
Structure of the HCMV UL16-MICB Complex Elucidates Select Binding of a Viral Immunoevasin to Diverse NKG2D Ligands
The activating immunoreceptor NKG2D promotes elimination of infected or malignant cells by cytotoxic lymphocytes through engagement of stress-induced MHC class I-related ligands. The human cytomegalovirus (HCMV)-encoded immunoevasin UL16 subverts NKG2D-mediated immune responses by retaining a select group of diverse NKG2D ligands inside the cell. We report here the crystal structure of UL16 in complex with the NKG2D ligand MICB at 1.8 Å resolution, revealing the molecular basis for the promiscuous, but highly selective, binding of UL16 to unrelated NKG2D ligands. The immunoglobulin-like UL16 protein utilizes a three-stranded β-sheet to engage the α-helical surface of the MHC class I-like MICB platform domain. Intriguingly, residues at the center of this β-sheet mimic a central binding motif employed by the structurally unrelated C-type lectin-like NKG2D to facilitate engagement of diverse NKG2D ligands. Using surface plasmon resonance, we find that UL16 binds MICB, ULBP1, and ULBP2 with similar affinities that lie in the nanomolar range (12–66 nM). The ability of UL16 to bind its ligands depends critically on the presence of a glutamine (MICB) or closely related glutamate (ULBP1 and ULBP2) at position 169. An arginine residue at this position however, as found for example in MICA or ULBP3, would cause steric clashes with UL16 residues. The inability of UL16 to bind MICA and ULBP3 can therefore be attributed to single substitutions at key NKG2D ligand locations. This indicates that selective pressure exerted by viral immunoevasins such as UL16 contributed to the diversification of NKG2D ligands.
Published in the journal:
. PLoS Pathog 6(1): e32767. doi:10.1371/journal.ppat.1000723
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1000723
Summary
The activating immunoreceptor NKG2D promotes elimination of infected or malignant cells by cytotoxic lymphocytes through engagement of stress-induced MHC class I-related ligands. The human cytomegalovirus (HCMV)-encoded immunoevasin UL16 subverts NKG2D-mediated immune responses by retaining a select group of diverse NKG2D ligands inside the cell. We report here the crystal structure of UL16 in complex with the NKG2D ligand MICB at 1.8 Å resolution, revealing the molecular basis for the promiscuous, but highly selective, binding of UL16 to unrelated NKG2D ligands. The immunoglobulin-like UL16 protein utilizes a three-stranded β-sheet to engage the α-helical surface of the MHC class I-like MICB platform domain. Intriguingly, residues at the center of this β-sheet mimic a central binding motif employed by the structurally unrelated C-type lectin-like NKG2D to facilitate engagement of diverse NKG2D ligands. Using surface plasmon resonance, we find that UL16 binds MICB, ULBP1, and ULBP2 with similar affinities that lie in the nanomolar range (12–66 nM). The ability of UL16 to bind its ligands depends critically on the presence of a glutamine (MICB) or closely related glutamate (ULBP1 and ULBP2) at position 169. An arginine residue at this position however, as found for example in MICA or ULBP3, would cause steric clashes with UL16 residues. The inability of UL16 to bind MICA and ULBP3 can therefore be attributed to single substitutions at key NKG2D ligand locations. This indicates that selective pressure exerted by viral immunoevasins such as UL16 contributed to the diversification of NKG2D ligands.
Introduction
Human cytomegalovirus (HCMV) is a β-herpesvirus that causes lifelong asymptomatic infections in healthy individuals but endangers the lives of immunocompromised individuals and very young children [1]. Cytotoxic lymphocytes such as CD8 T cells and natural killer (NK) cells are essential for the control of HCMV infection [1]–[4]. HCMV possesses a broad arsenal of immune evasive strategies that counteract cellular immunosurveillance and ensure long-term persistence in infected human hosts [2], [5]–[8]. One such strategy is the degradation of MHC class I molecules in order to subvert presentation of HCMV-derived peptide antigens to CD8 αβ T cells [2],[6],[8]. However, in line with the ‘missing-self-hypothesis’, impaired MHC class I expression results in a decreased engagement of MHC class I-specific inhibitory NK cell surface receptors and thus may facilitate NK cell-mediated lysis of the infected cells [9]. NK cell activity, however, is not solely controlled by receptors that inhibit NK cell activation, but rather is determined by the integration of signals from both inhibitory and activating NK cell receptors [10]–[12]. A potent activating receptor that mediates NK surveillance of stressed cells such as infected or malignant cells (‘induced-self’ or ‘stressed-self’ recognition) is NKG2D (natural-killer group 2, member D) [11],[13],[14]. NKG2D is a C-type lectin-like homodimer expressed on NK cells and cytotoxic T cells [15]. In humans, NKG2D transmits activating (NK cells) or co-stimulatory (CD8 αβ T cells and γδ T cells) signals via the associated DAP10 adaptor [16] and is triggered through engagement of cell stress-inducible MHC class I-related ligands belonging to the diverse MIC (MHC class I chain related molecule) and ULBP (UL16 binding protein) families. Two MIC (MICA and MICB) and six ULBP proteins (ULBP1-6) are currently known [2], [13], [14], [17]–[19]. The ULBP proteins are also sometimes referred to as ‘retinoic acid early transcript’ proteins (RAET; ULBP1/RAET1I, ULBP2/RAET1H, ULBP3/RAET1N, ULBP4/RAET1E, ULBP5/RAET1G and ULBP6/RAET1L). To thwart an antiviral NKG2D-mediated immune response, HCMV counteracts virally induced cellular expression of NKG2D ligands by means of several immunoevasins [2],[5],[8]. HCMV-encoded glycoproteins UL16 and UL142 selectively prevent the surface expression of MICB, ULBP1 and ULBP2 (UL16) and MICA (UL142), respectively, through intracellular retention [2], [20]–[23]. The significance of evasion from NKG2D-mediated immunosurveillance is further highlighted by the recent discovery that the HCMV gene UL112 is transcribed into a microRNA (miRNA) which specifically suppresses translation of MICB mRNA [24]. Although all NKG2D ligands share a MHC class I-like α1α2-platform domain [25]–[28] that binds NKG2D, UL16 does not bind to MICA, ULBP3, ULBP4 or ULBP5 [18], [29]–[31]. This selectivity is surprising since MICA and MICB are highly homologous in sequence (83% identical residues in the α1α2 region) but much more distantly related to the ULBP molecules (that share 21–29% identical residues in the α1α2 region with the MICs and 38–59% amino acid sequence identity among each other), which were originally discovered in a screen for UL16-binding proteins [18],[32].
In order to elucidate the structural basis for the ability of UL16 to engage highly diverse NKG2D ligands and to compare this promiscuous binding mode to that of NKG2D, we determined the structure of the UL16 ectodomain in complex with the α1α2-platform domain of MICB (MICBpf) at 1.8 Å resolution (Table 1). We also expected structural insights into the selective UL16 binding to MICB (but not MICA) and to some ULBP family members as selective pressure exerted by viral immunoevasins such as UL16 may have contributed to the diversification of NKG2D ligands [2],[17],[23]. We find that UL16, which possesses no structural homology to NKG2D, nevertheless employs a NKG2D-like binding mode to interact with MICB. Our results also offer structural explanations for the selective UL16 binding to some NKG2D ligands, and illustrate how the immunological arms race between a persistent pathogen and the human immune system may have driven the evolution of proteins of both, virus and host.

Results
Structure of UL16
UL16 is a heavily glycosylated 50 kDa type I transmembrane glycoprotein whose structure could not be predicted from its primary sequence [33]. In order to obtain soluble and homogeneously glycosylated protein for our structural and surface plasmon resonance (SPR) studies, we expressed the UL16 ectodomain in Chinese hamster ovary (CHO) Lec 3.2.8.1 cells [34]. UL16 was co-crystallized with MICBpf refolded from E. coli inclusion bodies (see Materials and Methods). The UL16 ectodomain folds into a modified version of the immunoglobulin (Ig)-like domain (Figure 1A). The presence of nine β-strands, arranged in two antiparallel β-sheets (formed by β-strands A, G, F, C, C′, C″ and β-strands D, E, B, respectively) and a central disulfide bond linking β-strands B and F clearly classifies it as a variable (V-type) Ig-like domain [35]–[37]. In contrast to classical V-type Ig domains, however, UL16 also has an additional N-terminal “plug” (amino acids 27–50), formed by a two-stranded antiparallel β-sheet (β-strands X1 and X2) and a short 310-helix (Figure 1A). The plug covers the concave side of the AGFCC′C″ β-sheet and is covalently linked to the Ig-like core with a disulfide bond between β-strands X2 and F. The UL16-MICBpf complex was partially deglycosylated prior to crystallization, leaving only single N-acetylglucosamine (NAG) molecules attached to glycosylation sites. While there is no evidence for O-linked glycosylation, our electron density maps provide clear evidence for the presence of NAGs at seven out of eight putative N-glycosylation sites (asparagines 35, 41, 68, 84, 95, 101 and 132). Modeling experiments show that native glycosylation would effectively shield much of the UL16 surface from solvent (Figure 1B). In particular, the outward-facing AGFCC′C″ β-sheet and the N-terminal plug are expected to be mostly covered with glycans in the fully glycosylated protein. By contrast, the solvent-exposed face of the DEB β-sheet is devoid of glycans and available for interactions with other proteins.

Structure of MICB
The extracellular region of MICB consists of two structural domains, the α1α2-platform domain (MICBpf) and the C-type Ig-like α3-domain [25]. The α3-domain is present only in the MIC family members of NKG2D ligands, but not among members of the ULBP family [17], [25]–[28]. Our SPR measurements (Figure 2 and Table S1) yielded almost identical dissociation constants (KD) for the complexes formed by UL16 with MICBpf (KD = 66 nM) or the complete MICB ectodomain (KD = 67 nM), respectively. Together with a previous report [29], this demonstrates that the α3-domain does not contribute to UL16 binding. Based on these results, only MICBpf was expressed and used for co-crystallization with UL16. As previously reported for the unliganded MICB [25], MICBpf folds into a structure that closely resembles MHC class I molecules, with two long parallel α-helices, contributed by domains α1 and α2, arranged above an eight-stranded antiparallel β-sheet (Figure 1B; for nomenclature of domains and secondary structure elements see Figure 3A). Comparison of MICBpf with the structure of the unliganded MICB ectodomain [25] shows that the platform domain remains essentially unchanged upon engagement of UL16 (root-mean-square deviation of 1.4 Å for 172 common Cα atoms). Although minor differences are seen within three surface-exposed loops and a short N-terminal helix (α0), the residues in these regions have elevated temperature factors and do not contact UL16.


The UL16-MICB interface
UL16 primarily engages MICBpf via a predominantly hydrophobic, glycan-free (see also Ref. [29]) surface comprised of its DEB β-sheet and the adjacent β-strand A, with additional contacts provided by the DE-loop (connecting β-strands D and E) and four amino acids (aa 160–163) at the C-terminus (Figures 3B and 4). This surface interacts with the two long parallel helices at the top of the MICB platform domain and the β5β6-loop connecting β-strands β5 and β6 of MICB (Figures 3A and 4), shielding an area of 2194 Å2 from solvent. With the exception of the MICB region that corresponds to the peptide-binding groove in MHC class I proteins, the contact area contains few interfacial solvent-filled cavities. The complex features good surface complementarity (Sc = 0.77) and is highly curved (planarity = 4.0) [38],[39]. Its overall organization resembles a saddle with two stirrups (UL16) that is mounted on horseback (MICBpf) (Figure 4A, see also Figure 1B). The saddle is formed by the DEB β-sheet, whereas the stirrups are contributed by the DE-loop and the C-terminus on either side of the sheet. To facilitate discussion of interactions, we have divided the UL16-MICB interface into three regions (A, B and C, Figure 4). Contact region A, which is located at the center of the complex and mostly hydrophobic in nature, contributes 54% of the total contact area. Interactions predominantly involve residues within the DEB β-sheet and β-strand A of UL16. Eight UL16 residues (Trp54, Leu56, Met59, Ile61, Ile63, Tyr125, Leu110 and Leu114) define a compact hydrophobic face that interacts with non-polar regions of MICBpf residues in its central α3-helix. These interactions are augmented with a salt bridge between UL16 Asp112 and MICB Lys152 and a number of mostly water-mediated hydrogen bonds (Figure 4B). Contact region B, with 23% of the total contact area, is located at one end of the DEB β-sheet and within the DE-loop of UL16. UL16 residues in this region contact several acidic residues (Glu64, Asp65 and Glu68) in the α1-helix of MICBpf, mostly via polar interactions (Figure 4C). Contact region C, which contributes 23% to the total contact area, is located on the other side of the UL16 saddle. Here, the C-terminus of the UL16 ectodomain interacts with the β5β6-loop and the N-terminus of MICBpf via a mixture of hydrophilic and hydrophobic contacts (Figure 4D). The overall architecture of the complex, with its large contact area and substantial number of interactions between contacting residues, indicates tight binding, which is in agreement with our SPR data that place the affinity of UL16 for MICBpf at 66 nM (Figure 2 and Table S1).
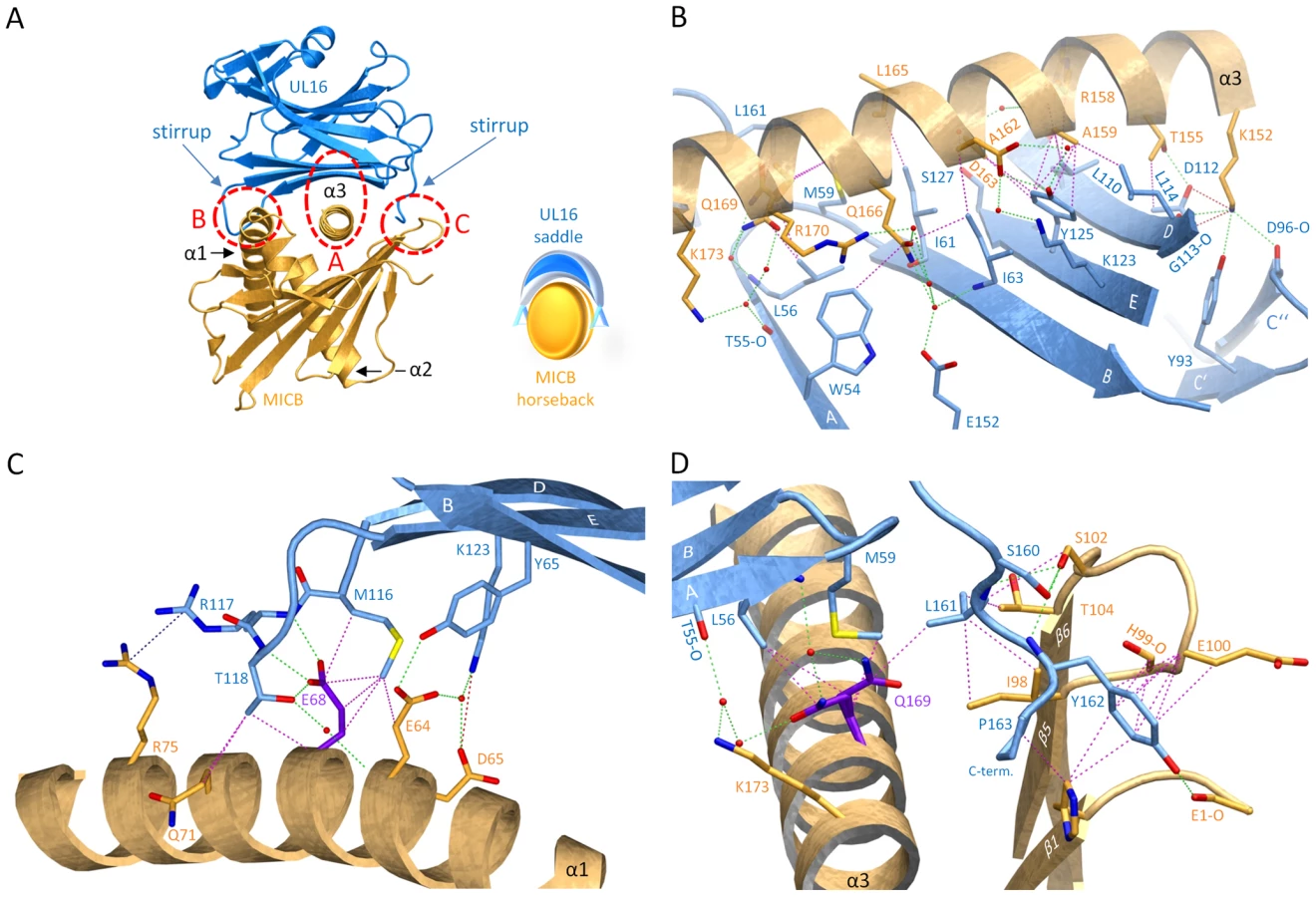
Comparison with the NKG2D-MICA complex
A crystal structure of the NKG2D homodimer bound to MICB is unavailable. However, the NKG2D structure in complex with the highly homologous MICA protein [26] shows that both NKG2D monomers make extensive contacts with the long helices at the top of the MICA α1α2-platform domain. The NKG2D-MICA complex buries a surface area of 2170 Å2, which is almost exactly the same area buried in the UL16-MICBpf complex. A superimposition of the two complexes demonstrates that contacts formed by UL16 overlap substantially with those made by one NKG2D monomer (Figures 5A, B). One could therefore envision a scenario in which UL16 acts as a direct competitor for NKG2D [18], perhaps even displacing it from its ligands. While the higher affinity of UL16 for MICB and ULBP1 (KD values of 66 and 12 nM, respectively) (Figures 2 and S1 and Table S1) compared with the respective affinities of NKG2D for the same ligands (KD values of 0.8 and 1 µM, respectively)[40] would support this scenario, most reports to date indicate that UL16 acts inside the cell and is therefore unlikely to compete with NKG2D for ligand binding [2],[17],[20],[21].

The detailed comparison of the central contact regions in each case reveals that, despite having entirely different folds, NKG2D and UL16 use an almost identical pattern of amino acid side chains to engage their ligands (Figure 5C, see also Figures 3B and S2). In UL16, this pattern includes the MICB-contacting residues Ile63, Lys123 and Tyr125, while NKG2D uses an identical pattern of residues, Ile182, Lys197, and Tyr199, to form very similar contacts with MICA. Remarkably, although the three side chains are contributed by different structural elements in each case, their position in space overlaps closely (Figure 5C). This is also true for two additional UL16 residues, Leu110 and Leu114, which are hydrophobic in nature and overlap with chemically related NKG2D residues Met184 and Tyr152 (Figure 5C). Together, the five residues constitute a predominantly hydrophobic binding motif that is common to NKG2D and UL16 (Figures 5B, C), and that forms the center of the interaction with the MIC molecules. This central binding motif is augmented by additional contacts, such as those mediated by UL16 residue Tyr65 and NKG2D residue Ser195, that perform similar functions in the UL16-MICBpf and NKG2D-MICA [26] complexes (Figures 3B and 5C). Since all MICA and MICB residues contacted by this central binding motif are identical, and since the structures of MICA and MICB superimpose well in this region, we conclude that UL16 mimics a key structural motif of NKG2D with an entirely different fold in order to engage MICB. Furthermore, we consider it likely that the central binding motif of UL16 also plays an important role in the recognition of other NKG2D ligands, for which structures of complexes with UL16 are not yet available.
Discussion
Bacterial and viral pathogens often interfere with cellular activities and immunosurveillance processes to enhance their survival and effectiveness [41]. This is typically achieved by virulence factors, which imitate the function of a host protein by mimicking its key structural features. In the majority of such cases, pathogens first hijack and then manipulate host genes to produce structurally homologous versions of host proteins [41]–[45]. Thus, virulence factors and host proteins are derived from the same origin and arise from divergent evolution. However, structural mimics can also be generated through convergent evolution. Although differing in evolutionary origin and three-dimensional structure, the virulence factors have in this case evolved to mimic key structural features of cellular proteins. Examples for the latter strategy, which can only be revealed through structural analyses, are still exceedingly rare and are limited to a small number of virulence factors [41],[46],[47]. The comparison of HCMV UL16 with human NKG2D, reveals a striking example of convergent evolution [41]. A set of five predominantly hydrophobic core residues on the UL16 surface precisely mimics a set of five equivalent residues in the central region of the interface used by the structurally unrelated immunoreceptor NKG2D to interact with its ligands.
As this central binding motif represents only a portion of the total interface between NKG2D and its ligands (Figure 5), one may wonder why UL16 mimics just this particular structural motif of NKG2D. McFarland et al. reported that residues constituting this motif (Tyr152, Met184 and Tyr199) form the basis for the highly degenerate ligand recognition mode of NKG2D [40],[48]. They proposed a “rigid adaptation” mechanism, in which a rigid binding site on NKG2D uses the same set of predominantly hydrophobic core residues to make diverse interactions with a series of chemically and structurally distinct ligand residues. As an example, Tyr199 and Tyr152 of NKG2D can accommodate residues as diverse as Ala, Met or Phe at ligand position 159 [40] (Figures 3A and S2). Mimicry of these core residues likely enables UL16 to employ this binding mechanism of NKG2D to contact a similar set of ligands. The “rigid adaptation” concept is furthermore supported by the finding that UL16 engages its ligands via a rigid β-sheet, which does not allow for much conformational flexibility. The ligand residues contacted by NKG2D and UL16 in MICA and MICB, respectively, are Asp65, Thr155, Ala159, Ala162, Asp163 and the hydrophobic portions of the Arg/His158 side chain (Figures 3A and 5C and S2) [26],[40],[48]. Since NKG2D and UL16 both evolved the same central binding motif in order to contact this specific set of ligand residues, the latter likely represent binding hot spots in MICA and MICB [48]. Furthermore, these residues probably are also of major importance for interactions with ULBP molecules (Figures 3A and S2). We note for instance that (1) based on the “rigid adaptation” concept the amino acid at ligand position 159 can be quite variable in size and chemical nature, (2) Asp163 is conserved in all NKG2D ligands, and (3) alanine and glycine dominate at position 162.
Unlike NKG2D, UL16 engages only MICB, ULBP1, ULBP2 and ULBP6, but not MICA, ULBP3, ULBP4 and ULBP5 [18]–[20], [29]–[31]. Our SPR measurements show that UL16 binds MICB with high affinity, whereas the affinity of UL16 for MICA is negligible (Table S1), in line with earlier studies [2],[21],[29]. Given the high degree of similarity between MICA and MICB at the sequence and structural level, the inability of UL16 to engage MICA is puzzling. In order to better understand the structural parameters that guide UL16 binding to MICB vs. MICA, Spreu et al. [29] assayed binding of soluble UL16-Fc to MICB chimeras in which they had exchanged domains, subdomains and single amino acids of MICB against equivalent regions of MICA. These experiments clearly demonstrated that recognition by UL16 is linked to residues projecting from the helical structures in the MICB α2-domain. However, the molecular mechanism by which these residues confer selectivity remained unclear.
The crystal structure of the UL16-MICB complex now allows us to identify the key determinants of NKG2D ligand binding to UL16. Our structural alignment of MICA and MICB identifies only seven MICB residues that contact UL16 in the complex and that are replaced by other amino acids in MICA (Figure 3A). Residues at positions 64, 71, 75, 102 and 158 can assume alternate conformations that would not interfere with binding, and could in some cases even mediate favorable contacts with UL16. Therefore, their effect on UL16 binding is likely to be negligible (see also Ref. [29]). Replacement of α1-domain Glu68 with glycine (Figure 4C) in MICA would eliminate several hydrophobic contacts and three hydrogen bonds with UL16 residues 117 and 118, and could therefore conceivably have a negative effect on UL16 binding. However, as complete replacement of the α1-domain of MICB by MICA (including residue Glu68) did not significantly affect UL16 binding [29], residue 68 is probably not a key determinant of UL16 binding.
On the other hand, however, Gln169 in the α2-domain of MICB is likely to be critical. Our structure shows that substitution of Gln169 with arginine, which is present at this position in MICA, would lead to steric clashes with UL16 residues Met59 and Leu161 (Figure 6A) that would prevent binding. This is in perfect agreement with previous experiments demonstrating that MICB carrying a Gln169Arg substitution no longer bound UL16 [29]. We consider it in fact likely that the side chain at position 169 is not only the key determinant of selective UL16 binding to the MIC molecules but all NKG2D ligands, which is based on the following reasons. (1) All NKG2D ligands that carry a glutamine or glutamate at position 169, i. e. MICB, ULBP1, ULBP2 and ULBP6, bind UL16, while all ligands that have an arginine at this position, i. e. MICA, ULBP3 and ULBP4, do not bind UL16 (Figure 3A). Although ULBP5 also carries a glutamate at position 169 and should therefore bind UL16, Wittenbrink et al. demonstrated by mutational studies that a substitution in the α2-domain, which is unique among all NKG2D ligands (Figure 3A), prevents binding of ULBP5 to UL16 [31]. (2) Arg169 has a similar conformation, stabilized by contacts with surrounding hydrophobic residues, in the unliganded [27] and liganded [26] MICA structures (Figure 6A). In this orientation, however, the Arg169 side chain would clash with UL16 residues. Modeling suggests that the arginine side chain could adopt only a single rotamer conformation, sandwiched between the hydrophobic side chain regions of Leu172 and Lys173, that would not result in steric clashes with UL16 (Figure 6A). However, such a rotamer is only seen in 2% of all observed arginines [49]. (3) The conformation of Arg169 in the ULBP3 structure [28], which is held in place by a salt bridge to Asp170, would also clash with UL16 (Figure 6A). A similar arrangement of Arg169 can be expected for ULBP4, where Asp170 is replaced with glutamate (Figure 3A). We note that Arg169 is not located near the NKG2D binding site and therefore does not play a role in the interaction of either MICA or ULBP3 with NKG2D.

A second important requirement for binding of NKG2D ligands to UL16 is the presence of a small hydrophobic side chain at position 162. In the UL16-MICBpf complex, Ala162 faces towards Tyr125, a UL16 footprint residue (Figures 5C and 6B). With the exception of ULBP3, which has an arginine at this position, all other NKG2D ligands have either an alanine or a glycine at position 162 (Figures 3A and S2). The long and positively charged Arg side chain of ULBP3 would clash with several UL16 residues (Figure 6B), likely contributing to the failure of UL16 to bind ULBP3 [2],[17],[18],[21] (Table S1). Interestingly, Arg162 would also clash with Met184 of the L2-loop of NKG2D in its MICA-liganded form. To allow for ULBP3 binding, NKG2D undergoes a conformational adjustment in which the L2-loop displaces Met184, resulting in sufficient space for the accommodation of Arg162 (Figure 6B). However, the rigid DEB β-sheet of UL16, which would not allow for such larger conformational adjustments, is unlikely to accommodate Arg162.
Taken together, these analyses suggest that some NKG2D ligands apparently bypass intracellular retention by UL16 through alteration of a small number of key residues at strategic locations of their potential UL16 binding interface. We therefore consider it likely that the selective pressure exerted by UL16 contributed to drive the diversification of NKG2D ligands, which eventually may have led to the emergence of non-UL16 binding ligands such as MICA and ULBP3 [2],[5],[17],[18],[23]. Further support for an HCMV-driven diversification of NKG2D ligands comes from studies by Cosman and colleagues showing that the HCMV immunoevasin UL142 targets most MICA allelic variants except MICA*08 [2],[5],[22]. Intriguingly, MICA*08 contains a truncated cytoplasmic domain and is by far the most frequent MICA variant in many populations [22]. As yet, no direct interaction of UL142 and MICA has been shown and the molecular mechanisms of MICA sequestration by UL142 are unknown. In contrast to UL16, UL142 and MCMV-encoded immunoevasins m145, m152, and m155 that suppress surface expression of mouse NKG2D ligands MULT-1, RAE-1, and H60, respectively, are predicted to have an MHC class I-like fold [2], [22], [23], [50]–[54]. It will be of great interest to determine the structural basis of NKG2D ligand engagement by MHC class I-like HCMV immunoevasins and to compare these interactions of two MHC class I-like molecules to those of the NKG2D-like ligand binding mode of UL16.
NK receptors binding to MHC class I or class I-like molecules belong to two structurally distinct families, the Ig superfamily and the C-type lectin superfamily [10]. While NKG2D belongs to the latter group, our structural analysis shows that UL16 assumes an Ig-like fold. Therefore, one may ask whether UL16 is related to the Ig-like NK receptors that bind MHC class I molecules, such as the leukocyte Ig-like receptors (LIRs) or the killer immunoglobulin-like receptors (KIRs). Structures of LIR-1 in complex with HLA-A2 [10] and with the HCMV MHC class I decoy UL18 [45] show that, in both cases, LIR-1 contacts β2-microglobulin and the α3-domain of the HLA-A2 and UL18 ligands via loops located at the interdomain hinge region of its two tandem Ig domains. In contrast KIRs, like UL16, engage the α-helical parts of the platform domain of MHC class I molecules, but, similar to LIRs, employ loops at the interdomain hinge region of their Ig domains for this interaction [10]. Therefore, LIRs and KIRs exhibit an MHC class I-binding mode that is distinct from that used by UL16. Since there is also no obvious sequence homology between these Ig-like NK receptors and UL16, we favor the view that UL16 evolved independently, mimicking a central binding motif of the structurally unrelated NKG2D immunoreceptor.
To the best of our knowledge, the structure presented here is the first structure of a viral immunoevasin in complex with a stimulatory NK receptor ligand as well as the first reported case of structural mimicry through convergent evolution of a human immunoreceptor by a viral immunoevasin. The results of our structural analyses revealed that HCMV and humans independently evolved two structurally distinct receptors, NKG2D and UL16, that share the same central ligand binding motif in order to achieve promiscuous binding to MIC and ULBP molecules. Our findings provide new insights into the structural basis of the evolutionary struggle between persistent viruses and cellular immune surveillance, exemplified by the promiscuous binding mode of the HCMV immunoevasin UL16 and the diversification of NKG2D ligands.
Materials and Methods
Expression and purification
Expression and purification of UL16
A recombinant cDNA fragment including the N-terminal signal peptide (residues 1–26) and the ectodomain (residues 27–184) of UL16 (GI: 9625700; UniProt P16757) was fused to a thrombin cleavage site followed by the human IgG1-Fc sequence and cloned into a pcDNA3.1©(−) vector (Invitrogen). CHO Lec 3.2.8.1 cells [34] were stably transfected with this construct using Lipofectamine 2000 (Invitrogen), and selected with 1.5 mg/ml G418 (Invitrogen) in α-MEM, supplemented with 10% Ultra-Low IgG FCS (Invitrogen), 100 U/ml penicillin (PAA), 100 µg/ml streptomycin (PAA), 2 mM L-glutamine (Invitrogen), and 1 mM pyruvate (PAA). A single cell clone was selected and grown in roller bottles at 37°C and 5% CO2. For each purification, 10 liters of cell culture supernatant were filtered through a 0.2 µm filter, adjusted to pH 9 with 5 M NaOH, and loaded overnight onto two serially connected Protein A HP columns (5 ml column volume each; GE Healthcare) using an Äkta FPLC system (GE Healthcare). The columns were extensively washed with Protein A binding-buffer (500 mM NaCl, 170 mM glycine pH 9 at 4°C) and the protein eluted with arginine-buffer (10 mM NaCl, 500 mM arginine pH 4.1 at 4°C) directly into reservoir-buffer (500 mM HEPES pH 9 at 4°C). Fractions containing UL16 were pooled, dialysed against TBS (pH 8 at 22°C), and concentrated. Thrombin (Sigma) cleavage was performed with 1 U/mg recombinant protein at 22°C for 18 hours. The cleaved samples were diluted five-fold in Protein A binding-buffer and run over two consecutive Protein A HP columns (1 ml column volume each; GE Healthcare) followed by a benzamidine column (1 ml column volume; GE Healthcare) to remove cleaved Fc tag and thrombin, respectively. Flow-through containing UL16 was concentrated and dialyzed against TBS (pH 7.4 at 4°C) for storage. UL16 migrated as a single band on reducing SDS gels, but appeared as two bands, corresponding to monomers and dimers, on non-reducing gels. Dimeric UL16 did not bind MICB and could not be converted to monomer by incubation with reducing agents. The dimer was separated from the monomer with hydrophobic interaction chromatography (HIC). Briefly, the UL16 monomer/dimer mixture was diluted in HIC-binding buffer (1 M NaCl, 0.05 M Na2HPO4 pH 7.4 at 4°C), loaded onto 3 serially connected phenyl sepharose columns (5 ml column volume each; GE Healthcare), and washed extensively with HIC-binding buffer. Phenyl-sepharose bound monomer was eluted with HIC-elution buffer (0.05 M Na2HPO4 pH 7.4 at 4°C) and dialysed against TBS pH 7.4 at 4°C. The yield of monomeric UL16 was 0.2 mg per 1 liter of cell culture supernatant.
Expression and purification of MICB
Gene sequences (GI:2454261; UniProt Q29980) encoding ectodomains α1 and α2 of MICB*02 (MICBpf, residues 1–181) and ectodomains α1, α2 and α3 of MICB*02 (residues 1–276) were fused to a thrombin cleavage site followed by (His)8 - and (His)6-tags, respectively, and cloned into the pET-21a(+) vector (Novagen). Both proteins were purified with the same strategy. E.coli Rosetta2 cells transformed with the appropriate expression construct were grown in LB medium, supplemented with 50 µg/ml ampicillin and 34 µg/ml chloramphenicol, to an optical density (OD)600 of 0.6 before induction with 1 mM IPTG at 37°C for 12 hours. Inclusion bodies containing MICB were refolded by stepwise arginine/urea dialysis. Soluble MICB in TBS (pH 7.4 at 4°C) was further purified via Ni-NTA affinity chromatography (1 ml column volume; GE Healthcare) followed by Superdex 75 size-exclusion chromatography (GE Healthcare).
Complex formation
UL16 and an excess amount of MICBpf were incubated for 16 hrs in TBS pH 7.4 at 4°C. The complex was separated from excess MICB through gel filtration (Superdex 75). In order to obtain diffracting crystals, UL16 was deglycosylated by incubation with EndoH after complex formation. Briefly, complex was diluted in EndoH-buffer (0.1 M NaAc pH 5.2 at 25°C), containing 0.5 U/µl EndoH (NEB) and incubated for 1 h at 37°C. Removal of cleaved glycans and EndoH was performed by size exclusion chromatography (Superdex 75). The complex was then concentrated to 15 mg/ml and used for crystallization.
Surface plasmon resonance
All SPR experiments were performed and evaluated as described previously [55]. Using two consecutive flow cells on a CM5 biosensor chip, MICBα1α2 (MICBpf) and MICBα1α3 ligands, respectively, were each covalently immobilized on the surface of the downstream (experimental) flow cell via amine-coupling chemistry (GE Healthcare) following manufacturer's instructions, while the surface of the upstream (reference) flow cell was subjected to the same coupling reaction in the absence of protein. For the Protein A-G chip preparation, an amount of 3500 RU (resonance units) of recombinant Protein A-G (BioVision) was covalently immobilized to the upstream (reference) and downstream (experimental) flow cells of a CM5 biosensor chip (GE Healthcare) by amine-coupling chemistry (GE Healthcare). Fc-tagged ULBP1, ULBP2, ULBP3 (all R&D Systems), ULBP4 and ULBP5 ligands [31] were diluted in HBS-EP (10 mM HEPES, 150 mM NaCl, 3 mM EDTA, 0.005% (v/v) Surfactant P20, pH 7.4 at 25°C) and noncovalenty bound to the experimental flow cell surface. In all experiments, untagged, monomeric UL16 analyte was serially diluted in running buffer and injected in series over the reference - and experimental biosensor surface at 50 µl/min. After each cycle using a Protein A-G chip, the biosensor surface was regenerated (stripped of any remaining analyte and ligand) with two 1 min injections of 10 mM glycine pH 1.7. CM5 chips were not regenerated.
Crystallization
For crystallization, complex at 15 mg/ml was mixed in a 1∶1 ratio with a reservoir solution containing 0.2 M ammonium sulfate, 0.1 M sodium cacodylate pH 6.5, and 25% PEG 8000. Crystals grew at 4°C over a time period of 4 months using the hanging drop vapor diffusion method. They were soaked in reservoir solution enriched with 15% ethylene glycol, and then flash frozen in liquid nitrogen prior to data collection.
Structure determination
The crystals belong to space group P212121 and contain two complexes in the asymmetric unit. All diffraction data were collected at 100 K and a wavelength of 1.0013 Å at the Swiss Light Source (SLS, Villigen, Switzerland) beamline X06SA using the PILATUS 6M detector. Data were indexed, integrated and scaled with XDS [56], and the structure was solved by molecular replacement as implemented in PHASER [57] using the unliganded MICB structure [25] (PDB code 1JE6) as search model. The initial density map already clearly showed the approximate location of the UL16 molecules. Phases were then improved through non-crystallographic symmetry averaging using RESOLVE [58]. Structural refinement was performed with PHENIX [59] and model building was done with Coot [49]. Refinement included TLS-refinement of 26 TLS groups assigned by the TLSMD Server [60]. A data set for Rfree calculation was generated with 5% randomly selected reflections, and refinement progress was monitored by the decrease of R and Rfree throughout. The final model has R and Rfree values of 17.7% and 21.5%, respectively, and was validated using PROCHECK [57] and WHAT_IF [61]. Secondary structure elements were assigned with DSSP [62]. Structural figures were created with PyMOL [63].
Glycan modeling
Eight potential N-linked glycosylation sites were identified in the UL16 ectodomain. Six of the possible eight asparagine residues (Asn41, 68, 84, 95, 101, 132) carry NAG residues that are clearly defined by electron density (Figure 1A). While extra density is present at the seventh residue, Asn35, this density is not well defined, and no NAG residue was built at this location. No extra electron density is observed at the final asparagine, Asn145, and thus this residue is either not glycosylated or carries an especially flexible glycan moiety. We note that Asn145 is close in space to Asn35, which is glycosylated. In order to produce a realistic estimate of size and distribution of the glycan structure of native UL16 (Figure 1B) we used the GlyProt [64] online server and modeled hybrid and complex glycans linked to the seven Asn residues with NAG electron density.
Accession numbers
Atomic coordinates and structure factors have been deposited with the Protein Data Bank under accession code 2wy3.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. ReddehaseMJ
2002 Antigens and immunoevasins: opponents in cytomegalovirus immune surveillance. Nat Rev Immunol 2 831 844
2. LanierLL
2008 Evolutionary struggles between NK cells and viruses. Nat Rev Immunol 8 259 268
3. LodoenMB
LanierLL
2006 Natural killer cells as an initial defense against pathogens. Curr Opin Immunol 18 391 398
4. BironCA
ByronKS
SullivanJL
1989 Severe herpesvirus infections in an adolescent without natural killer cells. N Engl J Med 320 1731 1735
5. JonjicS
BabicM
PolicB
KrmpoticA
2008 Immune evasion of natural killer cells by viruses. Curr Opin Immunol 20 30 38
6. PowersC
DeFilippisV
MalouliD
FruhK
2008 Cytomegalovirus immune evasion. Curr Top Microbiol Immunol 325 333 359
7. WilkinsonGW
TomasecP
StantonRJ
ArmstrongM
Prod'hommeV
2008 Modulation of natural killer cells by human cytomegalovirus. J Clin Virol 41 206 212
8. LodoenMB
LanierLL
2005 Viral modulation of NK cell immunity. Nat Rev Microbiol 3 59 69
9. LjunggrenHG
KarreK
1990 In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol Today 11 237 244
10. DengL
MariuzzaRA
2006 Structural basis for recognition of MHC and MHC-like ligands by natural killer cell receptors. Semin Immunol 18 159 166
11. LanierLL
2008 Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol 9 495 502
12. VivierE
TomaselloE
BaratinM
WalzerT
UgoliniS
2008 Functions of natural killer cells. Nat Immunol 9 503 510
13. GonzalezS
GrohV
SpiesT
2006 Immunobiology of human NKG2D and its ligands. Curr Top Microbiol Immunol 298 121 138
14. RauletDH
2003 Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol 3 781 790
15. BauerS
GrohV
WuJ
SteinleA
PhillipsJH
1999 Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 285 727 729
16. WuJ
SongY
BakkerAB
BauerS
SpiesT
1999 An activating immunoreceptor complex formed by NKG2D and DAP10. Science 285 730 732
17. EagleRA
TrowsdaleJ
2007 Promiscuity and the single receptor: NKG2D. Nat Rev Immunol 7 737 744
18. CosmanD
MullbergJ
SutherlandCL
ChinW
ArmitageR
2001 ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity 14 123 133
19. EagleRA
TraherneJA
HairJR
JafferjiI
TrowsdaleJ
2009 ULBP6/RAET1L is an additional human NKG2D ligand. Eur J Immunol
20. DunnC
ChalupnyNJ
SutherlandCL
DoschS
SivakumarPV
2003 Human cytomegalovirus glycoprotein UL16 causes intracellular sequestration of NKG2D ligands, protecting against natural killer cell cytotoxicity. J Exp Med 197 1427 1439
21. WelteSA
SinzgerC
LutzSZ
Singh-JasujaH
SampaioKL
2003 Selective intracellular retention of virally induced NKG2D ligands by the human cytomegalovirus UL16 glycoprotein. Eur J Immunol 33 194 203
22. ChalupnyNJ
Rein-WestonA
DoschS
CosmanD
2006 Down-regulation of the NKG2D ligand MICA by the human cytomegalovirus glycoprotein UL142. Biochem Biophys Res Commun 346 175 181
23. JonjicS
PolicB
KrmpoticA
2008 Viral inhibitors of NKG2D ligands: friends or foes of immune surveillance? Eur J Immunol 38 2952 2956
24. Stern-GinossarN
ElefantN
ZimmermannA
WolfDG
SalehN
2007 Host immune system gene targeting by a viral miRNA. Science 317 376 381
25. HolmesMA
LiP
PetersdorfEW
StrongRK
2002 Structural studies of allelic diversity of the MHC class I homolog MIC-B, a stress-inducible ligand for the activating immunoreceptor NKG2D. J Immunol 169 1395 1400
26. LiP
MorrisDL
WillcoxBE
SteinleA
SpiesT
2001 Complex structure of the activating immunoreceptor NKG2D and its MHC class I-like ligand MICA. Nat Immunol 2 443 451
27. LiP
WillieST
BauerS
MorrisDL
SpiesT
1999 Crystal structure of the MHC class I homolog MIC-A, a gammadelta T cell ligand. Immunity 10 577 584
28. RadaevS
RostroB
BrooksAG
ColonnaM
SunPD
2001 Conformational plasticity revealed by the cocrystal structure of NKG2D and its class I MHC-like ligand ULBP3. Immunity 15 1039 1049
29. SpreuJ
StehleT
SteinleA
2006 Human cytomegalovirus-encoded UL16 discriminates MIC molecules by their alpha2 domains. J Immunol 177 3143 3149
30. ChalupnyNJ
SutherlandCL
LawrenceWA
Rein-WestonA
CosmanD
2003 ULBP4 is a novel ligand for human NKG2D. Biochem Biophys Res Commun 305 129 135
31. WittenbrinkM
SpreuJ
SteinleA
2009 Differential NKG2D binding to highly related human NKG2D ligands ULBP2 and RAET1G is determined by a single amino acid in the alpha2 domain. Eur J Immunol
32. RobinsonJ
WallerMJ
FailSC
McWilliamH
LopezR
2009 The IMGT/HLA database. Nucleic Acids Res 37 D1013 1017
33. KayeJ
BrowneH
StoffelM
MinsonT
1992 The UL16 gene of human cytomegalovirus encodes a glycoprotein that is dispensable for growth in vitro. J Virol 66 6609 6615
34. StanleyP
1989 Chinese hamster ovary cell mutants with multiple glycosylation defects for production of glycoproteins with minimal carbohydrate heterogeneity. Mol Cell Biol 9 377 383
35. BorkP
HolmL
SanderC
1994 The immunoglobulin fold. Structural classification, sequence patterns and common core. J Mol Biol 242 309 320
36. HarpazY
ChothiaC
1994 Many of the immunoglobulin superfamily domains in cell adhesion molecules and surface receptors belong to a new structural set which is close to that containing variable domains. J Mol Biol 238 528 539
37. HalabyDM
PouponA
MornonJ
1999 The immunoglobulin fold family: sequence analysis and 3D structure comparisons. Protein Eng 12 563 571
38. BahadurRP
ZachariasM
2008 The interface of protein-protein complexes: analysis of contacts and prediction of interactions. Cell Mol Life Sci 65 1059 1072
39. LawrenceMC
ColmanPM
1993 Shape complementarity at protein/protein interfaces. J Mol Biol 234 946 950
40. McFarlandBJ
StrongRK
2003 Thermodynamic analysis of degenerate recognition by the NKG2D immunoreceptor: not induced fit but rigid adaptation. Immunity 19 803 812
41. StebbinsCE
GalanJE
2001 Structural mimicry in bacterial virulence. Nature 412 701 705
42. AlcamiA
2003 Viral mimicry of cytokines, chemokines and their receptors. Nat Rev Immunol 3 36 50
43. LambrisJD
RicklinD
GeisbrechtBV
2008 Complement evasion by human pathogens. Nat Rev Microbiol 6 132 142
44. van CleefKW
SmitMJ
BruggemanCA
VinkC
2006 Cytomegalovirus-encoded homologs of G protein-coupled receptors and chemokines. J Clin Virol 35 343 348
45. YangZ
BjorkmanPJ
2008 Structure of UL18, a peptide-binding viral MHC mimic, bound to a host inhibitory receptor. Proc Natl Acad Sci U S A 105 10095 10100
46. AlexanderJM
NelsonCA
van BerkelV
LauEK
StudtsJM
2002 Structural basis of chemokine sequestration by a herpesvirus decoy receptor. Cell 111 343 356
47. CarfiA
SmithCA
SmolakPJ
McGrewJ
WileyDC
1999 Structure of a soluble secreted chemokine inhibitor vCCI (p35) from cowpox virus. Proc Natl Acad Sci U S A 96 12379 12383
48. McFarlandBJ
KortemmeT
YuSF
BakerD
StrongRK
2003 Symmetry recognizing asymmetry: analysis of the interactions between the C-type lectin-like immunoreceptor NKG2D and MHC class I-like ligands. Structure 11 411 422
49. EmsleyP
CowtanK
2004 Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60 2126 2132
50. MansJ
ZhiL
RevillezaMJ
SmithL
RedwoodA
2009 Structure and function of murine cytomegalovirus MHC-I-like molecules: how the virus turned the host defense to its advantage. Immunol Res 43 264 279
51. SmithHR
HeuselJW
MehtaIK
KimS
DornerBG
2002 Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc Natl Acad Sci U S A 99 8826 8831
52. KrmpoticA
HasanM
LoewendorfA
SauligT
HaleniusA
2005 NK cell activation through the NKG2D ligand MULT-1 is selectively prevented by the glycoprotein encoded by mouse cytomegalovirus gene m145. J Exp Med 201 211 220
53. LodoenMB
AbenesG
UmamotoS
HouchinsJP
LiuF
2004 The cytomegalovirus m155 gene product subverts natural killer cell antiviral protection by disruption of H60-NKG2D interactions. J Exp Med 200 1075 1081
54. LenacT
ArapovicJ
TravenL
KrmpoticA
JonjicS
2008 Murine cytomegalovirus regulation of NKG2D ligands. Med Microbiol Immunol 197 159 166
55. PerssonBD
MullerS
ReiterDM
SchmittBB
MarttilaM
2009 An arginine switch in the species B adenovirus knob determines high-affinity engagement of cellular receptor CD46. J Virol 83 673 686
56. KabschW
1993 Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. Journal of Applied Crystallography 26 795 800
57. Collaborative Computational Project N 1994 The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50 760 763
58. TerwilligerT
2004 SOLVE and RESOLVE: automated structure solution, density modification and model building. J Synchrotron Radiat 11 49 52
59. AdamsPD
Grosse-KunstleveRW
HungLW
IoergerTR
McCoyAJ
2002 PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr 58 1948 1954
60. PainterJ
MerrittEA
2006 Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr D Biol Crystallogr 62 439 450
61. VriendG
1990 WHAT IF: a molecular modeling and drug design program. J Mol Graph 8 52 56, 29
62. KabschW
SanderC
1983 Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22 2577 2637
63. DeLanoWL
2002 The Pymol Molecular Graphics System.
64. Bohne-LangA
von der LiethCW
2005 GlyProt: in silico glycosylation of proteins. Nucleic Acids Res 33 W214 219
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 1
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Panton-Valentine Leukocidin Is a Very Potent Cytotoxic Factor for Human Neutrophils
- CD8+ T Cell Control of HIV—A Known Unknown
- Polyoma Virus-Induced Osteosarcomas in Inbred Strains of Mice: Host Determinants of Metastasis
- The Deadly Chytrid Fungus: A Story of an Emerging Pathogen
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy