were severed from mice at the tail base, immediately bagged and put on dry ice, frozen in liquid N2 and pulverized with a similarly chilled biopulverizer (Biospec Products Inc.). Pulverized tails were resuspended in 1 ml PBS supplemented with complete, EDTA-free, protease inhibitor cocktail, and left on ice for 1.5 h. Finally the suspensions were centrifuged (20.000g, 20 min, 4°C) twice to pellet debris, and supernatants were collected for analysis. Total protein content in each tail suspension was determined by Bradford analysis (Bio-Rad). The amount of indicated cytokines was measured by Luminex analysis (see below), and for each tail, the amount of cytokine detected (pg/ml) was normalized to the total amount of protein (mg/ml) in that tail suspension, as determined by Bradford analysis. For Western blot analysis of mature IL-1β, similar amounts of samples were separated by SDS-PAGE. Mature IL-1β was detected with purified hamster anti-mouse IL-1β Abs (1 µg/ml final concentration, BD Biosciences Pharmingen) followed by goat anti-hamster HRP-conjugated secondary Abs. As loading control, actin was analyzed with affinity purified rabbit anti-actin Abs (Sigma) followed by donkey anti-rabbit HRP-conjugated Abs. Membranes were developed using a ChemiDoc XRS system (Bio-Rad) and the relative amounts of mature IL-1β and actin were quantified using Quantity One software (Bio-Rad). For each tail analyzed, the amount of mature IL-1β was divided by the amount of actin detected, and all values were subsequently normalized to tail with the highest ratio (i.e. most mature IL-1β).
Luminex analysis
The concentration of indicated cytokines was determined using the Luminex 100 system (Luminex Corporation) run by the Bio-Plex Manager 5.0 software (Bio-Rad). All cytokines were measured using Bio-Plex reagent kits (Bio-Rad), and curve fitting was performed either by a Logistic-5 PL or 4-PL regression method.
Histological analysis of granulomatous lesions
Tails were fixed in 10% buffered formalin followed by decalcification in Immunocal (Decal Chemical Corp) for 48 hours. Five transverse 3 µm sections, which included soft tissue and coccygeal vertebrae, were evaluated for each animal (at least 2 animals were analyzed per group). Sections of tails were stained with hematoxylin and eosin (H&E) for routine histologic evaluation, or for immunohistochemical evaluation, with either rat anti-mouse F4/80 (Serotec, Raleigh NC) at 10 ug/ml or with rabbit anti-CD3 clone SP7 (Lab Vision, Fremont CA) at a dilution of 1∶200. Photomicrographs were captured using a Nikon DXM1200C digital camera and images shown are at either 10× or 40× magnification. We scored a granuloma as caseating if there was acellular, amorphous eosinophilic material centrally located in an inflammatory lesion. For quantification of CD3-positive cells, images were acquired using the Ariol SL-50 automated slide scanning platform (Genetix Ltd, Hampshire, UK) at 100× final magnification. Using these scans, lesions from wild type and ΔRD1 infected tails were selected and defined by a pathologist in a blinded manner. CD3-positive cells within the defined lesion areas were identified and counted using Ariol's proprietary cell counting algorithm.
Transmission electron microscope (TEM) analysis of granulomatous lesions
Cross sections (∼1 mm thickness) of formalin fixed tails were cut out. Sections were washed three times in 0.1 M sodium cacodylate buffer containing 3 µM calcium chloride for 15 min each, and then incubated with 1% osmium tetroxide, 0.8% potassium ferrocyanide, 3 µM calcium chloride in 0.1 M sodium cacodylate for 1 hour. After washing with distilled water three times for 15 min each, samples were stained and stabilized in ice-cold 2% uranyl acetate for 1 hour and dehydrated in an ethanol series of 20%, 50%, 70%, 90% and three times 100% successively for 3 min each. After washed with propylene oxide (EMS) two times for 3 min each, the samples were then infiltrated in well-mixed 50% propylene oxide, 50% Epon-812 (EMS) two times for 4 hours with agitation followed by 100% Epon-812 three times for 4 hours each with agitation, after which the samples were placed in an oven and allowed to polymerize at 60–80°C for 48 hours. Thick section (∼1 µm) were performed and stained with 1% toluidine blue for the selection of granulomas. The selected areas were trimmed for thin section. Thin sections (∼80 nm) were collected and pre-stained with 2% uranyl acetate and lead citrate before examination in an FEI CM12 TEM.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. WHO 2009 http://www.who.int/tb/publications/global_report/2009/en/
2. CosmaCL
ShermanDR
RamakrishnanL
2003 The secret lives of the pathogenic mycobacteria. Annu Rev Microbiol 57 641 676
3. StinearTP
SeemannT
HarrisonPF
JenkinGA
DaviesJK
2008 Insights from the complete genome sequence of Mycobacterium marinum on the evolution of Mycobacterium tuberculosis. Genome Res 18 729 741
4. TravisWD
TravisLB
RobertsGD
SuDW
WeilandLW
1985 The histopathologic spectrum in Mycobacterium marinum infection. Arch Pathol Lab Med 109 1109 1113
5. StanleySA
RaghavanS
HwangWW
CoxJS
2003 Acute infection and macrophage subversion by Mycobacterium tuberculosis require a specialized secretion system. Proc Natl Acad Sci U S A 100 13001 13006
6. GuinnKM
HickeyMJ
MathurSK
ZakelKL
GrotzkeJE
2004 Individual RD1-region genes are required for export of ESAT-6/CFP-10 and for virulence of Mycobacterium tuberculosis. Mol Microbiol 51 359 370
7. VolkmanHE
ClayH
BeeryD
ChangJC
ShermanDR
2004 Tuberculous granuloma formation is enhanced by a mycobacterium virulence determinant. PLoS Biol 2 e367 doi:10.1371/journal.pbio.0020367
8. GaoLY
GuoS
McLaughlinB
MorisakiH
EngelJN
2004 A mycobacterial virulence gene cluster extending RD1 is required for cytolysis, bacterial spreading and ESAT-6 secretion. Mol Microbiol 53 1677 1693
9. DiGiuseppe ChampionPA
CoxJS
2007 Protein secretion systems in Mycobacteria. Cell Microbiol 9 1376 1384
10. PymAS
BrodinP
BroschR
HuerreM
ColeST
2002 Loss of RD1 contributed to the attenuation of the live tuberculosis vaccines Mycobacterium bovis BCG and Mycobacterium microti. Mol Microbiol 46 709 717
11. GiacominiE
IonaE
FerroniL
MiettinenM
FattoriniL
2001 Infection of human macrophages and dendritic cells with Mycobacterium tuberculosis induces a differential cytokine gene expression that modulates T cell response. J Immunol 166 7033 7041
12. MonteroMT
MatillaJ
Gomez-MampasoE
LasuncionMA
2004 Geranylgeraniol regulates negatively caspase-1 autoprocessing: implication in the Th1 response against Mycobacterium tuberculosis. J Immunol 173 4936 4944
13. KooIC
WangC
RaghavanS
MorisakiJH
CoxJS
2008 ESX-1-dependent cytolysis in lysosome secretion and inflammasome activation during mycobacterial infection. Cell Microbiol 10 1866 1878
14. KurenumaT
KawamuraI
HaraH
UchiyamaR
DaimS
2009 The RD1 locus in the Mycobacterium tuberculosis genome contributes to activation of caspase-1 via induction of potassium ion efflux in infected macrophages. Infect Immun 77 3992 4001
15. KleinnijenhuisJ
JoostenLA
van de VeerdonkFL
SavageN
van CrevelR
2009 Transcriptional and inflammasome-mediated pathways for the induction of IL-1beta production by Mycobacterium tuberculosis. Eur J Immunol 39 1914 1922
16. MasterSS
RampiniSK
DavisAS
KellerC
EhlersS
2008 Mycobacterium tuberculosis prevents inflammasome activation. Cell Host Microbe 3 224 232
17. PerssonJ
VanceRE
2007 Genetics-squared: combining host and pathogen genetics in the analysis of innate immunity and bacterial virulence. Immunogenetics 59 761 778
18. ClarkHF
ShepardCC
1963 Effect of environmental temperatures on infection with Mycobacterium marinum (balnei) of mice and a number of poikilothermic species. J Bacteriol 86 1057 1069
19. BarckKH
LeeWP
DiehlLJ
RossJ
GriblingP
2004 Quantification of cortical bone loss and repair for therapeutic evaluation in collagen-induced arthritis, by micro-computed tomography and automated image analysis. Arthritis Rheum 50 3377 3386
20. EhlersS
1999 Immunity to tuberculosis: a delicate balance between protection and pathology. FEMS Immunol Med Microbiol 23 149 158
21. NorthRJ
JungYJ
2004 Immunity to tuberculosis. Annu Rev Immunol 22 599 623
22. DerrickSC
MorrisSL
2007 The ESAT6 protein of Mycobacterium tuberculosis induces apoptosis of macrophages by activating caspase expression. Cell Microbiol 9 1547 1555
23. StanleySA
JohndrowJE
ManzanilloP
CoxJS
2007 The Type I IFN response to infection with Mycobacterium tuberculosis requires ESX-1-mediated secretion and contributes to pathogenesis. J Immunol 178 3143 3152
24. NakanoN
NishiyamaC
KanadaS
NiwaY
ShimokawaN
2007 Involvement of mast cells in IL-12/23 p40 production is essential for survival from polymicrobial infections. Blood 109 4846 4855
25. MariathasanS
MonackDM
2007 Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat Rev Immunol 7 31 40
26. PozosTC
RamakrishnanL
2004 New models for the study of Mycobacterium-host interactions. Curr Opin Immunol 16 499 505
27. KooIC
OholYM
WuP
MorisakiJH
CoxJS
2008 Role for lysosomal enzyme beta-hexosaminidase in the control of mycobacteria infection. Proc Natl Acad Sci U S A 105 710 715
28. DionneMS
PhamLN
Shirasu-HizaM
SchneiderDS
2006 Akt and FOXO dysregulation contribute to infection-induced wasting in Drosophila. Curr Biol 16 1977 1985
29. ClayH
VolkmanHE
RamakrishnanL
2008 Tumor necrosis factor signaling mediates resistance to mycobacteria by inhibiting bacterial growth and macrophage death. Immunity 29 283 294
30. ShepardCC
HabasJA
1967 Relation of infection to tissue temperature in mice infected with Mycobacterium marinum and Mycobacterium leprae. J Bacteriol 93 790 796
31. AbdallahAM
Gey van PittiusNC
ChampionPA
CoxJ
LuirinkJ
2007 Type VII secretion–mycobacteria show the way. Nat Rev Microbiol 5 883 891
32. DavisJM
RamakrishnanL
2009 The role of the granuloma in expansion and dissemination of early tuberculous infection. Cell 136 37 49
33. LawK
WeidenM
HarkinT
Tchou-WongK
ChiC
1996 Increased release of interleukin-1 beta, interleukin-6, and tumor necrosis factor-alpha by bronchoalveolar cells lavaged from involved sites in pulmonary tuberculosis. Am J Respir Crit Care Med 153 799 804
34. KinjoY
KawakamiK
UezuK
YaraS
MiyagiK
2002 Contribution of IL-18 to Th1 response and host defense against infection by Mycobacterium tuberculosis: a comparative study with IL-12p40. J Immunol 169 323 329
35. SugawaraI
YamadaH
KanekoH
MizunoS
TakedaK
1999 Role of interleukin-18 (IL-18) in mycobacterial infection in IL-18-gene-disrupted mice. Infect Immun 67 2585 2589
36. SutterwalaFS
OguraY
FlavellRA
2007 The inflammasome in pathogen recognition and inflammation. J Leukoc Biol 82 259 264
37. VolkmanHE
PozosTC
ZhengJ
DavisJM
RawlsJF
2010 Tuberculous granuloma induction via interaction of a bacterial secreted protein with host epithelium. Science 327 466 469
38. StammLM
MorisakiJH
GaoLY
JengRL
McDonaldKL
2003 Mycobacterium marinum escapes from phagosomes and is propelled by actin-based motility. J Exp Med 198 1361 1368
39. van der WelN
HavaD
HoubenD
FluitsmaD
van ZonM
2007 M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell 129 1287 1298
40. MazzaccaroRJ
GeddeM
JensenER
van SantenHM
PloeghHL
1996 Major histocompatibility class I presentation of soluble antigen facilitated by Mycobacterium tuberculosis infection. Proc Natl Acad Sci U S A 93 11786 11791
41. TeitelbaumR
CammerM
MaitlandML
FreitagNE
CondeelisJ
1999 Mycobacterial infection of macrophages results in membrane-permeable phagosomes. Proc Natl Acad Sci U S A 96 15190 15195
42. LewinsohnDM
GrotzkeJE
HeinzelAS
ZhuL
OvendalePJ
2006 Secreted proteins from Mycobacterium tuberculosis gain access to the cytosolic MHC class-I antigen-processing pathway. J Immunol 177 437 442
43. RussellDG
2007 Who puts the tubercle in tuberculosis? Nat Rev Microbiol 5 39 47
44. UlrichsT
KaufmannSH
2006 New insights into the function of granulomas in human tuberculosis. J Pathol 208 261 269
45. FlynnJL
2006 Lessons from experimental Mycobacterium tuberculosis infections. Microbes Infect 8 1179 1188
46. MakinoM
MaedaY
MukaiT
KaufmannSH
2006 Impaired maturation and function of dendritic cells by mycobacteria through IL-1beta. Eur J Immunol 36 1443 1452
47. WolfAJ
LinasB
Trevejo-NunezGJ
KincaidE
TamuraT
2007 Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo. J Immunol 179 2509 2519
48. CosmaCL
HumbertO
RamakrishnanL
2004 Superinfecting mycobacteria home to established tuberculous granulomas. Nat Immunol 5 828 835
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 5
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Quorum Sensing Inhibition Selects for Virulence and Cooperation in
- The Role of Intestinal Microbiota in the Development and Severity of Chemotherapy-Induced Mucositis
- Crystal Structure of HIV-1 gp41 Including Both Fusion Peptide and Membrane Proximal External Regions
- Susceptibility to Anthrax Lethal Toxin-Induced Rat Death Is Controlled by a Single Chromosome 10 Locus That Includes
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy