
Internalin B Activates Junctional Endocytosis to Accelerate Intestinal Invasion
Listeria monocytogenes (Lm) uses InlA to invade the tips of the intestinal villi, a location at which cell extrusion generates a transient defect in epithelial polarity that exposes the receptor for InlA, E-cadherin, on the cell surface. As the dying cell is removed from the epithelium, the surrounding cells reorganize to form a multicellular junction (MCJ) that Lm exploits to find its basolateral receptor and invade. By examining individual infected villi using 3D-confocal imaging, we uncovered a novel role for the second major invasin, InlB, during invasion of the intestine. We infected mice intragastrically with isogenic strains of Lm that express or lack InlB and that have a modified InlA capable of binding murine E-cadherin and found that Lm lacking InlB invade the same number of villi but have decreased numbers of bacteria within each infected villus tip. We studied the mechanism of InlB action at the MCJs of polarized MDCK monolayers and find that InlB does not act as an adhesin, but instead accelerates bacterial internalization after attachment. InlB locally activates its receptor, c-Met, and increases endocytosis of junctional components, including E-cadherin. We show that MCJs are naturally more endocytic than other sites of the apical membrane, that endocytosis and Lm invasion of MCJs depends on functional dynamin, and that c-Met activation by soluble InlB or hepatocyte growth factor (HGF) increases MCJ endocytosis. Also, in vivo, InlB applied through the intestinal lumen increases endocytosis at the villus tips. Our findings demonstrate a two-step mechanism of synergy between Lm's invasins: InlA provides the specificity of Lm adhesion to MCJs at the villus tips and InlB locally activates c-Met to accelerate junctional endocytosis and bacterial invasion of the intestine.
Published in the journal:
. PLoS Pathog 6(5): e32767. doi:10.1371/journal.ppat.1000900
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1000900
Summary
Listeria monocytogenes (Lm) uses InlA to invade the tips of the intestinal villi, a location at which cell extrusion generates a transient defect in epithelial polarity that exposes the receptor for InlA, E-cadherin, on the cell surface. As the dying cell is removed from the epithelium, the surrounding cells reorganize to form a multicellular junction (MCJ) that Lm exploits to find its basolateral receptor and invade. By examining individual infected villi using 3D-confocal imaging, we uncovered a novel role for the second major invasin, InlB, during invasion of the intestine. We infected mice intragastrically with isogenic strains of Lm that express or lack InlB and that have a modified InlA capable of binding murine E-cadherin and found that Lm lacking InlB invade the same number of villi but have decreased numbers of bacteria within each infected villus tip. We studied the mechanism of InlB action at the MCJs of polarized MDCK monolayers and find that InlB does not act as an adhesin, but instead accelerates bacterial internalization after attachment. InlB locally activates its receptor, c-Met, and increases endocytosis of junctional components, including E-cadherin. We show that MCJs are naturally more endocytic than other sites of the apical membrane, that endocytosis and Lm invasion of MCJs depends on functional dynamin, and that c-Met activation by soluble InlB or hepatocyte growth factor (HGF) increases MCJ endocytosis. Also, in vivo, InlB applied through the intestinal lumen increases endocytosis at the villus tips. Our findings demonstrate a two-step mechanism of synergy between Lm's invasins: InlA provides the specificity of Lm adhesion to MCJs at the villus tips and InlB locally activates c-Met to accelerate junctional endocytosis and bacterial invasion of the intestine.
Introduction
Listeria monocytogenes (Lm) is a potentially deadly food-borne pathogen that colonizes the gastrointestinal tract of several mammalian species, and can also cause invasive disease and systemic spread if it crosses the intestinal epithelial barrier [1]. Lm evolved two major molecular invasion proteins, referred to here as invasins: Internalin A (InlA, Internalin) and Internalin B (InlB) [2], [3]. These proteins promote internalization into nonphagocytic cells where Lm can grow in the cytosol as a facultative intracellular pathogen and directly spread to neighboring cells through actin-based motility [2]–[5]. Listerial invasion of the gastrointestinal tract requires InlA since deletion of the inlA gene makes Lm avirulent when given through the enteric route [6]. By contrast, inlA is dispensable for simulation of late-stage pathogenesis when bacteria are administered intravenously [6]. InlA binds the most distal extracellular domain of E-cadherin, a transmembrane epithelial cell-cell junction protein [7]–[9]. InlB, the second Lm surface protein involved in invasion, binds c-Met, a receptor tyrosine kinase (RTK) and the natural receptor for Hepatocyte Growth Factor (HGF) [2], [10]. InlB promotes invasion of multiple mammalian cell types, and has been implicated in liver colonization after intravenous infection of mice [2], [10]–[24]. Although InlB is not essential for fetoplacental infection, it was recently shown to act synergistically with InlA to promote fetoplacental infection of intravenously inoculated pregnant gerbils and transgenic mice expressing a humanized E-cadherin [20], [25], [26]. InlB is also known to function synergistically with InlA during invasion of cultured epithelial cells through an unknown mechanism [2], [13], [20], [24], [27]–[30]. Paradoxically, neither E-cadherin or c-Met are available on the apical or lumenal side of epithelia, thus it was puzzling to understand where Lm finds its receptors for invasion of the intestine [31]–[33].
We identified the cell extrusion zone at the tips of the intestinal villi as a novel site for gastrointestinal invasion where Lm uses InlA to bind E-cadherin for attachment and entry [30]. The intestinal epithelium is in a constant state of rapid renewal in a process that begins with stem cell division within the crypts, followed by maturation and migration of cells up to the tips of the intestinal villi. Once the oldest cells reach the villus tip, programmed cell death is triggered and individual dying cells are extruded into the lumen [34], [35]. It has been estimated that 1400 cells are shed from each villus tip per day, which is ∼1011 cells per day from the human small intestine [34]. Surprisingly, this occurs without disruption of epithelial continuity because the surrounding cells constrict the dying cell and meet to form a new multicellular junction (MCJ) below the extruding cell [30], [35]–[38]. In the process, the cells that form the MCJ may also remove and recycle the old junctions and adhesive contacts by endocytosis [35]. We showed that Lm takes advantage of extrusion for adhesion and invasion because MCJs transiently expose basolateral E-cadherin to the lumen of the intestine and at analogous sites in tissue culture [30]. Although a reasonable hypothesis, it is not known whether other basolateral proteins, like c-Met, are exposed to the apical side at MCJs.
In contrast to what has been observed during infection of cultured cells, a role for InlB in the intestinal phase of infection could not be demonstrated previously [20], [24]. However, several observations suggest that InlA and InlB may both function during infection of the gastrointestinal tract. First, the inlB gene is immediately downstream of inlA and is translated bicistronically with inlA [3]. The inlAB operon is upregulated when bacteria are in the intestinal lumen or under conditions simulating the gastrointestinal environment, indicating that InlB expression is temporally upregulated prior to bacterial invasion of intestinal tissue [39]–[42]. Finally, InlB promotes invasion of isolated intestinal epithelial cells when InlA-E-cadherin interactions are functional [20]. Thus, we hypothesized that InlB functions synergistically with InlA to promote Lm invasion of MCJs of the villus tip extrusion zone and that c-Met may be exposed to lumenal surfaces during cell extrusion.
Until recently, it was not technically feasible to study the functions of InlA and InlB together in commonly utilized animal models since both proteins are ‘species specific’: InlA binds rabbit and guinea pig E-cadherin, but not rat and mouse E-cadherin; InlB activates mouse c-Met, but not guinea pig or rabbit c-Met [24]. The mouse is the predominant animal model for studying systemic Listeriosis and host immune responses following intraperitoneal or intravascular infection [43]. However, an understanding of the intestinal phase of infection has lagged behind, since mice are very resistant to enteric infection with Lm due to the absence of the InlA-E-cadherin interaction [6], [9]. To study the intestinal phase of Listeriosis in the mouse, one strategy has been to develop transgenic mice that express a permissive E-cadherin [6], [20]. Alternately, InlA was recently engineered to bind murine E-cadherin (InlAm) and is sufficient to reconstitute intestinal invasion after intragastric infection of mice [44], [45].
In this study we constructed Lm strains that express InlAm with or without InlB to dissect the role of InlB in a mouse model of enteric infection. We also made strains that express green fluorescent protein (GFP) in order to perform co-infection studies where two strains that are differentially marked are mixed and inoculated together. Using this method we confirm that InlA is essential to invade the extrusion zone of the intestinal villus tips after oral infection, and establish a role for InlB working synergistically with InlA in colonization of the intestinal villi.
Based on published cell biological experiments in non-polarized epithelial cells, we considered three nonexclusive hypotheses for InlB action at MCJs of villus tips. One is that InlB acts directly as an adhesion protein (adhesin) to promote Lm uptake, as suggested by experiments with endothelial cells [14]. A second is that InlB activates c-Met to promote cell-cell dissociation, as seen with recombinant HGF or InlB applied to small islands of cultured epithelial cells, thereby allowing access of Lm to E-cadherin at the basolateral surface [10]. Finally, we considered that InlB might promote Lm invasion by increasing endocytosis of junctional E-cadherin through c-Met activation as shown for HGF action on nonpolarized cells [46], [47]. To study these possibilities on polarized epithelia we used Madin-Darby canine kidney (MDCK) cell monolayers grown on Transwell filters, a well-characterized model epithelium that is permissive for all aspects of the Listeria's intracellular life-cycle including InlA - and InlB-mediated invasion [30], [48]. We discovered that InlB promotes invasion of the MCJs in polarized MDCK monolayers, but not by acting as an adhesin or increasing Lm attachment to E-cadherin across the junctions. Instead we find that InlB locally activates c-Met from the lumenal side to modulate the kinetics of invasion. Using endocytosis assays combined with confocal microscopy analysis, we show that both MCJs in tissue culture and the villus tip extrusion zone are naturally more endocytic than other regions of the epithelium and that InlB modulates this process. We propose that Lm has evolved a two-step mechanism to hijack and alter junctional remodeling for epithelial attachment and invasion. First Lm specifically target and adhere to the MCJs of the villus tips through apically exposed E-cadherin, and then they use InlB to accelerate the recycling of junction components to increase invasion at MCJs.
Results
InlB Promotes Invasion of the Villus Tip Extrusion Zone
In order to study InlB and InlA in the same animal model we had to overcome the species specificity of each molecule. We chose to use an InlA mutation that is capable of binding murine E-cadherin (InlAm) [44]. In contrast to Lm expressing wild type InlA, Lm expressing InlAm are pathogenic to mice by enteric inoculation [44]. In the small intestine, InlAm promotes invasion through villous tissue but has no effect on passive bacterial uptake by Peyer's Patches [44]. We infected mice intragastrically with Lm that express InlAm and GFP (WTm GFP) to study Lm invasion of the intestinal villous epithelium. By culturing fecal pellets at different times after infection, we noted that peak shedding of the inoculum occurs by 3 hours. We therefore chose to examine the small intestine for evidence of bacterial invasion by direct visualization of tissue whole mounts within 4–6 h of infection.
We find that WTm GFP invade the extrusion zone at the tips of the murine intestinal villi, similar to what we previously reported for Lm in a rabbit ileal loop model, and in accord with the observation by Wollert et al. that a similarly modified strain invades the murine intestinal epithelium [30], [44]. Infected villus tips were most abundantly observed in tissue from the terminal ileum, in agreement with previous observations of enteric infection of permissive animals [49], [50]. We found that that ingestion of Lm does not result in generalized invasion of all intestinal villi. Rather, we find that infection occurs at sporadic villus tips (Figure 1). We used 3D confocal microscopy analysis to characterize Lm invasion of villus tips within ∼1 cm2 tissue sections from the terminal ileum (Figure 1, Figure S1, Video S1). Intracellular Lm with polymerized actin comet tails are observed in villus tips by 4 hours after intragastric inoculation (Figure 1A), only slightly longer than the time needed to generate actin-based motility in tissue culture (∼3 h) [30], [48], [51]. Thus Lm rapidly traffic through the murine bowel and establish initial infection of villus tips.

To examine the role of InlB in intestinal infection, we inoculated mice with either WTm GFP or an isogenic strain lacking inlB (ΔinlBm GFP) and examined the small intestine using confocal microscopy to determine the frequency of infected villi and the number of Listeria per infected villus tip. Both strains preferentially invade the terminal ileum and invade approximately the same number of villi (N) within a section of tissue by 5 hours post inoculation (Figure S1). However, mice infected with WTm GFP have approximately twice the number of Lm per villus tip than mice infected with ΔinlBm GFP (Figure 1B–C, 3D rendered top panels, Figure S1). Both strains are able to escape the endosome and replicate in the cytosol of enterocytes since they induce actin polymerization on the bacterial surface, as observed in Z-planes located below the apical brush border (Figure 1B–C, lower panels). To control for variability between mice in intestinal transit, and thus more stringently examine whether InlB is involved in early colonization of the villus tips, we mixed the two strains at a 1∶1 ratio and performed co-infection experiments. In order to distinguish the two strains, we tagged them differentially with GFP and then counterstained them with anti-L. monocytogenes antibodies in red. Thus, the GFP expressing strain appears yellow (or a combination of red and green) in a merged image and the non-GFP expressing strain appears red (Figure 1D–H).
As shown in Figure 1D, in co-infections with WTm GFP and ΔinlBm, scattered villi are infected. In all co-infections, the villus tips infected with WTm GFP have significantly more intracellular bacteria than villus tips infected with ΔinlBm at 6 hours, even though the number of infected villi by each strain (N) was similar (Figure 1E). We switched the strains in which GFP was expressed to control for possible variations in antibody staining or possible effects of GFP on bacterial colonization (Figure 1F–H). As with the converse experiment, the presence of inlB significantly increases villus tip infection (Figure 1H). The majority of bacterial plaques within each infected villus are probably clonal since we found only 1 villus tip with both red and yellow bacteria (Figure 1G) among 175 infected villi analyzed (Figure 1E, 1H).
InlB Accelerates Apical Invasion at Multicellular Junctions but Does Not Act as an Adhesin
To better understand how InlB promotes invasion of the villus tip extrusion zone, we studied the kinetics and mechanisms of Lm invasion in polarized epithelial cells (Figure 2). We used MDCK cells grown on Transwell supports to visualize and study events at multicellular junctions (MCJs). Several clues of InlB function have been derived from studies using recombinant InlB, a genetically modified InlB that is covalently linked to the bacterial cell wall (InlB-SPA), or InlB-coated beads interacting with non-polarized epithelia [10], [11], [19], [24], [52]–[61]. These studies indicate that InlB can bind and activate the basolateral c-Met receptor leading to clathrin-mediated internalization of c-Met. It is not known whether InlB functions for Lm invasion as a soluble or a bacterium-associated factor or how InlB reaches this receptor in an intact epithelium since c-Met is not usually exposed on the apical membrane of polarized epithelia [62], [63]. Additionally, there are conflicting data regarding the role of InlB in intracellular replication [64], [65].

We infected MDCK monolayers polarized on Transwell filters from the apical side with GFP-expressing wild type Lm (WT) or GFP-expressing inlB-mutant Lm (ΔinlB) and analyzed attachment, invasion and intracellular replication. Attachment to the apical surface is not affected by the absence of InlB as determined by recovered colony forming units (CFUs) from a 10-minute attachment assay (Figure 2A). This is in agreement with our previous finding that InlA, rather than InlB, is the dominant adhesin for polarized cells [30]. Microscopic examination of the sites of attachment shows that ΔinlB also bind exclusively at intercellular junctions and preferentially at MCJs with the same specificity and frequency as WT ([30] and see below).
Since attachment was not affected by InlB, we studied its role in invasion following attachment by incubating adhered WT or ΔinlB with the epithelium for a period of 1 h, treating with gentamicin for 30 minutes to kill extracellular bacteria, and determining the number of viable intracellular bacteria. We find that InlB is important for efficient invasion since intracellular ΔinlB are significantly reduced compared to WT (∼35%, p<0.0001, Figure 2A). At various time points during the 1 h infection, polarized MDCK monolayers were fixed and analyzed by confocal immunofluorescence microscopy with an inside-outside staining protocol that distinguishes attached extracellular bacteria from internalized bacteria. Both WT and ΔinlB invade polarized MDCK monolayers almost exclusively through MCJs, which represent only ∼2% of all available junctions (Figure 2B, Figure S2 and [30]), however invasion by ΔinlB is delayed. By 20 minutes after adhesion, internalized WT bacteria are observed, while all ΔinlB remain extracellular. At each time point after attachment a greater proportion of WT than ΔinlB are internalized (Figure 2B, 2C). Thus, InlB is dispensable for cell attachment in polarized epithelia but increases invasion once bacteria are associated with the cell surface.
We also investigated the role of InlB in intracellular replication to determine whether the increase in internalized bacteria is solely due to an accelerated entry of WT bacteria or also due to increased replication within the cell. Polarized MDCK monolayers were infected with WT or ΔinlB at a multiplicity of infection (MOI) of 10 bacteria/cell. At various time points, the monolayers were fixed and analyzed by confocal immunofluorescence microscopy to quantify the replication rate of the intracellular bacteria (Figure S2). At each time point during infection WT plaques are greater in area and bacterial number than ΔinlB (Figure S2, Figure 2D). However, intracellular doubling times are essentially identical between the two strains (WT Td = 1.25 h and ΔinlB Td = 1.26 h; comparison of fits (k), p = 0.97; Figure 2D). Thus, InlB influences the rate of epithelial invasion at MCJs but is not involved in intracellular growth.
InlB Accelerates Invasion of Polarized MDCK Cells by Activating c-MET from the Apical Compartment
Soluble InlB activates c-Met signaling when added to nonconfluent epithelia with exposed basolateral surfaces [10], [11], [19]. However, since c-Met is a basolateral protein not exposed on the apical side it is unclear whether the same occurs in polarized epithelia [31], [66]. To test the role of c-Met on apical invasion of polarized epithelial cells, we pretreated the confluent polarized monolayers with SU11274 to inhibit c-Met signaling or DMSO as a control, then infected them with WT or ΔinlB through the apical compartment [67]. The kinase inhibitor reduces WT invasion to the level of ΔinlB invasion but has no significant effect on the invasion of ΔinlB (Figure 2E). Thus, c-Met activation is required for InlB activity during apical invasion of the MCJs.
Since c-Met is not readily available in the apical surface, we wondered whether InlB acts as a soluble factor or whether c-Met is activated locally at the MCJs after bacterial attachment. It has been suggested that InlB may function as a soluble and diffuse c-Met agonist since InlB is only loosely associated with the bacterial surface, and since recombinant InlB can mimic HGF by inducing cell membrane ruffling or cell scattering [10], [19], [52], [62], [68]. On the other hand, Lm invade cells through tight membrane invaginations without apparent changes of cell surfaces where bacteria are absent, suggesting that InlB associated with the bacterial surface mediates c-Met activation within close proximity to each individual bacterium [14], [63], [69]. We performed co-infections of polarized MDCK monolayers with a mixture of WT and ΔinlB and hypothesized that WT would rescue the defect of ΔinlB invasion if InlB acts as a soluble factor acting on all cells within the epithelium. We find that InlB does not act globally on the epithelium, since ΔinlB continue to exhibit a defect in invasion in the presence of WT in a mixed infection. The magnitude of the defect is the same in mixed as in separate infections (Figure 2A, 2F) and we obtained the same competition defect for ΔinlB at MOI ratios of 100∶1, 10∶1, or 1∶1 (Figure 2F, Figure S3).
To further address whether c-Met activation is restricted to the immediate surrounding of individual bacteria, we tested whether the c-Met kinase inhibitor used in a mixed infection would reduce both WT and ΔinlB invasion, or alternately whether c-Met inhibition would selectively affect WT invasion. As in single infections, the c-Met kinase inhibitor reduced WT invasion to the level of ΔinlB in a mixed infection (Figure 2F, Figure S3). These results indicate that local c-Met activation by InlB at the MCJ is responsible for the increased invasion.
InlB and HGF Accelerate Endocytosis at Multicellular Junctions
Since InlB increases the rate of Lm internalization through activation of c-Met at MCJs, we also wondered whether MCJs are intrinsically different in their endocytic activity as compared to the rest of the apical surface. MCJs represent sites of recent or ongoing cell extrusion where the tight junctions (TJs) are being rapidly remodeled [30], [35]. Additionally, we find that E-cadherin is remodeled through endocytosis during cell extrusion and MCJ formation (Figure S4A). Thus, we hypothesized that MCJs may be more permissive to bacterial entry than other junctional sites because of greater endocytic potential. This is also suggested by the observation that Lm invasion through MCJs is more likely than invasion through other junctional sites of attachment: 26% of Lm associated with a polarized MDCK epithelium attach to epithelial junctions that are not a MCJ but invasion occurs almost exclusively at MCJs since 97% of intracellular foci of Lm originate at these sites, even in the absence of InlB (Figure 2B, Figure S2 and [30]).
To test whether endocytosis is naturally increased at MCJs, we added fluorescent dextran to the apical side of uninfected polarized MDCK monolayers for 30 minutes and determined whether uptake is greater at MCJs than through the rest of the apical surface (Figure 3). Puncta of internalized dextran are readily found in the cells making MCJs and the fluorescence intensity of dextran is higher than at non-multicellular junction (Non-MCJ) regions of the polarized monolayer (Figure 3A, 3E). Interestingly, some internalized dextran at MCJs colocalizes with internalized E-cadherin as well as ZO-1, a scaffolding protein associated with the TJs in polarized cells (Figure 3E) [70]. We observe similar puncta of endocytosed E-cadherin at MCJs in vivo at villus tips (Figure S4B). Thus, significant endocytosis occurs specifically at MCJ sites in a polarized epithelium. In addition, E-cadherin, the receptor for Lm internalization, is naturally endocytosed at MCJs.
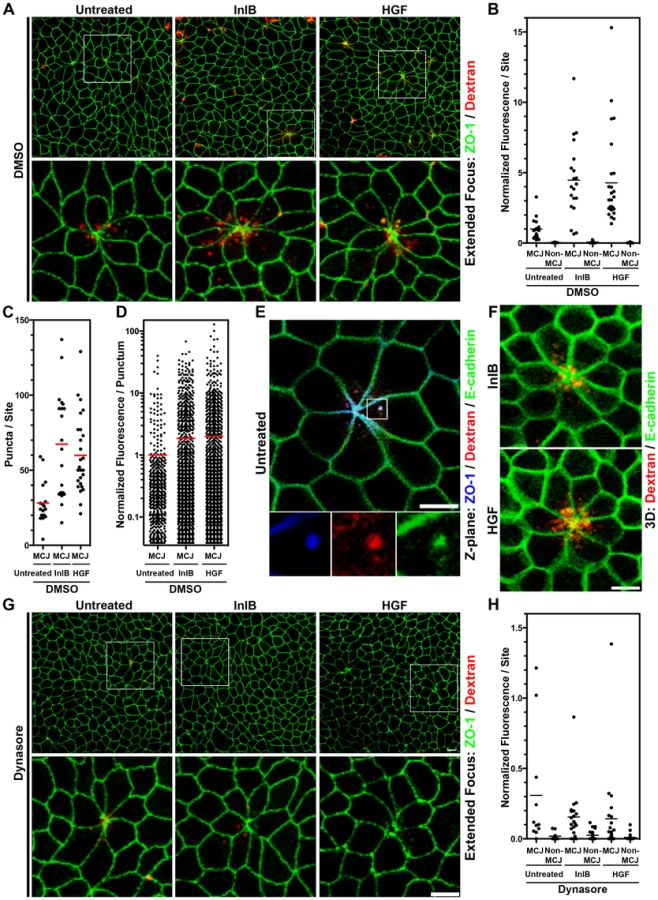
We asked whether c-Met activation at MCJs could locally accelerate endocytosis since growth factor activation of RTKs has been shown to induce endocytosis of E-cadherin through either macropinocytosis or clathrin-mediated endocytosis [46], [71], [72]. We pretreated polarized MDCK cells from the apical side for 1 h with HGF or InlB prior to the addition of fluorescent dextran to the apical compartment. We find that both HGF and InlB significantly increase the amount of dextran endocytosed at MCJs (p<0.001), but not at non-MCJ regions compared to untreated cells (Figure 3A–B, Figure S5). To control for the specificity of this process we used a truncated InlB consisting of only the C-terminal GW domains (GW[2]–[3]) and this has no effect on endocytosis compared to untreated monolayers (Figure S5) [15]. These results suggest that basolateral c-Met is made transiently accessible through the rapid junctional remodeling at MCJs. Puncta of endocytosed dextran were also co-localized with junctional proteins at MCJs in HGF and InlB treated monolayers (Figure 3A, 3F). Increased endocytosis of dextran after HGF and InlB treatment is the product of an increase in the number of puncta of internalized dextran per MCJ (Untreated versus InlB or HGF p<0.001, Figure 3C) and an increase in the amount of dextran internalized as determined by fluorescence intensity per punctum (Untreated versus InlB p<0.05, Untreated versus HGF p<0.01, Figure 3D). This suggests that both the rate of endocytosis as well as the capacity of individual endocytic vesicles is increased by HGF or InlB.
Endocytosis and L. monocytogenes Invasion at Multicellular Junctions Require Common Endocytic Machinery
In nonpolarized cells, Lm invasion requires molecular machinery associated with clathrin-mediated endocytosis, including dynamin [59], [73]. To test whether invasion of the MCJs is also dynamin-dependent, we pretreated polarized MDCK monolayers with either DMSO as a control or dynasore, an inhibitor of dynamin, and infected them with Lm (Figure 4) [74]. Using inside-outside confocal microscopy analysis of monolayers infected for 1 h, we find that Lm invade control cells at MCJs, but cannot invade cells treated with dynasore (Figure 4A). A second assay using gentamicin protection also confirmed this result. Lm were allowed to invade for a period of 1 h, the infected monolayers were treated with gentamicin for 30 minutes and the number of viable intracellular bacteria was determined. Compared to control cells, polarized cells treated with dynasore are significantly less permissive for Lm invasion (Figure 4B–C; ∼13% DMSO, WT p<0.0001; ∼16% DMSO, ΔinlB GFP p<0.0001)

To test whether the increased rate of apical endocytosis at MCJs is also a dynamin-dependent process, we pretreated polarized MDCK monolayers with dynasore or DMSO as a control prior to addition of fluorescent dextran. Pretreatment of polarized cells with dynasore inhibits nearly all endocytosis of dextran at multicellular junctions regardless of HGF or InlB treatment (Figure 3F, 3G). Indeed, uptake at multicellular junctions is not significantly higher than uptake at non-multicellular junctions within monolayers treated with dynasore (Figure 3G). These data suggest that InlB accelerates dynamin-dependent endocytosis at MCJs leading to an increase the rate of Lm uptake at these sites.
InlB Enhances Apical Endocytosis at the Villus Tip Extrusion Zone
Our tissue culture results suggested that the villus tip extrusion zone might also be permissive to Lm invasion because of an increased rate of endocytosis in vivo. We incubated fluorescent dextran in mouse ileal loops for 45 minutes and examined villus tips by confocal microscopy to test this hypothesis (Figure 5). Puncta of fluorescent dextran are readily found in the villus tip epithelium, but not the epithelium along the lateral sides of villi or crypt epithelium (Figure 5A, 5B and data not shown). To test whether InlB promotes endocytosis at the villus tips, we incubated InlB with dextran in mouse ileal loops (Figure 5D, 5E). Internalized puncta of dextran at MCJs are found associated with E-cadherin in both untreated and InlB treated villi (Figure 5C, 5F). However, InlB significantly increases the amount of dextran endocytosis at the villus tips (p<0.05, Figure 5G) by increasing the number of puncta of dextran per villus tip (p<0.05, Figure 5H) and the amount of dextran per punctum (p<0.005, Figure 5I).

Discussion
Epithelia are the first site of interaction between the host and a wide variety of invading pathogens and the intercellular junctions are crucial to maintain a tight seal between epithelial cells to prevent microbial invasion. It is interesting that diverse microbes have evolved strategies to usurp the epithelial junctions to mediate extracellular colonization, intracellular invasion or paracellular breach (reviewed in [75]–[79]).
Microbes that invade epithelial cells often use receptors for internalization that are part of the junctions or are basolateral proteins. For example, reoviruses bind JAM-A, coxsackie and adenovirus bind CAR, hepatitis C virus binds claudins and occludin, rotaviruses, Shigella flexneri and enteropathogenic Yersiniae bind integrins, α-herpesviruses bind Nectins, and Listeria monocytogenes (Lm) binds E-cadherin [7], [80]–[97]. Although targeting of junction or basolateral proteins by invasive pathogens is a successful strategy, it is also seemingly paradoxical since these receptors are not normally localized at the apical surface.
The study of Listeria pathogenesis in the gastrointestinal tract reveals that Lm has evolved to target a subset of intercellular junctions that have a natural and transient defect in cell polarity generated during the process of cell extrusion. First, we noted that Lm uses InlA to access E-cadherin as it becomes exposed at multicellular junctions (MCJs, Figure 6A) [30]. Our studies here of InlB suggest that the MCJ's are not only a natural site of local loss of polarity, but also that the normal process of junction renewal involves accelerated endocytic processes that can be hijacked and modulated by additional bacterial invasive factors (Figure 6B).
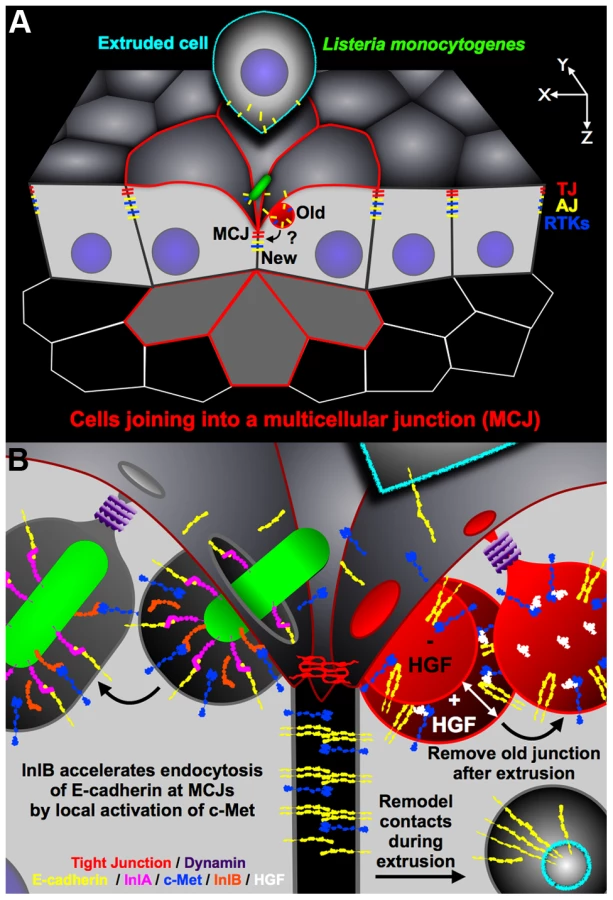
Why are MCJs inherently endocytic? The formation and resolution of MCJs by cell extrusion requires junctional reorganization, changes in cell position, and changes in cell morphology [30], [35], [38], [98]. There is increasing evidence that remodeling of adhesive contacts, including modification of junctional length or cell position within epithelia, requires endocytosis of adhesion molecules such as E-cadherin [99]–[104]. Furthermore, it was found that in cells neighboring extruding cells, large endosome-like structures contain tight junction (TJ) strands [35]. We also find that cells neighboring extruding cells internalize E-cadherin, a component of the adherens junction (AJ), from the extruding cell while forming a MCJ (Figure S4, Figure 5B). Thus endocytosis at MCJs may be important to release adhesive contacts between the extruding cell and the rest of the epithelium, for removal of lumenally exposed basolateral and junctional proteins, and for redistribution of cell shape and position during cell extrusion [35], [99]–[101], [103]–[105]. It has been suggested that Lm adherence and invasion via E-cadherin is analogous to AJ assembly because of the similarity of their molecular requirements [28], [63], [106], [107]. However, our model suggests that Lm invasion subverts junction disassembly, rather than assembly (Figure 6). This concept is supported by the fact that InlA binding results in tyrosine phosphorylation, ubiquitination and endocytosis of E-cadherin [27].
InlA binding to E-cadherin is sufficient for Listeria invasion, however modulation of endocytosis by InlB accelerates this process (Figure 6B). We show that while InlB is dispensable for attachment, it synergistically promotes invasion of MCJs through activation of c-Met kinase signaling. Activation of cell signaling that results in endocytosis is a strategy utilized by other invasive microbes. For example, viruses like coxsackievirus, HIV, caposi's sarcoma-associated herpesvirus and adenovirus, and bacteria like Salmonella, Shigella, Brucella, Neisseria, Mycobacteria, Haemophilus and Legionella can trigger macropinocytosis or macropinocytosis-like processes [80], [108]–[123]. In contrast, Lm utilizes a so-called ‘zipper-like’ mechanism of invasion/endocytosis distinct from macropinocytosis [14], [69]. Other investigators have shown that Lm requires dynamin and other molecular components of clathrin-mediated endocytosis for efficient invasion of nonpolarized cells [55], [59], [73]. Furthermore, macropinocytosis is thought to be independent of dynamin and requiring an alternate pinchase [124], [125]. It has been suggested that Lm hijacks the actin - and dynamin-dependent internalization of clathrin-coated paques, which are larger than clathrin-coated pits [126], [127]. We also find that Lm invasion of a polarized epithelium through the MCJs requires functional dynamin even in the absence of InlB. Similarly, L. innocua expressing InlA, but not InlB, requires functional dynamin for invasion [128]. This further supports the notion that Lm subverts junction disassembly since both Lm invasion via E-cadherin and junction regulation via E-cadherin endocytosis require functional dynamin [102], [129].
InlB has been shown to promote dynamin-dependent internalization of Listeria when the bacteria have access to the basolateral surface, and HGF similarly promotes internalization of E-cadherin when added to basal surfaces [59], [71]. Although c-Met is not exposed on the apical surface of epithelia, we hypothesized that InlB could activate c-Met because of the local loss of cell polarity that occurs at MCJs. We find that apical treatment of polarized epithelia with either HGF or InlB increases apical endocytosis of dextran at MCJs (Figure 6B). Interestingly HGF and InlB do not increase endocytosis at non-MCJ regions of the epithelia suggesting that c-Met, a basolateral protein like E-cadherin, is also only accessible from the apical side through the process of cell extrusion and MCJ formation [30]. We confirmed these results in vivo showing that purified InlB added from the lumenal side increases endocytosis of fluorescent dextran at the extrusion zone of the intestinal villus tip.
We provide here the first evidence that InlB is involved in intestinal invasion. Other studies have failed to identify a role for InlB in the intestinal phase of infection [20], [24]. However, the contribution of InlB to infection may have been difficult to discern at late time points because most studies utilize severe systemic disease as an endpoint of infection, or because of high variation in animal to animal infections. Additionally, other studies of enteric Listeriosis have used treatments that neutralize stomach acid. This may suppress expression of inlA and inlB, which are upregulated by an acid stress response [39]–[42]. In contrast, we did not alter the acid environment and also developed a coinfection assay that allows for precise quantification of Lm in the villus tips of the same animal at early time points. Although the effect of InlB for promoting invasion of the villus tips is not large, it is comparable in magnitude of the role of InlB for invasion of cultured epithelial cells. In addition, it is comparable in magnitude to the recently discovered role for InlB in placental invasion after intravenous infection, an experimental route that bypasses the gastrointestinal tract and prior cell invasion [20]. Our study has focused only on the role of InlB in modulating Lm invasion of a very specific site, the MCJ. Future investigation will address whether InlB affects the pathophysiology of gastrointestinal colonization and of invasive Listeriosis after oral infection.
In summary, we have explored the mechanisms of Lm invasion of polarized epithelia, the first stage of an infection that can range from asymptomatic colonization, to self-limiting enteritis, to potentially deadly invasive and disseminated disease. Our mechanistic model demonstrates how two microbial invasins with different receptors and different adhesin properties can function cooperatively to promote invasion of the intestinal villus tips (Figure 6). The process of cell extrusion requires junctional remodeling and removal of adhesive contacts that allows the dying cell to detach from the epithelium (Figure 6A). After the cell has been extruded, basolateral proteins from the old junction must be removed from above the newly formed TJ on the surrounding cells at the MCJ (Figure 6A, 6B). As an evolutionary strategy, it is interesting that Lm targets junction remodeling and dynamin-dependent removal of E-cadherin from the cell surface as a mechanism of internalization rather than binding a more accessible, but more stable, apical receptor. This concept should be relevant to the study of other microbes that target junctional receptors. Without InlB, Lm invasion is less efficient. Without InlA, InlB does not provide adhesive strength for Lm to bind to the epithelium. Since activation of c-Met results in the co-endocytosis of both receptors, InlB has evolved to provide a local increase in junctional remodeling that allows for enhanced dynamin-dependent Lm internalization (Figure 6B).
Materials and Methods
Ethics Statement
All animal experiments were performed in accordance to NIH guidelines, the Animal Welfare Act, and US federal law. Such experiments were approved by Stanford University's Administrative Panel on Laboratory Animal Care (A-PLAC), which has been accredited by the Association of Assessment and Accreditation of Laboratory Animal Care International (AAALAC). All animals were housed in a centralized and AAALAC-accredited research animal facility that is fully staffed with trained husbandry, technical, and veterinary personnel.
Chemicals and Reagents
A stock of 5 µg/ml HGF in H2O 0.1% BSA was stored at −80°C until dilution at use (Sigma-Aldrich, St. Louis, MO). InlB-His6 and a truncated variant containing only the terminal GW domains, GW[2]–[3]-His6 at ∼25 mg/ml in 10 mM sodium acetate pH 4.5, 1 mM DTT, 0.5 mM EDTA were purified as described in [15], [16] and stored −80°C until dilution at use. c-Met Inhibitor SU11274 and dynamin inhibitor dynasore ([67], [74]; Calbiochem, San Diego, California) were stored in DMSO at −20°C until dilution at use. A stock of Neutral fixable Texas Red 10 kDa dextran (Molecular Probes, Eugene, Oregon) was stored at 25 mg/ml in DMEM at −20°C until dilution at use.
Cloning and Generation of L. monocytogenes Strains Expressing inlAm, inlAmB and sGFP
The tRNAARG site-specific shuttle integration vectors pPL3 and pPL3e, which respectively confer chloramphenicol and erythromycin resistance to Listeria, and the L. monocytogenes (Lm) strain DH-L1039, which expresses sGFP under the control of the Hyper-SPO1 promoter fused to the 5′ UTR of hly (pHyperSPO1-hly5′UTR-sGFP), were the kind gifts of Dr. Darren E. Higgins (Harvard University, Boston, Massachusetts) [130]. pHyperSPO1-hly5′UTR-sGFP was PCR amplified from DH-L1039 genomic DNA with primers 37/33 (Table 1). SalI digested pHyperSPO1-hly5′UTR-sGFP was ligated with SalI digested pPL3 or pPL3e to generate pMP74 or pMP76, respectively.

inlA and inlAB were PCR amplified from WT Lm 10403S genomic DNA with primers 1/2 and 1/3, respectively, as in [25]. inlA or inlAB were ligated with pCR4-BluntTOPO (Qiagen, Valencia, CA) and subjected to two rounds of Quickchange site-directed mutagenesis (Stratagene, La Jolla, CA) with primer pairs 47/48 and 49/50 to introduce S192N and Y369S mutations into inlA and generate the murinized variants inlAm or inlAmB (Table 1). inlAm or inlAmB were digested with BamHI and ligated with BamHI digested pPL3, pPL3e, pMP74 or pMP76. These constructs were transformed into SM10 (λpir), and introduced to Lm by conjugative mating as described in [131] (Table 2). Integration was confirmed with primers NC16/PL95 as described in [131] (Table 1).
Bacterial Strains and Culture Conditions
Lm strains are listed in Table 2. Lm were grown on BHI agar or in BHI broth (BD/Difco, San Jose, California) supplemented with streptomycin at 200 µg/ml, chloramphenicol at 7.5 µg/ml or erythromycin at 5 µg/ml, when appropriate. One-shot Top10 E. coli (Invitrogen, Carlsbad, California), used for general cloning steps, was cultured in LB broth and on LB agar supplemented with kanamycin at 50 µg/ml or choramphenicol at 25 µg/ml, when appropriate. E. coli strain SM10 (λpir) was kindly provided by Dr. Denise Monack (Stanford University, Stanford, California). E. coli SM10 (λ pir), as the donor for bacterial conjugation, was cultured in LB supplemented with kanamycin at 30 µg/ml and chloramphenicol at 25 µg/ml, when appropriate.
Cell Culture and Infection
MDCK II, MDCK II E-cadherin-GFP and MDCK II E-cadherin-RFP cells were kindly provided by W. James Nelson (Stanford University, Stanford, California) [132], [133]. Cells were maintained at 37°C in 5% CO2 atmosphere in DMEM (Gibco, San Diego, California) supplemented with 5% fetal bovine serum (FBS, Gibco). For infection experiments, cells were trypsinized and seeded on 12 well polycarbonate tissue culture dishes or 12 mm polycarbonate tissue culture inserts (Transwell filters; Costar, Cambridge, Massachusetts) at a density of 106 cells/cm2 and supplemented with fresh media daily for 4 days. For experiments with inhibitors, DMEM 2.5 µM c-Met Inhibitor SU11274/0.15% DMSO was added to the monolayers 12 h prior to infection or DMEM 80 µM dynasore/0.1% DMSO was added to the monolayers 30 min or 1 h prior to infection. Lm infections (multiplicity of infections, MOIs, of 1∶1 to 100∶1) and assays of attachment invasion were performed essentially as described in [30]. To assay for intracellular replication, polarized MDCK monolayers were infected with an MOI of 10 bacteria/cell for a 10 minutes to allow attachment and were then washed 4X with DMEM to remove unadhered bacteria. Six to ten plaques per time point were randomly found and imaged by 3D confocal microscopy without regard to size or bacterial number and subsequently analyzed for bacterial number from all acquired images. Prism software (GraphPad, San Diego, California) was utilized for construction of graphs and for statistical analysis of data. Student's t-test was used to compare two sample groups. ANOVA with Bonferroni's post-tests was used to analyze 3 or more sample groups. The competitive index (C.I.) of two strains was determined as C.I. = (Stain A output/Strain B output)/(Stain A input/Strain B input).
Listeria Infection of Mice
Lm cultures were grown at 30°C overnight in BHI without agitation, pelleted and resuspended in phosphate buffered saline (PBS). Female 8-week old BALB/c mice (obtained at 6–7 weeks from The Jackson Laboratory, Bar Harbor, Maine) were food restricted overnight but allowed free access to water and inoculated with a feeding needle intragrastrically with a maximum volume of 200 µl. Mice were then immediately allowed free access to food and water.
Dextran Endocytosis in Tissue Culture
MDCK II or MDCK II E-cadherin-GFP cells were trypsinized and seeded on 12 mm polycarbonate Transwell tissue culture inserts at a density of 106 cells/cm2 and supplemented with fresh basal media daily for 5 days. The media was changed to plain DMEM 80 µM Dynasore/0.1% DMSO or DMEM 0.1% DMSO at −1 : 30 hours. A final concentration of 1 µg/ml InlB or GW[2]–[3], or 0.1 µg/ml HGF was added at −1 : 00 h to the apical side and at time 0 : 00 1 mg/ml neutral fixable Texas Red 10 kDa dextran was added to the apical side for 30 minutes. Monolayers were washed 4X to remove extracellular dextran and monolayers were fixed and processed for immunofluoresence microscopy, as described in [30]. Confocal images were analyzed using Volocity software (Improvision, Lexington, Massachusetts). To quantify and quantitatively describe intracellular fluorescent dextran puncta, an analysis script was designed to find objects within 5–100% fluorescence intensity, exclude objects less than 0.5 µm3 or greater than 100 µm3 and separate touching objects with an object size guide of 0.1 µm3. The data were clipped to a square region of interest 50 µm×50 µm centered at a multicellular junction (MCJ) or at non-MCJ regions.
Dextran Endocytosis in Mouse Ileal Loops
BALB/c mice (The Jackson Laboratory) were fasted overnight prior to surgery but allowed free access to water. Anesthesia was induced by intraperitoneal injection with a mixture of ketamine (40 mg/kg) and xylazine (4–5 mg/kg) in water and the animal was kept on a 37°C pad for the duration of the procedure. For each mouse a midline laparotomy was performed to expose the bowel. The ileocecal junction was identified, and the ileum was ligated with a silk tie just proximal to the cecum. A second circumferential ligature was placed ∼4 cm proximal. A suspension of 2.5 mg/ml neutral fixable Texas Red 10 kDa dextran with or without 10 µg/ml InlB in dPBS was inoculated via a hypodermic needle into the loop (∼50 µl/cm). The intestine was returned to the abdominal cavity and the incision was closed with surgical staples. The mouse was kept under anesthetic for 45 minutes at which time the animal was euthanized and intestines were removed and fixed for whole-mount confocal microscopy imaging, as described in [30]. Confocal images were analyzed using Volocity software (Improvision). To quantify and quantitatively describe intracellular fluorescent dextran puncta, an analysis script was designed to find objects within 5–100% fluorescence intensity, exclude objects less than 1 µm3 or greater than 20 µm3. The data were clipped to region of interest surrounding each villus tip analyzed.
Microscopy and Antibodies
Live-cell time-lapse microscopy was performed essentially as described in [134]. Confocal immunofluorescence microscopy was performed as described in [30]. Lm were detected by incubation of samples with biotin-conjugated rabbit anti-L. monocytogenes, all antigens (YVS4207, Accurate Chemical & Scientific Corp., Westbury, NY; 1∶100 for tissue, 1∶600 for tissue culture). Tight junctions were detected by incubating samples with mouse anti-ZO-1 antibodies (Zymed, South San Francisco, California; 1∶300 dilution). E-cadherin was detected with mAb anti-E-cadherin (BD Transduction Labs, San Jose, California; 1∶600 dilution). Alexa-fluor conjugated streptavidin or Anti-IgG Alexa-fluor conjugated antibodies of appropriate species reactivity and fluorescence spectra were used for secondary detection (Molecular Probes). An immunofluorescence inside/outside staining that distinguishes extracellular from intracellular L. monocytogenes was modified from [135] with appropriate antibodies for this study. All nuclei were visualized by incubating samples with TOPRO-3 (Molecular Probes). F-actin was visualized by incubating samples with Alexa-fluor conjugated phalloidins (Molecular Probes).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Vazquez-BolandJA
KuhnM
BercheP
ChakrabortyT
Dominguez-BernalG
2001 Listeria pathogenesis and molecular virulence determinants. Clin Microbiol Rev 14 584 640
2. DramsiS
BiswasI
MaguinE
BraunL
MastroeniP
1995 Entry of Listeria monocytogenes into hepatocytes requires expression of inIB, a surface protein of the internalin multigene family. Mol Microbiol 16 251 261
3. GaillardJL
BercheP
FrehelC
GouinE
CossartP
1991 Entry of L. monocytogenes into cells is mediated by internalin, a repeat protein reminiscent of surface antigens from gram-positive cocci. Cell 65 1127 1141
4. Temm-GroveCJ
JockuschBM
RohdeM
NiebuhrK
ChakrabortyT
1994 Exploitation of microfilament proteins by Listeria monocytogenes: microvillus-like composition of the comet tails and vectorial spreading in polarized epithelial sheets. J Cell Sci 107 (Pt 10) 2951 2960
5. RayK
MarteynB
SansonettiPJ
TangCM
2009 Life on the inside: the intracellular lifestyle of cytosolic bacteria. Nat Rev Microbiol 7 333 340
6. LecuitM
Vandormael-PourninS
LefortJ
HuerreM
GounonP
2001 A transgenic model for listeriosis: role of internalin in crossing the intestinal barrier. Science 292 1722 1725
7. MengaudJ
OhayonH
GounonP
MegeRM
CossartP
1996 E-cadherin is the receptor for internalin, a surface protein required for entry of L. monocytogenes into epithelial cells. Cell 84 923 932
8. SchubertWD
UrbankeC
ZiehmT
BeierV
MachnerMP
2002 Structure of internalin, a major invasion protein of Listeria monocytogenes, in complex with its human receptor E-cadherin. Cell 111 825 836
9. LecuitM
DramsiS
GottardiC
Fedor-ChaikenM
GumbinerB
1999 A single amino acid in E-cadherin responsible for host specificity towards the human pathogen Listeria monocytogenes. Embo J 18 3956 3963
10. ShenY
NaujokasM
ParkM
IretonK
2000 InIB-dependent internalization of Listeria is mediated by the Met receptor tyrosine kinase. Cell 103 501 510
11. LiN
XiangGS
DokainishH
IretonK
ElferinkLA
2005 The listeria protein internalin B mimics hepatocyte growth factor-induced receptor trafficking. Traffic 6 459 473
12. GreiffenbergL
GoebelW
KimKS
WeigleinI
BubertA
1998 Interaction of Listeria monocytogenes with human brain microvascular endothelial cells: InlB-dependent invasion, long-term intracellular growth, and spread from macrophages to endothelial cells. Infect Immun 66 5260 5267
13. LingnauA
DomannE
HudelM
BockM
NichterleinT
1995 Expression of the Listeria monocytogenes EGD inlA and inlB genes, whose products mediate bacterial entry into tissue culture cell lines, by PrfA-dependent and -independent mechanisms. Infect Immun 63 3896 3903
14. ParidaSK
DomannE
RohdeM
MullerS
DarjiA
1998 Internalin B is essential for adhesion and mediates the invasion of Listeria monocytogenes into human endothelial cells. Mol Microbiol 28 81 93
15. BanerjeeM
CoppJ
VugaD
MarinoM
ChapmanT
2004 GW domains of the Listeria monocytogenes invasion protein InlB are required for potentiation of Met activation. Mol Microbiol 52 257 271
16. MarinoM
BanerjeeM
JonquieresR
CossartP
GhoshP
2002 GW domains of the Listeria monocytogenes invasion protein InlB are SH3-like and mediate binding to host ligands. Embo J 21 5623 5634
17. CoppJ
MarinoM
BanerjeeM
GhoshP
van der GeerP
2003 Multiple regions of internalin B contribute to its ability to turn on the Ras-mitogen-activated protein kinase pathway. J Biol Chem 278 7783 7789
18. MarinoM
BraunL
CossartP
GhoshP
1999 Structure of the lnlB leucine-rich repeats, a domain that triggers host cell invasion by the bacterial pathogen L. monocytogenes. Mol Cell 4 1063 1072
19. IretonK
PayrastreB
CossartP
1999 The Listeria monocytogenes protein InlB is an agonist of mammalian phosphoinositide 3-kinase. J Biol Chem 274 17025 17032
20. DissonO
GrayoS
HuilletE
NikitasG
Langa-VivesF
2008 Conjugated action of two species-specific invasion proteins for fetoplacental listeriosis. Nature 455 1114 1118
21. NiemannHH
JagerV
ButlerPJ
van den HeuvelJ
SchmidtS
2007 Structure of the human receptor tyrosine kinase met in complex with the Listeria invasion protein InlB. Cell 130 235 246
22. GaillardJL
JaubertF
BercheP
1996 The inlAB locus mediates the entry of Listeria monocytogenes into hepatocytes in vivo. J Exp Med 183 359 369
23. DramsiS
BourdichonF
CabanesD
LecuitM
FsihiH
2004 FbpA, a novel multifunctional Listeria monocytogenes virulence factor. Mol Microbiol 53 639 649
24. KhelefN
LecuitM
BierneH
CossartP
2006 Species specificity of the Listeria monocytogenes InlB protein. Cell Microbiol 8 457 470
25. BakardjievAI
StacyBA
FisherSJ
PortnoyDA
2004 Listeriosis in the pregnant guinea pig: a model of vertical transmission. Infect Immun 72 489 497
26. RobbinsJR
SkrzypczynskaKM
ZeldovichVB
KapidzicM
BakardjievAI
2010 Placental syncytiotrophoblast constitutes a major barrier to vertical transmission of Listeria monocytogenes. PLoS Pathog 6 e1000732
27. BonazziM
VeigaE
Pizarro-CerdaJ
CossartP
2008 Successive post-translational modifications of E-cadherin are required for InlA-mediated internalization of Listeria monocytogenes. Cell Microbiol 10 2208 2222
28. SousaS
CabanesD
BougneresL
LecuitM
SansonettiP
2007 Src, cortactin and Arp2/3 complex are required for E-cadherin-mediated internalization of Listeria into cells. Cell Microbiol 9 2629 2643
29. BergmannB
RaffelsbauerD
KuhnM
GoetzM
HomS
2002 InlA - but not InlB-mediated internalization of Listeria monocytogenes by non-phagocytic mammalian cells needs the support of other internalins. Mol Microbiol 43 557 570
30. PentecostM
OttoG
TheriotJA
AmievaMR
2006 Listeria monocytogenes invades the epithelial junctions at sites of cell extrusion. PLoS Pathog 2 e3
31. CrepaldiT
PollackAL
PratM
ZborekA
MostovK
1994 Targeting of the SF/HGF receptor to the basolateral domain of polarized epithelial cells. J Cell Biol 125 313 320
32. CossartP
1998 Interactions of the bacterial pathogen Listeria monocytogenes with mammalian cells: bacterial factors, cellular ligands, and signaling. Folia Microbiol (Praha) 43 291 303
33. BollerK
VestweberD
KemlerR
1985 Cell-adhesion molecule uvomorulin is localized in the intermediate junctions of adult intestinal epithelial cells. J Cell Biol 100 327 332
34. PottenCS
LoefflerM
1990 Stem cells: attributes, cycles, spirals, pitfalls and uncertainties. Lessons for and from the crypt. Development 110 1001 1020
35. MadaraJL
1990 Maintenance of the macromolecular barrier at cell extrusion sites in intestinal epithelium: physiological rearrangement of tight junctions. J Membr Biol 116 177 184
36. GordonJI
HermistonML
1994 Differentiation and self-renewal in the mouse gastrointestinal epithelium. Curr Opin Cell Biol 6 795 803
37. CorfeBM
DiveC
GarrodDR
2000 Changes in intercellular junctions during apoptosis precede nuclear condensation or phosphatidylserine exposure on the cell surface. Cell Death Differ 7 234 235
38. RosenblattJ
RaffMC
CramerLP
2001 An epithelial cell destined for apoptosis signals its neighbors to extrude it by an actin - and myosin-dependent mechanism. Curr Biol 11 1847 1857
39. SueD
FinkD
WiedmannM
BoorKJ
2004 sigmaB-dependent gene induction and expression in Listeria monocytogenes during osmotic and acid stress conditions simulating the intestinal environment. Microbiology 150 3843 3855
40. McGannP
WiedmannM
BoorKJ
2007 The alternative sigma factor sigma B and the virulence gene regulator PrfA both regulate transcription of Listeria monocytogenes internalins. Appl Environ Microbiol 73 2919 2930
41. Toledo-AranaA
DussurgetO
NikitasG
SestoN
Guet-RevilletH
2009 The Listeria transcriptional landscape from saprophytism to virulence. Nature 459 950 956
42. SleatorRD
WatsonD
HillC
GahanCG
2009 The interaction between Listeria monocytogenes and the host gastrointestinal tract. Microbiology 155 2463 2475
43. PamerEG
2004 Immune responses to Listeria monocytogenes. Nat Rev Immunol 4 812 823
44. WollertT
PascheB
RochonM
DeppenmeierS
van den HeuvelJ
2007 Extending the host range of Listeria monocytogenes by rational protein design. Cell 129 891 902
45. WollertT
HeinzDW
SchubertWD
2007 Thermodynamically reengineering the listerial invasion complex InlA/E-cadherin. Proc Natl Acad Sci U S A 104 13960 13965
46. KameiT
MatozakiT
SakisakaT
KodamaA
YokoyamaS
1999 Coendocytosis of cadherin and c-Met coupled to disruption of cell-cell adhesion in MDCK cells–regulation by Rho, Rac and Rab small G proteins. Oncogene 18 6776 6784
47. FujitaY
KrauseG
ScheffnerM
ZechnerD
LeddyHE
2002 Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat Cell Biol 4 222 231
48. RobbinsJR
BarthAI
MarquisH
de HostosEL
NelsonWJ
1999 Listeria monocytogenes exploits normal host cell processes to spread from cell to cell. J Cell Biol 146 1333 1350
49. LecuitM
SonnenburgJL
CossartP
GordonJI
2007 Functional genomic studies of the intestinal response to a foodborne enteropathogen in a humanized gnotobiotic mouse model. J Biol Chem 282 15065 15072
50. RaczP
TennerK
MeroE
1972 Experimental Listeria enteritis. I. An electron microscopic study of the epithelial phase in experimental listeria infection. Lab Invest 26 694 700
51. TilneyLG
PortnoyDA
1989 Actin filaments and the growth, movement, and spread of the intracellular bacterial parasite, Listeria monocytogenes. J Cell Biol 109 1597 1608
52. JonquieresR
BierneH
FiedlerF
GounonP
CossartP
1999 Interaction between the protein InlB of Listeria monocytogenes and lipoteichoic acid: a novel mechanism of protein association at the surface of gram-positive bacteria. Mol Microbiol 34 902 914
53. JonquieresR
Pizarro-CerdaJ
CossartP
2001 Synergy between the N - and C-terminal domains of InlB for efficient invasion of non-phagocytic cells by Listeria monocytogenes. Mol Microbiol 42 955 965
54. BraunL
NatoF
PayrastreB
MazieJC
CossartP
1999 The 213-amino-acid leucine-rich repeat region of the listeria monocytogenes InlB protein is sufficient for entry into mammalian cells, stimulation of PI 3-kinase and membrane ruffling. Mol Microbiol 34 10 23
55. VeigaE
GuttmanJA
BonazziM
BoucrotE
Toledo-AranaA
2007 Invasive and adherent bacterial pathogens co-Opt host clathrin for infection. Cell Host Microbe 2 340 351
56. SeveauS
ThamTN
PayrastreB
HoppeAD
SwansonJA
2007 A FRET analysis to unravel the role of cholesterol in Rac1 and PI 3-kinase activation in the InlB/Met signalling pathway. Cell Microbiol 9 790 803
57. BierneH
GouinE
RouxP
CaroniP
YinHL
2001 A role for cofilin and LIM kinase in Listeria-induced phagocytosis. J Cell Biol 155 101 112
58. SeveauS
BierneH
GirouxS
PrevostMC
CossartP
2004 Role of lipid rafts in E-cadherin–and HGF-R/Met–mediated entry of Listeria monocytogenes into host cells. J Cell Biol 166 743 753
59. VeigaE
CossartP
2005 Listeria hijacks the clathrin-dependent endocytic machinery to invade mammalian cells. Nat Cell Biol
60. BierneH
MikiH
InnocentiM
ScitaG
GertlerFB
2005 WASP-related proteins, Abi1 and Ena/VASP are required for Listeria invasion induced by the Met receptor. J Cell Sci 118 1537 1547
61. BraunL
OhayonH
CossartP
1998 The InIB protein of Listeria monocytogenes is sufficient to promote entry into mammalian cells. Mol Microbiol 27 1077 1087
62. CossartP
Pizarro-CerdaJ
LecuitM
2003 Invasion of mammalian cells by Listeria monocytogenes: functional mimicry to subvert cellular functions. Trends Cell Biol 13 23 31
63. IretonK
2007 Entry of the bacterial pathogen Listeria monocytogenes into mammalian cells. Cell Microbiol 9 1365 1375
64. GoetzM
BubertA
WangG
Chico-CaleroI
Vazquez-BolandJA
2001 Microinjection and growth of bacteria in the cytosol of mammalian host cells. Proc Natl Acad Sci U S A 98 12221 12226
65. GregorySH
SagnimeniAJ
WingEJ
1997 Internalin B promotes the replication of Listeria monocytogenes in mouse hepatocytes. Infect Immun 65 5137 5141
66. BalkovetzDF
PollackAL
MostovKE
1997 Hepatocyte growth factor alters the polarity of Madin-Darby canine kidney cell monolayers. J Biol Chem 272 3471 3477
67. WangX
LeP
LiangC
ChanJ
KiewlichD
2003 Potent and selective inhibitors of the Met [hepatocyte growth factor/scatter factor (HGF/SF) receptor] tyrosine kinase block HGF/SF-induced tumor cell growth and invasion. Mol Cancer Ther 2 1085 1092
68. BierneH
CossartP
2002 InlB, a surface protein of Listeria monocytogenes that behaves as an invasin and a growth factor. J Cell Sci 115 3357 3367
69. KarunasagarI
SenghaasB
KrohneG
GoebelW
1994 Ultrastructural study of Listeria monocytogenes entry into cultured human colonic epithelial cells. Infect Immun 62 3554 3558
70. HartsockA
NelsonWJ
2008 Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta 1778 660 669
71. IzumiG
SakisakaT
BabaT
TanakaS
MorimotoK
2004 Endocytosis of E-cadherin regulated by Rac and Cdc42 small G proteins through IQGAP1 and actin filaments. J Cell Biol 166 237 248
72. BryantDM
KerrMC
HammondLA
JosephSR
MostovKE
2007 EGF induces macropinocytosis and SNX1-modulated recycling of E-cadherin. J Cell Sci 120 1818 1828
73. Pizarro-CerdaJ
PayrastreB
WangYJ
VeigaE
YinHL
2007 Type II phosphatidylinositol 4-kinases promote Listeria monocytogenes entry into target cells. Cell Microbiol 9 2381 2390
74. MaciaE
EhrlichM
MassolR
BoucrotE
BrunnerC
2006 Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell 10 839 850
75. GuttmanJA
FinlayBB
2009 Tight junctions as targets of infectious agents. Biochim Biophys Acta 1788 832 841
76. VogelmannR
AmievaMR
FalkowS
NelsonWJ
2004 Breaking into the epithelial apical-junctional complex–news from pathogen hackers. Curr Opin Cell Biol 16 86 93
77. SousaS
LecuitM
CossartP
2005 Microbial strategies to target, cross or disrupt epithelia. Curr Opin Cell Biol
78. O'HaraJR
BuretAG
2008 Mechanisms of intestinal tight junctional disruption during infection. Front Biosci 13 7008 7021
79. HauckCR
AgererF
MuenznerP
SchmitterT
2006 Cellular adhesion molecules as targets for bacterial infection. Eur J Cell Biol 85 235 242
80. CoyneCB
ShenL
TurnerJR
BergelsonJM
2007 Coxsackievirus entry across epithelial tight junctions requires occludin and the small GTPases Rab34 and Rab5. Cell Host Microbe 2 181 192
81. BergelsonJM
CunninghamJA
DroguettG
Kurt-JonesEA
KrithivasA
1997 Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science 275 1320 1323
82. AntarAA
KonopkaJL
CampbellJA
HenryRA
PerdigotoAL
2009 Junctional adhesion molecule-A is required for hematogenous dissemination of reovirus. Cell Host Microbe 5 59 71
83. BartonES
ForrestJC
ConnollyJL
ChappellJD
LiuY
2001 Junction adhesion molecule is a receptor for reovirus. Cell 104 441 451
84. LiuS
YangW
ShenL
TurnerJR
CoyneCB
2009 Tight junction proteins claudin-1 and occludin control hepatitis C virus entry and are downregulated during infection to prevent superinfection. J Virol 83 2011 2014
85. YangW
QiuC
BiswasN
JinJ
WatkinsSC
2008 Correlation of the tight junction-like distribution of Claudin-1 to the cellular tropism of hepatitis C virus. J Biol Chem 283 8643 8653
86. EvansMJ
von HahnT
TscherneDM
SyderAJ
PanisM
2007 Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446 801 805
87. YoonM
SpearPG
2002 Disruption of adherens junctions liberates nectin-1 to serve as receptor for herpes simplex virus and pseudorabies virus entry. J Virol 76 7203 7208
88. SpearPG
2004 Herpes simplex virus: receptors and ligands for cell entry. Cell Microbiol 6 401 410
89. GrahamKL
HalaszP
TanY
HewishMJ
TakadaY
2003 Integrin-using rotaviruses bind alpha2beta1 integrin alpha2 I domain via VP4 DGE sequence and recognize alphaXbeta2 and alphaVbeta3 by using VP7 during cell entry. J Virol 77 9969 9978
90. CiarletM
CrawfordSE
ChengE
BluttSE
RiceDA
2002 VLA-2 (alpha2beta1) integrin promotes rotavirus entry into cells but is not necessary for rotavirus attachment. J Virol 76 1109 1123
91. GuerreroCA
MendezE
ZarateS
IsaP
LopezS
2000 Integrin alpha(v)beta(3) mediates rotavirus cell entry. Proc Natl Acad Sci U S A 97 14644 14649
92. HewishMJ
TakadaY
CoulsonBS
2000 Integrins alpha2beta1 and alpha4beta1 can mediate SA11 rotavirus attachment and entry into cells. J Virol 74 228 236
93. ClarkMA
HirstBH
JepsonMA
1998 M-cell surface beta1 integrin expression and invasin-mediated targeting of Yersinia pseudotuberculosis to mouse Peyer's patch M cells. Infect Immun 66 1237 1243
94. WataraiM
FunatoS
SasakawaC
1996 Interaction of Ipa proteins of Shigella flexneri with alpha5beta1 integrin promotes entry of the bacteria into mammalian cells. J Exp Med 183 991 999
95. IsbergRR
LeongJM
1990 Multiple beta 1 chain integrins are receptors for invasin, a protein that promotes bacterial penetration into mammalian cells. Cell 60 861 871
96. TaylorJM
LinE
SusmarskiN
YoonM
ZagoA
2007 Alternative entry receptors for herpes simplex virus and their roles in disease. Cell Host Microbe 2 19 28
97. KoppSJ
BanisadrG
GlajchK
MaurerUE
GrunewaldK
2009 Infection of neurons and encephalitis after intracranial inoculation of herpes simplex virus requires the entry receptor nectin-1. Proc Natl Acad Sci U S A
98. PincusZ
TheriotJA
2007 Comparison of quantitative methods for cell-shape analysis. J Microsc 227 140 156
99. LeTL
YapAS
StowJL
1999 Recycling of E-cadherin: a potential mechanism for regulating cadherin dynamics. J Cell Biol 146 219 232
100. JarrettO
StowJL
YapAS
KeyB
2002 Dynamin-dependent endocytosis is necessary for convergent-extension movements in Xenopus animal cap explants. Int J Dev Biol 46 467 473
101. ShayeDD
CasanovaJ
LlimargasM
2008 Modulation of intracellular trafficking regulates cell intercalation in the Drosophila trachea. Nat Cell Biol 10 964 970
102. de BecoS
GueudryC
AmblardF
CoscoyS
2009 Endocytosis is required for E-cadherin redistribution at mature adherens junctions. Proc Natl Acad Sci U S A
103. TroyanovskyRB
SokolovEP
TroyanovskySM
2006 Endocytosis of cadherin from intracellular junctions is the driving force for cadherin adhesive dimer disassembly. Mol Biol Cell 17 3484 3493
104. GeorgiouM
MarinariE
BurdenJ
BaumB
2008 Cdc42, Par6, and aPKC regulate Arp2/3-mediated endocytosis to control local adherens junction stability. Curr Biol 18 1631 1638
105. PatersonAD
PartonRG
FergusonC
StowJL
YapAS
2003 Characterization of E-cadherin endocytosis in isolated MCF-7 and chinese hamster ovary cells: the initial fate of unbound E-cadherin. J Biol Chem 278 21050 21057
106. SeveauS
Pizarro-CerdaJ
CossartP
2007 Molecular mechanisms exploited by Listeria monocytogenes during host cell invasion. Microbes Infect 9 1167 1175
107. Pizarro-CerdaJ
CossartP
2006 Subversion of cellular functions by Listeria monocytogenes. J Pathol 208 215 223
108. LiuNQ
LossinskyAS
PopikW
LiX
GujuluvaC
2002 Human immunodeficiency virus type 1 enters brain microvascular endothelia by macropinocytosis dependent on lipid rafts and the mitogen-activated protein kinase signaling pathway. J Virol 76 6689 6700
109. AmstutzB
GastaldelliM
KalinS
ImelliN
BouckeK
2008 Subversion of CtBP1-controlled macropinocytosis by human adenovirus serotype 3. Embo J 27 956 969
110. RaghuH
Sharma-WaliaN
VeettilMV
SadagopanS
ChandranB
2009 Kaposi's sarcoma-associated herpesvirus utilizes an actin polymerization-dependent macropinocytic pathway to enter human dermal microvascular endothelial and human umbilical vein endothelial cells. J Virol 83 4895 4911
111. MeierO
BouckeK
HammerSV
KellerS
StidwillRP
2002 Adenovirus triggers macropinocytosis and endosomal leakage together with its clathrin-mediated uptake. J Cell Biol 158 1119 1131
112. ZenniMK
GiardinaPC
HarveyHA
ShaoJ
KettererMR
2000 Macropinocytosis as a mechanism of entry into primary human urethral epithelial cells by Neisseria gonorrhoeae. Infect Immun 68 1696 1699
113. WataraiM
MakinoS
FujiiY
OkamotoK
ShirahataT
2002 Modulation of Brucella-induced macropinocytosis by lipid rafts mediates intracellular replication. Cell Microbiol 4 341 355
114. WataraiM
DerreI
KirbyJ
GrowneyJD
DietrichWF
2001 Legionella pneumophila is internalized by a macropinocytotic uptake pathway controlled by the Dot/Icm system and the mouse Lgn1 locus. J Exp Med 194 1081 1096
115. GruenheidS
FinlayBB
2003 Microbial pathogenesis and cytoskeletal function. Nature 422 775 781
116. FinlayBB
FryJ
RockEP
FalkowS
1989 Passage of Salmonella through polarized epithelial cells: role of the host and bacterium. J Cell Sci Suppl 11 99 107
117. FinlayBB
RuschkowskiS
DedharS
1991 Cytoskeletal rearrangements accompanying salmonella entry into epithelial cells. J Cell Sci 99 (Pt 2) 283 296
118. FrancisCL
StarnbachMN
FalkowS
1992 Morphological and cytoskeletal changes in epithelial cells occur immediately upon interaction with Salmonella typhimurium grown under low-oxygen conditions. Mol Microbiol 6 3077 3087
119. DehioC
PrevostMC
SansonettiPJ
1995 Invasion of epithelial cells by Shigella flexneri induces tyrosine phosphorylation of cortactin by a pp60c-src-mediated signalling pathway. Embo J 14 2471 2482
120. Garcia-PerezBE
Mondragon-FloresR
Luna-HerreraJ
2003 Internalization of Mycobacterium tuberculosis by macropinocytosis in non-phagocytic cells. Microb Pathog 35 49 55
121. Garcia-PerezBE
Hernandez-GonzalezJC
Garcia-NietoS
Luna-HerreraJ
2008 Internalization of a non-pathogenic mycobacteria by macropinocytosis in human alveolar epithelial A549 cells. Microb Pathog 45 1 6
122. GinocchioCC
OlmstedSB
WellsCL
GalanJE
1994 Contact with epithelial cells induces the formation of surface appendages on Salmonella typhimurium. Cell 76 717 724
123. KettererMR
ShaoJQ
HornickDB
BuscherB
BandiVK
1999 Infection of primary human bronchial epithelial cells by Haemophilus influenzae: macropinocytosis as a mechanism of airway epithelial cell entry. Infect Immun 67 4161 4170
124. BonazziM
SpanoS
TuracchioG
CericolaC
ValenteC
2005 CtBP3/BARS drives membrane fission in dynamin-independent transport pathways. Nat Cell Biol 7 570 580
125. SwansonJA
WattsC
1995 Macropinocytosis. Trends Cell Biol 5 424 428
126. SaffarianS
CocucciE
KirchhausenT
2009 Distinct dynamics of endocytic clathrin-coated pits and coated plaques. PLoS Biol 7 e1000191
127. CuretonDK
MassolRH
SaffarianS
KirchhausenTL
WhelanSP
2009 Vesicular stomatitis virus enters cells through vesicles incompletely coated with clathrin that depend upon actin for internalization. PLoS Pathog 5 e1000394
128. VeigaE
CossartP
2007 Listeria InlB takes a different route to met. Cell 130 218 219
129. PalaciosF
SchweitzerJK
BoshansRL
D'Souza-SchoreyC
2002 ARF6-GTP recruits Nm23-H1 to facilitate dynamin-mediated endocytosis during adherens junctions disassembly. Nat Cell Biol 4 929 936
130. ShenA
HigginsDE
2005 The 5′ untranslated region-mediated enhancement of intracellular listeriolysin O production is required for Listeria monocytogenes pathogenicity. Mol Microbiol 57 1460 1473
131. LauerP
ChowMY
LoessnerMJ
PortnoyDA
CalendarR
2002 Construction, characterization, and use of two Listeria monocytogenes site-specific phage integration vectors. J Bacteriol 184 4177 4186
132. YamadaS
PokuttaS
DreesF
WeisWI
NelsonWJ
2005 Deconstructing the cadherin-catenin-actin complex. Cell 123 889 901
133. PerezTD
TamadaM
SheetzMP
NelsonWJ
2008 Immediate-early signaling induced by E-cadherin engagement and adhesion. J Biol Chem 283 5014 5022
134. TanS
TompkinsLS
AmievaMR
2009 Helicobacter pylori usurps cell polarity to turn the cell surface into a replicative niche. PLoS Pathog 5 e1000407
135. AmievaMR
SalamaNR
TompkinsLS
FalkowS
2002 Helicobacter pylori enter and survive within multivesicular vacuoles of epithelial cells. Cell Microbiol 4 677 690
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 5
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Quorum Sensing Inhibition Selects for Virulence and Cooperation in
- The Role of Intestinal Microbiota in the Development and Severity of Chemotherapy-Induced Mucositis
- Crystal Structure of HIV-1 gp41 Including Both Fusion Peptide and Membrane Proximal External Regions
- Susceptibility to Anthrax Lethal Toxin-Induced Rat Death Is Controlled by a Single Chromosome 10 Locus That Includes
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy