
Isolates with Antimony-Resistant but Not -Sensitive Phenotype Inhibit Sodium Antimony Gluconate-Induced Dendritic Cell Activation
The inability of sodium antimony gluconate (SAG)-unresponsive kala-azar patients to clear Leishmania donovani (LD) infection despite SAG therapy is partly due to an ill-defined immune-dysfunction. Since dendritic cells (DCs) typically initiate anti-leishmanial immunity, a role for DCs in aberrant LD clearance was investigated. Accordingly, regulation of SAG-induced activation of murine DCs following infection with LD isolates exhibiting two distinct phenotypes such as antimony-resistant (SbRLD) and antimony-sensitive (SbSLD) was compared in vitro. Unlike SbSLD, infection of DCs with SbRLD induced more IL-10 production and inhibited SAG-induced secretion of proinflammatory cytokines, up-regulation of co-stimulatory molecules and leishmanicidal effects. SbRLD inhibited these effects of SAG by blocking activation of PI3K/AKT and NF-κB pathways. In contrast, SbSLD failed to block activation of SAG (20 µg/ml)-induced PI3K/AKT pathway; which continued to stimulate NF-κB signaling, induce leishmanicidal effects and promote DC activation. Notably, prolonged incubation of DCs with SbSLD also inhibited SAG (20 µg/ml)-induced activation of PI3K/AKT and NF-κB pathways and leishmanicidal effects, which was restored by increasing the dose of SAG to 40 µg/ml. In contrast, SbRLD inhibited these SAG-induced events regardless of duration of DC exposure to SbRLD or dose of SAG. Interestingly, the inhibitory effects of isogenic SbSLD expressing ATP-binding cassette (ABC) transporter MRPA on SAG-induced leishmanicidal effects mimicked that of SbRLD to some extent, although antimony resistance in clinical LD isolates is known to be multifactorial. Furthermore, NF-κB was found to transcriptionally regulate expression of murine γglutamylcysteine synthetase heavy-chain (mγGCShc) gene, presumably an important regulator of antimony resistance. Importantly, SbRLD but not SbSLD blocked SAG-induced mγGCS expression in DCs by preventing NF-κB binding to the mγGCShc promoter. Our findings demonstrate that SbRLD but not SbSLD prevents SAG-induced DC activation by suppressing a PI3K-dependent NF-κB pathway and provide the evidence for differential host-pathogen interaction mediated by SbRLD and SbSLD.
Published in the journal:
. PLoS Pathog 6(5): e32767. doi:10.1371/journal.ppat.1000907
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1000907
Summary
The inability of sodium antimony gluconate (SAG)-unresponsive kala-azar patients to clear Leishmania donovani (LD) infection despite SAG therapy is partly due to an ill-defined immune-dysfunction. Since dendritic cells (DCs) typically initiate anti-leishmanial immunity, a role for DCs in aberrant LD clearance was investigated. Accordingly, regulation of SAG-induced activation of murine DCs following infection with LD isolates exhibiting two distinct phenotypes such as antimony-resistant (SbRLD) and antimony-sensitive (SbSLD) was compared in vitro. Unlike SbSLD, infection of DCs with SbRLD induced more IL-10 production and inhibited SAG-induced secretion of proinflammatory cytokines, up-regulation of co-stimulatory molecules and leishmanicidal effects. SbRLD inhibited these effects of SAG by blocking activation of PI3K/AKT and NF-κB pathways. In contrast, SbSLD failed to block activation of SAG (20 µg/ml)-induced PI3K/AKT pathway; which continued to stimulate NF-κB signaling, induce leishmanicidal effects and promote DC activation. Notably, prolonged incubation of DCs with SbSLD also inhibited SAG (20 µg/ml)-induced activation of PI3K/AKT and NF-κB pathways and leishmanicidal effects, which was restored by increasing the dose of SAG to 40 µg/ml. In contrast, SbRLD inhibited these SAG-induced events regardless of duration of DC exposure to SbRLD or dose of SAG. Interestingly, the inhibitory effects of isogenic SbSLD expressing ATP-binding cassette (ABC) transporter MRPA on SAG-induced leishmanicidal effects mimicked that of SbRLD to some extent, although antimony resistance in clinical LD isolates is known to be multifactorial. Furthermore, NF-κB was found to transcriptionally regulate expression of murine γglutamylcysteine synthetase heavy-chain (mγGCShc) gene, presumably an important regulator of antimony resistance. Importantly, SbRLD but not SbSLD blocked SAG-induced mγGCS expression in DCs by preventing NF-κB binding to the mγGCShc promoter. Our findings demonstrate that SbRLD but not SbSLD prevents SAG-induced DC activation by suppressing a PI3K-dependent NF-κB pathway and provide the evidence for differential host-pathogen interaction mediated by SbRLD and SbSLD.
Introduction
Kala-azar, caused by Leishmania donovani (LD), is regarded as the most severe form of leishmanial infection, which can be fatal in patients when left untreated. In the absence of an effective vaccine, treatment with pentavalent antimonial compounds such as sodium antimony gluconate (SAG) remains as the first-choice therapy for kala-azar. However, therapeutic utility of SAG is now jeopardized by the emergence of antimony-resistant strains of LD [1], which is becoming a major concern of the World Health Organization (www.who.int/infections-disease-report/2000).
Resistance to antimonial drugs, as observed in leishmanial infection, is marked by two independent “checkpoints”. The first is associated with the impaired biological reduction of the pentavalent antimony (SbV) prodrug to a toxic trivalent (SbIII) form, although the site (macrophage (Mφ) and/or parasite) and mechanism of reduction (enzymatic or nonezymatic) are undefined. The second checkpoint involves a regulatory mechanism promoting reduced influx and/or enhanced efflux/sequestration of active drug that lowers its intracellular accumulation [2], [3]. Importantly, these two events are largely dependent on the intracellular level of thiol compounds such as glutathione (γglutamylcysteinylglycine, GSH) and parasite-specific trypanothione, which in turn are regulated by both host - and LD-γglutamylcysteine synthetase, a rate-limiting enzyme in glutathione biosynthesis [2]–[5]. Although the increased expression of γglutamylcysteine synthetase (γGCS) gene in antimony-resistant strains of LD is controversial [3]–[6], inhibition of γGCS by buthionine sulfoxamine (BSO) reverses SbIII resistance in LD [7]. Therefore, γGCS expression contributes to antimony resistance in LD by regulating intracellular thiol level.
In addition to the mechanisms noted above, the unresponsiveness of kala-azar patients to treatment with SAG is also believed to be a consequence of skewed type-2 immune response that suppresses interferon (IFN) γ-mediated protective immunity [8]. Nonetheless, IFNγ production by T cells is reduced in non-responders compared to SAG-responders [8], [9]. The endogenous production of IFNγ and importantly, its principal inducer IL-12, determine the anti-leishmanial efficacy of SAG in a fully immunocompetent host infected with LD [10], [11]. Following LD infection, the early production of IL-12 is exclusively mediated by dendritic cells (DCs) [12]. This tempted us to speculate a possible involvement of DCs in regulating “SAG responsiveness” versus “unresponsiveness” in kala-azar patients.
DCs normally play a key role in initiating and regulating Leishmania-specific T cell reactivity [13], [14]. However, the T cell stimulatory capacity of DCs depends on their state of activation and maturation. In contrast to mature DCs, immature DCs exhibit a reduced capacity to stimulate T cells due to low expression of MHC and co-stimulatory molecules, and the lack of production of proinflammatory cytokines. Gene expression associated with the development, activation, maturation and antigen-presenting cell (APC) function of DCs is largely regulated by the transcription factor NF-κB [15]–[18]. For instance, inhibition of NF-κB activation suppresses DC maturation and APC function [19], [20]. NF-κB is a hetero - or homo-dimeric complex of structurally related proteins p50, p52, p65 (RelA), cRel and RelB. In resting cells, NF-κB is sequestered in the cytoplasm by the inhibitory proteins IκBα, IκBβ and IκBε [21]. However, cellular activation with wide range of stimuli such as LPS, TNFα and IL-1phosphorylates and thereby activates a multisubunit complex IκB kinase (IKK) consisting of IKKα/IKK1, IKKβ/IKK2 and IKKγ/NEMO [22]. Subsequently, activated IKK promotes downstream events, for example, phosphorylation followed by polyubiquitination and 26S proteasome-mediated degradation of IκB proteins [21]. NF-κB dimers then translocate to the nucleus, and bind to consensus sequences to induce gene transcription. Notably, the phosphatidylinositol 3-kinase (PI3K)/AKT pathway has been demonstrated in a variety of models to regulate NF-κB activation [20], [23], [24].
Studies demonstrated that stimulation with SAG induces the PI3K/AKT pathway and enhances production of proinflammatory cytokines and leishmanicidal effector molecules in Mφ [25]. Furthermore, SAG stimulates NF-κB activation in different cell types, such as CD4+ T cells and peripheral blood mononuclear cells [26]. Importantly, blockade of NF-κB activation is shown to impair γGCS expression in murine Mφ-like cell line [27]. However, direct role of NF-κB in transcriptional regulation of murine γGCS promoter is undefined. With this in mind, the current study was initiated to define the role of SAG in murine DC activation and its regulation by LD isolates with SAG-resistant (SbRLD) and SAG-sensitive (SbSLD) phenotype. We demonstrate that SbRLD but not SbSLD infection suppresses SAG-induced activation/maturation and γGCS expression of DCs by inhibiting NF-κB activation in a PI3K/AKT-dependent manner.
Results
SbSLD - and SbRLD-infected DCs respond differentially to SAG treatment
Although Mφs are regarded as a “primary target” for leishmanial infection, recent studies indicate DC infection with various Leishmania spp. including LD [28], [29]. Indeed, LD infection was observed in both immature bone marrow-derived DC (BMDC) (CD11c+CD8α-) and splenic DC (sDC) (Figure 1A). To determine the leishmanicidal effect of SAG on intracellular SbRLD and SbSLD in DCs, BMDCs and sDCs were infected with GFP expressing promastigotes of SbSLD strain 2001 (GFP-2001) or SbRLD strain R5 (GFP-R5) for 3 hours, stimulated with SAG (20 µg/ml) for 24 hours, and the frequency of infected DCs measured via flow cytometry. A comparable level of DC infection was observed with both GFP-2001 and GFP-R5 (Figure 1B). However, the percentage of BMDCs or sDCs infected with GFP-2001 (SbSLD) was reduced by 5 to 9-fold following SAG treatment (Figure 1B). In marked contrast, SAG treatment failed to exhibit any significant effect on GFP-R5 (SbRLD) infection in DCs (Figure 1B). Furthermore, analyses via Giemsa staining demonstrated that intracellular amastigotes of other SbRLD strains exhibit similar resistance to the SAG-induced leishmanicidal effect in DCs. For instance, a significant reduction in both percentage of infected BMDCs and intracellular parasite number were observed in AG83 (SbSLD) - and to a lesser extent in 39 (SbRLD)-infected BMDCs after SAG treatment (10 and 20 µg/ml) for 24 and 48 hours (Figure S1). Titration of parasites demonstrated that parasite to DC ratio (multiplicity of infection; MOI) of 10∶1 was the optimum ratio for maximum LD infection in DCs (data not shown). Therefore, this MOI was used for all subsequent experiments unless otherwise stated.

Next, the regulation of SAG-induced activation and maturation of DCs by SbRLD and SbSLD was investigated. For this purpose, BMDCs and sDCs were infected with SbRLD and SbSLD promastigotes for 3 hours, washed and cultured with or without SAG (20 µg/ml) for an additional 48 hours. The activation and maturation of DCs were determined by analyzing MHC and co-stimulatory molecule expression and secretion of cytokines. Infection of BMDCs and sDCs with SbRLD strains 39 or GE1F8R induced more IL-10 production as compared with SbSLD strains AG83 or 2001 (Figures 1C and S2A). Interestingly, IL-10 secretion from SbRLD-infected DCs was not affected by SAG treatment (Figures 1C and S2A). In contrast, SAG treatment significantly inhibited IL-10 secretion by SbSLD-infected DCs (Figures 1C and S2A). Furthermore, SAG-stimulated secretion of proinflammatory cytokines such as IL-12p70 and TNFα from DCs were inhibited by SbRLD and not SbSLD infection (Figures 1D-E and S2B-C). Finally, SAG treatment up-regulated CD40, CD80, CD86, MHC-I (H2Kd) and MHC-II (IAd) expression in BMDCs infected with SbSLD but not SbRLD (Figure 1F). Consistent with work by other groups [30], [31], co-stimulatory molecule and MHC expression of untreated BMDCs remained unaltered following SbRLD or SbSLD infection (Figure 1F). Together, these results demonstrate that SAG treatment protects DCs from SbSLD but not SbRLD infection. Moreover, stimulation with SAG fails to activate and mature SbRLD-infected DCs, while SbSLD-infected DCs are still capable of activation and maturation upon SAG treatment.
SAG treatment induces NF-κB activation in DCs
Since NF-κB is a key regulator of maturation and APC function of DCs, the effect of SAG treatment on NF-κB activation was investigated. BMDCs were treated with SAG for varying times and DNA binding activity of nuclear NF-κB was determined via electrophoretic mobility shift assay (EMSA). Relative to untreated BMDCs, a 23-fold increase in NF-κB DNA binding activity was initially observed by 0.3 hours, which persisted up to 1 hour after SAG treatment (Figure 2A). Notably, OCT-1 DNA binding was unaltered despite SAG treatment indicating that SAG-induced enhancement of nuclear DNA binding was NF-κB-specific (Figure 2A). Furthermore, supershift analysis using antibodies specific for each Rel family member demonstrated that SAG stimulation of BMDCs induced DNA binding of NF-κB complexes consisting of the p50, p65 and RelB subunits (Figure 2B). In contrast to SAG, BMDC treatment with varying concentrations (25 to 200 µg/ml) of sodium gluconate for 0.3 hours, or 200 µg/ml of sodium gluconate for various times failed to induce DNA binding activity of NF-κB (Figure 2C-D).

Consistent with the EMSA data, SAG treatment for 0.3, 0.5 and 1 hour induced degradation of IκB proteins in BMDCs (Figure 3A). Importantly, SAG-induced IκB degradation corresponded to enhanced IκBα phosphorylation (Figure 3B), which could be due to increased activity of upstream IKK complex. To test this hypothesis, BMDCs were stimulated with SAG for 0.3, 0.5 and 1 hour. IKK signalosome was immunoprecipitated from cytoplasmic extracts and kinase activity of the complex determined by measuring phosphorylation of an IκBα-GST substrate in vitro. BMDCs stimulated with SAG for 0.3, 0.5 and 1 hour exhibited approximately 3.6 to 4.7-fold increase in IKK activity compared to untreated BMDCs (Figure 3C). In comparison, the level of IKK1 and IKK2 proteins were similar in all BMDC extracts (Figure 3C). The SAG-induced NF-κB DNA binding, IκBα degradation and IKK activity were also observed in sDCs (Figure S3).

Of note, SAG treatment failed to activate the mitogen-activated protein kinase (MAPK) pathway in BMDCs. In contrast to LPS stimulation, phosphorylation of p38MAPK, ERK1/ERK2 and JNK was not detected in SAG-treated DCs (Figure 3D-F). Collectively, these findings demonstrate that stimulation with SAG induces activation of the IKK complex, phosphorylation and degradation of IκB proteins and downstream nuclear DNA binding of NF-κB in both BMDCs and sDCs, and that the induction of these events in DC is contributed by antimonial moiety of SAG. Furthermore, among different signaling pathways, which are known to regulate DC activation/maturation, NF-κB signaling is selectively induced by SAG treatment.
SbRLD but not SbSLD infection inhibits SAG-induced NF-κB activation in DCs
The effect(s) of SbRLD and SbSLD infection on SAG-stimulated NF-κB activation in DCs was determined. BMDCs were infected with either promastigotes or amastigotes of 39 (SbRLD) or 2001 (SbSLD) for varying times, stimulated with SAG for 0.3 hours and nuclear NF-κB DNA binding activity to H2K-specific probe measured via EMSA. SAG-induced NF-κB activity was completely inhibited in BMDCs upon infection with either 39 promastigotes (39Pm) or amastigotes (39Am) (SbRLD) at all times of LD infection analyzed (Figure 4A-B). In contrast, BMDC infection with 2001 promastigotes (2001Pm) or amastigotes (2001Am) (SbSLD) for up to 6 and 3 hours, respectively, failed to inhibit SAG-stimulated NF-κB DNA binding (Figure 4A-B). SAG-induced NF-κB DNA binding activity was also inhibited in 39Pm (SbRLD)-infected sDCs (Figure S4A). The ability of SbRLD to block SAG-stimulated NF-κB DNA binding in DCs was not LD strain specific. For example, BMDC infection with promastigotes of SbRLD strain GE1F8R (GE1F8RPm), unlike SbSLD strain AG83 (AG83Pm), completely prevented SAG-induced NF-κB DNA binding (Figure S4B).

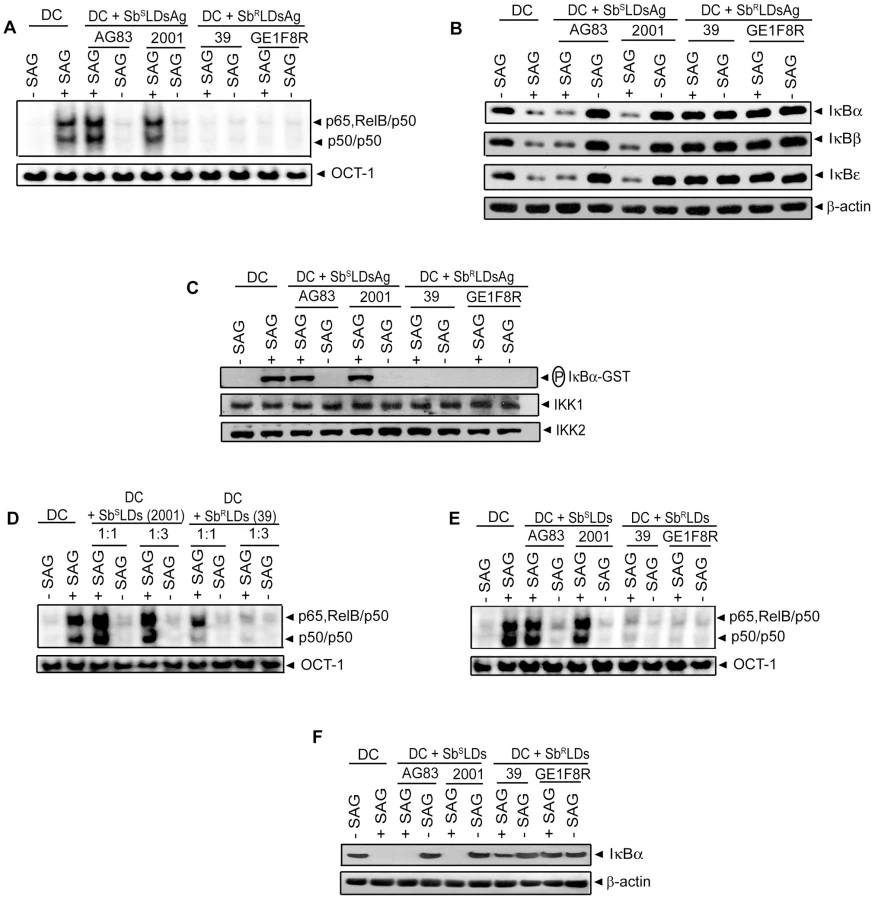
Our finding that SAG-induced NF-κB activity is inhibited selectively in SbRLD-infected BMDCs was further confirmed by temporal analysis of IκB protein degradation and activation of IKK. BMDC infection with either 2001Pm or 2001Am (SbSLD) for up to 6 or 3 hours, respectively, had no significant effect on SAG-induced IκB degradation (Figure 4C-D). In contrast, IκB degradation stimulated by SAG was persistently inhibited by 39 (SbRLD) regardless of the duration of BMDC infection and form of parasite (Figure 4C-D). SAG-induced IκBα degradation was similarly inhibited in sDCs infected with 39Pm (SbRLD) and BMDCs infected with promastigotes of a different SbRLD strain (Figure S4C-D). Interestingly, inhibition of SAG-induced IκB degradation corresponded with a reduction in IκBα phosphorylation in BMDCs infected with SbRLD promastigotes (Figure 4C, E). Furthermore, SAG-induced IKK activation as determined by in vitro IKK activity or phosphorylation of IKK1 and IKK2 was inhibited in extracts prepared from BMDCs and sDCs infected with SbRLD but not SbSLD promastigotes (Figures 4F and S4E-F). BMDC infection with SbRLD and not SbSLD amastigotes also blocked SAG-stimulated IKK activity (Figure 4G). Additionally, pretreatment of BMDCs with parasite antigen(s) (SbRLDsAg) or culture supernatant (SbRLDs) of SbRLD inhibited SAG-induced NF-κB DNA binding activity, IκB degradation and IKK activity; whereas culture supernatant of SbSLD (SbSLDs) or SbSLD-derived antigen(s) (SbSLDsAg) did not (Figure 5).
As demonstrated in Figure 4A-D; SAG-induced NF-κB activation was inhibited in BMDCs infected at a MOI 10∶1 (promastigote or amastigote to DC) with SbSLD promastigotes or amastigotes for 24 or 6 hours, respectively, similar to SbRLD-infected BMDCs. Accordingly, we tested whether the increased intracellular parasite number at these time points of infection rendered 20 µg/ml of SAG insufficient to activate NF-κB. In fact, number of intracellular 2001 (SbSLD) or 39 (SbRLD) was significantly increased if BMDCs were infected with promastigotes for 24 hours rather than 6 hours (Figure S5A). Likewise, BMDCs infected with amastigotes of the above LD strains for 6 hours exhibited increased intracellular parasite number compared to BMDCs infected for 3 hours (Figure S5B). Notably, SAG (20 µg/ml)-induced NF-κB DNA binding and IκBα degradation were detected despite the presence of intracellular parasites in BMDCs infected with 2001Pm and 2001Am (SbSLD) for 6 and 3 hours, respectively (Figures 4A-D and S5A-B). Therefore, these two time points of BMDC infection were selected as a “reference” to analyze the basis of defective SAG-induced NF-κB activation in BMDCs infected with 2001Pm or 2001Am (SbSLD) for 24 or 6 hours, respectively. Initially, the association of increased intracellular parasite number with impairment of SAG-induced NF-κB activation was verified in both BMDCs infected with 2001Pm or 2001Am (SbSLD) for 24 or 6 hours, respectively. For this purpose, BMDCs were infected with 2001Pm (SbSLD) or 39Pm (SbRLD) for 24 hours at varying MOIs and stimulated with SAG (20 µg/ml) for 0.3 hours. Despite LD infection for fixed duration (24 hours), this approach established varying levels of intracellular parasite number, which was elevated in BMDCs infected at MOI 10∶1 compared to other MOIs (Figure S6A). BMDC infection with both 2001Pm (SbSLD) and 39Pm (SbRLD) at a MOI 10∶1 inhibited SAG-induced NF-κB DNA binding activity and IκBα degradation (Figure 6A-B). However, SAG-induced NF-κB DNA binding activity and IκBα degradation were observed only in 2001Pm (SbSLD)-infected BMDCs but not 39Pm (SbRLD)-infected BMDCs when BMDC infection was done at MOIs 2.5∶1 and 5∶1 (Figure 6A-B). Notably, at each of these MOIs both 2001Pm (SbSLD)-infected BMDCs and 39Pm (SbRLD)-infected BMDCs had comparable level of intracellular parasite number (Figure S6A). Using identical MOIs for BMDC infection, similar results were obtained when the intracellular parasite number and SAG-induced NF-κB activation were analyzed in BMDCs infected for 6 hours with 39Am (SbRLD) and 2001Am (SbSLD) (Figures 6C-D and S6B). These findings suggest that irrespective of the form of parasite, 2001 (SbSLD) and 39 (SbRLD) differentially regulate SAG-induced NF-κB signaling in DCs with low intracellular parasite number.

It is possible that with increased intracellular parasite number 2001(SbSLD), similar to 39 (SbRLD), developed the capacity to inhibit SAG-induced NF-κB activation and that occurred when BMDCs were infected at a MOI 10∶1 with 2001Pm or 2001Am (SbSLD) for 24 and 6 hours, respectively. However, this possibility was ruled out when NF-κB activation in response to 20 and 40 µg/ml of SAG treatment was analyzed in BMDCs infected for 6 and 24 hours with 2001Pm (SbSLD) or 39Pm (SbRLD) at a MOI of 10∶1. The effect of SAG (40 µg/ml) stimulation was also verified in BMDCs infected similarly with 2001Am or 39Am for 3 and 6 hours. Stimulation with both 20 and 40 µg/ml of SAG induced NF-κB DNA binding and IκBα degradation in BMDCs infected with 2001Pm (SbSLD) for 6 hours (Figure 6E-F). In contrast, BMDCs infected for 24 hours with 2001Pm (SbSLD) exhibited enhanced NF-κB DNA binding and IκBα degradation only when stimulated with 40 µg/ml of SAG (Figure 6E-F). Similarly, the inhibition of SAG-induced NF-κB DNA binding and IκBα degradation due to BMDC infection with 2001Am (SbSLD) for 6 hours was overcome by increasing the dose of SAG from 20 to 40 µg/ml (Figure 6G-H). On the contrary, 39Pm/39Am (SbRLD) continued to suppress NF-κB DNA binding and IκBα degradation at various durations of infection tested irrespective of dose of SAG used for stimulation (Figure 6E-H). Together, these results demonstrate that SAG-induced NF-κB signaling is impaired by SbRLD infection of DCs.
SbRLD inhibits SAG-induced NF-κB signaling, DC activation and leishmanicidal effects by suppressing the PI3K/AKT pathway in an IL-10-independent manner
Stimulation with SAG induces PI3K/AKT activation in Mφ [25]. Furthermore, PI3K/AKT regulates the NF-κB pathway in DCs via IKK [19], [20]. Therefore, the possibility that SAG-induced activation of NF-κB in DCs is PI3K/AKT-dependent was investigated. Initially, the effect of SAG stimulation on AKT activation was assessed by measuring phosphorylation of AKT. Compared to unstimulated BMDCs, SAG treatment induced a 5 to 7-fold increase in AKT phosphorylation in BMDCs (Figure 7A). Notably, SAG-induced AKT phosphorylation was not observed in BMDCs pretreated with PI3K inhibitors wortmannin (Wort) or Ly294002 (Ly) (Figure 7A). Next, the effect of PI3K inhibitors on SAG-induced NF-κB signaling was determined. Pretreatment with Wort or Ly effectively blocked SAG-induced IKK activity, IκBα degradation and nuclear NF-κB DNA binding in BMDCs (Figure 7B-D). Importantly, infection of BMDCs and sDCs with 39Pm (SbRLD) but not 2001Pm (SbSLD) for 3 hours inhibited SAG (20 µg/ml)-induced AKT phosphorylation (Figures 7E and S7). SAG (20 µg/ml)-induced AKT phosphorylation was also inhibited due to BMDCs infection for 1 and 3 hours with 39Am (SbRLD) but not 2001Am (SbSLD) (Figure 7F). Similar to 39Pm (SbRLD)-infected BMDCs, SAG (20 µg/ml)-induced AKT phosphorylation, however, was not observed if BMDCs were infected with 2001Pm (SbSLD) for 24 hours (Figure 7G). Interestingly, AKT phosphorylation was observed in these 2001Pm (SbSLD)-infected BMDCs but not 39Pm (SbRLD)-infected BMDCs upon stimulation with of 40 µg/ml of SAG (Figure 7G).

Since SbRLD infection stimulated high IL-10 secretion by DCs (Figures 1C and S2A), the possibility that SbRLD inhibited SAG-induced PI3K/AKT (Figures 7E-F and S7) and NF-κB pathways (Figures 4 and S4) in an IL-10-dependent manner was investigated by using a neutralizing αIL-10 Ab. Temporal analyses demonstrated that significant IL-10 production by BMDCs was initially detected after an infection for 12 hours with SbRLD but not SbSLD promastigotes (Figure 7H). Compared to SbSLD-infected BMDCs, IL-10 production was significantly increased in BMDCs infected with SbRLD for 24 and 48 hours (Figure 7H). Strikingly, SAG-induced AKT phosphorylation and NF-κB DNA binding activity were not restored in BMDCs infected with SbRLD for 3 and 24 hours despite αIL-10 Ab treatment (Figure 7I-J). However, αIL-10 Ab treatment effectively prevented inhibition of LPS-induced NF-κB DNA binding in BMDCs pretreated with IL-10 (Figure S8). Therefore, this finding ruled out the involvement of IL-10 in suppression of SAG-induced PI3K/AKT and NF-κB pathways in SbRLD-infected BMDCs.
Consistent with previous report [5], an overexpression of ATP-binding cassette (ABC) transporter MRPA (PGPA) was observed in SbRLD strains 39Pm and GE1F8RPm (Figure S9A). Whether MRPA plays any role in mediating the inhibitory effects of SbRLD on SAG-induced PI3K/AKT and NF-κB activation in DC was then investigated. Accordingly, SAG-induced AKT phosphorylation and NF-κB activation in BMDCs infected for 3 hours with 39Pm (SbRLD), 2001Pm (SbSLD) and isogenic 2001Pm expressing MRPA (2001Pm-MRPA) (Figure S9B) were compared. The latter was developed by transfecting 2001Pm (SbSLD) with a DNA construct expressing MRPA. A complete blockade of SAG-induced AKT phosphorylation and NF-κB DNA binding activity was observed in 39Pm (SbRLD)-infected BMDCs (Figure 8A-B). In contrast, SAG-induced AKT phosphorylation and NF-κB DNA binding activity were detected in 2001Pm (SbSLD)-infected BMDCs (Figure 8A-B). However, infection of BMDCs with 2001Pm-MRPA inhibited SAG-induced AKT phosphorylation and DNA binding activity of NF-κB in BMDCs, albeit partially (Figure 8A-B).

Next, a direct role for PI3K in SbRLD and SbSLD regulation of SAG-induced proinflammatory cytokine secretion and leishmanicidal effects in DCs was investigated using Wort or Ly. In contrast to 39Pm (SbRLD)-infected BMDCs, SAG-stimulated IL-12 and TNFα production were observed in both uninfected and 2001Pm (SbSLD)-infected BMDCs (Figure 9A-B). However, pretreatment of uninfected and 2001Pm (SbSLD)-infected BMDCs with Wort or Ly significantly inhibited SAG-induced secretion of IL-12 and TNFα (Figure 9A-B). Furthermore, PI3K inhibitors prevented SAG (20 µg/ml)-induced reduction of percentage of infected BMDCs and intracellular parasite number in BMDCs infected for 3 hours with 2001Pm (SbSLD) (Figure 9C-D). Importantly, BMDCs infected with 2001Pm (SbSLD) for 24 hours exhibited a significant reduction in both percentage of infected BMDCs and intracellular parasite number only when treated with 40 but not 20 µg/ml of SAG (Figure S10). Treatment of these 2001Pm (SbSLD)-infected BMDCs with Wort or Ly blocked the leishmanicidal effects of SAG (40 µg/ml) (Figure S10). In contrast to 2001Pm (SbSLD)-infected BMDCs, SAG-induced leishmanicidal effects were not observed in 39Pm (SbRLD)-infected BMDCs regardless of duration of infection and dose of SAG (Figures 9C-D and S10). In addition, BMDC infection for 3 hours with 2001Pm-MRPA, unlike 2001Pm (SbSLD), partly but significantly suppressed SAG-induced leishmanicidal effects (Figure 9E-F). Collectively, these data demonstrate that blockade of PI3K/AKT pathway by SbRLD impairs SAG-induced NF-κB signaling, DC activation and leishmanicidal function and that is IL-10-independent. Furthermore, these inhibitory effects of SbRLD are partly contributed by MRPA.

Inhibition of NF-κB activation by SbRLD suppresses SAG-stimulated murine γGCS heavy-chain gene expression in DC
Previous studies have demonstrated an association of antimony resistance of leishmanial parasite with γGCS heavy-chain (γGCShc) gene expression of host [4]. The latter encodes the catalytic subunit of γGCS [32]. In fact, a comparative analysis demonstrated that SAG-induced murine γGCShc (mγGCShc) expression was unaffected in 2001Pm (SbSLD)-infected BMDCs but selectively inhibited in 39Pm (SbRLD)-infected BMDCs (Figure 10A). The molecular basis for SbRLD-mediated suppression of mγGCShc expression in DC was then explored. Despite SAG stimulation, the inhibition of mγGCShc expression could be due to suppression of NF-κB activation in SbRLD-infected BMDC. To investigate this possibility, the regulatory role of NF-κB in mγGCShc promoter activity was initially ascertained. An approximately 1.0 kb DNA sequence upstream of the transcriptional start site of mγGCShc gene (Mus musculus chromosome 9 genomic contig, NT_039474.7; GI:149260095) was selected as the promoter region using Ensembl and UCSC browsers. The mγGCShc promoter was found to contain a putative NF-κB binding site -904GGGGAAACTT-895 that differs from the consensus sequence GGGRNNYYCC at positions 7, 9 and 10 (Figure 10B). ChIP analysis demonstrated that SAG treatment of BMDCs induced NF-κB binding to −991/−673 region of mγGCShc promoter that includes the sequence -904GGGGAAACTT-895 (Figure 10B-C). Furthermore, DNA binding of NF-κB complexes consisting of p50, RelB and p65 subunits specifically to the sequence

-904GGGGAAACTT-895 in SAG-treated BMDCs was confirmed via EMSA using mγGCShc probes containing wild-type sequence -904GGGGAAACTT-895 (WT-mγGCShc probe) or mutant sequence -904CTCTAAGAAT-895 (Mut-mγGCShc probe) (Figure 10D) and supershift analysis (Figure 10E).
Next, the role of NF-κB binding site -904GGGGAAACTT-895 in regulation of promoter activity of mγGCShc gene was tested via luciferase reporter assay using p987-luc and Mut p987-luc, the reporter constructs of mγGCShc promoter fragment containing wild-type and mutant NF-κB binding site, respectively. Compared to control vector (pEGFP-C1) transfected cells, expression of NF-κB subunit p65 strongly induced luciferase activity of p987-luc (Figure 10F). This enhanced luciferase activity of p987-luc was completely blocked upon co-transfection with pEGFP-dominant negative IκBα (pEGFP-IκBαΔN) encoding the NF-κB-specific inhibitor, IκBαΔN (Figure 10F). The lack of luciferase activity of Mut p987-luc despite p65 expression (Figure 10F) further indicated that the sequence -904GGGGAAACTT-895 is required for NF-κB-mediated transcriptional activation of mγGCShc gene. Interestingly, SAG-induced NF-κB DNA binding to WT-mγGCShc probe was inhibited in 39Pm (SbRLD)-infected BMDCs (Figure 10G). In contrast, SAG-induced NF-κB DNA binding to WT-mγGCShc probe was readily detected in 2001Pm (SbSLD)-infected BMDCs (Figure 10G). These findings suggest that SbRLD suppresses SAG-induced mγGCShc expression in DC by inhibiting NF-κB DNA binding to the mγGCShc promoter.
Discussion
Antimonial drugs activate innate effector cells to promote an anti-leishmanial effect [25]. However, regulation of antimonial drug-mediated immune activation by SbRLD and SbSLD is ill-defined. This is of particular interest in view of the lack of efficacy of antimonial compounds reported for SAG-unresponsive kala-azar patients. Recent studies indicated that DCs play a key role in regulating anti-leishmanial immune response [12]–[14], [33]. Accordingly, the role of SAG in activation of DCs, its regulation by SbRLD and SbSLD and the molecular mechanism involved therein were investigated. Here we provide evidence that SbRLD and SbSLD differentially regulate activation of DCs. Furthermore, SAG-induced signaling pathway associated with DC activation is selectively targeted by SbRLD infection.
In an agreement with an earlier report [28], both BMDCs and ex vivo sDCs were infected in vitro with LD promastigotes (Figure 1A). The “SAG-resistant” phenotype did not significantly affect the efficiency of LD infection, but did impact the susceptibility of LD to the leishmanicidal effects of SAG. In contrast, SAG treatment significantly impaired DC infectivity of SbSLD including reduction in both intracellular parasite number and percentage of infected DCs (Figures 1B and S1). The differential response of SbRLD and SbSLD towards SAG treatment was also noted in their ability to regulate activation and maturation of DCs. In contrast to SbSLD, SbRLD infection inhibited SAG-induced proinflammatory cytokine secretion and up-regulation of co-stimulatory molecule and MHC expression in DCs (Figures 1D-F and S2B-C). Noteworthy is that SbRLD induced increased IL-10 secretion by DCs compared to SbSLD (Figures 1C and S2A). This finding reinforces the inherent ability of SbRLD and SbSLD to differentially immunoregulate DC activation. Previous studies demonstrated that IL-10, a potent suppressor of anti-leishmanial immunity, minimizes responsiveness to SAG [34], [35]. Therefore, increased IL-10 production may play a critical role in disease pathogenesis in the host infected with SbRLD.
The second important finding arising from this study is that both promastigotes and amastigotes of SbRLD and SbSLD differentially regulate SAG-induced NF-κB activation in DCs. Indeed, SbRLD but not SbSLD infection blocks SAG-induced NF-κB signaling by suppressing IKK activation, and IκB protein phosphorylation and degradation (Figures 4 and S4). In this regard, it should be noted that SAG stimulation of uninfected DCs induced concomitant degradation of all three IκB proteins (Figure 3A), although degradation of IκBε generally occurs with delayed kinetics upon cellular activation [36], [37]. This finding, however, is consistent with a number of studies reporting rapid degradation of IκBβ and IκBε depending on the type of cell and the nature of the stimulation [19], [20], [36]–[39]. Nevertheless, the suppression of SAG-induced IκB protein degradation by SbRLD ultimately impaired nuclear NF-κB DNA binding activity (Figures 4 and S4). Surprisingly, BMDC infection at a MOI 10∶1 with SbSLD promastigotes or amastigotes for 24 or 6 hours, respectively, also inhibited SAG (20 µg/ml)-induced NF-κB activation (Figure 4). SAG (20 µg/ml)-induced NF-κB activation was restored in these BMDCs but not SbRLD-infected BMDCs upon lowering the MOIs (<10∶1) for BMDC infection, which established reduced levels of intracellular parasite number compared to MOI 10∶1(Figures 6A-D and S6). This finding suggests that intracellular parasite number plays a critical role for differential regulation of SAG-induced NF-κB activation by SbRLD and SbSLD. The early inhibition of NF-κB activation in SbSLD amastigote versus promastigote-infected BMDCs (Figure 4) is in agreement with the fact that DCs internalize amastigotes more efficiently than promastigotes [40]–[42]. However, SbRLD and SbSLD still retain their ability to differentially regulate SAG-induced NF-κB activation in BMDCs with high intracellular parasite number. This conclusion is supported by results demonstrating that despite stimulation with 40 µg/ml of SAG, NF-κB activation was blocked in BMDCs infected for above durations with SbRLD amastigotes or promastigotes at a MOI 10∶1 but readily observed in BMDCs infected similarly with SbSLD (Figure 6). Here, the SAG dose was increased from 20 to 40 µg/ml keeping in mind that SAG therapy requires multiple dosing schedules to ensure enough antimony accumulation in tissues of kala-azar patients [43]. Furthermore, equivalent and/or increased concentrations of SAG have previously been used by other groups [44], [45]. Under identical conditions, the recurrence of NF-κB activation in SbSLD-infected BMDCs by increasing the dose of SAG from 20 to 40 µg/ml (Figure 6) further suggested that 20 µg/ml of SAG was insufficient to activate NF-κB in these BMDCs due to high intracellular parasite number.
Noteworthy is that the inhibitory effect on NF-κB activation was not dependent on live SbRLD. For instance, parasite antigens derived from SbRLD (SbRLDsAg) and SbRLD culture supernatant (SbRLDs) but not the SbSLD-derived antigens (SbSLDsAg) or culture supernatant of SbSLD (SbSLDs) efficiently inhibited SAG-induced NF-κB activation (Figure 5). These findings suggest that the inhibition of NF-κB activation is specific for SbRLD/SbRLD-derived antigen(s)/factor(s) secreted by SbRLD. Strikingly, the inhibition of NF-κB activation correlated with suppression of SAG-induced DC activation by SbRLD infection (Figures 1, 4 and S2). Studies involving gene transfer of a modified IκBα recombinant into immature DC demonstrated that blockade of NF-κB activation alone prevents up-regulation of co-stimulatory molecule expression and production of proinflammatory cytokines [16], [18]. Based on these reports coupled with our own observations, we conclude that the SbRLD blocks SAG-induced NF-κB signaling to prevent DC activation and maturation.
Our results further suggest that SbRLD inhibits IKK and NF-κB activation by blocking SAG-induced PI3K/AKT signaling. SAG stimulation of DCs induced PI3K activation as measured by phosphorylation of AKT, a downstream signaling mediator of PI3K (Figures 7A and S7). Blockade of PI3K/AKT activation by Wort or Ly completely suppressed SAG-stimulated IKK activity and NF-κB signaling (Figure 7A-D), indicating a direct involvement of the PI3K/AKT pathway in NF-κB activation by SAG in DCs. Importantly, PI3K/AKT activation is negatively regulated by Src homology phosphotyrosine phosphatase (SHP)-1, which dephosphorylates PI3K [46]. Furthermore, SHP-1activity is inhibited by SAG [47]. Therefore, blockade of SHP-1activity by SAG may indirectly promote PI3K phosphorylation and activation of downstream AKT, IKK and NF-κB in DCs. Similar to uninfected DCs, SAG treatment induced PI3K/AKT activation in DCs infected with SbSLD promastigotes for 3 hours and SbSLD amastigotes for 1 and 3 hours (Figures 7E-F and S7). Importantly, PI3K inhibitors impaired SAG (20 µg/ml)-induced NF-κB pathway, DC activation and leishmanicidal effects in BMDCs infected with SbSLD promastigotes for 3 hours (Figures 9 and S11). These results suggest the inability of SbSLD to regulate SAG-induced PI3K/AKT and NF-κB pathways. Consequently, SAG continues to exhibit leishmanicidal effects in SbSLD-infected DCs. The inability of SbSLD to regulate SAG-induced leishmanicidal effects was maintained despite BMDC infection for 24 hours with SbSLD promastigotes. This was apparent when increased dose of SAG (40 µg/ml) was used for treatment (Figure S10). In fact, treatment with 40 µg/ml of SAG restored AKT phosphorylation and therefore exhibited leishmanicidal effects in a PI3K-dependent manner in these SbSLD-infected BMDCs (Figures 7G and S10). Interestingly, SbRLD infection mimicked the effects of the PI3K inhibitors in that all SAG-induced events as mentioned above were also blocked by SbRLD regardless of duration of infection, form of parasite and dose of SAG (Figures 7, 9, S10 and S11). One intriguing possibility is that IL-10 produced by SbRLD-infected DCs mediated SbRLD-induced suppression of PI3K/AKT and NF-κB pathways (Figures 1C, 4, 7, S2A and S7). This correlation can be made because IL-10 inhibits AKT activation and NF-κB pathway in DCs [19]. Furthermore, the inhibitory effects of IL-10 on DCs can be mediated in an autocrine manner [48]. However, this is unlikely since SAG-induced NF-κB activation was inhibited even in BMDCs infected with SbRLD for 1 and 3 hours, when IL-10 production was not detected (Figures 4 and 7H). Moreover, neutralization of IL-10 produced by BMDCs upon SbRLD infection for 24 hours failed to block SbRLD-induced inhibition of PI3K/AKT and NF-κB pathways (Figure 7H-J). Therefore, SbRLD infection of BMDCs for up to 24 hours inhibits SAG-induced PI3K/AKT and NF-κB pathways in an IL-10-independent manner and eventually impairs DC activation.
Another key finding is that the suppression of NF-κB activation by SbRLD but not SbSLD inhibits not only DC activation but also SAG-induced mγGCShc expression in DC (Figure 10). Importantly, regulation of host γGCShc expression and therefore host GSH level by SbRLD plays a key role for antimony resistance in LD infection [4]. Although regulation of γGCShc expression by NF-κB was shown in a murine Mφ-like cell line by blocking NF-κB activation [27], our observation establishes a direct role for NF-κB in mediating the promoter activity of the mγGCShc gene (Figure 10C-F). Interestingly, SbRLD blocked SAG-induced mγGCShc expression in DC by preventing NF-κB binding to the mγGCShc gene promoter (Figure 10G). This suggests a key role for NF-κB in SbRLD-mediated suppression of mγGCShc expression in DC. Our findings (Figures 10A and S12) are consistent with the recent work by Carter and colleagues demonstrating that SbRLD transcriptionally down-regulate host γGCShc expression and up-regulate their own γGCShc (LD-γGCShc) expression [4]. Whereas SbRLD-induced inhibition of host γGCS expression reduces host GSH level and impairs reduction of SbV to toxic SbIII form; elevated expression of LD γGCS by SbRLD restores GSH level that promotes efflux of SAG and confers protection against oxidative stress [4].
The mechanism of antimony resistance in clinical LD isolates is unknown and may differ from laboratory-derived resistant parasites [3]. The true markers of clinical antimony resistance in LD isolates are still lacking [2]. A gene, PG1, is reported to confer antimony resistance in clinical isolates of LD [45], [49]. In addition, enhanced expression of several other genes including MRPA and proteophosphoglycans (PPG) was demonstrated in antimony-resistant compared to antimony-sensitive field isolates (Salotra P, Singh R, Nakhasi H. 2005. Clinical Microbiology and Infection. Vol 11, Suppl 2 : 47) [5], [50]. As an initial effort to determine whether any SbRLD-specific factor(s) mediated the suppression of SAG-induced NF-κB signaling and leishmanicidal effects in DCs, the role for MRPA was investigated. Results obtained using 2001Pm and its isogenic strain expressing MRPA (2001Pm-MRPA) showed that the inhibitory effect of 2001Pm-MRPA on SAG-induced PI3K/AKT and NF-κB pathways and leishmanicidal activities mimics that of SbRLD to some extent (Figures 8 and 9). However, the real bearing of this observation in naturally occurring antimony-resistant LD isolates is still questionable and needs detailed investigation further. Moreover, the effects of SAG are likely to be inhibited in DCs by other SbRLD-specific factor(s) also. The relative contribution of these parasite-specific factors in SbRLD-mediated suppression of DC activation is currently under investigation. Furthermore, an association of antimony resistance with genetic variation among LD strains has been proposed [51]. Recent studies demonstrated that due to high genetic polymorphism, strain 39 is remarkably distinct not only from antimony-sensitive strain 2001 but also from other antimony-resistant clinical LD isolates exhibiting homology with antimony-sensitive parasites [51]. On the other hand, the antimony-sensitive strains 2001 and Dd8 exhibit significant genetic similarity [51]. The extreme genetic polymorphism might be a potential cause of antimony resistance in strain 39 [51]. These reports together with our findings emphasize the notion that antimony resistance of clinical LD isolates is “multifactorial” [3].
SAG unresponsiveness in kala-azar patients also entails a number of host-regulated events including dominance of a type-2 T cell response and altered host gene expression [3], [4], [8], [9]. Our findings demonstrate that SAG treatment induces PI3K-dependent NF-κB activation in DCs, which is blocked by SbRLD but not SbSLD infection. Dysregulation of SAG-induced NF-κB activation favors persistent survival of SbRLD in DCs despite SAG treatment by: 1) inhibiting NF-κB-dependent mγGCS expression, a key mediator of SbV reduction to SbIII, and 2) preventing DC activation and maturation required for the initiation of the anti-leishmanial immune response. Notably, a heterogeneous response to SAG treatment may be observed in humans. This possibility is raised by a report demonstrating that SAG treatment of monocyte-derived DCs restores their capacity to respond to LPS in ∼60% of type 1diabetes patients [52]. Importantly, some variability in SAG responsiveness is also reported in kala-azar patients [53]. It is speculated that the genetic differences among individuals may influence the response following SAG therapy in the patients infected with same Leishmania species and living in the same endemic area [54]. Further studies are needed to define how genetic variation, if any, influences the outcome of SAG treatment in kala-azar patients.
Materials and Methods
Animals
BALB/c mice and golden hamsters (Mesocricetus auratus) were maintained and bred under pathogen-free conditions.
Ethics statement
Use of both mice and hamsters was approved by the Institutional Animal Ethics Committees of Institute of Microbial Technology and Indian Institute of Chemical Biology, India. All animal experimentations were performed according to the National Regulatory Guidelines issued by CPSEA (Committee for the Purpose of Supervision of Experiments on Animals), Ministry of Environment and Forest, Govt. of India.
Parasite cultures and preparation of soluble antigen from LD
SbRLD [GE1F8R (MHOM/IN/89/GE1), 39, R5] and SbSLD [AG83 (MHOM/IN/83/AG83), 2001] strains are gifts from Dr. Neeloo Singh (Central Drug Research Institute, India) and Dr. Shyam Sundar (Banaras Hindu University, India) and were maintained in golden hamsters as described [45], [55]–[57]. Amastigotes were obtained from spleens of infected hamsters as described [58]. Subsequently, amastigotes were transformed into promastigotes and maintained as described [59]. GFP-2001 and GFP-R5 are gifts from Dr. Neeloo Singh and were maintained in M199 complete medium (10% FBS, penicillin/streptomycin) with 720 µg/ml geneticin disulfate (G418) (Sigma, St. Louis, MO) [60]. Soluble antigens were prepared from SbSLD and SbRLD promastigotes (109/ml) as described [59].
Transfection of 2001Pm
2001Pm (SbSLD) at mid log or stationary phase were washed twice with electroporation buffer (21 mM HEPES, pH 7.05; 137 mM NaCl; 5 mM KCl; 0.7 mM NaH2PO4; 6 mM glucose). The promastigotes were resuspended in ice-cold electroporation buffer to a final concentration of 107/ml. An aliquot (400 µl) of parasite suspension was mixed with 35–40 µg of chilled pGEM 7ZF α-neo-α L. tarentolae MRPA or pGEM 7ZF α-neo-α DNA (kind gifts from Dr. Marc Ouellette, Laval University, Canada), transferred to a 2-mm gap cuvette and electroporated using BIO-RAD Gene-Pulser X cell instrument at 450 V and 500 µF (3.5 to 4 milli-seconds pulse time). After electroporation, parasites were immediately placed on ice for 10 minutes and cultured at 22°C for 24 hours in Schneider's insect medium with 1500 µg/ml paromomycin and 10 µM Biopterin. Subsequently, 40 µg/ml of G418 was added to the parasite culture. After 24 hours, 1 ml of this promastigote culture was transferred to a new flask containing 5 ml of fresh medium with 10 µM biopterin and 120 µg/ml G418. The culture was maintained for one month under drug pressure (once per week) to obtain the stable transfectants. The mRNA expression of MRPA in these stable transfectants was verified via RT-PCR.
DC preparation
BMDCs and sDCs were prepared from male or female BALB/c mice between 8–12 weeks of age as described [19]. Flow cytometric analyses indicated >85% and ∼90% purity of BMDCs and sDCs, respectively, based on CD11c expression.
DC infection with promastigotes or amastigotes of LD
DCs (5×106/well) were infected in vitro at specified MOIs either with amastigotes of SbRLD or SbSLD; or promastigotes of stationary phase SbRLD, SbSLD or respective GFP-LD for indicated times in a 6-well plate in RPMI 1640 complete medium (10% FBS, penicillin/streptomycin, L-glutamine, sodium pyruvate, non-essential amino acids, 2-mercaptoethanol). Subsequently, DCs were washed, resuspended in RPMI 1640 complete medium and stimulated with SAG (10, 20 and 40 µg/ml) of clinical grade or sodium gluconate (25, 50, 100 and 200 µg/ml) for specified times. The doses of SAG mentioned here and for all experiments represent the concentration of SbV. DC infection with GFP-SbRLD or GFP-SbSLD was determined via flow cytometry. SAG containing 20 µg/ml of SbV, and 35 µg/ml of sodium gluconate had equivalent molar concentration. Sodium gluconate was purchased from Acros Organics, New Jersey, USA. SAG was obtained as kind gift from Albert David Ltd., Kolkata, India. For other experiments, DCs (2.5×105/ml) were adhered on 22×22 mm cover slips, infected with promastigotes/amastigotes of SbSLD or SbRLD and treated with SAG for indicated times. The number of intracellular parasites in DCs was determined via Giemsa staining. In some experiments, DCs (5×106/well) were infected with SbSLD or SbRLD promastigotes for 3 hours or left uninfected. Alternatively, infection of DCs (5×106/well) with LD promastigotes was done for specified times. DCs were then treated or not with Wort (200 nM) or Ly (50 µM) (Cell Signaling Technology, Beverly, MA) 1 hour prior to SAG stimulation as described [19]. In some cases, DCs (5×106/well) were infected with SbSLD or SbRLD promastigotes for 3 and 24 hours or left uninfected. Neutralizing αIL-10 monoclonal Ab (10 µg/ml)/isotype control rat IgG2b Ab (10 µg/ml) (BD Biosciences, San Jose, CA) were added at 2nd hour of BMDC infection. After infection, DCs were washed and stimulated with SAG for 0.3 hours.
DC pretreatment with soluble antigens/culture supernatant of LD or IL-10
DCs (5×106/well) were pretreated for 3 hours with 50 µg/ml soluble antigens derived from SbRLD (SbRLDsAg) or SbSLD (SbSLDsAg) and stimulated with SAG for 0.3 hours in a 6-well plate. Alternatively, SbRLD or SbSLD promastigotes (106/ml) were grown in M199 complete medium till the parasite concentration reached ≥107/ml. The respective culture supernatants of SbRLD (SbRLDs) and SbSLD (SbSLDs) were then collected. Subsequently, DCs (5×106/well) were cultured for 3 hours in RPMI 1640 complete medium containing SbRLDs or SbSLDs at specified complete medium to supernatant ratios and stimulated with SAG for 0.3 hours as above. In some experiments, BMDCs (5×106/well) were pretreated with murine IL-10 (50 ng/ml) (PeproTech, Rocky Hill, NJ) for 24 hours as described [19], in the presence or absence of 10 µg/ml of αIL-10Ab/isotype control Ab and stimulated with LPS (500 ng/ml) for 0.5 hours.
EMSA and Western blot
Nuclear and cytoplasmic extracts were prepared from DC as described [61]. EMSA was performed as described [62] using 32P-labeled DNA probes containing NF-κB binding sites derived from MHC-I H2K promoter:
5′-CAGGGCTGGGGATTCCCCATCTCCACAGTTTCACTTC-3′ [20]. In some experiments, DNA probes specific for murine γGCS heavy-chain (mγGCShc) promoter containing wild-type or mutant NF-κB binding sites as represented by WT-mγGCShc probe, ′-CGGTTCTGAAGGTGGGGAAACTTCTGAAGAAACTT-3′5and Mut-mγGCShc probe, 5′-CGGTTCTGAAGGTCTCTAAGAATCTGAAGAAACTT-3′ (NF-κB binding site is underlined and mutated bases are in italics) respectively, were used. A double stranded OCT-1 DNA probe, 5′-TGTCGAATGCAAATCACTAGAA-3′ was used as control. Supershift EMSAs were carried out as described [19] using following Abs: αp50, αRelB (Active Motif, CA); αp65 (Abcam plc. Cambridge, UK); αp52, αcRel, rabbit IgG (Santa Cruz Biotechnology, Santa Cruz, CA). Bands were visualized using a phosphoimager (Bio-Rad Molecular Imager FX, Hercules, CA).
Western blotting was carried out as described [20]. Blots were probed with Abs specific for: IκBα, IκBβ, IκBε, IKK1, IKK2 (Santa Cruz Biotechnology); pIκBα, pIKK1 (Ser180)/pIKK2 (Ser181), pAKT (Ser473), AKT, pSAPK/JNK (Thr183/Tyr185), SAPK/JNK, phospho-p38MAPK (Thr180/Tyr182), p38MAPK, pERK1/pERK2 (Thr202/Tyr204), ERK1/ERK2 (Cell Signaling Technology); β-actin (Sigma). Binding of secondary HRP-labeled goat-αrabbit (Santa Cruz Biotechnology) or goat-αmouse Abs (Sigma) was analyzed using SuperSignalR West Pico or West Dura Chemiluminescent Substrate (Pierce, Rockland, IL).
IKK assay
IKK signalosome was immunoprecipitated from 700 µg of a whole DC lysate using Protein A/G agarose beads (Santa Cruz Biotechnology) and rabbit polyclonal αIKK1 Ab. In vitro kinase reaction was performed and kinase activity of immunoprecipitated IKK complex determined as described [20].
Flow cytometry
The following monoclonal antibodies used for flow cytometry were purchased from eBioscience (San Diego, CA): PE-αmouse CD11c, FITC-αmouse CD11b, FITC-αmouse CD40, FITC-αmouse CD86, FITC-αmouse CD80, FITC-αmouse H2Kd and FITC-αmouse IAd. The fluorescence of stained cells was analyzed on a FACSCalibur (BD Biosciences) using Cell Quest Pro software.
Measurement of IL-12, TNFα and IL-10 secretions from DCs
DCs (1×106/ml) were infected with promastigotes of SbRLD or SbSLD for 3 hours or left uninfected, washed and stimulated with SAG for additional 48 hours in a 24-well plate. Alternatively, DCs (1×106/ml) were infected with promastigotes of SbRLD or SbSLD for varying times or left uninfected. The culture supernatants were analyzed for IL-12, TNFα and IL-10 productions in triplicate using ELISA kits (BD Biosciences) following the manufacturer's instructions.
Analysis of murine γGCS heavy-chain (mγGCShc) and LD γGCS heavy-chain (LD-γGCShc) expression in SbRLD - and SbSLD-infected BMDCs
BMDCs (5×106/well) were infected with SbRLD or SbSLD for 3 hours or left uninfected, stimulated with SAG for 3 hours and RNA prepared using RNeasy minikit reagent (QIAGEN). The mRNA expression of mγGCShc, LD-γGCShc, murine glyceraldehyde-3-phosphate dehydrogenase (mGAPDH) and LD-tubulin genes were determined via reverse transcription-PCR using platinum quantitative RT-PCR Thermoscript One Step system kit (Invitrogen, Carlsbad, CA), and primers: LD-γGCShc 5′-TATCAAGTCTCGCTACGACT-3′, 5′-CGGAGTCCTTCAGAAGTT-3′; LD-tubulin 5′-ACATCACGAACTCGGTGTTT-3′, 5′-TTCGTCTTGATCGTCGCAAT-3′; mγGCShc 5′-AGAACAATCGCTTTAGGATCA-3′, 5′-AGAAGATGATCGATGCCTTC-3′; mGAPDH 5′-AGATTGTTGCCATCAACGAC 3′, 5′-ATGACAAGCTTCCCATTCTC-3′ [4].
Detection of MRPA expression in LD
RNA was isolated from LD promastigotes and mRNA expression of MRPA was detected by analyzing the amplification of 179 bp cDNA fragment of MRPA via reverse transcription-PCR using primers: 5′-GCGCAGCCGTTTGTGCTTGTGG-3′, 5′-TTGCCGTACGTCGCGATGGTGC-3′ [5]. mRNA expression of LD-tubulin was used as loading control.
Chromatin immunoprecipitation (ChIP)
BMDCs (5×106/well) were stimulated with SAG for 0.3 hours or left untreated. ChIP was performed using ChIP-IT kit (Active Motif) following the manufacturer's instructions. After immunoprecipitation using rabbit IgG or NF-κB Abs such as αp50, αRelB and αp65, followed by DNA extraction; PCR was performed to amplify −991/−673 region of mγGCShc promoter using primers: P1, 5′-CCAGTTCCCAGAGCCTTCCG-3′ and P2, 5′-TTGTACGACTCCACATGGCATG-3′. For a negative control, mGAPDH promoter was amplified by using primers: 5′-CACCCTGGCATTTTCTTCCA-3 and 5′-GACCCAGAGACCTGAATGCTG-3′ [63].
Reporter assay
An approximately 1.0 kb (−987/+25) long 5′-flanking sequence of mγGCShc gene was amplified by PCR using murine genomic DNA (Promega, Madison, WI) and cloned into pGL3-Basic vector. Using this resulting construct, p987-luc, and a QuickChangeII PCR-based site-directed mutagenesis kit (Stratagene, Cedar Creek, TX); the construct (Mut p987-luc) containing a mutant NF-κB binding site, similar to that described in EMSA studies, in −987/+25 region of mγGCShc promoter was generated. Both constructs were confirmed by sequencing. NIH3T3 cells were transiently transfected with a DNA mixture containing p987-luc or Mut p987-luc (0.266 µg), pRL-CMV (0.200 µg), pEGFP-p65 (0.266 µg) and/or pEGFP-IκBαΔN (IκBα with amino acids 1–36 deleted) (0.266 µg) using lipofectamine LTX (Invitrogen). The latter two expression vectors are gifts from Dr. Johannes Schmid (Medical University of Vienna, Austria) and Dr. Susan Kandarian (Boston University, USA) respectively [64], [65]. The DNA amount in each transfection was kept constant. Cells were grown for 24 hours after transfection. The luciferase activity of the cell lysates was determined using Dual Luciferase Reporter Assay System (Promega) and GLOMAX luminometer (Promega) following the manufacturer's instructions. The level of luciferase activity was normalized to the level of Renilla luciferase activity.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. LiraR
SundarS
MakhariaA
KenneyR
GamA
1999 Evidence that the high incidence of treatment failures in Indian kala-azar is due to the emergence of antimony-resistant strains of Leishmania donovani. J Infect Dis 180 564 567
2. CroftSL
SundarS
FairlambAH
2006 Drug resistance in leishmaniasis. Clin Microbiol Rev 19 111 126
3. Ashutosh
SundarS
GoyalN
2007 Molecular mechanisms of antimony resistance in Leishmania. J Med Microbiol 56 143 153
4. CarterKC
HutchisonS
HenriquezFL
LegareD
OuelletteM
2006 Resistance of Leishmania donovani to sodium stibogluconate is related to the expression of host and parasite gamma-glutamylcysteine synthetase. Antimicrob Agents Chemother 50 88 95
5. MukherjeeA
PadmanabhanPK
SinghS
RoyG
GirardI
2007 Role of ABC transporter MRPA, gamma-glutamylcysteine synthetase and ornithine decarboxylase in natural antimony-resistant isolates of Leishmania donovani. J Antimicrob Chemother 59 204 211
6. DecuypereS
RijalS
YardleyV
De DonckerS
LaurentT
2005 Gene expression analysis of the mechanism of natural Sb(V) resistance in Leishmania donovani isolates from Nepal. Antimicrob Agents Chemother 49 4616 4621
7. CarterKC
SundarS
SpickettC
PereiraOC
MullenAB
2003 The in vivo susceptibility of Leishmania donovani to sodium stibogluconate is drug specific and can be reversed by inhibiting glutathione biosynthesis. Antimicrob Agents Chemother 47 1529 1535
8. ThakurCP
MitraDK
NarayanS
2003 Skewing of cytokine profiles towards T helper cell type 2 response in visceral leishmaniasis patients unresponsive to sodium antimony gluconate. Trans R Soc Trop Med Hyg 97 409 412
9. NarayanS
BimalS
SinghSK
GuptaAK
SinghVP
2009 Leishmania donovani vs immunity: T-cells sensitized from Leishmania of one donor may modulate their cytokines pattern on re-stimulation with Leishmania from different donor in visceral leishmaniasis. Exp Parasitol 121 69 75
10. MurrayHW
Delph-EtienneS
2000 Roles of endogenous gamma interferon and macrophage microbicidal mechanisms in host response to chemotherapy in experimental visceral leishmaniasis. Infect Immun 68 288 293
11. MurrayHW
MontelibanoC
PetersonR
SypekJP
2000 Interleukin-12 regulates the response to chemotherapy in experimental visceral Leishmaniasis. J Infect Dis 182 1497 1502
12. GorakPM
EngwerdaCR
KayePM
1998 Dendritic cells, but not macrophages, produce IL-12 immediately following Leishmania donovani infection. Eur J Immunol 28 687 695
13. SuzueK
KobayashiS
TakeuchiT
SuzukiM
KoyasuS
2008 Critical role of dendritic cells in determining the Th1/Th2 balance upon Leishmania major infection. Int Immunol 20 337 343
14. BennettCL
MisslitzA
ColledgeL
AebischerT
BlackburnCC
2001 Silent infection of bone marrow-derived dendritic cells by Leishmania mexicana amastigotes. Eur J Immunol 31 876 883
15. RescignoM
MartinoM
SutherlandCL
GoldMR
Ricciardi-CastagnoliP
1998 Dendritic cell survival and maturation are regulated by different signaling pathways. J Exp Med 188 2175 2180
16. PoligoneB
WeaverDJJr
SenP
BaldwinASJr
TischR
2002 Elevated NF-kappaB activation in nonobese diabetic mouse dendritic cells results in enhanced APC function. J Immunol 168 188 196
17. OuaazF
ArronJ
ZhengY
ChoiY
BegAA
2002 Dendritic cell development and survival require distinct NF-kappaB subunits. Immunity 16 257 270
18. WeaverDJJr
PoligoneB
BuiT
Abdel-MotalUM
BaldwinASJr
2001 Dendritic cells from nonobese diabetic mice exhibit a defect in NF-kappaB regulation due to a hyperactive IkappaB kinase. J Immunol 167 1461 1468
19. BhattacharyyaS
SenP
WalletM
LongB
BaldwinASJr
2004 Immunoregulation of dendritic cells by IL-10 is mediated through suppression of the PI3K/Akt pathway and of IkappaB kinase activity. Blood 104 1100 1109
20. SenP
WalletMA
YiZ
HuangY
HendersonM
2007 Apoptotic cells induce Mer tyrosine kinase-dependent blockade of NF-kappaB activation in dendritic cells. Blood 109 653 660
21. GhoshS
KarinM
2002 Missing pieces in the NF-kappaB puzzle. Cell 109 Suppl S81 96
22. KarinM
1999 How NF-kappaB is activated: the role of the IkappaB kinase (IKK) complex. Oncogene 18 6867 6874
23. MadridLV
WangCY
GuttridgeDC
SchotteliusAJ
BaldwinASJr
2000 Akt suppresses apoptosis by stimulating the transactivation potential of the RelA/p65 subunit of NF-kappaB. Mol Cell Biol 20 1626 1638
24. GuhaM
MackmanN
2002 The phosphatidylinositol 3-kinase-Akt pathway limits lipopolysaccharide activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J Biol Chem 277 32124 32132
25. Mookerjee BasuJ
MookerjeeA
SenP
BhaumikS
BanerjeeS
2006 Sodium antimony gluconate induces generation of reactive oxygen species and nitric oxide via phosphoinositide 3-kinase and mitogen-activated protein kinase activation in Leishmania donovani-infected macrophages. Antimicrob Agents Chemother 50 1788 1797
26. BaratC
ZhaoC
OuelletteM
TremblayMJ
2007 HIV-1 replication is stimulated by sodium stibogluconate, the therapeutic mainstay in the treatment of leishmaniasis. J Infect Dis 195 236 245
27. KurozumiR
KojimaS
2005 Increase of intracellular glutathione by low-level NO mediated by transcription factor NF-kappaB in RAW 264.7 cells. Biochim Biophys Acta 1744 58 67
28. TejleK
LindrothM
MagnussonKE
RasmussonB
2008 Wild-type Leishmania donovani promastigotes block maturation, increase integrin expression and inhibit detachment of human monocyte-derived dendritic cells–the influence of phosphoglycans. FEMS Microbiol Lett 279 92 102
29. MarovichMA
McDowellMA
ThomasEK
NutmanTB
2000 IL-12p70 production by Leishmania major-harboring human dendritic cells is a CD40/CD40 ligand-dependent process. J Immunol 164 5858 5865
30. AtoM
StagerS
EngwerdaCR
KayePM
2002 Defective CCR7 expression on dendritic cells contributes to the development of visceral leishmaniasis. Nat Immunol 3 1185 1191
31. DonovanMJ
JayakumarA
McDowellMA
2007 Inhibition of groups 1 and 2 CD1 molecules on human dendritic cells by Leishmania species. Parasite Immunol 29 515 524
32. CaiJ
HuangZZ
LuSC
1997 Differential regulation of gamma-glutamylcysteine synthetase heavy and light subunit gene expression. Biochem J 326 167 172
33. QiH
PopovV
SoongL
2001 Leishmania amazonensis-dendritic cell interactions in vitro and the priming of parasite-specific CD4(+) T cells in vivo. J Immunol 167 4534 4542
34. MurphyML
WilleU
VillegasEN
HunterCA
FarrellJP
2001 IL-10 mediates susceptibility to Leishmania donovani infection. Eur J Immunol 31 2848 2856
35. MurrayHW
LuCM
MauzeS
FreemanS
MoreiraAL
2002 Interleukin-10 (IL-10) in experimental visceral leishmaniasis and IL-10 receptor blockade as immunotherapy. Infect Immun 70 6284 6293
36. DobrovolskaiaMA
MedvedevAE
ThomasKE
CuestaN
ToshchakovV
2003 Induction of in vitro reprogramming by Toll-like receptor (TLR)2 and TLR4 agonists in murine macrophages: effects of TLR “homotolerance” versus “heterotolerance” on NF-kappaB signaling pathway components. J Immunol 170 508 519
37. WhitesideST
EpinatJC
RiceNR
IsraelA
1997 IkappaB epsilon, a novel member of the IkappaB family, controls RelA and cRel NF-kappaB activity. EMBO J 16 1413 1426
38. KearnsJD
BasakS
WernerSL
HuangCS
HoffmannA
2006 IkappaB epsilon provides negative feedback to control NF-kappaB oscillations, signaling dynamics, and inflammatory gene expression. J Cell Biol 173 659 664
39. LaskovR
BergerN
HorwitzMS
2005 Differential effects of tumor necrosis factor-alpha and CD40L on NF-kappaB inhibitory proteins IkappaB alpha, beta and epsilon and on the induction of the Jun amino-terminal kinase pathway in Ramos Burkitt lymphoma cells. Eur Cytokine Netw 16 267 276
40. ZhaoC
CantinR
BretonM
PapadopoulouB
TremblayMJ
2005 DC-SIGN-mediated transfer of HIV-1 is compromised by the ability of Leishmania infantum to exploit DC-SIGN as a ligand. J Infect Dis 191 1665 1669
41. PepeM
AltamuraM
SpinelliR
CalvelloR
SacciaM
2006 Toll-like receptor-positive cells and recognition of pathogens: how human myeloid dendritic cells respond to in vitro infection with Leishmania infantum. Curr Pharm Des 12 4255 4262
42. GargR
BaratC
OuelletM
LodgeR
TremblayMJ
2009 Leishmania infantum Amastigotes Enhance HIV-1 Production in Cocultures of Human Dendritic Cells and CD4 T Cells by Inducing Secretion of IL-6 and TNF-alpha. PLoS Negl Trop Dis 3 e441
43. ChulayJD
FleckensteinL
SmithDH
1988 Pharmacokinetics of antimony during treatment of visceral leishmaniasis with sodium stibogluconate or meglumine antimoniate. Trans R Soc Trop Med Hyg 82 69 72
44. SinghR
KumarD
RameshV
NegiNS
SinghS
2006 Visceral leishmaniasis, or kala azar (KA): high incidence of refractoriness to antimony is contributed by anthroponotic transmission via post-KA dermal leishmaniasis. J Infect Dis 194 302 306
45. SinghN
SinghRT
SundarS
2003 Novel mechanism of drug resistance in kala azar field isolates. J Infect Dis 188 600 607
46. CuevasB
LuY
WattS
KumarR
ZhangJ
1999 SHP-1 regulates Lck-induced phosphatidylinositol 3-kinase phosphorylation and activity. J Biol Chem 274 27583 27589
47. PathakMK
YiT
2001 Sodium stibogluconate is a potent inhibitor of protein tyrosine phosphatases and augments cytokine responses in hemopoietic cell lines. J Immunol 167 3391 3397
48. CorintiS
AlbanesiC
la SalaA
PastoreS
GirolomoniG
2001 Regulatory activity of autocrine IL-10 on dendritic cell functions. J Immunol 166 4312 4318
49. KothariH
KumarP
SundarS
SinghN
2007 Possibility of membrane modification as a mechanism of antimony resistance in Leishmania donovani. Parasitol Int 56 77 80
50. SamantM
SahasrabuddheAA
SinghN
GuptaSK
SundarS
2007 Proteophosphoglycan is differentially expressed in sodium stibogluconate-sensitive and resistant Indian clinical isolates of Leishmania donovani. Parasitology 134 1175 1184
51. KumarA
BoggulaVR
SundarS
ShasanyAK
DubeA
2009 Identification of genetic markers in sodium antimony gluconate (SAG) sensitive and resistant Indian clinical isolates of Leishmania donovani through amplified fragment length polymorphism (AFLP). Acta Trop 110 80 85
52. MollahZU
PaiS
MooreC
O'SullivanBJ
HarrisonMJ
2008 Abnormal NF-kappaB function characterizes human type 1 diabetes dendritic cells and monocytes. J Immunol 180 3166 3175
53. ThakurCP
ThakurS
NarayanS
SinhaA
2008 Comparison of treatment regimens of kala-azar based on culture & sensitivity of amastigotes to sodium antimony gluconate. Indian J Med Res 127 582 588
54. AmeenM
2009 Cutaneous leishmaniasis: disease susceptibility and pharmacogenetic implications. Pharmacogenomics 10 451 461
55. MukhopadhyayR
MadhubalaR
1994 Antileishmanial activity and modification of hepatic xenobiotic metabolizing enzymes in golden hamster by 2(3)-tert-butyl-4-hydroxyanisole following infection with Leishmania donovani. Biochem Pharmacol 47 253 256
56. BasuR
BhaumikS
BasuJM
NaskarK
DeT
2005 Kinetoplastid membrane protein-11 DNA vaccination induces complete protection against both pentavalent antimonial-sensitive and -resistant strains of Leishmania donovani that correlates with inducible nitric oxide synthase activity and IL-4 generation: evidence for mixed Th1 - and Th2-like responses in visceral leishmaniasis. J Immunol 174 7160 7171
57. SinghN
AlmeidaR
KothariH
KumarP
MandalG
2007 Differential gene expression analysis in antimony-unresponsive Indian kala azar (visceral leishmaniasis) clinical isolates by DNA microarray. Parasitology 134 777 787
58. HartDT
VickermanK
CoombsGH
1981 A quick, simple method for purifying Leishmania mexicana amastigotes in large numbers. Parasitology 82 345 355
59. ChakrabortyD
BanerjeeS
SenA
BanerjeeKK
DasP
2005 Leishmania donovani affects antigen presentation of macrophage by disrupting lipid rafts. J Immunol 175 3214 3224
60. DubeA
SinghN
SundarS
2005 Refractoriness to the treatment of sodium stibogluconate in Indian kala-azar field isolates persist in in vitro and in vivo experimental models. Parasitol Res 96 216 223
61. BegAA
FincoTS
NantermetPV
BaldwinASJr
1993 Tumor necrosis factor and interleukin-1 lead to phosphorylation and loss of IkappaB alpha: a mechanism for NF-kappaB activation. Mol Cell Biol 13 3301 3310
62. MurphyTL
ClevelandMG
KuleszaP
MagramJ
MurphyKM
1995 Regulation of interleukin 12 p40 expression through an NF-kappaB half-site. Mol Cell Biol 15 5258 5267
63. LeeCH
MelchersM
WangH
TorreyTA
SlotaR
2006 Regulation of the germinal center gene program by interferon (IFN) regulatory factor 8/IFN consensus sequence-binding protein. J Exp Med 203 63 72
64. SchmidJA
BirbachA
Hofer-WarbinekR
PenggM
BurnerU
2000 Dynamics of NF-kappaB and IkappaB alpha studied with green fluorescent protein (GFP) fusion proteins. Investigation of GFP-p65 binding to DNA by fluorescence resonance energy transfer. J Biol Chem 275 17035 17042
65. JudgeAR
KoncarevicA
HunterRB
LiouHC
JackmanRW
2007 Role for IkappaBalpha, but not c-Rel, in skeletal muscle atrophy. Am J Physiol Cell Physiol 292 C372 382
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 5
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Quorum Sensing Inhibition Selects for Virulence and Cooperation in
- The Role of Intestinal Microbiota in the Development and Severity of Chemotherapy-Induced Mucositis
- Crystal Structure of HIV-1 gp41 Including Both Fusion Peptide and Membrane Proximal External Regions
- Susceptibility to Anthrax Lethal Toxin-Induced Rat Death Is Controlled by a Single Chromosome 10 Locus That Includes
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy