
Noise Cancellation: Viral Fine Tuning of the Cellular Environment for Its Own Genome Replication
Productive replication of DNA viruses elicits host cell DNA damage responses, which cause both beneficial and detrimental effects on viral replication. In response to the viral productive replication, host cells attempt to attenuate the S-phase cyclin-dependent kinase (CDK) activities to inhibit viral replication. However, accumulating evidence regarding interactions between viral factors and cellular signaling molecules indicate that viruses utilize them and selectively block the downstream signaling pathways that lead to attenuation of the high S-phase CDK activities required for viral replication. In this review, we describe the sophisticated strategy of Epstein-Barr virus to cancel such “noisy” host defense signals in order to hijack the cellular environment.
Published in the journal:
. PLoS Pathog 6(12): e32767. doi:10.1371/journal.ppat.1001158
Category:
Review
doi:
https://doi.org/10.1371/journal.ppat.1001158
Summary
Productive replication of DNA viruses elicits host cell DNA damage responses, which cause both beneficial and detrimental effects on viral replication. In response to the viral productive replication, host cells attempt to attenuate the S-phase cyclin-dependent kinase (CDK) activities to inhibit viral replication. However, accumulating evidence regarding interactions between viral factors and cellular signaling molecules indicate that viruses utilize them and selectively block the downstream signaling pathways that lead to attenuation of the high S-phase CDK activities required for viral replication. In this review, we describe the sophisticated strategy of Epstein-Barr virus to cancel such “noisy” host defense signals in order to hijack the cellular environment.
Introduction
Cellular DNA damage responses initiate with activation and rapid recruitment of repair proteins to DNA damage sites [1], [2]. Until the damage is repaired, cells are prevented from transitioning to the next stage of the cell cycle. The tumor suppressor p53 is phosphorylated by DNA damage–responsive kinases, resulting in stabilization of p53 and an increase in its protein level. This leads to activation of target gene transcription including p53 itself, which subsequently causes cell cycle arrest or apoptosis [3], [4]. The replicated viral genomes of DNA viruses, including adenoviruses, the polyomavirus, and herpesviruses, are recognized by cellular DNA damage sensors, triggering activation of DNA damage responses [5], [6], [7], [8], [9]. Several lines of evidence revealed viral approaches to create an optimal environment for viral replication by manipulating the host defense systems. In this review, we describe the elegant strategies used by Epstein-Barr virus (EBV) to cancel “noisy” cellular signaling in order to manipulate the cellular environment for its own genome replication.
Life Cycle of the Epstein-Barr Virus
EBV, a human lymphotropic herpesvirus, infects more than 90% of world's population and is now known to contribute to a variety of human disorders, including infectious mononucleosis, nasopharyngeal carcinoma, Burkitt's lymphoma, and lymphoproliferative diseases occurring in immune-compromised individuals [10]. The lifecycle of EBV is quite distinctive, featuring two alternative infection cycles: “latent” and “lytic.” Primary EBV infection targets resting B lymphocytes, inducing their continuous proliferation. In the resultant B lymphoblastoid cell lines that express a limited number of EBV gene products, the viral genomes are maintained as circular plasmids forming nucleosomal structures with histones [11], and there is no production of virus particles, this being called “latent” infection. In the latent state, viral DNA is replicated only once during S phase, just as host chromosomal DNA [11]. Only a small percentage of infected cells switch their states from the latent stage into the “lytic” cycle to produce progeny viruses. EBV DNA replication occurs at discrete sites in nuclei called “replication compartments,” where all of the viral replication proteins are assembled [12]. During lytic replication, the circular genome becomes a ready template for amplification by the viral replication machinery, generating thousands of copies per cell. This reactivation is correlated with the emergence of human cancers [13], [14]. The switch from latent to lytic replication is triggered by expression of the EBV BZLF1 gene product (also called Zta or ZEBRA) [15]. The BZLF1 protein is a lytic replication origin binding protein and acts to transactivate various viral promoters [16], leading to an ordered cascade of viral gene expression: activation of early genes followed by viral genome replication and late gene expression. Using the EBV system, the alteration in cellular conditions, from latent to virus-productive infection without overlapping signals triggered by virus entry, can be monitored [17], [18].
Regulation of p53 during the Latent Phase of EBV Infection
In uninfected cells, p53 is hypophosphorylated [9] and its level is regulated by cellular E3 ubiquitin ligase MDM2 and cellular ubiquitin–specific protease USP7 (Figure 1A) [19]. The EBV latent protein, EBNA1, contributes to repression of p53-dependent DNA damage signaling by competition of the USP7 binding site with p53 (Figure 1A) [20] (Figure 1A). Furthermore, this interaction between EBNA1 and USP7 leads to the disruption of PML bodies, the nuclear structures important for p53 activation and DNA repair [21]. These findings suggest that EBNA1 expression protects cells from DNA damage–induced apoptosis by destabilizing p53 protein. This possible mechanism points to an important role of EBNA1 in cancer development by allowing uncontrolled cellular proliferation without inducing apoptosis in latent EBV-infected cells.
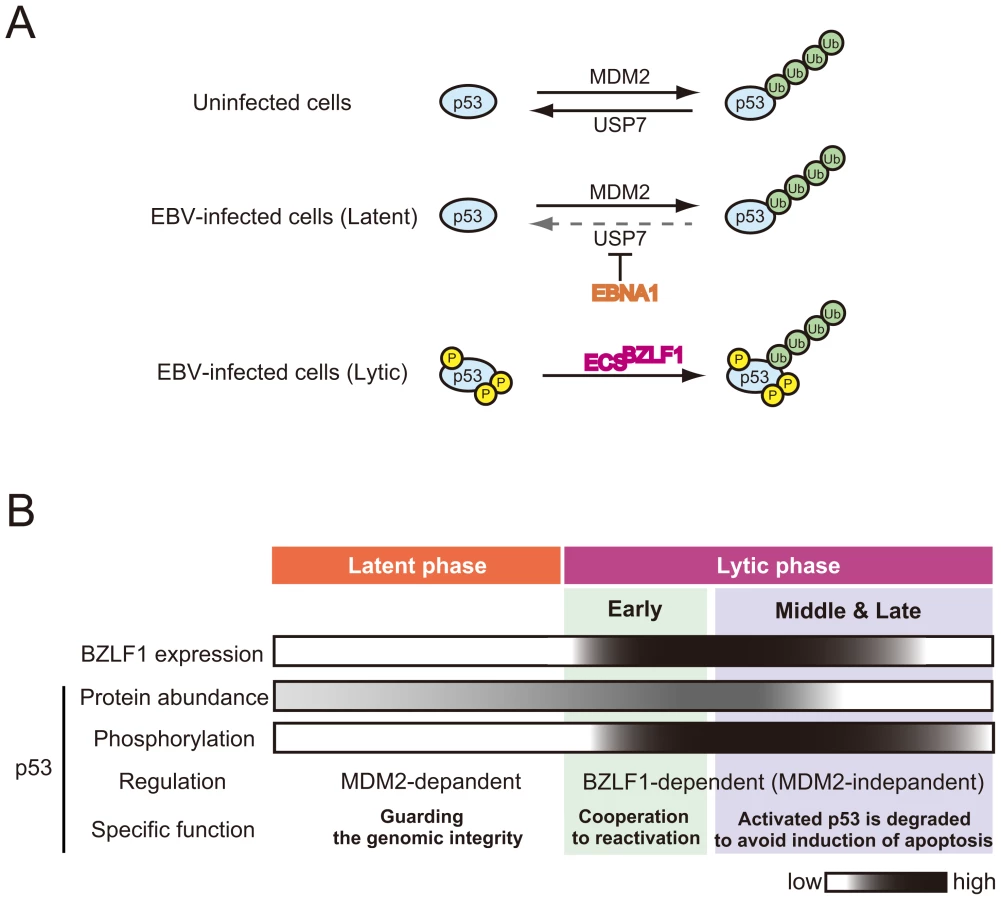
The Important Role of p53 in the Early Stages of EBV Lytic Replication
We propose that, during the course of lytic replication, BZLF1 protein plays two distinct roles in the regulation of p53-mediated transactivation, which depend on the progression of lytic replication: the early stage and the middle to late stages (described below) (Figure 1B). Previous studies demonstrated that the EBV immediate-early protein BZLF1, which was either conditionally expressed [22], [23] or overexpressed by a recombinant adenovirus [24], [25], could induce G1 arrest in some cell lines. The BZLF1 protein causes the accumulation of both mRNA and protein of CDK inhibitor p21Cip1/Waf1 [23], a well-known p53-target gene product. BZLF1 protein accelerates the rate of p53-DNA complex formation through physical interaction with p53 [26]. In the early phases of lytic replication, p53 is hypophosphorylated and therefore exhibits weak DNA binding ability to its recognition sequences [9]. The BZLF1 protein helps the hypophosphorylated p53 to bind to its recognition sequences, leading to the enhancement of p53-dependent transcription [26]. Levels of p53 and p21Cip1/Waf1 are transiently elevated in the early stages of lytic replication, and then decline with the progression of lytic infection [26], probably reflecting the effects of BZLF1 expression.
Recently, we and other groups have shown that p53 is involved in reactivation of EBV [26], [27]. Tsai and his colleagues have reported that induction of viral lytic proteins by a chemical inducer, sodium butylate, does not occur in p53-negative H1299A and Saos2A cells [27], although the ability of BZLF1 or BRLF1 protein to transactivate its downstream genes is not notably affected by the lack of p53 [28], [29]. This implies that p53 might instead be required for a switch from the latent to the lytic cycle. Indeed, we found that overexpression of p53 in the early stages of lytic replication enhances subsequent viral genome replication [26].
In the case of human cytomegalovirus (HCMV), the level of p53 is elevated upon viral infection, but its downstream transcriptional targets remain inactivated [30], [31]. It has been reported that cells infected with HCMV in the absence of p53 produce fewer infectious viral particles and cause delays in viral protein production and trafficking [30]. The HCMV genome has 21 potential p53-responsive sites [32]. HCMV gene expression is thought to be influenced by p53 molecules bound to HCMV genome at immediate-early and early stages of the infection, which could explain the mechanism for reduced and delayed production of virions in p53-negative cells. Similarly, potential p53 recognition sequences are present on the EBV genome (T. Murata et al., unpublished results). Indeed, we have found that p53 is associated with EBV replication compartments [9]. Thus, in the early stages of EBV lytic infection, p53 could be recruited to the EBV genomic regions through its direct binding to the recognition sequences. The BZLF1-mediated enhancement of p53-DNA binding may therefore contribute to the expression of viral genes (Figure 1B).
Newly Synthesized Viral DNA Elicits Host DNA Damage Responses
Herpesviruses such as HSV, HCMV, and EBV modulate the cell cycle to promote a transition through G1-S phase and achieve the cellular environment with high S-phase CDK activities, called the S-phase-like condition, for virus-productive replication (reviewed in [18]). During the EBV lytic replication, the levels of cyclin E and cyclin A continue to be elevated, and cyclin E - and cyclin A-associated CDK activities actually increase [9]. Moreover, this elevation of S-phase CDK activities drives accumulation of the hyperphosphorylated form of retinoblastoma protein (Rb) and an increase in the level of E2F-1 transcription factor [9]. The observation that chemical CDK inhibitors, such as purvalanol A and roscovitine, block viral lytic replication through prevention of viral immediate-early and early gene expression [33] suggests that a cellular environment featuring high CDK activities is required for efficient viral replication. It is conceivable that expression of proteins involved in DNA metabolism may be promoted under S-phase conditions, when energy generation and other resources support viral replication [18]. However, cellular DNA synthesis is almost entirely blocked during the lytic phase of EBV DNA replication, despite S-phase-like cellular conditions with high CDK activities [17]. The EBV-encoding protein kinase (PK) BGLF4 phosphorylates MCM complex to inhibit its replicative helicase activity (Figure 2) [34]. Although the precise mechanism remains unclear, it might be one of the reasons for inhibition of chromosomal DNA replication and for the blockage of the cell-cycle progression from S to G2 phase.

The host cell DNA damage-sensing machinery recognizes the newly synthesized viral DNA in the lytic phase as “abnormal” DNA, activating ATM-dependent DNA damage signaling [9] (Figure 2). ATM phosphorylates histone H2AX (H2AX), which initiates the DNA damage response. The EBV BGLF4 PK might further amplify this response through phosphorylation of H2AX [35]. ATM phosphorylates p53 at Ser-15, which liberates p53 from MDM2-mediated degradation. The downstream kinases of ATM, Chk1, and Chk2 also phosphorylate p53 at various sites. Therefore, elicitation of DNA damage responses in general activates the transcriptional functions of p53.
Ubiquitin-Mediated Degradation of p53 in the Middle and Late Stages of Lytic Infection
Paradoxically, reactivation of EBV induces cellular DNA damage responses that causes phosphorylation of p53, which could lead to accumulation of p53 and subsequent activation of p53 downstream signaling (Figure 1), at the same time it establishes the S-phase-like cellular environment. At the middle to late stages of the lytic replication, the p53 target gene products are indeed maintained at low levels [9], [17], [26], [36]. An explanation for this comes from the observation that p53 is degraded via the ubiquitin-proteasome pathway in the middle and late stages of lytic infection, allowing EBV to exploit cellular environments with high CDK activities for efficient viral replication (Figure 2).
A series of recent studies have shown that induction of the EBV lytic program leads to degradation of p53 via a ubiquitin-proteasome pathway independently of MDM2 [36]. The BZLF1 protein functions as an adaptor component of the ECS (Elongin B/C-Cul2/5-SOCS-box protein) ubiquitin ligase complex that targets both unphosphorylated and hyperphosphorylated p53 for degradation (Figure 2) [37]. The BZLF1 M3 mutant, which lacks the ability to bind to Cullin 2 and 5, degrades p53 very little in EBV-positive cells, and the yield of infectious viruses is poorer than in wild-type BZLF1-expressing cells [37]. The BZLF1 M3 mutant includes a mutation at residue E54, previously reported to prevent activation of the EBV lytic cycle, but the underlying mechanism was unknown [38]. These findings suggest that the deficiency of viral replication is due to the failure of p53 degradation.
Chk2 is known to mediate phosphorylation of p53 at Ser-366 and Ser-378 in response to genotoxic stresses [39]. Indeed, p53 is phosphorylated at least at Ser-15, Ser-20, Ser-366, and Ser-378 with progression of EBV lytic infection [37]. Intriguingly, C-terminal phosphorylation of p53 at both Ser-366 and Ser-378 enhances the association with BZLF1 protein and subsequent ubiquitination of p53 [37], possibly through the phosphorylation-induced conformational change of p53. These results suggest that DNA damage responses play a pivotal role in lytic infection.
In addition, inhibition of p53 degradation by the BZLF1 M3 mutant induces apoptotic cellular changes [37]. The maintenance of p53 at very low levels, therefore, is required not only for establishing S-phase-like conditions [9], [17], [36], but also for inhibiting apoptosis for efficient viral propagation. In fact, caspase activity is not induced during lytic infection [40]. Similarly, a body of evidence in the herpesvirus family shows that p53 is inactivated in lytic replication, although its molecular mechanism is controversial [41], [42], [43].
Thus, studies on the relationship between p53 alteration and viral DNA replication have demonstrated that BZLF1 enables hypophosphorylated p53 to transactivate the p53 target genes in the initial phase of lytic replication. In the middle and late stages, activated p53 is subjected to BZLF1-dependent degradation to maintain an S-phase-like environment for efficient viral propagation (Figure 1B).
Several groups have reported that BZLF1 is transiently expressed as an immediate-early gene following EBV primary infection of resting B lymphocytes, although early and late lytic gene expression is very low or undetectable [44], [45], [46]. A transient BZLF1 expression at the primary infection may contribute to establishing a latent infection, as speculated by Wen and colleagues [44]. This could be driven by degradation of p53, which blocks reprogramming of B lymphocyte proliferation. Interestingly, p53 serves as a negative regulator for reprogramming of somatic cells into induced pluripotent stem (iPS) cells [47], [48], [49], [50], [51]. Thus, it is possible that the degradation of p53 by BZLF1 protein-associated ECS ubiquitin ligases contributes to the efficient transformation of B lymphocytes.
Regulation of CDK Inhibitors during Lytic Replication
The large body of evidence implicating Cullin-based E3 ubiquitin ligase in the regulation of diverse cellular processes [52], [53] provides us with new insights into their significance as potential targets of viruses manipulating the host cellular system. Post-translational modifications, especially phosphorylation and ubiquitination, play a crucial role in cell-cycle progression. Phosphorylation controls the activity of proteins involved in G1-S and G2-M transitions. Ubiquitination and its mediated proteolysis are commonly facilitated to maintain threshold levels of cell-cycle regulators. Two distinct classes of E3 ubiquitin ligase regulate cell-cycle progression [52], possessing an adaptor protein to determine substrate specificity [54], [55], [56]. E3 ligase activity of the anaphase-promoting complex is required for the G2-M transition [57]. The SCF (Skp1-Cul1-F-box protein) family of E3 ligase promotes ubiquitination of phosphorylated substrates and typically targets the mediators of G1-S transition [58]. For instance, ubiquitin-mediated degradation of p27Kip1 is regulated by the SCFSkp2 complex only when p27 Kip1 is phosphorylated at Thr-187 by the cyclin E-CDK2 complex, which induces S phase conditions [59], [60], [61].
The EBV lytic program promotes specific cell cycle–associated activity involved in progression from G1 to S phase, since virus-productive replication occurs under S-phase-like circumstances [18]. Similar to p53, CDK inhibitors are also regulated during lytic replication, contributing to establishment of an S-phase-like cellular environment with high-CDK activities [9], [33]. γ-Herpesviruses possess their own strategies to degrade p27Kip1. For example, Kaposi's sarcoma-associated herpesvirus (KSHV)-encoding cyclin (v-cyclin), a latent viral protein, forms a complex with CDK6 to phosphorylate Thr-187 on p27Kip1, leading to down-regulation at the protein level [62], [63]. Also, the viral cyclin encoded by murine herpesvirus 68 preferentially associates with CDK2 to phosphorylate Thr-187 on p27Kip1 [64]. While EBV does not encode any v-cyclin homologue in its genome, our recent study revealed that the EBV protein kinase BGLF4 can phosphorylate the Thr-187 residue of p27Kip1, resulting in its ubiquitination and degradation in an SCFSkp2 ubiquitin ligase-dependent manner [65] (Figure 2).
Manipulating the ubiquitin system by EBV involves two aspects of its regulation: attachment of ubiquitin to a substrate and removal from its substrate. As an EBV-encoding deubiquitinating enzyme, BPLF1 deubiquitinates and reduces activity of EBV ribonucleotide reductase [66]. In this case, deubiquitination influences the function of the protein rather than targeting it for proteasomal degradation. A recent paper documented that BPLF1 also act as a deneddylase [67]. Neddylation, which is a conjugation of ubiquitin-like modifier NEDD8 to its substrate, is an important mechanism for regulating Cullin-based E3 ubiquitin ligase [68]. EBV BPLF1 binds to Cullins and attenuates the activity of the Cullin-RING ligases, resulting in accumulation of the licensing factor Cdt1 and induction of DNA re-replication. Inhibition of BPLF1 during the lytic infection prevents viral replication in the cells that carries a recombinant EBV [67]. These findings support the idea that manipulating ubiquitin system by virus promotes viral productive replication. Furthermore, two lytic proteins (BSLF1 and BXLF1) are found as deubiquitinases by a bioinformatic search on the EBV genome [69], although their functions in viral replication remain obscure. Further investigations are needed to determine the exact role of deubiquitination in the context of EBV lytic infection.
The level of another CDK inhibitor protein p21Cip1/Waf1, of course, becomes low during lytic replication [36]. Although the detailed mechanisms remain unknown, one reason is that p53 is actively degraded during lytic infection and another is that the SCFSkp2 ubiquitin ligase complex directs p21Cip1/Waf1 for degradation through S-phase CDK-mediated phosphoryration [70]. Recent study showed that KSHV-encoding microRNA, miR-K1 represses expression of p21Cip1/Waf in latent infection [71]. As an additional mechanism, an EBV-encoding miRNA that has yet to be discovered might regulate p21Cip1/Waf1 for maintaining S-phase-like conditions.
On the other hand, maintaining low levels of CDK inhibitors results in accumulation of the hyperphosphorylated Rb protein due to high S-phase CDK activities and causes accumulation of active E2F-1 as lytic replication progresses [9]. E2F-1 in turn activates the transcription of many proteins involved in cellular DNA synthesis and cell-cycle progression [72], and probably transcription of the EBV DNA polymerase gene as well [73]. The available data suggest that E2F activity is required for lytic viral DNA replication. Alternatively, the EBV immediate-early transactivator BZLF1 and BRLF1 proteins are reported to increase the level of E2F-1 [74], [75]. Furthermore, since activated ATM or Chk2 phosphorylates and activates E2F-1 in response to DNA damage [76], [77], the DNA damage response induced by EBV lytic replication could activate E2F-1. To achieve effective viral lytic replication, EBV therefore possesses a variety of strategies to maintain the S-phase-like cellular environment.
Beneficial Aspects of DNA Damage Signaling on EBV DNA Replication
During EBV lytic replication, phosphorylated ATM and Mre11/Rad50/Nbs1 (MRN) complexes are targeted to replication compartments in nuclei. Simultaneously, homologous recombinational repair (HRR) factors such as replication protein A (RPA), Rad51, and Rad52, as well as MRN complex, are recruited and loaded onto the newly synthesized viral genome in replication compartments [40]. The 32 kDa subunit of RPA is extensively phosphorylated at sites in accordance with these events [40]. Hyperphosphorylation of RPA32 causes a change in RPA conformation, resulting in a switch from catalysis of DNA replication to participation in DNA repair. RNAi knockdown of RPA32 and Rad51 prevents viral DNA synthesis, suggesting that homologous recombination and/or repair of the viral DNA genome might occur, coupled with viral DNA replication to facilitate viral genome synthesis (Figure 2). Thus, the host DNA damage response induced by productive viral replication is essential for efficient EBV lytic genomic replication.
Conclusions
Replication of DNA viruses in host cells triggers a variety of cellular signaling cascades, including the DNA damage response. Recent studies indicate that such cellular responses to viral genomic replication paradoxically play a crucial role in EBV lytic replication by establishing cellular conditions appropriate for efficient viral replication. To achieve these conditions, EBV manipulates host ubiquitin-proteasome systems, and thereby cancels host antivirus signals. During lytic infection, the interaction between BZLF1 protein and ECS E3 ligase complexes leads to p53 degradation, and the SCF E3 complex recognizes and ubiquitinates phosphorylated p27Kip1 through viral protein kinase. Therefore, by skipping the induction of checkpoint signaling and apoptosis, virus-producing cells stay in a persistent S-phase-like environment with high CDK activity.
Accession numbers
The Entrez Gene (http://www.ncbi.nlm.nih.gov/gene) accession numbers for genes and gene products discussed in this study are as follows: p53 (7157), p21Cip1/Waf1 (1026), p27Kip1 (1027), USP7 (7874), MDM2 (4193), E2F-1 (1869), ATM (472), Chk2 (11200), H2AX (3014), PARP (142), Skp2 (6502), Cdt1 (81620), ubiquitin (7314), NEDD8 (4738), Rb (5925), Cyclin E (898), Cyclin A (890), CDK2 (1017), CDK6 (1021), RPA32 (6118), Rad51 (5888), Rad52 (5893), KSHV v-cyclin (4961471), and EBV EBNA1 (3783709), BGLF4 (3783704) BPLF1 (3783726), BSLF1 (3783730), BXLF1 (3783741), and BZLF1 (3783744).
Zdroje
1. SancarA
Lindsey-BoltzLA
Unsal-KacmazK
LinnS
2004 Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 73 39 85
2. RouseJ
JacksonSP
2002 Interfaces between the detection, signaling, and repair of DNA damage. Science 297 547 551
3. HaffnerR
OrenM
1995 Biochemical properties and biological effects of p53. Curr Opin Genet Dev 5 84 90
4. KoLJ
PrivesC
1996 p53: puzzle and paradigm. Genes Dev 10 1054 1072
5. StrackerTH
CarsonCT
WeitzmanMD
2002 Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature 418 348 352
6. DahlJ
YouJ
BenjaminTL
2005 Induction and utilization of an ATM signaling pathway by polyomavirus. J Virol 79 13007 13017
7. ShirataN
KudohA
DaikokuT
TatsumiY
FujitaM
2005 Activation of ataxia telangiectasia-mutated DNA damage checkpoint signal transduction elicited by herpes simplex virus infection. J Biol Chem 280 30336 30341
8. GasparM
ShenkT
2006 Human cytomegalovirus inhibits a DNA damage response by mislocalizing checkpoint proteins. Proc Natl Acad Sci U S A 103 2821 2826
9. KudohA
FujitaM
ZhangL
ShirataN
DaikokuT
2005 Epstein-Barr virus lytic replication elicits ATM checkpoint signal transduction while providing an S-phase-like cellular environment. J Biol Chem 280 8156 8163
10. YoungLS
RickinsonAB
2004 Epstein-Barr virus: 40 years on. Nat Rev Cancer 4 757 768
11. AdamsA
1987 Replication of latent Epstein-Barr virus genomes in Raji cells. J Virol 61 1743 1746
12. DaikokuT
KudohA
FujitaM
SugayaY
IsomuraH
2005 Architecture of replication compartments formed during Epstein-Barr virus lytic replication. J Virol 79 3409 3418
13. JoabI
NicolasJC
SchwaabG
de-TheG
ClausseB
1991 Detection of anti-Epstein-Barr-virus transactivator (ZEBRA) antibodies in sera from patients with nasopharyngeal carcinoma. Int J Cancer 48 647 649
14. FengWH
CohenJI
FischerS
LiL
SnellerM
2004 Reactivation of latent Epstein-Barr virus by methotrexate: a potential contributor to methotrexate-associated lymphomas. J Natl Cancer Inst 96 1691 1702
15. HammerschmidtW
SugdenB
1988 Identification and characterization of oriLyt, a lytic origin of DNA replication of Epstein-Barr virus. Cell 55 427 433
16. FlemingtonEK
GoldfeldAE
SpeckSH
1991 Efficient transcription of the Epstein-Barr virus immediate-early BZLF1 and BRLF1 genes requires protein synthesis. J Virol 65 7073 7077
17. KudohA
FujitaM
KiyonoT
KuzushimaK
SugayaY
2003 Reactivation of lytic replication from B cells latently infected with Epstein-Barr virus occurs with high S-phase cyclin-dependent kinase activity while inhibiting cellular DNA replication. J Virol 77 851 861
18. TsurumiT
FujitaM
KudohA
2005 Latent and lytic Epstein-Barr virus replication strategies. Rev Med Virol 15 3 15
19. LiM
ChenD
ShilohA
LuoJ
NikolaevAY
2002 Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 416 648 653
20. SaridakisV
ShengY
SarkariF
HolowatyMN
ShireK
2005 Structure of the p53 binding domain of HAUSP/USP7 bound to Epstein-Barr nuclear antigen 1 implications for EBV-mediated immortalization. Mol Cell 18 25 36
21. SivachandranN
SarkariF
FrappierL
2008 Epstein-Barr nuclear antigen 1 contributes to nasopharyngeal carcinoma through disruption of PML nuclear bodies. PLoS Pathog 4 e1000170
22. CayrolC
FlemingtonE
1996 G0/G1 growth arrest mediated by a region encompassing the basic leucine zipper (bZIP) domain of the Epstein-Barr virus transactivator Zta. J Biol Chem 271 31799 31802
23. CayrolC
FlemingtonEK
1996 The Epstein-Barr virus bZIP transcription factor Zta causes G0/G1 cell cycle arrest through induction of cyclin-dependent kinase inhibitors. EMBO J 15 2748 2759
24. MauserA
Holley-GuthrieE
SimpsonD
KaufmannW
KenneyS
2002 The Epstein-Barr virus immediate-early protein BZLF1 induces both a G(2) and a mitotic block. J Virol 76 10030 10037
25. MauserA
SaitoS
AppellaE
AndersonCW
SeamanWT
2002 The Epstein-Barr virus immediate-early protein BZLF1 regulates p53 function through multiple mechanisms. J Virol 76 12503 12512
26. SatoY
ShirataN
MurataT
NakasuS
KudohA
2010 Transient increases in p53-responsible gene expression at early stages of Epstein-Barr virus productive replication. Cell Cycle 9 807 814
27. ChangSS
LoYC
ChuaHH
ChiuHY
TsaiSC
2008 Critical role of p53 in histone deacetylase inhibitor-induced Epstein-Barr virus Zta expression. J Virol 82 7745 7751
28. Chevallier-GrecoA
ManetE
ChavrierP
MosnierC
DaillieJ
1986 Both Epstein-Barr virus (EBV)-encoded trans-acting factors, EB1 and EB2, are required to activate transcription from an EBV early promoter. EMBO J 5 3243 3249
29. HardwickJM
LiebermanPM
HaywardSD
1988 A new Epstein-Barr virus transactivator, R, induces expression of a cytoplasmic early antigen. J Virol 62 2274 2284
30. CasavantNC
LuoMH
RosenkeK
WinegardnerT
ZurawskaA
2006 Potential role for p53 in the permissive life cycle of human cytomegalovirus. J Virol 80 8390 8401
31. JaultFM
JaultJM
RuchtiF
FortunatoEA
ClarkC
1995 Cytomegalovirus infection induces high levels of cyclins, phosphorylated Rb, and p53, leading to cell cycle arrest. J Virol 69 6697 6704
32. RosenkeK
SamuelMA
McDowellET
ToerneMA
FortunatoEA
2006 An intact sequence-specific DNA-binding domain is required for human cytomegalovirus-mediated sequestration of p53 and may promote in vivo binding to the viral genome during infection. Virology 348 19 34
33. KudohA
DaikokuT
SugayaY
IsomuraH
FujitaM
2004 Inhibition of S-phase cyclin-dependent kinase activity blocks expression of Epstein-Barr virus immediate-early and early genes, preventing viral lytic replication. J Virol 78 104 115
34. KudohA
DaikokuT
IshimiY
KawaguchiY
ShirataN
2006 Phosphorylation of MCM4 at sites inactivating DNA helicase activity of the MCM4-MCM6-MCM7 complex during Epstein-Barr virus productive replication. J Virol 80 10064 10072
35. TarakanovaVL
Leung-PinedaV
HwangS
YangCW
MatatallK
2007 Gamma-herpesvirus kinase actively initiates a DNA damage response by inducing phosphorylation of H2AX to foster viral replication. Cell Host Microbe 1 275 286
36. SatoY
ShirataN
KudohA
IwahoriS
NakayamaS
2009 Expression of Epstein-Barr virus BZLF1 immediate-early protein induces p53 degradation independent of MDM2, leading to repression of p53-mediated transcription. Virology 388 204 211
37. SatoY
KamuraT
ShirataN
MurataT
KudohA
2009 Degradation of Phosphorylated p53 by Viral Protein-ECS E3 Ligase Complex. PLoS Pathog 5 e1000530
38. DengZ
ChenCJ
ZerbyD
DelecluseHJ
LiebermanPM
2001 Identification of acidic and aromatic residues in the Zta activation domain essential for Epstein-Barr virus reactivation. J Virol 75 10334 10347
39. OuYH
ChungPH
SunTP
ShiehSY
2005 p53 C-terminal phosphorylation by CHK1 and CHK2 participates in the regulation of DNA-damage-induced C-terminal acetylation. Mol Biol Cell 16 1684 1695
40. KudohA
IwahoriS
SatoY
NakayamaS
IsomuraH
2009 Homologous recombinational repair factors are recruited and loaded onto the viral DNA genome in Epstein-Barr virus replication compartments. J Virol 83 6641 6651
41. FortunatoEA
SpectorDH
1998 p53 and RPA are sequestered in viral replication centers in the nuclei of cells infected with human cytomegalovirus. J Virol 72 2033 2039
42. HsuCH
ChangMD
TaiKY
YangYT
WangPS
2004 HCMV IE2-mediated inhibition of HAT activity downregulates p53 function. EMBO J 23 2269 2280
43. WilcockD
LaneDP
1991 Localization of p53, retinoblastoma and host replication proteins at sites of viral replication in herpes-infected cells. Nature 349 429 431
44. WenW
IwakiriD
YamamotoK
MaruoS
KandaT
2007 Epstein-Barr virus BZLF1 gene, a switch from latency to lytic infection, is expressed as an immediate-early gene after primary infection of B lymphocytes. J Virol 81 1037 1042
45. HalderS
MurakamiM
VermaSC
KumarP
YiF
2009 Early events associated with infection of Epstein-Barr virus infection of primary B-cells. PLoS One 4 e7214
46. KallaM
SchmeinckA
BergbauerM
PichD
HammerschmidtW
2010 AP-1 homolog BZLF1 of Epstein-Barr virus has two essential functions dependent on the epigenetic state of the viral genome. Proc Natl Acad Sci U S A 107 850 855
47. HongH
TakahashiK
IchisakaT
AoiT
KanagawaO
2009 Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature 460 1132 1135
48. LiH
ColladoM
VillasanteA
StratiK
OrtegaS
2009 The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature 460 1136 1139
49. KawamuraT
SuzukiJ
WangYV
MenendezS
MoreraLB
2009 Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature 460 1140 1144
50. UtikalJ
PoloJM
StadtfeldM
MaheraliN
KulalertW
2009 Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature 460 1145 1148
51. MarionRM
StratiK
LiH
MurgaM
BlancoR
2009 A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature 460 1149 1153
52. NakayamaKI
NakayamaK
2006 Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer 6 369 381
53. PetroskiMD
DeshaiesRJ
2005 Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol 6 9 20
54. KraftC
VodermaierHC
Maurer-StrohS
EisenhaberF
PetersJM
2005 The WD40 propeller domain of Cdh1 functions as a destruction box receptor for APC/C substrates. Mol Cell 18 543 553
55. SpruckC
StrohmaierH
WatsonM
SmithAP
RyanA
2001 A CDK-independent function of mammalian Cks1: targeting of SCF(Skp2) to the CDK inhibitor p27Kip1. Mol Cell 7 639 650
56. HaoB
ZhengN
SchulmanBA
WuG
MillerJJ
2005 Structural basis of the Cks1-dependent recognition of p27(Kip1) by the SCF(Skp2) ubiquitin ligase. Mol Cell 20 9 19
57. HarperJW
BurtonJL
SolomonMJ
2002 The anaphase-promoting complex: it's not just for mitosis any more. Genes Dev 16 2179 2206
58. SkowyraD
CraigKL
TyersM
ElledgeSJ
HarperJW
1997 F-box proteins are receptors that recruit phosphorylated substrates to the SCF ubiquitin-ligase complex. Cell 91 209 219
59. CarranoAC
EytanE
HershkoA
PaganoM
1999 SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol 1 193 199
60. SutterlutyH
ChatelainE
MartiA
WirbelauerC
SenftenM
1999 p45SKP2 promotes p27Kip1 degradation and induces S phase in quiescent cells. Nat Cell Biol 1 207 214
61. TsvetkovLM
YehKH
LeeSJ
SunH
ZhangH
1999 p27(Kip1) ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Curr Biol 9 661 664
62. MannDJ
ChildES
SwantonC
LamanH
JonesN
1999 Modulation of p27(Kip1) levels by the cyclin encoded by Kaposi's sarcoma-associated herpesvirus. EMBO J 18 654 663
63. EllisM
ChewYP
FallisL
FreddersdorfS
BoshoffC
1999 Degradation of p27(Kip) cdk inhibitor triggered by Kaposi's sarcoma virus cyclin-cdk6 complex. EMBO J 18 644 653
64. YarmishynA
ChildES
ElphickLM
MannDJ
2008 Differential regulation of the cyclin-dependent kinase inhibitors p21(Cip1) and p27(Kip1) by phosphorylation directed by the cyclin encoded by Murine Herpesvirus 68. Exp Cell Res 314 204 212
65. IwahoriS
MurataT
KudohA
SatoY
NakayamaS
2009 Phosphorylation of p27Kip1 by Epstein-Barr Virus Protein Kinase Induces Its Degradation through SCFSkp2 Ubiquitin Ligase Actions during Viral Lytic Replication. J Biol Chem 284 18923 18931
66. WhitehurstCB
NingS
BentzGL
DufourF
GershburgE
2009 The Epstein-Barr virus (EBV) deubiquitinating enzyme BPLF1 reduces EBV ribonucleotide reductase activity. J Virol 83 4345 4353
67. GastaldelloS
HildebrandS
FaridaniO
CallegariS
PalmkvistM
2010 A deneddylase encoded by Epstein-Barr virus promotes viral DNA replication by regulating the activity of cullin-RING ligases. Nat Cell Biol 12 351 361
68. RabutG
PeterM
2008 Function and regulation of protein neddylation. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep 9 969 976
69. SompallaeR
GastaldelloS
HildebrandS
ZininN
HassinkG
2008 Epstein-barr virus encodes three bona fide ubiquitin-specific proteases. J Virol 82 10477 10486
70. BornsteinG
BloomJ
Sitry-ShevahD
NakayamaK
PaganoM
2003 Role of the SCFSkp2 ubiquitin ligase in the degradation of p21Cip1 in S phase. J Biol Chem 278 25752 25757
71. GottweinE
CullenBR
2010 A human herpesvirus microRNA inhibits p21 expression and attenuates p21-mediated cell cycle arrest. J Virol 84 5229 5237
72. AdamsPD
KaelinWGJr
1995 Transcriptional control by E2F. Semin Cancer Biol 6 99 108
73. LiuC
SistaND
PaganoJS
1996 Activation of the Epstein-Barr virus DNA polymerase promoter by the BRLF1 immediate-early protein is mediated through USF and E2F. J Virol 70 2545 2555
74. MauserA
Holley-GuthrieE
ZanationA
YarboroughW
KaufmannW
2002 The Epstein-Barr virus immediate-early protein BZLF1 induces expression of E2F-1 and other proteins involved in cell cycle progression in primary keratinocytes and gastric carcinoma cells. J Virol 76 12543 12552
75. SwensonJJ
MauserAE
KaufmannWK
KenneySC
1999 The Epstein-Barr virus protein BRLF1 activates S phase entry through E2F1 induction. J Virol 73 6540 6550
76. LinWC
LinFT
NevinsJR
2001 Selective induction of E2F1 in response to DNA damage, mediated by ATM-dependent phosphorylation. Genes Dev 15 1833 1844
77. StevensC
SmithL
La ThangueNB
2003 Chk2 activates E2F-1 in response to DNA damage. Nat Cell Biol 5 401 409
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 12
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- HIV-1 Envelope Subregion Length Variation during Disease Progression
- Coming of Age—Sexual Reproduction in Species
- Evidence That Intracellular Stages of Utilize Amino Sugars as a Major Carbon Source
- Compartmentation of Redox Metabolism in Malaria Parasites
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy