
Structural Basis for Apoptosis Inhibition by Epstein-Barr Virus BHRF1
Epstein-Barr virus (EBV) is associated with human malignancies, especially those affecting the B cell compartment such as Burkitt lymphoma. The virally encoded homolog of the mammalian pro-survival protein Bcl-2, BHRF1 contributes to viral infectivity and lymphomagenesis. In addition to the pro-apoptotic BH3-only protein Bim, its key target in lymphoid cells, BHRF1 also binds a selective sub-set of pro-apoptotic proteins (Bid, Puma, Bak) expressed by host cells. A consequence of BHRF1 expression is marked resistance to a range of cytotoxic agents and in particular, we show that its expression renders a mouse model of Burkitt lymphoma untreatable. As current small organic antagonists of Bcl-2 do not target BHRF1, the structures of it in complex with Bim or Bak shown here will be useful to guide efforts to target BHRF1 in EBV-associated malignancies, which are usually associated with poor clinical outcomes.
Published in the journal:
. PLoS Pathog 6(12): e32767. doi:10.1371/journal.ppat.1001236
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1001236
Summary
Epstein-Barr virus (EBV) is associated with human malignancies, especially those affecting the B cell compartment such as Burkitt lymphoma. The virally encoded homolog of the mammalian pro-survival protein Bcl-2, BHRF1 contributes to viral infectivity and lymphomagenesis. In addition to the pro-apoptotic BH3-only protein Bim, its key target in lymphoid cells, BHRF1 also binds a selective sub-set of pro-apoptotic proteins (Bid, Puma, Bak) expressed by host cells. A consequence of BHRF1 expression is marked resistance to a range of cytotoxic agents and in particular, we show that its expression renders a mouse model of Burkitt lymphoma untreatable. As current small organic antagonists of Bcl-2 do not target BHRF1, the structures of it in complex with Bim or Bak shown here will be useful to guide efforts to target BHRF1 in EBV-associated malignancies, which are usually associated with poor clinical outcomes.
Introduction
To combat invading viruses, altruistic suicide of the infected host cells may be initiated to rapidly and efficiently eliminate the pathogen [1], [2]. Often, this response is a critical component of host defences [1], [2]. Consequently, many viruses have co-evolved adaptive mechanisms to subvert apopt osis, thereby ensuring their own survival and propagation. Some viruses, such as Epstein-Barr virus (EBV), encode homologs of the mammalian pro-survival protein Bcl-2 [3], [4], [5]. EBV was first identified in association with Burkitt lymphoma and it is also linked to other lymphoid malignancies (Hodgkin's lymphoma, post-transplant lymphoproliferative disorders) and nasopharyngeal carcinoma [6]. Whereas increased expression of Bcl-2 promotes malignancies such as human follicular lymphoma [7], the precise role of the EBV encoded Bcl-2 homolog BHRF1 in EBV-associated malignancies is less well defined.
However, more recent studies link BHRF1 to the transformation of primary B lymphocytes [8] and to lymphomagenesis [9]. Since overactivity of the oncogene myc is obligatory for Burkitt lymphoma [10], [11], [12], expression of BHRF1 may be necessary to block myc-induced apoptosis, akin to the striking synergy observed between Bcl-2 and myc during B cell transformation [13], [14]. Of note, the constitutive expression of BHRF1 permits lymphoblastoid immortalization by EBV and their prolonged survival [9], and together with expression of BHRF1 during normal B cells transformation [8] suggests a role for BHRF1 in post-transplant lymphoproliferative disease. Although confirmed BHRF1 expression has been shown in only a subset of Burkitt lymphomas [9], [15], it is plausible that BHRF1 plays a central role in the maintenance of this subset of Burkitt lymphomas as Bcl-2 overexpression is rare in this disease.
As BHRF1 may be central for developing and maintaining certain EBV-associated lymphomas, we investigated if BHRF1 can modulate responses to therapy in experimental models. If so, BHRF1 represents an attractive drug target since normal cells may well be spared by its selective antagonism. Here, we show that BHRF1 potently confers chemoresistance, and importantly, it adversely impacts upon survival in a mouse model of Burkitt lymphoma. BHRF1 acts by sequestering a subset of the pro-apoptotic Bcl-2 family proteins; we show here the 3D structures of it in complex with BH3 domains of two, Bim and Bak, which may provide the basis for developing small molecule inhibitors of BHRF1 to improve the generally poor prognosis in EBV-associated malignancies.
Results/Discussion
BHRF1 counters apoptosis induced by multiple chemotherapeutic agents
Using cultured cell lines, we tested the ability of BHRF1 to confer resistance against a range of apoptotic stimuli, especially those used for cancer chemotherapy. Stable expression of BHRF1 in FDC-P1 mouse myelomonocytic cells conferred resistance to etoposide or γ-irradiation (Fig. 1A) comparable to that observed in cells expressing similar levels of Bcl-2, Bcl-xL or Bcl-w (Fig. 1D). It also inhibited apoptosis induced by other cytotoxics including cytosine arabinoside (Ara-C), doxorubicin, etoposide and staurosporine in other cell lines (Figs. 1B, 1C, S1 and data not shown), comparable to that observed in cells expressing similar levels of Bcl-2 (Fig. 1E). Thus, BHRF1, like its mammalian counterparts, inhibits apoptosis induced in multiple cell types by diverse chemotherapeutic agents.

BHRF1 preserves mitochondrial function by inhibiting the activation of Bax and Bak
To ascertain precisely how BHRF1 interferes with apoptosis signaling, we examined mitochondrial outer membrane permeabilization (MOMP) [16] in FDC-P1 cells after treatment with staurosporine. Whereas MOMP occurred rapidly in the parental cells, as indicated by the reduced uptake of the mitochondrial dye DiOC6(3), it was inhibited in cells expressing BHRF1 or Bcl-2 (Fig. 2A), implicating an anti-apoptotic effect upstream of mitochondrial damage. When the mediators of mitochondrial damage (Bax and Bak) were examined, we found that BHRF1 inhibited translocation of Bax from the cytosol (Fig. 2B) and activation of Bax and Bak was abrogated, as their conformational alteration associated with activation is blocked (Fig. 2C). Consistent with these observations, BHRF1 also inhibited the release of cytochrome c from within the mitochondria (Fig. 2B).
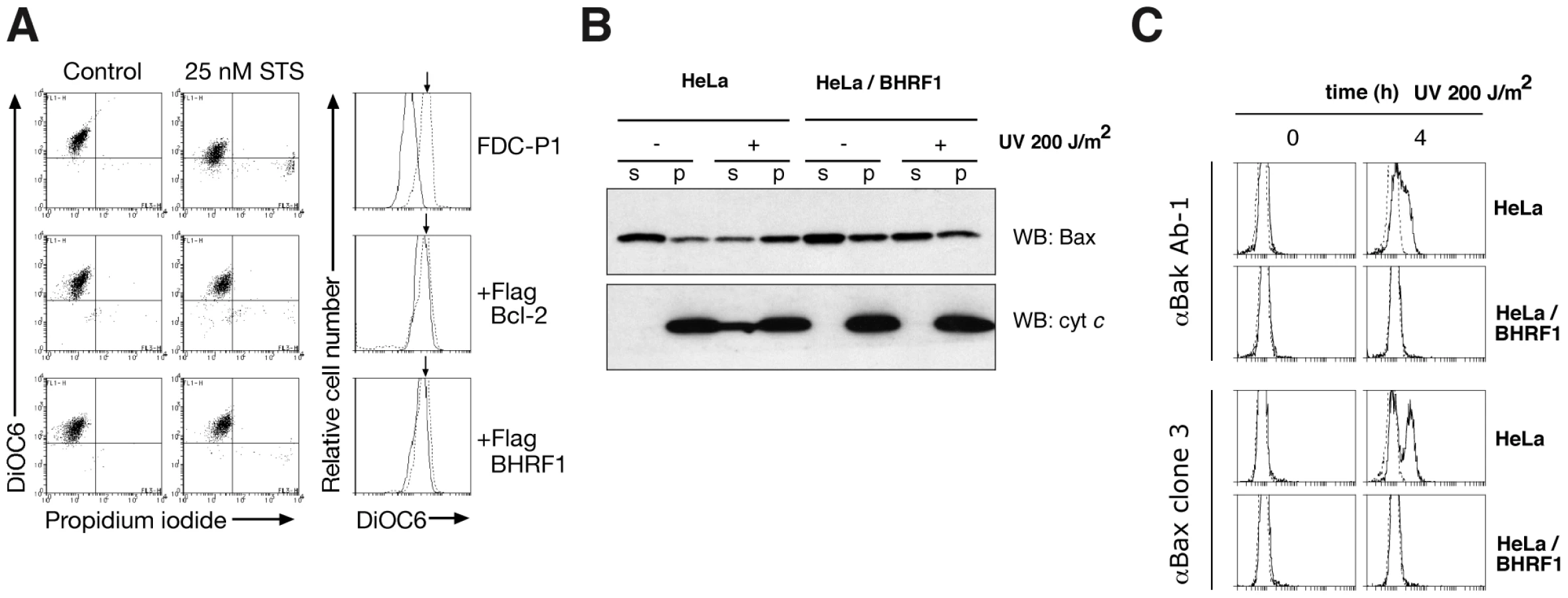
We conclude that BHRF1 must exert its anti-apoptotic effect at the level of, or prior to, Bax and Bak activation, consistent with a report that BHRF1 prevented Bax and Bak conformational change, oligomerization and activation of the initiator caspase, caspase-9 [17]. How then might BHRF1 inhibit activation of these essential cell death mediators?
BHRF1 interacts with a sub-set of pro-apoptotic Bcl-2 family proteins
It is most likely that BHRF1 acts to abort cell death initiation by sequestering the endogenous pro-apoptotic mammalian Bcl-2 family members. Thus, we assessed the ability of a recombinant C-terminally truncated form of BHRF1 to directly bind peptides spanning the BH3 domains of pro-apoptotic Bcl-2 proteins using isothermal titration calorimetry (ITC). Binding was observed with peptides from the BH3-only proteins Bim (KD = 18 nM), Puma (70 nM) or Bid (110 nM) (Fig. 3A). No detectable binding was observed with peptides from other BH3-only proteins, or with Mule and Beclin-1, other proteins harboring a BH3 domain. We confirmed the interaction of intact BHRF1 in mammalian cells with selected full-length BH3-only proteins in co-immunoprecipitation assays (Fig. 3B). These results closely mirror those obtained in solution competition assays, using either fluorescence polarization [18] or surface plasmon resonance [19]. Thus, BHRF1 probably antagonizes a subset of the BH3-only proteins by direct sequestration. Interestingly, the ones targeted (Bid, Bim, Puma) are potent inducers of apoptosis, either because they neutralize most, if not all, the mammalian pro-survival proteins [20], [21] or because they can directly activate Bax or Bak [22], [23], [24].

We also investigated if BHRF1, in a manner similar to some mammalian [21], [25] and viral Bcl-2 proteins [26], [27], might directly bind Bax and Bak, the downstream mediators of mitochondrial damage. Recombinant BHRF1 bound a 26-mer Bak BH3 peptide (KD = 150 nM), but only weakly (>1 µM) to Bax BH3 (Fig. 3A). Consistent with the binding data, we observed that BHRF1 could directly counter Bak (Fig. 3C), but not Bax, when these proteins were expressed in yeast [28]. This heterologous model system is suited for studying the functional interactions by circumventing potential complications due to the presence of endogenous Bcl-2 family proteins in mammalian cells and avoids the use of detergents that may artificially induce or disrupt interactions between Bcl-2 family proteins [29]. Our assay is based on the observation that overexpression of Bax or Bak in yeast is lethal, even though yeasts do not express Bcl-2 family members and do not undergo apoptosis. Nonetheless, co-expression of Bcl-2, Bcl-xL, Mcl-1, or A1 with Bax and Bak can suppress death induced in yeast [30], thus reconstituting key aspects of the mammalian apoptotic machinery. Our observation that BHRF1 could counter Bak (Fig. 3C), but not Bax-induced yeast death when these proteins were expressed in yeast suggests that BHRF1 is only able to directly neutralize Bak, but not Bax.
Taken together with the previous reports that BHRF1 interacts with the full-length Bak, but not with Bax [17], [31], [32], we conclude that BHRF1 can keep Bak inactive by direct binding (Fig. 3A, 3C, [19]), but must inhibit Bax indirectly, presumably by its ability to sequester BH3-only proteins such as Bim (Fig. 3A, 3B, [31]). It will therefore be interesting to investigate which pro-apoptotic protein is the critical target for BHRF1 in diverse cell types, especially those targeted by EBV during oncogenesis. It is noteworthy that in some lymphoid cells, the pro-survival action of BHRF1 tracked with its ability to bind Bim [31], which is critical for apoptosis induced by multiple stimuli in this cell type [33] and plays a role in suppressing myc-driven lymphomagenesis [34].
Structural basis for the engagement of BH3 domains by BHRF1
Our data (Fig. 3) and previously published studies [18] suggest that BHRF1, like its mammalian counterparts, inhibits cell death by sequestering endogenous pro-apoptotic Bcl-2 proteins. However, the structural basis for this is unclear since a previously published structure of C-terminally truncated BHRF1 lacked the characteristic hydrophobic surface groove responsible for interaction with the BH3 domains [35]. We have therefore determined crystal structures of BHRF1 in complex with its key targets, the BH3 domains of Bim (Fig. 4A–B, Table 1) and Bak (Fig. 4C, Table 1).


The Bim BH3 peptide binds into a surface groove formed by helices α2-5 of BHRF1 (Fig. 4A), in a similar manner to that previously observed for mammalian pro-survival Bcl-2 members such as Bcl-xL [36] or the unrelated viral Bcl-2 protein M11L [27]. Bak BH3 binds in an equivalent manner (Fig. 4C), and the two complexes superimpose with an RMSD of only 0.6 Å over the entire BHRF1 backbone indicating their similarities. As the characteristic hydrophobic surface groove was absent in unliganded BHRF1 due to the close proximity of helices α3 and α4 (Fig. 4E; [35]), significant structural changes are required in order to accommodate Bim or Bak BH3 domains. These changes affect mainly α4, which is at a 120° angle to α3 in the BH3-bound form (Fig. 4A), compared to the near anti-parallel alignment in the free form (Fig. 4E). Overall, the free and bound BHRF1 structures (comparing Fig. 4A with 4B) superimpose with an RMSD of 3.5 Å, with most differences found within the BH3 binding groove. This is reminiscent of the movement observed in Bcl-xL, which in the ligand-free state displays a narrow binding groove [37]. However, upon ligand binding, both α3 and 4 helices move to widen the hydrophobic groove and allow binding [36]. In contrast, the movement of α3 that enables groove opening in BHRF1 upon ligand binding is much less pronounced.
The side-chain interactions contributing to the BHRF1-BH3 complexes are equivalent to those observed for mammalian pro-survival proteins such as Bcl-xL, with the four conserved hydrophobic BH3 residues of Bim (I58, L62, I65 and F69; numbering based on human BimL) protruding into pockets within the BHRF1 hydrophobic binding groove (Fig. 4G). Similarly, Bak residues V74, L78, I81 and I85 (numbering based on human Bak) interact with the BHRF1 binding groove (Fig. 4H). In addition to the hydrophobic interactions, BHRF1 R100 forms a salt bridge with Bim D67 or with Bak D83 (Fig. 4G, 4H). This electrostatic interaction is also observed in complexes of Mcl-1 and Bcl-xL with pro-apoptotic BH3 domains [36], [38] and even in a complex of Bcl-xL with a peptide foldamer [39]. The conservation of aspartic acid in the mammalian BH3 ligands suggests that this interaction is of particular importance for complex formation, and indeed a BHRF1 R100A mutation reduces Bim binding and abolishes interaction with Bak [31].
BHRF1 confers potent chemoresistance in vivo
Since BHRF1 engages BH3 domains using a hydrophobic groove (Fig. 4) in a manner equivalent to that of its mammalian counterparts, conserving key interactions, we asked whether ABT-737, a BH3 mimetic compound known to inhibit Bcl-2, Bcl-xL, and Bcl-w [40], [41], could also target BHRF1. In the absence of pro-survival Mcl-1, ABT-737 is a potent cytotoxic agent. However, cells expressing BHRF1 were completely insensitive to ABT-737, even at the highest dose tested (10 µM) (Fig. S2), and survived long-term (Fig. 5A). Similarly, recombinant BHRF1 did not bind ABT-737 in biosensor assays (IC50>20 µM, data not shown). As ABT-737 is ineffective and as BHRF1 can potently confer chemoresistance when tested in cultured cell lines (Fig. 1), we evaluated the impact of its expression in a transgenic mouse model of Burkitt lymphoma [11]. In the Eµ-myc mouse, myc is overexpressed in the B cell and drives an aggressive B cell leukemia/lymphoma syndrome that is very similar to human Burkitt lymphoma.

Malignant cells derived from sick Eµ-myc mice are readily transplantable into syngeneic wild-type recipients which succumb within four weeks to a disseminated disease if left untreated [42]. Treatment with Ara-C (Fig. 5C, black line), an agent used in the clinic for treating patients with Burkitt lymphoma, resulted in a sustained disease remission and survival with all mice alive and disease-free at 100 days with normal peripheral blood counts and spleen weights. In striking contrast, only a handful of treated mice that were inoculated with tumor cells overexpressing BHRF1 or Bcl-2 survive long-term (Fig. 5C). Comparable results were observed when two other cytotoxic agents, cyclophosphamide and etoposide, were used in similar efficacy studies (data not shown). Therefore BHRF1, like Bcl-2 [42], can potently confer chemoresistance in a mouse model of Burkitt lymphoma. Thus, it is highly likely that expression of BHRF1 will attenuate the response during treatment for EBV-driven malignancies.
Concluding remarks
In this study, we have confirmed that EBV BHRF1 exerts its pro-survival function by directly inhibiting a sub-set of pro-apoptotic Bcl-2 family proteins Bid, Bim, Puma and Bak, presumably ones most critical for the virus. Our three-dimensional structures show that these interactions closely resemble those seen with mammalian pro-survival proteins such as Bcl-xL. In light of the importance of BHRF1 in certain Burkitt lymphomas [9], and its detection in other EBV-associated malignancies including nasopharyngeal carcinoma [43] and B cell lymphomas [44], the development of therapeutic inhibitors of BHRF1 may be highly desirable. The inability of current small molecule inhibitors of mammalian pro-survival proteins such as ABT-737 to inhibit BHRF1 will require novel small molecule antagonists to be developed. The successful development of small molecule inhibitors of mammalian pro-survival Bcl-2 family proteins [40] suggests that similar approaches might be applied to the development of BHRF1 inhibitors.
Materials and Methods
Recombinant proteins and binding experiments
BHRF1ΔC31 was cloned into pET DUET (Invitrogen) using BamHI and EcoRI, and expressed in E.coli BL21 DE3 pLysS. Cells were homogenized using an Avestin EmulsiFlex homogenizer in lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 10 mM BME). His-tagged BHRF1ΔC31 was purified using a nickel charged Hi-Trap chelating column (Amersham), eluted in lysis buffer with 250 mM imidazole and subjected to gel-filtration chromatography in 25 mM Hepes pH 7.5, 150 mM NaCl using a Superdex 200 column (GE Healthcare). Calorimetry data were collected on a VP-ITC (MicroCal) with BHRF1ΔC31 as previously described [45]. All peptides were purified by reverse-phase HPLC and dissolved as 2–5 mM stock solutions in water. The accession numbers for the peptides were: human BimL (AAC39594), human Puma (AAK39542), mouse Bmf (AAK38747), human Bad (NP_004313), human Bik (NP_001188), human Hrk (NP_003797), human Bid (NP_001187), human Noxa (NP_066950), human Bax (NP_620119), human Bak (NP_001179), mouse Mule (UniProt Accession code Q7TMY8; residues 1969–1994; PGGTTQEVGQLLQDMGDDVYQQYRSL) and mmBeclin (UniProt Accession code O88597, residue 103–128; DGGTMENLSRRLKVTGDLFDIMSGQT).
Crystallization and structure determination
BHRF1 complexes with Bim or Bak BH3 were obtained by mixing BHRF1 with human Bim or Bak 26-mer peptide in a 1∶1.25 molar ratio and concentrating using a centricon (Millipore) to 10 mg/mL. Crystals were grown in sitting drops at 20°C in 1.2 M NaBr, 50 mM malic acid pH 4.0 (BHRF1:Bim) or 1.6 M NaNO3, 50 mM malic acid pH 4.4 (BHRF1:Bak). The crystals belong to space group P3221 with a = b = 62.75 Å, c = 92.38 Å, α = β = 90°, γ = 120° (BHRF1:Bim) or a = b = 62.39 Å, c = 93.73 Å, α = β = 90°, γ = 120° (BHRF1:Bak). The asymmetric units contain 1 BHRF1:peptide complex. Diffraction data were collected from flash frozen crystals at 100 K at the Australian Synchrotron (beamline 3BM1) or at the Swiss Light Source (beamlines X06SA, X10SA) and processed with HKL2000. For the BHRF1:Bim complex, a heavy atom derivative was obtained by soaking crystals in mother liquor supplemented with 1 mM MeHgCl for 2 h. Hg sites were found and refined with Sharp. Clear and continuous electron density was obtained for residues 2–158 of BHRF1 and 51–72 of Bim. The final model was built with Coot [46], refined with Refmac5 [47] to a resolution of 1.5 Å and has a final R-factor of 0.198 (R-free 0.205). 95.0% of the residues are in the core regions of the Ramachandran plot, and no residues are in disallowed regions.
The BHRF1:Bak structure was solved by molecular replacement with PHASER [48] using the BHRF1:Bim structure as a search model. The final model was built with Coot and refined with Refmac5 to a resolution of 2.05 Å and has a final R-factor of 0.184 (R-free 0.217). 97.4% of the residues are in the core regions of the Ramachandran plot, and no residues are in disallowed regions. All data collection and refinement statistics are summarized in Table 1. The coordinates have been deposited in the Protein Data Bank (accession codes 2v6q, 2wh6, 2xpx). Figures were prepared using PyMol (DeLano Scientific).
Mouse tumor model
Eµ-myc transgenic mice on an inbred C57BL/6 background with clinical evidence of lymphoma were culled by CO2 asphyxiation and lymphomatous tissue excised. Single-cell suspensions were obtained by manual sieving and stable Eµ-myc tumor cell cultures were established in FMA medium. Retroviral transduction using a pMSCV-IRES-GFP vector containing BHRF1 or Bcl-2 and sorting of GFP positive cells were conducted as previously described [11]. 1×106 viable tumor cells in 100 µL PBS were injected intraperitoneally into syngeneic C57BL/6 mice. Ara-C or PBS was injected intraperitoneally in 100 µL total volume on days 4, 5 and 6. Mice were culled when sick (hindleg paralysis, tremor, lethargic, tumor nodule >1 cm diameter, >5% weight loss) and leukemia/lymphoma confirmed by the presence of peripheral blood leucocytosis and enlarged spleen and/or lymph nodes. Survival of cohorts of 5 mice was compared by log-rank test and Kaplan-Meier analysis using GraphPad Prism statistical software. All experiments were approved by an institutional ethics committee.
Cell lines and tissue culture
MEF, HEK-293T and Phoenix packaging 293 cells were cultured in Dulbecco's Modified Eagles medium (DMEM) supplemented with 10% FCS. FDC-P1 cells were additionally supplemented with mouse IL-3 (1000 U/mL). Eµ-myc tumor cells were harvested from a symptomatic Eµ-myc transgenic mouse and cultured in FMA, a high glucose version of DMEM supplemented with 10% FCS, 50 µM 2-mercaptoethanol and 250 µM asparagine.
Mammalian expression constructs
Epitope-tagged mammalian expression vectors for human Bcl-2 family proteins have been described previously [20], [49], [50], [51], [52]. All constructs were verified by sequencing. Details and constructs are available from the authors.
Retrovirus production and transduction
To produce retroviral supernatant, 2.5×106 ecotropic Phoenix packaging cells were seeded overnight in 10 cm tissue culture plates. Media was replaced with 5 ml serum-free DMEM containing 5 µg MSCV-based retroviral plasmid with 15 µL Lipofectamine (Invitrogen). After 24 h media was replaced with medium supplemented with 20% FCS and incubated for a further 24 h at 32°C. Viral supernatant was cleared of cell debris by centrifugation for 5 min at 1500 rpm. 500 µL filtered virus (0.45 µm, Millipore) was spin-infected onto target cells in a total volume of 1 mL media containing 4 µg/mL polybrene (Sigma) in 24 well plates at 32°C with 2500 rpm radial centrifugation for 45 min. Infection efficiency of MEFs were generally >90% and 20–30% for FDC-P1 cells.
Cell survival assays and cytotoxic drugs
Cell death was induced by 0–100 µM etoposide (Pharmacia-Upjohn), 0–30 Gy γ-irradiation, 0–10 µM Ara C (Pharmacia-Upjohn), 0–100 nM staurosprorine (Sigma-Aldrich) or 0–10 µM ABT-737 (Abbott Laboratories). Cell viability was quantified by flow cytometric analysis of cells excluding 5 µg/mL propidium iodide (PI) (Sigma-Aldrich) using a FACScan (Becton Dickinson). Each time point was performed at least three times. For long-term colony assays using MEFs, cells were infected with GFP-expressing retroviral constructs, then treated with qVD.OPH (Enzyme Systems) to prevent cell death. After culture for 24 h, 200 GFP+ve cells were sorted into 6-well plates. Colonies were stained and counted 6 d later.
Immunofluorescence
Cells expressing mammalian FLAG-tagged pro-survival Bcl-2 proteins, BHRF1 or empty vector were washed in PBS, fixed in 1% paraformaldehyde/PBS (10 min, 4°C) and washed twice in KDS-BSS. Cells were incubated with 1∶1,000 primary anti-FLAG M2 (Sigma) antibody for 20 minutes, washed in KDS-BSS/0.02% saponin and then incubated with 1∶100 goat anti-mouse FITC or PE antibody (Southern Biotechnology) for 20 minutes before analysis on a FACScan (BD) using Cell Quest software (BD).
Cytofluorometric determination of mitochondrial transmembrane potential and Bax/Bak activation
To assess mitochondrial transmembrane potential (Δψm), cells were incubated for 15 min at 37°C in buffer containing 40 nM 3,3′ - dihexyloxacarbocyanine iodide (DiOC6[3]; Molecular Probes) before adding 10 µg/mL of PI. The cells were kept on ice until flow cytometric analysis. To assess the activation of Bax and Bak, HeLa cells were left untreated or pretreated with a proteasome inhibitor (10 µM MG-132; Calbiochem) or a wide-spectrum caspase inhibitor (100 µM zVAD.fmk; Bachem) for 1 h before treatment with 200 J/m2 UV-irradiation. Following UV irradiation, cells were fixed with 1% paraformaldehyde (5 min at room temperature) and then washed with buffer supplemented with 2% fetal bovine serum. Fixed cells were then incubated with the primary antibodies: 2 µg/mL anti-Bak Ab-1 (Calbiochem) or 5 µg/mL anti-Bax clone 3 (BD) diluted in FACS buffer supplemented with 0.3% saponin for 30 min on ice. Cells were then washed, before incubation with the appropriate secondary antibody, either FITC-conjugated goat-anti-mouse IgG (10 µg/ml; SouthernBiotech) to detect Bax activation or a biotin-conjugated anti-mouse (diluted 1∶200; SouthernBiotech) followed by Streptavidin-conjugated PE (diluted 1∶300; Caltag) to detect Bak activation. The samples were analyzed using a FACScan (BD).
Subcellular fractionation
Fractionation of whole cell lysates into the soluble and pellet fractions has been previously described [54]. In brief, cells lysed in HMKEE buffer (20 mM Hepes, pH 7.2, 5 mM MgCl2, 10 mM KCl, 1 mM EDTA, 1 mM EGTA, and protease inhibitors) containing 250 mM sucrose and 0.025% digitonin (Calbiochem) were left on ice for 10 min, and then the organelles, cytoskeleton, and membranes were pelleted by centrifugation (13,000 rpm, 5 min at 4°C). The pellet was solubilized in RIPA buffer (150 mM NaCl, 1% Triton X-100, 0.5% deoxycholic acid, 0.1% SDS, 50 mM Tris-HCl, pH 8.0, and protease inhibitors). The protease inhibitors used include Pefabloc SC, soybean trypsin inhibitor, leupeptin, aprotinin, E64, and pepstatin (Sigma-Aldrich or Roche).
Transient transfection, immunoprecipitation and immunoblotting
The transfection and metabolic labeling of HEK-293T cells with 35S-methionine/cysteine (NEN) as well as co-immunoprecipitation have been described [49], [50], [52]. Briefly, equivalent TCA-precipitable lysates were immunoprecipiated using the mouse monoclonal antibodies to FLAG (M2; Sigma), Glu-Glu (CRP) and control HA (HA.11; CRP) tags. The proteins were resolved by SDS:PAGE, transferred onto nitrocellulose membranes and detected by autoradiography after 20 h at −80°C. Immunoblotting was performed using mouse monoclonal antibodies to Bax (5B7; Sigma-Aldrich) cytochrome c (7H8.2C12; BD Pharmingen) and detected using HRP-conjugated secondary antibodies (Southern Biotechnology) revealed by enhanced chemiluminescence (ECL; Amersham Biosciences).
Yeast colony assays
Yeast expression vectors were made by subcloning the cDNAs for full-length human Bcl-xL and BHRF1, or human Bax and human Bak, respectively, into the pGALL(TRP1) and pGALS(LEU2) vectors [55]. Saccharomyces cerevisiae W303a cells were co-transformed with indicated plasmids and grown under selection. For the survival assays, the cells were spotted as 5-fold serial dilutions onto glucose (repressing, “OFF”) or galactose (inducing, “ON”) plates as previously described [28]. Plates were incubated for 48 h at 30°C and then photographed.
Ethics statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Health and Medical Research Council. The protocol was approved by the Committee on the Ethics of Animal Experiments of the Walter and Eliza Hall Institute of Medical Research (Permit Number: NKOT_07_008). All surgery was performed under sodium pentobarbital anesthesia, and all efforts were made to minimize suffering.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. GalluzziL
BrennerC
MorselliE
TouatZ
KroemerG
2008 Viral control of mitochondrial apoptosis. PLoS Pathog 4 e1000018
2. CuconatiA
WhiteE
2002 Viral homologs of BCL-2: role of apoptosis in the regulation of virus infection. Genes and Development 16 2465 2478
3. CuconatiA
DegenhardtK
SundararajanR
AnschelA
WhiteE
2002 Bak and Bax function to limit adenovirus replication through apoptosis induction. J Virol 76 4547 4558
4. MarchiniA
TomkinsonB
CohenJI
KieffE
1991 BHRF1, the Epstein-Barr virus gene with homology to Bc12, is dispensable for B-lymphocyte transformation and virus replication. J Virol 65 5991 6000
5. HendersonS
HuenD
RoweM
DawsonC
JohnsonG
1993 Epstein-Barr virus-coded BHRF1 protein, a viral homologue of Bcl-2, protects human B cells from programmed cell death. Proc Natl Acad Sci U S A 90 8479 8483
6. Thorley-LawsonDA
GrossA
2004 Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N Engl J Med 350 1328 1337
7. TsujimotoY
CossmanJ
JaffeE
CroceCM
1985 Involvement of the bcl-2 gene in human follicular lymphoma. Science 228 1440 1443
8. AltmannM
HammerschmidtW
2005 Epstein-Barr virus provides a new paradigm: a requirement for the immediate inhibition of apoptosis. PLoS Biol 3 e404
9. KellyGL
LongHM
StylianouJ
ThomasWA
LeeseA
2009 An Epstein-Barr virus anti-apoptotic protein constitutively expressed in transformed cells and implicated in burkitt lymphomagenesis: the Wp/BHRF1 link. PLoS Pathog 5 e1000341
10. TaubR
KirschI
MortonC
LenoirG
SwanD
1982 Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human Burkitt lymphoma and murine plasmacytoma cells. Proc Natl Acad Sci U S A 79 7837 7841
11. AdamsJM
HarrisAW
PinkertCA
CorcoranLM
AlexanderWS
1985 The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 318 533 538
12. MagrathI
1990 The pathogenesis of Burkitt's lymphoma. Adv Cancer Res 55 133 270
13. StrasserA
HarrisAW
BathML
CoryS
1990 Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature 348 331 333
14. BeverlyLJ
VarmusHE
2009 MYC-induced myeloid leukemogenesis is accelerated by all six members of the antiapoptotic BCL family. Oncogene 28 1274 1279
15. KellyG
BellA
RickinsonA
2002 Epstein-Barr virus-associated Burkitt lymphomagenesis selects for downregulation of the nuclear antigen EBNA2. Nat Med 8 1098 1104
16. GreenDR
KroemerG
2004 The pathophysiology of mitochondrial cell death. Science 305 626 629
17. CrossJR
PostigoA
BlightK
DownwardJ
2008 Viral pro-survival proteins block separate stages in Bax activation but changes in mitochondrial ultrastructure still occur. Cell Death Differ 15 997 1008
18. FlanaganAM
LetaiA
2008 BH3 domains define selective inhibitory interactions with BHRF-1 and KSHV BCL-2. Cell Death Differ 15 580 588
19. UrenRT
DewsonG
ChenL
CoyneSC
HuangDC
2007 Mitochondrial permeabilization relies on BH3 ligands engaging multiple prosurvival Bcl-2 relatives, not Bak. J Cell Biol 177 277 287
20. ChenL
WillisSN
WeiA
SmithBJ
FletcherJI
2005 Differential targeting of pro-survival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Molecular Cell 17 393 403
21. WillisSN
FletcherJI
KaufmannT
van DelftMF
ChenL
2007 Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 315 856 859
22. CartronPF
GallenneT
BougrasG
GautierF
ManeroF
2004 The first alpha helix of Bax plays a necessary role in its ligand-induced activation by the BH3-only proteins Bid and PUMA. Molecular Cell 16 807 818
23. KuwanaT
Bouchier-HayesL
ChipukJE
BonzonC
SullivanBA
2005 BH3 Domains of BH3-Only Proteins Differentially Regulate Bax-Mediated Mitochondrial Membrane Permeabilization Both Directly and Indirectly. Molecular Cell 17 525 535
24. LetaiA
BassikM
WalenskyL
SorcinelliM
WeilerS
2002 Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2 183 192
25. OltvaiZN
MillimanCL
KorsmeyerSJ
1993 Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 74 609 619
26. WhiteE
SabbatiniP
DebbasM
WoldWSM
KusherDI
1992 The 19-kilodalton adenovirus E1B transforming protein inhibits programmed cell death and prevents cytolysis by tumor necrosis factor α. Molecular and Cellular Biology 12 2570 2580
27. KvansakulM
van DelftMF
LeeEF
GulbisJM
FairlieWD
2007 A structural viral mimic of prosurvival bcl-2: a pivotal role for sequestering proapoptotic bax and bak. Molecular Cell 25 933 942
28. JabbourAM
PuryerMA
YuJY
LithgowT
RiffkinCD
2006 Human Bcl-2 cannot directly inhibit the Caenorhabditis elegans Apaf-1 homologue CED-4, but can interact with EGL-1. J Cell Sci 119 2572 2582
29. HsuYT
YouleRJ
1997 Nonionic detergents induce dimerization among members of the Bcl-2 family. J Biol Chem 272 13829 13834
30. TaoW
KurschnerC
MorganJI
1997 Modulation of cell death in yeast by the Bcl-2 family of proteins. J Biol Chem 272 15547 15552
31. DesbienAL
KapplerJW
MarrackP
2009 The Epstein-Barr virus Bcl-2 homolog, BHRF1, blocks apoptosis by binding to a limited amount of Bim. Proc Natl Acad Sci U S A 106 5663 5668
32. TheodorakisP
D'Sa-EipperC
SubramanianT
ChinnaduraiG
1996 Unmasking of a proliferation-restraining activity of the anti-apoptosis protein EBV BHRF1. Oncogene 12 1707 1713
33. BouilletP
MetcalfD
HuangDCS
TarlintonDM
KayTWH
1999 Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science 286 1735 1738
34. EgleA
HarrisAW
BouilletP
CoryS
2004 Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc Natl Acad Sci U S A 101 6164 6169
35. HuangQ
PetrosAM
VirginHW
FesikSW
OlejniczakET
2003 Solution structure of the BHRF1 protein from Epstein-Barr virus, a homolog of human Bcl-2. Journal of Molecular Biology 332 1123 1130
36. LiuX
DaiS
ZhuY
MarrackP
KapplerJW
2003 The structure of a Bcl-xL/Bim fragment complex: Implications for Bim function. Immunity 19 341 352
37. MuchmoreSW
SattlerM
LiangH
MeadowsRP
HarlanJE
1996 X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature 381 335 341
38. CzabotarPE
LeeEF
van DelftMF
DayCL
SmithBJ
2007 Structural insights into the degradation of Mcl-1 induced by BH3 domains. Proc Natl Acad Sci U S A 104 6217 6222
39. LeeEF
SadowskyJD
SmithBJ
CzabotarPE
Peterson-KaufmanKJ
2009 High-resolution structural characterization of a helical alpha/beta-peptide foldamer bound to the anti-apoptotic protein Bcl-xL. Angew Chem Int Ed Engl 48 4318 4322
40. OltersdorfT
ElmoreSW
ShoemakerAR
ArmstrongRC
AugeriDJ
2005 An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435 677 681
41. van DelftMF
WeiAH
MasonKD
VandenbergCJ
ChenL
2006 The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 10 389 399
42. SchmittCA
RosenthalCT
LoweSW
2000 Genetic analysis of chemoresistance in primary murine lymphomas. Nat Med 6 1029 1035
43. LiuMY
ShihYY
LiLY
ChouSP
SheenTS
2000 Expression of the Epstein-Barr virus BHRF1 gene, a homologue of Bcl-2, in nasopharyngeal carcinoma tissue. J Med Virol 61 241 250
44. OudejansJJ
van den BruleAJ
JiwaNM
de BruinPC
OssenkoppeleGJ
1995 BHRF1, the Epstein-Barr virus (EBV) homologue of the BCL-2 protooncogene, is transcribed in EBV-associated B-cell lymphomas and in reactive lymphocytes. Blood 86 1893 1902
45. KvansakulM
YangH
FairlieWD
CzabotarPE
FischerSF
2008 Vaccinia virus anti-apoptotic F1L is a novel Bcl-2-like domain-swapped dimer that binds a highly selective subset of BH3-containing death ligands. Cell Death Differ 15 1564 1571
46. EmsleyP
CowtanK
2004 Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60 2126 2132
47. MurshudovGN
VaginAA
DodsonEJ
1997 Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53 240 255
48. StoroniLC
McCoyAJ
ReadRJ
2004 Likelihood-enhanced fast rotation functions. Acta Crystallogr D Biol Crystallogr 60 432 438
49. O'ConnorL
StrasserA
O'ReillyLA
HausmannG
AdamsJM
1998 Bim: a novel member of the Bcl-2 family that promotes apoptosis. Embo J 17 384 395
50. MoriishiK
HuangDC
CoryS
AdamsJM
1999 Bcl-2 family members do not inhibit apoptosis by binding the caspase activator Apaf-1. Proc Natl Acad Sci U S A 96 9683 9688
51. FletcherJI
MeusburgerS
HawkinsCJ
RiglarDT
LeeEF
2008 Apoptosis is triggered when prosurvival Bcl-2 proteins cannot restrain Bax. Proc Natl Acad Sci U S A 105 18081 18087
52. HuangDC
O'ReillyLA
StrasserA
CoryS
1997 The anti-apoptosis function of Bcl-2 can be genetically separated from its inhibitory effect on cell cycle entry. Embo J 16 4628 4638
53. DewsonG
SnowdenRT
AlmondJB
DyerMJ
CohenGM
2003 Conformational change and mitochondrial translocation of Bax accompany proteasome inhibitor-induced apoptosis of chronic lymphocytic leukemic cells. Oncogene 22 2643 2654
54. Wilson-AnnanJ
O'ReillyLA
CrawfordSA
HausmannG
BeaumontJG
2003 Proapoptotic BH3-only proteins trigger membrane integration of prosurvival Bcl-w and neutralize its activity. J Cell Biol 162 877 887
55. HawkinsCJ
WangSL
HayBA
1999 A cloning method to identify caspases and their regulators in yeast: identification of Drosophila IAP1 as an inhibitor of the Drosophila caspase DCP-1. Proc Natl Acad Sci U S A 96 2885 2890
56. ZhaJ
HaradaH
OsipovK
JockelJ
WaksmanG
1997 BH3 domain of BAD is required for heterodimerization with BCL-XL and pro-apoptotic activity. J Biol Chem 272 24101 24104
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 12
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- HIV-1 Envelope Subregion Length Variation during Disease Progression
- Coming of Age—Sexual Reproduction in Species
- Evidence That Intracellular Stages of Utilize Amino Sugars as a Major Carbon Source
- Compartmentation of Redox Metabolism in Malaria Parasites
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy