
Persistent ER Stress Induces the Spliced Leader RNA Silencing Pathway (SLS), Leading to Programmed Cell Death in
Trypanosomes are parasites that cycle between the insect host (procyclic form) and mammalian host (bloodstream form). These parasites lack conventional transcription regulation, including factors that induce the unfolded protein response (UPR). However, they possess a stress response mechanism, the spliced leader RNA silencing (SLS) pathway. SLS elicits shut-off of spliced leader RNA (SL RNA) transcription by perturbing the binding of the transcription factor tSNAP42 to its cognate promoter, thus eliminating trans-splicing of all mRNAs. Induction of endoplasmic reticulum (ER) stress in procyclic trypanosomes elicits changes in the transcriptome similar to those induced by conventional UPR found in other eukaryotes. The mechanism of up-regulation under ER stress is dependent on differential stabilization of mRNAs. The transcriptome changes are accompanied by ER dilation and elevation in the ER chaperone, BiP. Prolonged ER stress induces SLS pathway. RNAi silencing of SEC63, a factor that participates in protein translocation across the ER membrane, or SEC61, the translocation channel, also induces SLS. Silencing of these genes or prolonged ER stress led to programmed cell death (PCD), evident by exposure of phosphatidyl serine, DNA laddering, increase in reactive oxygen species (ROS) production, increase in cytoplasmic Ca2+, and decrease in mitochondrial membrane potential, as well as typical morphological changes observed by transmission electron microscopy (TEM). ER stress response is also induced in the bloodstream form and if the stress persists it leads to SLS. We propose that prolonged ER stress induces SLS, which serves as a unique death pathway, replacing the conventional caspase-mediated PCD observed in higher eukaryotes.
Published in the journal:
. PLoS Pathog 6(1): e32767. doi:10.1371/journal.ppat.1000731
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1000731
Summary
Trypanosomes are parasites that cycle between the insect host (procyclic form) and mammalian host (bloodstream form). These parasites lack conventional transcription regulation, including factors that induce the unfolded protein response (UPR). However, they possess a stress response mechanism, the spliced leader RNA silencing (SLS) pathway. SLS elicits shut-off of spliced leader RNA (SL RNA) transcription by perturbing the binding of the transcription factor tSNAP42 to its cognate promoter, thus eliminating trans-splicing of all mRNAs. Induction of endoplasmic reticulum (ER) stress in procyclic trypanosomes elicits changes in the transcriptome similar to those induced by conventional UPR found in other eukaryotes. The mechanism of up-regulation under ER stress is dependent on differential stabilization of mRNAs. The transcriptome changes are accompanied by ER dilation and elevation in the ER chaperone, BiP. Prolonged ER stress induces SLS pathway. RNAi silencing of SEC63, a factor that participates in protein translocation across the ER membrane, or SEC61, the translocation channel, also induces SLS. Silencing of these genes or prolonged ER stress led to programmed cell death (PCD), evident by exposure of phosphatidyl serine, DNA laddering, increase in reactive oxygen species (ROS) production, increase in cytoplasmic Ca2+, and decrease in mitochondrial membrane potential, as well as typical morphological changes observed by transmission electron microscopy (TEM). ER stress response is also induced in the bloodstream form and if the stress persists it leads to SLS. We propose that prolonged ER stress induces SLS, which serves as a unique death pathway, replacing the conventional caspase-mediated PCD observed in higher eukaryotes.
Introduction
The endoplasmic reticulum (ER) is the site for folding of proteins that traverse the ER on the way to the outer membrane or internal organelles of the vesicular transport machinery. The ER is also the site of lipid synthesis and it maintains the cellular calcium homeostasis [1]. The ER performs these functions using resident chaperones, high levels of calcium and an oxidating environment. Proteins that translocate to the ER undergo proper folding and those can exit the ER. However, if the protein fails to fold properly, it is exported back to the cytoplasm and is degraded by the proteasome [2],[3]. ER function is sensitive to changes in calcium level, inhibition of glycosylation, oxidative stress, and exposure to reducing agents. These conditions induce the ER stress response, which triggers specific signaling pathways including the unfolded proteins response (UPR), which leads to reduction in the load of proteins to be translocated, enhanced degradation of misfolded proteins, and increased folding capacity of the ER [3]–.
In yeast, UPR is dependent on IRE1, which is a bi-functional kinase and endonuclease that cleaves a non-conventional intron from HAC1 mRNA, which encodes a transcription factor (bZIP) [6],[7]. Homologues proteins exist in mammals; IRE1 cleaves an intron in XBP1 encoding a potent bZIP transcription factor. This transcription factor activates genes essential for executing the UPR [8]. In mammals, two additional ER transmembrane proteins, PERK and ATF6, participate in UPR. PERK phosphorylates eIF2α to mediate translational attenuation. ATF6 is cleaved in the Golgi and translocates to the nucleus to activate the transcription of chaperones, such as BiP [9]–[12]. When the ER stress is persistent, the cells activate the apoptotic pathway via the activation of Caspase 12 [13],[14].
Trypanosomatids are known for their non-conventional gene expression mechanisms, including trans-splicing [15],[16] and RNA editing [17],[18]. Trans-splicing is required for the maturation of all mRNAs in these parasites. In this process, a small exon, the spliced leader (SL), encoded by a small RNA, the SL RNA, is donated to all pre-mRNA by trans-splicing [16]. No promoters upstream to protein coding genes transcribed by polymerase II were identified. Protein coding genes are arrayed in long polycistronic transcription units which are processed post-transcriptionally by the concerted action of trans-splicing and polyadenylation [15]. It is believed that gene expression in these parasites is regulated primarily post-transcriptionally at the level of mRNA degradation and translation; for most genes, the signals that dictate this regulation are situated at the 3′ UTR [19]. However, trypanosomes do not completely lack transcriptional regulation. The SL RNA has a defined promoter that binds specific transcription factors such as tSNAPc [20]–[22].
Our collective knowledge of the regulation of trypanosome gene expression is consistent with the novel stress-induced regulatory mechanism that controls the transcriptome under variety of stresses; this mechanism was named SLS, for SL RNA silencing [23]. SLS was discovered in cells silenced for the signal recognition particle receptor, SRα. Depletion of SRα results in a dramatic decrease in the level of SL RNA, leading to major reduction in mRNAs. Nascent transcription in permeable cells showed that SL RNA transcription is specifically extinguished under these conditions. ChIP assay further demonstrated that under SRα depletion, the SL RNA transcription factor, tSNAP42, fails to bind to the SL RNA promoter. The hallmark of SLS is therefore shut-off of SL RNA transcription, followed by massive accumulation of tSNAP42 in the nucleus [23].
Most recently, a very detailed study on the trypanosome transcriptome was performed and revealed significant changes between procyclic and bloodstream form trypanosomes. However, the same study failed to detect changes generally elicited by classical UPR inducers or changes that are generally elicited under serum starvation [24]. It is currently unknown whether trypanosomes lack the UPR mechanism, which is replaced by SLS in these organisms, or whether both mechanisms exist, and SLS is activated to induce PCD when UPR fails. Accumulated data suggest that PCD exists in trypanosomatids, and plays a major role in maintaining and regulating the parasite population [25],[26]. In metazoa, type I PCD is induced by caspases. Effector caspases activate proteases and nucleases, eventually leading to apoptosis. Trypanosomes lack homologues to metazoan caspases, but have five metacaspases [27]–[29]. However, up till now, it is believed that these metacaspases are not involved in apoptosis [27],[29]. In recent years, PCD pathways that are caspase-independent, such as autophagy were shown to exist. Autophagy functions in protein degradation and organelle turnover during stresses, such as nutrient and growth factor deprivation, to assure cell survival. Autophagy machinery can be also recruited to kill cells under certain conditions, generating a caspase-independent form of PCD [30]. Recent studies indicate that trypanosomatids possess such an autophagic pathway [31],[32].
In this study, we demonstrate that despite the lack of IRE1/XBP1 homologues that mediate the UPR in all eukaryotes, trypanosomes change their transcriptome in response to ER stress much like yeast and metazoa. The mechanism of ER stress response is unique, and is based on stress-induced mRNA stabilization of mRNAs, which are essential for executing the response. SLS is triggered under prolonged ER stress induced either by chemicals or by silencing of SEC63 and SEC61 - two major components of ER translocation machinery. Typical hallmarks of PCD such as DNA fragmentation, exposure of phosphatidyl serine, increase in cytosolic calcium, reduction in mitochondria membrane potential and ROS production, were observed in SLS-induced cells. We propose that SLS is the apoptotic branch induced under persistent ER stress that leads to programmed cell death in both procyclic and bloodstream form trypanosomes.
Results
The ER stress response in trypanosomes: Transcriptome changes in T. brucei treated with Dithiothreitol (DTT) resemble UPR in other eukaryotes
To examine whether procyclic stage trypanosomes possess an ER stress response, the transcriptome was examined for up and down-regulation upon Dithiothreitol (DTT) treatment. RNA was prepared from cells treated with 4 mM DTT for 1 and 3 hours and the transcriptome was compared to untreated cells. The Lowess-normalized data were used to identify genes whose expression appeared to be significantly (P<0.05) up-regulated with an arbitrary cutoff of 1.5 fold. A list of these genes is provided in Table S2. In order to visualize the difference in gene expression between normal and under ER stress, we compared the changes of transcriptome after 1 and 3 hours of treatment. Hierarchical clustering was performed on the regulated genes, and a heat map was constructed (Fig. 1-A). The results demonstrate that the changes in the transcriptome between 1 and 3 hrs of treatment were similar in both up - and down-regulated genes (r = 0.86), indicating that the changes were treatment-specific (Fig. 1-B). Moreover, the amplitude of the up and down-regulation was increased by 1.24 fold when comparing the differential expression from 1 to 3 hours of treatment.

Inspection of the up-regulated genes (Table S2) suggests that many genes which are involved in the core processes of classic UPR, namely, protein folding, degradation, translocation, sorting, and lipid metabolism, were up-regulated. Additionally, genes involved in mitochondrial functions, redox balance, metabolism, cytoskeleton, and movement were also up-regulated. Interestingly, a large number of genes involved in signal transduction and gene expression were affected, as well.
To compare the response of trypanosomes under ER stress to that of other eukaryotes, microarray data from UPR induced cells of Saccharomyces cerevisiae [3], Caenorhabditis elegans [33], Drosophila melanogaster [34], and Homo sapiens [35] were downloaded and analyzed. Genes found to be up-regulated were classified into the same functional categories (Table S3) that were already defined for the trypanosome transcriptome (Table S2). Note that for S. cerevisiae, C. elegans, and D. melanogaster, the list of up-regulated genes was refined by filtering out genes whose expression under ER stress was not XBP-1 or IRE-1 dependent and therefore are not directly related to the UPR response. In trypanosomes and humans, no such filtering was possible.
The results presented as pie diagrams in Figure 1-C, demonstrate that in trypanosomes, as in all other organisms, the largest category of up-regulated genes in response to ER stress is the category of genes that function in protein secretion. Moreover, except movement, all the other functions that were shown to be up-regulated in trypanosomes are also up-regulated in some if not all other organisms. Changes in motility are specific to trypanosomes. Of note is also that a significantly high fraction of the down-regulated genes (35%, P<10−6) encode for proteins destined to traverse the ER (i.e., harboring either a signal-peptide or trans-membrane domains), similarly to D. melanogaster [34].
The response to ER stress is mediated by stress-induced mRNA stabilization
Trypanosomes lack conventional transcription regulation, and therefore it is expected that their genome lacks homologues of IRE1/XBP1. Since mRNA stability is a very dominant mechanism of regulation, we examined the possibility that up-regulation results from mRNA stabilization during ER stress. To this end, procyclic stage trypanosomes (untreated) and treated with DTT for 90 minutes, were treated with sinefungin and actinomycin D, inhibitors of trans-splicing and transcription, respectively [36]. Incubation was for increasing time points to measure possible changes in the half life of mRNAs during ER stress. RNA was extracted and subjected to Northern analysis with probes specific to genes whose level was significantly changed upon DTT treatment and that were shown to also be induced in other organisms. The results in Figure 2 demonstrate that the half-life of DNAJ, protein disulfide isomerase (PDI), thioredoxin and syntaxin were all increased (from 45 to 90 min, 60 to 90 min, 13 to 65 min and 10 to 20, respectively) under DTT treatment, suggesting that the up-regulation of these transcripts during ER stress can be explained by stress-induced mRNA stabilization. The level of mRNAs that were unchanged during DTT treatment (based on microarray analysis) was also examined, and no changes in their stability was observed upon the ER stress (results not shown), indicating the specificity of this regulation.

ER stressors elicit ER dilation and over expression of the ER chaperone, BiP
One of the hallmarks of UPR in yeast and metazoa is the ability to induce the production of ER chaperones to increase the folding capacity of the ER [1],[3],[4],[37]. Induction of chaperones is not only mediated by increase in the transcription but also by preferential translation and proteolysis under stress [38]. Although BiP mRNA is not among the mRNAs whose level is most highly increased under ER stress ([3],[39] and this study) its elevation at the protein level serves as a hallmark for UPR. To examine if in trypanosomes BiP is increased upon induction of ER stress, its level was examined after treating cells with DTT or 2-deoxy D - glucose (2-DG), which affects glycosylation [40],[41]. Although 2-DG inhibits glycosylation and is known to induce the UPR response in other eukaryotes, it may also have other effects on carbon source sensing and metabolism [41]. The results presented in Figure 3-Ai and Figure 3-Aii suggest that both treatments result in increased levels of BiP in a time-dependent manner. Next, we examined if ER stress response can be induced also in the bloodstream form. The results (Fig. 3-Aiii) suggest that under the conditions used, BiP level was increased. These results are contrast to those previously published [24]. However, note that we used 4mM DTT, whereas the previous study used only 1 mM DTT. The results suggest that ER stress response operates in both stages of the parasite.

To observe cellular changes that may accompany response to ER stress, ultrastructure analysis using electron microscopy was performed. After 1 hour of DTT treatment, the most prominent change observed by transmission electron microscopy (TEM) is the expansion of the ER (compare Fig. 3 Bi to Bii). ER expansion was previously observed in yeast and mammals, and it was suggested that the increase in the ER may help to accommodate newly synthesized ER proteins and inhibit aggregation of unfolded proteins by reducing their concentration [42],[43]. The ER lumen was not only dilated, but seemed to contain dense material that might correspond to aggregated proteins (Fig. 3-Bii).
ER stress induces irreversible growth arrest and SLS
To examine if trypanosomes can recover following removal of ER stress, procyclic stage parasites were exposed to DTT for different time periods ranging from 30 to 240 minutes. After incubation with DTT, the DTT was washed out, and growth was monitored. The results (Figure 4-A) suggest that after exposure to DTT for 120 minutes or more, cells were unable to resume growth upon withdrawal of DTT, suggesting that prolonged exposure to DTT leads to cell death.

SLS was shown to be induced in procyclic stage trypanosomes upon silencing of SRP receptor or low pH [23]. We previously proposed that SLS might replace the conventional ER stress response because of the lack of conventional transcriptional regulation that would be required to induce the production of mRNAs needed for this process. The results in Figures 1–3 suggest that trypanosomes possess an ER stress response, and it was therefore of great interest to examine if this response is associated with SLS. SLS is characterized by two hallmarks, reduction in SL RNA level and changes in the amount and localization of tSNAP42. The results in Figure 4-Bi and Figure 4-Bii suggest that SLS is induced 180 and 120 minutes after exposure to DTT or 2-DG, respectively. SLS is induced after BiP induction, since induction of BiP was detected 120 minutes after DTT addition (Fig. 3-Ai) and 60 minutes after 2-DG addition (Fig. 3-Aii), suggesting that the ER stress response is induced before SLS. To examine if prolonged ER stress induces SLS also in bloodstream trypanosomes, SL RNA level was examined upon treatment with DTT. The results in (Fig. 4-Biii) demonstrate the induction of SLS in bloodstream trypanosomes after 180 minutes exposure to DTT.
The induction of SLS is clearly observed when comparing the localization of the SL RNA transcription factor, tSNAP42. Under normal conditions, tSNAP42 is confined to the nuclear domain where SL RNA is transcribed and assembled with the Sm core proteins [44],[45]. Under prolonged ER stress, the level of tSNAP42 increases, filling up the entire nucleus (Fig. 4-C). These data suggest that SLS is activated after the response to ER stress was initiated; 180 minutes (in DTT-treated cells) or 120 minutes (in 2-DG-treated cells).
Silencing of functions involved in ER translocation induce SLS in both stages of the parasite
We previously demonstrated that SLS is induced following silencing of the SRP receptor in the procyclic stage [23]; it was therefore of interest to examine whether SLS is induced following other perturbations that are connected to ER translocation functions. For this analysis, we chose to investigate and compare three functions that control translocation across the ER membrane, SRα, SEC63 and SEC61. We have previously demonstrated that SEC63 is essential for translocation of proteins to the ER via both pathways - the co-translational pathway mediated by SRP and the post-translational pathway mediated by chaperones [46]. The SEC61 protein constitutes the translocon. SEC61 was silenced using a stem-loop construct as described in the Materials and Methods. The results in Figure 5-Ai demonstrate that upon silencing of SEC61 or SEC63, the level of SL RNA was reduced, with no change in the level of 7SL RNA. The reduction elicited a major decrease in the level of mRNAs due to inhibition of trans-splicing (Fig. 5-Aii). The reduction in SL RNA was accompanied by the classical nuclear accumulation of tSNAP42 (Fig. 5-B). Indeed the level of tSNAP42 was increased upon SEC63 silencing as observed in Figure 5-C. The results suggest the perturbation in ER translocation due to elimination of SEC61 or SEC63 elicits SLS, much like that observed in SRα silenced cells [23]. To examine if SEC63 silencing also induces SLS in bloodstream form, transgenic parasites expressing a T7 opposing construct to silence the SEC63 gene (Figure S2) were generated as described in Materials and Methods. The results (Figure 5-D) demonstrate that silencing of SEC63 induces SLS as evident by marked reduction in the level of SL RNA.

DNA is fragmented in SLS-induced cells
Is SLS a programmed cell death pathway that is induced under prolonged ER stress much like apoptosis in mammalian cells? To examine this intriguing possibility, the hallmarks of programmed cell death were investigated in SLS-induced procyclic stage trypanosomes. First, the DNA fragmentation during SLS was measured by TUNEL. TUNEL identifies nicks in DNA by measuring the incorporation of fluorescently labeled dUTP using terminal deoxynucleotidyl transferase (TdT). The results presented in Figure 6-A demonstrate an increase in free ends of DNA, marking DNA fragmentation in SRα silenced cells. Two controls were used to demonstrate the specificity of the assay; in the absence of TdT no signal was observed (Fig. 6Ai), and maximal signal was obtained upon treatment with micrococcal nuclease, which induces DNA breaks (Fig. 6Aii). Another assay to monitor DNA fragmentation is by propidium iodide (PI) staining and flow cytometry. Apoptotic cells display a broad hypodiploid (sub-G1) peak, which can be easily discriminated from the narrow peak of cells with normal (diploid) DNA content. SRα silenced cells were examined for the content of sub-G1 DNA upon silencing. The results (Fig. 6-B) demonstrate an increase in the number of sub-G1 cells, suggesting that many cells in the population undergo DNA fragmentation and lose DNA fragments as response to SRα silencing. The same experiment was performed for all silencing treatments that were shown to induce SLS, and the results in Figure 6-C indicate increase in sub-G1 population in cells silenced for SRα, SEC61 and SEC63. The TUNEL and sub-G1 assays can also detect DNA fragmentation in necrotic cells. The inter-nucleosomal DNA digestion by endogenous nucleases to yield a characteristic laddering pattern is one of the hallmarks of apoptotic cells that does not take place in necrotic cells [47]. Laddering of DNA is one of the final steps in the apoptotic process, together with chromatin condensation, nuclear picnosis and the formation of apoptotic bodies. The results in Figure 6-D demonstrate DNA laddering in SRα silenced cells, and that laddering is increased during SLS induction. The maximal laddering is observed 4 days after the induction of silencing. Identical DNA laddering was observed under SEC63 silencing (data not shown). In sum, the results demonstrate that SLS induction leads to DNA fragmentation and laddering, suggesting that SLS-induced procyclic stage trypanosomes undergo PCD.

Phosphatidyl serine (PS) exposure in SLS induced procyclic stage trypanosomes
Induction of apoptosis causes externalization of phosphatidyl serine (PS) on the surface of the apoptotic cells. AnnexinV binds to the exposed PS of apoptotic cells. The dual staining with propidium iodide (PI) enables the distinction between necrotic and apoptotic cells [48]. As indicated in Figure 7-A, necrotic cells lose their membrane integrity and become permeable to PI (Figure 7-A, upper left panel). Live cells are stained neither with AnnexinV nor with PI (bottom left panel). During early apoptosis, PS is exposed on the surface but because of the integrity of the plasma membrane the cells are not be stained with PI (bottom right panel). Late apoptotic or necrotic cells lose their membrane integrity and are stained with both PI and AnnexinV (upper right panel). SLS induced cells - elicited by silencing of SRα, SEC63 and SEC61 or after treatment with DTT - were stained with AnnexinV before the cells became permeable to PI, indicating that PS was externalized. These data suggest that apoptosis is induced 2 days after induction of silencing of each of the factors (Fig. 7-Bi-iii, middle column), or 12 hrs after DTT treatment (Fig. 7-Biv). After 3 days of silencing, the apoptotic cells also became permeable to PI (Fig. 7-Bi-iii, right column). Cells treated with DTT for 20 or 24 hrs also became permeable to PI (Fig. 7-Biv). To visualize cells externally stained by AnnexinV, cells were incubated with AnnexinV and PI, and visualized by fluorescence microscopy. The results in Figure 7-C demonstrate that although the cells were not stained by PI, AnnexinV staining was observed only in the silenced cells and not in control, uninduced cells. The results suggest that at early time points of silencing, PS became externally exposed on the cells, most probably as a result of membrane flipping, a signature of apoptosis. Note that, PS exposure was not observed in cells silenced either for SmD1 [49] or PTB2 [50] that are dying as a result of silencing (Figure S1-A).

Cytoplasmic Ca2+ increases in SLS induced cells
Recent studies have shown that an increase in cytoplasmic [Ca2+] occurs both at early and late stages of the apoptotic pathway. Ca2+ release from the ER induced by milder insults promotes cell death through apoptosis [51]. Trypanosomes, like other eukaryotes, maintain a low intracellular concentration of free Ca2+, and changes in the concentration of this ion were shown to affect the virulence of these organisms [52]. Several organelles were demonstrated to transport Ca2+ in an energy dependent manner, including the plasma membrane [53], endoplasmic reticulum [54], mitochondrion [55] and the acidocalcisome [56],[57]. In trypanosomes, the mitochondrion maintains a low resting level of Ca2+, but transiently accumulates large quantities of Ca2+ from the cytosol following Ca2+ influx across the plasma membrane or after release from the acidocalcisome [58]. Indeed, death in T. brucei was shown to be associated with changes in the ability of the mitochondrion to modulate Ca2+ levels [58]. To examine changes in cytoplasmic [Ca2+] during SLS, the level of cytoplasmic Ca2+ was examined using fluo-4-AM [59]. The results, shown in Figure 8-A, demonstrate an increase of cytoplasmic [Ca2+] mostly 42 to 48 hours following induction of SEC63 silencing. An increase in cytoplasmic [Ca2+] was also observed following treatment with DTT (Fig. 8-B). The results suggest that SLS is associated with perturbations of Ca2+ homoeostasis. Note that changes in the cytoplasmic [Ca2+] originate from internal pools, since these changes were unaffected by the presence of external EGTA (Fig. 8-C).

Mitochondria depolarization and reactive oxygen species (ROS) production during SLS
A reduction in mitochondrial membrane potential (ΔΨm) has been observed during apoptosis. Tetramethyl rhodamine methyl-ester (TMRM) is a cationic lipophilic dye that enters cells and reversibly accumulates in the negatively charged mitochondrial matrix, depending on mitochondrial membrane potential [60]. Cells (uninduced) or after 3 days of silencing of SRα, SEC61 or SEC63 were incubated with TMRM, and the fluorescence of the dye was measured by FACS. The results (Fig. 9-Ai) indicate that up to 80% of the cells showed a decrease in membrane potential as a result of the silencing. To examine if membrane depolarization also takes place during DTT treatment, cells were treated with DTT for different time periods, and ΔΨm was measured. The results in Figure 9-Aii suggest that changes in ΔΨm were already observed after 1 hr, and after 3 hours, depolarization was observed in all cells.
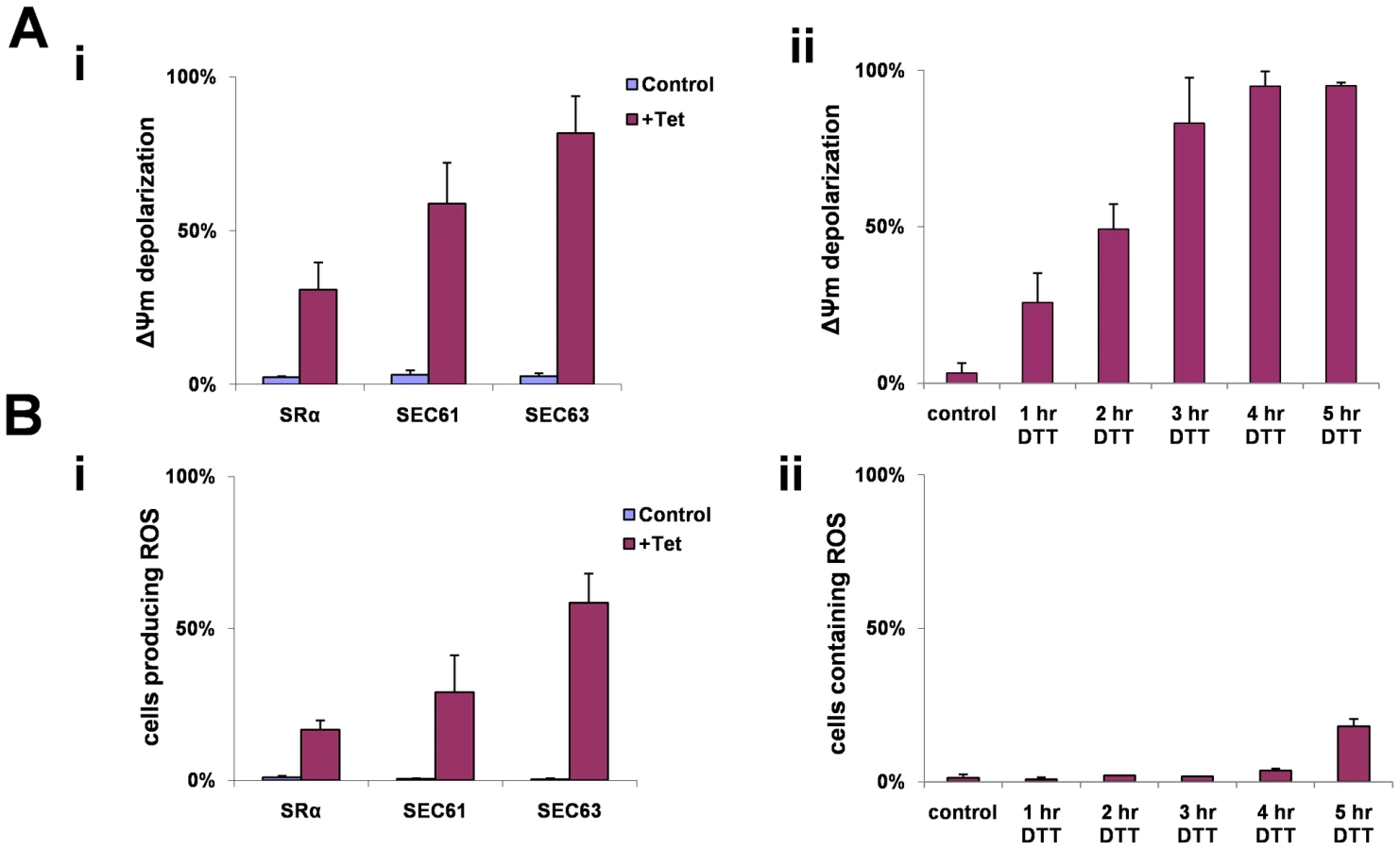
During apoptosis, the inner mitochondrial membrane loses its integrity, and oxidative phosphorylation is uncoupled. When this occurs, oxidation of metabolites by O2 proceeds with electron flux not coupled to proton pumping, resulting in dissipation of the transmembrane proton gradient and ATP production, leading to the production of ROS. To measure ROS production that may result in part from the dissipation of the mitochondrial proton gradient, we used the sensitive probe 2′–7′ dichlorodihydrofluorescein (DCFH-DA). This non-fluorescent dye diffuses across cell membrane and is hydrolyzed to DCFH intracellularly. In the presence of ROS, DCFH is rapidly oxidized to highly fluorescent, dichlorofluorescein [61]. Indeed, ROS production was observed in SRα, SEC61 and SEC63 silenced cells (Fig. 9-Bi). ROS production was also monitored in DTT treated cells (Fig. 9-Bii). Interestingly, ROS production only appeared 5 hrs after DTT addition, although ΔΨm was already decreased after 3 hrs of DTT treatment (Fig. 9-Aii). This delay in observing ROS production after mitochondria permeabilization may reflect the different sensitivity of the assays used to measure ΔΨm and ROS. Note that DTT treatment could also affect ROS measurements because of its reducing capabilities.
One of the characteristic of changes involving the mitochondria during apoptosis is the leakage of proteins such as cytochrome C, which in metazoa, induces the activation of caspases [62]. Recently, endonuclease G (endoG) was described in trypanosomatid species [63]. This enzyme resides in the mitochondria and is released when cell death is triggered. The release of endoG from the mitochondria may represent a caspase-independent cell death mechanism. Since DNA fragmentation was observed in SLS-induced procyclic stage trypanosomes it was of great interest to examine if endoG is released in these cells. Indeed, endoG “leaked” to the cytosol in the SEC63 silenced cells (data not shown).
TEM of SLS induced cells demonstrate morphological changes characteristic of apoptotic cells
Transmission electron microscopy (Fig. 10-A) of procyclic stage trypanosomes after 2 days of SEC63 silencing (v-x) or after 5 and 12 hrs of DTT treatment (xi-xiv, xv-xvii, respectively) show characteristic features of apoptotic cells. Untreated cells show nuclei with prominent central nucleolus (a) and equally distributed chromatin, mitochondria with no dilations (ii, iii), and a narrow ER (iv). In contrast, the SLS induced cells show condensed chromatin (v, vi, xv, xvi), expanded ER (xii, xiii, xiv, xv), and expanded, dilated mitochondria (vii, ix, x, xii, xvi). To verify that the expended organelle is indeed the ER, the procyclic cells slices were reacted with anti-BiP and visualized using gold-labeled antibodies by TEM. The results (xiii, xiv) demonstrate that the expanded compartment contained BiP and is therefore the ER lumen.

Note that chromatin condensation typical to apoptotic cells was not observed in SmD1 silenced cells, although these cells show major morphological perturbations such as accumulation of large vesicles in the cytoplasm (Figure S1-B). Autophagosomes appearing as membrane-surrounded bodies were observed in the cytosol (vii, xi, xvi), as well as inside the mitochondria (viii, xvii). Autophagy is induced as a survival response under starvation conditions [64], but autophagy was shown to regulate the endoplasmic reticulum during UPR [42],[65]. Autophagy involves the massive degradation of organelles such as mitochondria (mitophagy). Autophagy was also shown to induce PCD [66]. To examine if autophagy is induced during SLS, the formation of autophagosome was monitored using tagged ATG8. ATG8 is conserved from yeast to mammals and is commonly used as an autophagosome marker. ATG8 was tagged by in situ insertion of yellow fluorescent protein (YFP) to the authentic site both in parental cells and in cells expressing the SEC63 silencing construct. The results in Figure 10-B demonstrate that upon silencing of SEC63 (Fig. 10-Bi), or prolonged treatment with DTT (Fig. 10-Bii), clear autophagosome formation was observed. The most obvious change was the increase in autophagosomes number and size.
Discussion
In this study, two major questions were addressed. We wished to determine whether trypanosomes are able to change their transcriptome as a result of ER stress like other eukaryotes, and whether this process is mediated or governed by the stress-induced mechanism, SLS [23]. Our results suggest that ER stress induces a very distinct response; the transcriptome is changed in a manner similar to changes observed in other eukaryotes during UPR. The changes in the transcriptome do not result from transcriptional programming as in other eukaryotes, but by stabilizing mRNAs that are needed to execute the ER stress response, resembling the heat-shock response in trypanosomes [19]. However, when ER stress is persistent, SLS is induced, suggesting that SLS does not replace the UPR but triggers programmed cell death under severe ER stress in both stages of the parasite. We therefore suggest that the biological role of SLS is to serve as the apoptotic branch of UPR and induce a very rapid and aggressive PCD response to eliminate unfit parasites from the population.
Comparison of transcriptome during ER stress in trypanosomes to UPR in other eukaryotes
In this study, the changes of the transcriptome upon DTT treatment were compared to similar changes in yeast and metazoa. The data show that procyclic stage trypanosomes are able to change their transcriptome in response to ER stress. The most obvious change is the up-regulation of genes that are involved in protein secretory functions and the down-regulation of genes harboring signal-peptide and transmembrane domains. In yeast, the majority of the up-regulated genes play a role in secretion, phospholipid metabolism, protein glycosylation, vesicular transport, cell wall biosynthesis, vacuolar targeting and ER-associated degradation [3]. All together the UPR increased the level of proteins that lead to enlargement of the ER lumen, enhance export of the misfolded proteins for degradation [endoplasmic reticulum associated degradation (ERAD)] or enhance their transport from the ER via the vesicular transport. Very similar data was observed during UPR in C. elegans. [33]. In human cells, genes involved in targeting of proteins to the ER are induced, but most of these transcripts are only slightly induced, suggesting that modest changes in the level of their transcripts are sufficient for an adaptive response to ER stress [35]. In organisms such as S. cerevisiae, D. melanogaster and C. elegans, the specificity of the transcriptome changes during UPR was refined by examining the effect in IRE/XBP-1 mutants and their homologues [3],[33],[34]. In trypanosomes the specificity of the response to ER stress was refined only by kinetic analysis of the induced genes and therefore not all the affected genes might be directly linked to the ER stress response. However, the results indicate that in trypanosomes, the largest group of up-regulated genes, function in protein secretion and ER related functions similar to the proportion of such genes in yeast, fly, worm and human under UPR. Note, that as opposed to yeast, in trypanosomes, a large number of genes involved in signaling (17% vs. 8%) were up-regulated during the ER stress response, suggesting that signaling pathways are activated to execute this response. Of interest are also genes involved in movement, which resembles the signaling of chemotaxis in bacteria, enabling the microorganism to escape from chemical stressors by moving to a different microenvironment. Another important response of the UPR described in D. melanogaster suggest that IRE1 mediates the rapid degradation of a specific subset of mRNAs, encoding for genes harboring signal-peptide or transmembrane domains. This response helps to reduce the burden on the translocation and folding machinery [34]. Indeed, among the trypanosome genes that are down-regulated under ER stress, significant numbers contain signal-peptide or transmembrane domains.
ER stress in trypanosomes
A recent study investigated the changes of T. brucei transcriptome during development as well as in response to tunicamycin or DTT treatments in bloodstream form trypanosomes [24]. This study reached the conclusion that while trypanosomes regulate mRNA abundance during the developmental cycle, they do not show significant changes in response to ER stress. The study failed to detect changes in BiP in response to ER stress and concluded that UPR does not exist in trypanosomes, primarily because these parasites live in homeostatic conditions, especially in the mammalian host. Our study reached an opposite conclusion. First, we observed major changes in the transcriptome, and, in addition, we observed changes in BiP expression during ER stress in both procyclic and bloodstream form trypanosomes. Moreover, we observed ER dilation at the same time at which we observed BiP up-regulation (1 and 5 hours) [(Fig. 3Bii, Fig. 10A-(xii, xiii, xiv)]. Note that the discrepancy between these studies can be easily reconciled given the fact that different DTT concentrations were used (1mM DTT vs. 4 mM DTT in our study). As noted in the previous study and as presented here, prolonged exposure to ER stress induces death. Here, we show that this death is associated with SLS during the entire life cycle of the parasite.
Another recent study on the enzyme UDP-glucose:glycoprotein glucosyltransferase (UGGT) demonstrated that null mutations of this gene in T. brucei bloodstream form do not induce BiP production. These results suggest that proteins involved in ER glycosylation are not tightly regulated in the trypanosome bloodstream form which might reflect the need for an extremely high flux of glycoprotein synthesis and export of VSG [67]. Interestingly, deletion of UGGT in T. cruzi has been reported to increase BiP expression, suggesting that in T. cruzi there is an ER stress response that senses the misfolded proteins and compensates for the lack of UGGT by up-regulating BiP [68]. All these data suggest that the response to ER stress may vary during the life cycle and among trypanosomatid species. However, the SLS induction in both procyclic and bloodstream from parasites as presented in this study, suggest that induction of ER stress by DTT or by major perturbation to the ER integrity and other cellular membranes, induce a systematic response. The response might be induced only under very severe stress that perturbs the membranes, since RNAi silencing of the post-translational pathway by depletion of SEC71 that does not harm membrane protein biogenesis did not induce SLS [46].
How the response to ER stress is induced in trypanosomes, and how the mechanism is related to UPR in other eukaryotes
Conventional transcription regulation is absent in trypanosomes and therefore it was accepted that these organisms lack IRE1 and XBP1. In this study, we demonstrate a unique novel mechanism to induce UPR based on differential stabilization of mRNAs. The ER stress response resembles the heat shock regulation in trypanosomes, which is regulated mostly by mRNA stability and preferential translation [69]. mRNA stability is considered the major post-transcriptional mechanism that determines the trypanosome transcriptome [19]. The mechanism of the preferential stabilization under ER stress observed in this study is currently unknown. Stability of particular mRNAs can be regulated by two different mechanisms. RNA binding proteins can either stabilize the mRNAs under ER stress or a protein that normally destabilizes the mRNA can be inactivated during stress. Future challenges will be to identify the target site on the mRNAs that are the target of this mechanism, and further, to identify the cognate RNA binding protein(s) that mediates this regulation.
PERK is one of the key factors regulating UPR in metazoa. As opposed to yeast, which lack PERK, trypanosomes possess three potential PERK-homologues (TbITF2K1-K3) [70]. However, only one of these kinases, TbeIF2K2, carries a transmembrane domain and is able to phosphorylate eIF2α on Thr169, homologues to Ser51 of other eukaryotes [70]. This kinase level is reduced in high density culture but there is no evidence that this kinase participates in the ER stress response. Unexpectedly, the protein was localized to the flagellar pocket of the parasite [70]. No change in protein synthesis arrest as a result of DTT treatment was observed in cells silenced for this factor using RNAi (our unpublished results). Thus, TbeIF2K2 is most probably not involved in ER stress. Note that heat-shock in trypanosomes causes polysome collapse and translational shut-off independently of eIF2α phosphorylation, which takes place during the heat-shock response in other eukaryotes [69]. It remains possible that the other TbeIF2K kinases [70] function in ER stress. However, these kinases lack a transmembrane domain, and to function in the ER stress response these kinases must somehow associate with the ER membrane. Studies are in progress to decipher the possible role, if any, of these two kinases (TbeIF2K 1, 3) in ER stress and/or SLS.
UPR was shown to induce autophagy in yeast and mammals [65]. It was therefore interesting to observe autophagy in SLS-induced cells. The autophagy pathway functions as backup mechanism for ERAD, when substrates destined for degradation overwhelm the ERAD capacity [65]. This could also be the function of autophagy under ER stressors or silencing of SEC63 observed in this study.
SLS with respect to other reported PCD inducers in trypanosomes
The presence of PCD in unicellular organisms and in trypanosomatids was initially viewed with skepticism [26]. The major argument against the presence of PCD in these organisms is the absence in trypanosomes of caspases that function in PCD. However, PCD was shown to be induced by prostaglandin D2 (PGD2), a molecule secreted primarily by the stumpy form of the parasite [71]. Trypanosomes control their density within the mammalian host by secreting a quorum sensing molecule (SIF) [72]. Slender parasites secrete SIF, which induces differentiation to the stumpy form. Stumpy parasites are pre-adapted for transmission to the fly but respond to PGD2 by PCD in the blood. Thus, the altruistic removal of stumpy forms assures persistent infection [71],[73]. The PCD associated with the prostaglandins involves an increase in ROS. It was suggested that trypanosomes might exhibit a primitive form of apoptosis that does not depend on proteases but instead depends on ROS formation [26]. This ROS signal can lead to stabilization of mRNAs encoding for proteases and nucleases that are needed to execute PCD. The death pathway executed by ER stress via SLS might be another altruistic pathway present in these parasites that was developed to remove the unfit parasites from the population under stress.
Is SLS essential for PCD or is it stimulated to enhance PCD?
One can envision a mechanism by which ER stress induces imbalance of Ca2+ homeostasis. The increase in cytoplasmic [Ca2+] is most probably due to leakage from the malfunctioning ER, resulting from the reduced capacity of the ER to store Ca2+. This decreased capacity might stem from reduced levels of calreticulin and the ER-resident SERCA calcium pump, as well as the acidocalcisome Ca2+ transporters [74],[75]. These proteins either carry a signal-peptide or are polytopic membrane proteins. Indeed, major defects in biogenesis of both polytopic membrane proteins and signal peptide-containing proteins were observed in SEC63 and SEC61 silenced cells [46]. The ER stress induced by DTT and 2-DG may also affect the activity of the ER chaperone and the Ca2+ pumps.
In eukaryotes, Ca2+ from the ER or cytosol moves to the mitochondrial outer membrane through voltage dependent ion channel (VDAC) [76]. This leads to induced opening of the mitochondrial permeability transition pore (PTP) resulting in matrix swelling [51],[77]. Such changes cause the rupture of the outer membrane of the mitochondria, and release of apoptotic factors such as cytochrome C [78]. The rise in the mitochondrial Ca2+ stimulates the generation of ROS, and the opening of PTP causes dissipation of the ΔΨm, as was also observed in SLS-induced cells.
The PCD observed by silencing of ER functions (SRα, SEC61, SEC63) or by ER stressors can be explained by perturbations induced by Ca2+, leading to ROS production [79]. If so, what is the role of SLS and why is it required? We propose that SLS function is to speed up the death process. SLS is induced when response to ER stress fails to restore homeostasis, and it resembles apoptosis that takes place in mammalian cells under persistent ER stress [13],[80].
Very little is known about how the switch between protective and destructive pathways is regulated in eukaryotes. In metazoa a defined regulatory circuit that controls the switch from protective UPR to the pro-apoptotic pathway exists [81]. It was suggested that transient PERK signaling protects cells by temporarily dampening cellular protein synthesis and thus reducing the misfolded protein level in the ER. However, persistent PERK signaling could ultimately impair cell viability if extended translational inhibition interrupted the generation of proteins vital for cellular homeostasis [81]. An analogous situation may exist in trypanosomes. The severe inhibition of protein synthesis emerging from the drastic decrease in all mRNAs due to elimination of trans-splicing by SLS may lead to drastic reduction in the synthesis of proteins that are also required for the response to ER stress, and may resemble the physiological effects elicited by prolonged PERK induction in metazoa. SLS may accelerate cell death, rapidly eliminating unfit organisms from the population. One of the most intriguing and yet unresolved question is the nature of the signaling pathway that senses ER stress and transmits it to the nucleus to initiate the SLS response.
Materials and Methods
Cell growth and transfection
Procyclic forms of T. brucei strain 29–13 which carries integrated genes for T7 polymerase and the tetracycline repressor [82], were grown in SDM-79 [83] supplemented with 10% fetal calf serum in the presence of 50 µg/ml hygromycin B and 15 µg/ml G418.
The bloodstream form (BSFs) of T. brucei strain 427, cell line 1313–514 (a gift from C. Clayton, ZMBH, Heidelberg, Germany) [84] were aerobically cultivated at 37°C under 5% CO2 in HMI-9 medium [85] supplemented with 10% fetal calf serum containing 2 µg ml−1 G418 and 2.5 µg ml−1 phleomycin.
The oligonucleotides used in this study are listed in Table S1. Generation of the stem-loop silencing construct for SEC61α (Tb11.02.4100) using the oligonucleotides 104–106 was performed as previously described [86]. The construction of SEC63 and SRα silencing constructs was previously described [23],[46]. The construct to silence SEC63 in BSF was prepared by cloning a polymerase chain reaction (PCR) product generated using oligonucleotides 1111–1112 to the p2T7TA-177 vector. The transfection was performed as previously described [87]. Transgenic cell lines were selected on hygromycin B. To generate the YFP–ATG8 (Tb927.7.5910) fused protein, a PCR fragment was amplified using the oligonucleotides 829–830, covering positions 1 (first ATG) to the end of the protein. The vector, p2829 YFP-Dhh1, was digested with HindIII and NotI, and the fragment coding for ATG8 was cloned using the same sites [88]. The cloning creates an N-terminal fusion, and the HindIII site was used for fusing the gene and YFP. For integration, the plasmid was linearized with XcmI at position 276 from the ATG. Cloned transgenic cell lines were selected on blasticidin.
Microarray analysis
Total RNA was isolated from untreated cells and cells treated with DTT for 1 or 3 hours. RNA quality was determined using the Eukaryote Total RNA Nano 6000 assay kit (Agilent Technologies) on the Agilent Technologies 2100 Bioanalyzer. To generate the fluorescently labeled cRNA, total RNA was labeled using the Ambion Amino Allyl MessageAmp II aRNA kit (Ambion). A classical balanced block design with dye swap was used for comparing between RNA extracted following 1 hr DTT treatment, or 3 hr DTT treatment, and RNA extracted from uninduced cells. Arrays were blocked with 1% BSA, 0.1% SDS, 53 SSC at 42°C for 1 h, rinsed with double-distilled water, and air-dried. Next, 1.5 µg of cyanine 3-labeled and 1.5 µg of cyanine 5-labeled aRNA were fragmented and hybridized to the T. brucei DNA microarrays obtained through NIAID's Pathogen Functional Genomics Resource Center (managed and funded by the Division of Microbiology and Infectious Diseases, NIAID, NIH, DHHS, and operated by the J. Craig Venter Institute) using the Gene Expression hybridization kit (Agilent Technologies) for 16 h at 60°C in a hybridization oven. After hybridization, each array was subjected to three washes: first wash with 2× SSC, 0.1% SDS for 1 min at 55°C, and then 0.1× SSC, 0.1% SDS for 1 min, and finally with 0.1× SSC for 1 min, at room temperature. Arrays were scanned using the dual laser scanner (Agilent G2505B Microarray scanner). The data were then extracted from images using Spotreader software (Niles Scientific). Flawed features identified by visual inspection of the array images were flagged, and the data from these features were removed prior to further analysis.
Northern blot analysis
Total RNA was prepared with Trizol reagent and 20 µg/lane was separated on a 1.2% agarose gel, containing 2.2 M formaldehyde. Small RNAs were fractionated on a 6% (w/v) polyacrylamide gel containing 7M urea. The RNA blots were hybridized to oligonucleotides or [α-32P]-random labeled probes (Random Primer DNA Labeling Mix, Biological Industries, Co.). The oligonucleotides used to prepare the probes are detailed in Table S1.
mRNA stability analysis
Untreated cells and cells after 1.5 hour of DTT treatment (107 cells/ml) were divided into 50 ml aliquots for each time point, concentrated and resuspended in 5 ml of the remaining growth medium. The concentrated cells were incubated for 30 min at 27°C, treated with 2 µg/ml sinefungin (Sigma) for 10 min, and then 30 µg/ml of actinomycin D were added (Sigma). Aliquots were taken at different time points (0–90 min). For each time point, total RNA was prepared and the samples were subjected to Northern analysis. Quantization by densitometry was performed using ImageJ software, and the results were used for calculating the half-life of the mRNAs.
Western blot analysis
Whole cell lysates (106 cell equivalents per lane) were fractionated by SDS-PAGE, transferred to PROTRAN membranes (Whatman), and probed with anti BiP (diluted 1∶5000), kindly provided by Prof. James Bangs (University of Wisconsin-Madison, Madison, USA); anti tSNAP42 antisera (diluted 1∶7500) kindly provided by Prof. Vivian Bellofatto (UMDNJ - NJ, USA); and anti p58 antibodies raised by our group against a cytoplasmic protein Tb11.46.0009 (1∶4000). The bound antibodies were detected with goat anti-rabbit immunoglobulin G (IgG) or anti-mouse IgG coupled to horseradish peroxidase, and were visualized by ECL (Amersham Biosciences).
Immunofluorescence assay
Cells were washed with PBS, mounted on poly-L-lysine-coated slides, fixed in 4% formaldehyde, and immunofluorescence was performed as described in [23] using anti tSNAP42 antisera. The cells were visualized with a Zeiss LSM 510 META inverted microscope.
Propidium Iodide (PI) staining for FACS analysis
T. brucei cells, silenced for various genes, were fixed in 70% Ethanol-30% PBS and stored at 4°C overnight. Cells were then washed once with PBS and incubated on ice for 30 minutes to enable rehydration. The samples were resuspended in PBS containing 50 µg/ml RNase A (Roche Diagnostics) for 30 minutes at 4°C and stained with 50 µg/ml Propidium Iodide (Sigma). Samples were analyzed by FACS using the FACStar plus (Becton Dickinson, San Jose, CA) and the CellQuest list mode analysis software.
Measurement of mitochondrial membrane potential
T. brucei cells following the different treatments were harvested and loaded with 150 nM TMRM (Invitrogen) in serum-free medium. The samples were incubated in the dark for 15 min at 27°C and then analyzed by FACS.
Detection of intracellular reactive oxygen species
Reactive oxygen species (ROS) were measured by flow cytometry using DCFH-DA (Sigma). Following the different treatments cells were harvested and incubated at 27°C for 30 min with 10 µM DCFH-DA. The cells were then washed once with PBS and analyzed by FACS using the CellQuest list mode analysis software.
Measurement of intracellular calcium
For calcium measurements, 106 cells were loaded with 1 µM Fluo-4-AM (Invitrogen) for 1 hour at 27°C. The cells were washed three times with PBS and resuspended in PBS. Staining was evaluated by FACS. Results were analyzed using FlowJo software.
DNA laddering assay
Cells were pelleted by centrifugation, washed with PBS, resuspended in extraction buffer (10 mMTris pH 8.0, 0.1 mM EDTA pH 8.0, 20 µg/ml RNase, 0.5% SDS), incubated at 37°C for 1 hour, and for an additional 3 hours at 50°C in the presence of 4 µg/ml proteinase K. After phenol:chloroform (1∶1 V/V) and chloroform extractions, and ethanol precipitation, the DNA was treated with 10 µg/ml RNaseA for 1 hour at 37°C and 10 µg was electroporated on 1.8% Agarose gel containing 0.5 µg/ml ethidium bromide and visualized under UV light.
TUNEL assay
Detection of cleavage of genomic DNA was performed by using the In-Situ Cell Death Detection Kit, FITC (Roche Molecular Biochemicals) according to manufacturer's instructions.
Phosphatidylserine exposure assay
Trypanosomes were reacted with fluorescein isothiocyanate-labeled Annexin V antibodies (MBL©) and stained with propidium iodide according to the manufacturer's instructions. The cells were analyzed by FACS or visualized under a Zeiss LSM 510 META inverted microscope.
Transmission electron microscopy
Trypanosomes were fixed with Karnovsky (4% paraformaldehyde, 2.5% glutaraldehyde) solution for 1 h at room temperature, and then at 4°C over night. Thereafter, the cells were incubated with 1% OsO4 for 1 h at 4°C. Samples were dehydrated in ethanol and embedded in Epon-812 according to standard procedures. Ultrathin sections were prepared using a LKB Ultratome III, stained with uranyl acetate and contrasted with lead citrate. The samples were visualized using a transmission electron microscope FEI Tecnai 120 kV.
Immuno-electron microscopy
Trypanosomes were fixed with Karnovsky (2% paraformaldehyde, 0.5% glutaraldehyde) solution for 1 h at room temperature, and then at 4°C over night. Thereafter, the cells were incubated with 1% OsO4 for 1 h at 4°C. Samples were dehydrated in ethanol and embedded in Epon-812 according to standard procedures. Ultrathin sections were prepared using a LKB Ultratome III. The sections were floated in blocking buffer (10mM Tris pH 8.5, 0.05% Tween-20, 5% non-fat dry milk) for 30 minutes and then incubated with rabbit anti BiP antibodies, kindly provided by Prof. James Bangs (University of Wisconsin-Madison, Madison, USA) diluted (1∶20) in the blocking buffer (4°C, O.N) washed with wash buffer (10 mM Tris pH 8.5, 0.05% Tween 20) and incubated with Donkey anti rabbit IgG (H+L) conjugated to 18 nm gold particles (Jackson immune-research cat.# 711-215-152, diluted 1∶100) and washed again with wash buffer. The grids were stained with uranyl acetate. The samples were visualized using a transmission electron microscope FEI Tecnai 120 kV.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. MalhotraJD
KaufmanRJ
2007 The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol 18 716 731
2. VembarSS
BrodskyJL
2008 One step at a time: endoplasmic reticulum-associated degradation. Nat Rev Mol Cell Biol 9 944 957
3. TraversKJ
PatilCK
WodickaL
LockhartDJ
WeissmanJS
2000 Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 101 249 258
4. RonD
WalterP
2007 Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8 519 529
5. BernalesS
PapaFR
WalterP
2006 Intracellular signaling by the unfolded protein response. Annu Rev Cell Dev Biol 22 487 508
6. KawaharaT
YanagiH
YuraT
MoriK
1997 Endoplasmic reticulum stress-induced mRNA splicing permits synthesis of transcription factor Hac1p/Ern4p that activates the unfolded protein response. Mol Biol Cell 8 1845 1862
7. CoxJS
ChapmanRE
WalterP
1997 The unfolded protein response coordinates the production of endoplasmic reticulum protein and endoplasmic reticulum membrane. Mol Biol Cell 8 1805 1814
8. CalfonM
ZengH
UranoF
TillJH
HubbardSR
2002 IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415 92 96
9. YoshidaH
MatsuiT
HosokawaN
KaufmanRJ
NagataK
2003 A time-dependent phase shift in the mammalian unfolded protein response. Dev Cell 4 265 271
10. HazeK
YoshidaH
YanagiH
YuraT
MoriK
1999 Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell 10 3787 3799
11. LeeK
TirasophonW
ShenX
MichalakM
PrywesR
2002 IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev 16 452 466
12. YeJ
RawsonRB
KomuroR
ChenX
DaveUP
2000 ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell 6 1355 1364
13. MorishimaN
NakanishiK
TakenouchiH
ShibataT
YasuhikoY
2002 An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J Biol Chem 277 34287 34294
14. RaoRV
Castro-ObregonS
FrankowskiH
SchulerM
StokaV
2002 Coupling endoplasmic reticulum stress to the cell death program. An Apaf-1-independent intrinsic pathway. J Biol Chem 277 21836 21842
15. LiangXH
HaritanA
UlielS
MichaeliS
2003 trans and cis splicing in Trypanosomatids: mechanism, factors, and regulation. Eukaryot Cell 2 830 840
16. AgabianN
1990 Trans splicing of nuclear pre-mRNAs. Cell 61 1157 1160
17. FeaginJE
1990 RNA editing in kinetoplastid mitochondria. J Biol Chem 265 19373 19376
18. StuartKD
SchnauferA
ErnstNL
PanigrahiAK
2005 Complex management: RNA editing in trypanosomes. Trends Biochem Sci 30 97 105
19. ClaytonC
ShapiraM
2007 Post-transcriptional regulation of gene expression in trypanosomes and leishmanias. Mol Biochem Parasitol 156 93 101
20. DasA
ZhangQ
PalencharJB
ChatterjeeB
CrossGA
2005 Trypanosomal TBP functions with the multisubunit transcription factor tSNAP to direct spliced-leader RNA gene expression. Mol Cell Biol 25 7314 7322
21. SchimanskiB
NguyenTN
GunzlA
2005 Characterization of a multisubunit transcription factor complex essential for spliced-leader RNA gene transcription in Trypanosoma brucei. Mol Cell Biol 25 7303 7313
22. CampbellDA
ThomasS
SturmNR
2003 Transcription in kinetoplastid protozoa: why be normal? Microbes Infect 5 1231 1240
23. LustigY
SheinerL
VagimaY
GoldshmidtH
DasA
2007 Spliced-leader RNA silencing: a novel stress-induced mechanism in Trypanosoma brucei. EMBO Rep 8 408 413
24. KoumandouVL
NatesanSK
SergeenkoT
FieldMC
2008 The trypanosome transcriptome is remodelled during differentiation but displays limited responsiveness within life stages. BMC Genomics 9 298
25. DuszenkoM
FigarellaK
MacleodET
WelburnSC
2006 Death of a trypanosome: a selfish altruism. Trends in Parasitology 22 536 542
26. WelburnSC
MacleodE
FigarellaK
DuzenskoM
2006 Programmed cell death in African trypanosomes. Parasitology 132 Suppl S7 S18
27. GonzalezIJ
DespondsC
SchaffC
MottramJC
FaselN
2007 Leishmania major metacaspase can replace yeast metacaspase in programmed cell death and has arginine-specific cysteine peptidase activity. Int J Parasitol 37 161 172
28. HelmsMJ
AmbitA
AppletonP
TetleyL
CoombsGH
2006 Bloodstream form Trypanosoma brucei depend upon multiple metacaspases associated with RAB11-positive endosomes. J Cell Sci 119 1105 1117
29. LeeN
GannavaramS
SelvapandiyanA
DebrabantA
2007 Characterization of metacaspases with trypsin-like activity and their putative role in programmed cell death in the protozoan parasite Leishmania. Eukaryot Cell 6 1745 1757
30. AbrahamMC
ShahamS
2004 Death without caspases, caspases without death. Trends Cell Biol 14 184 193
31. BesteiroS
WilliamsRA
MorrisonLS
CoombsGH
MottramJC
2006 Endosome sorting and autophagy are essential for differentiation and virulence of Leishmania major. J Biol Chem 281 11384 11396
32. WilliamsRA
WoodsKL
JulianoL
MottramJC
CoombsGH
2009 Characterization of unusual families of ATG8-like proteins and ATG12 in the protozoan parasite Leishmania major. Autophagy 5 159 172
33. ShenX
EllisRE
SakakiK
KaufmanRJ
2005 Genetic interactions due to constitutive and inducible gene regulation mediated by the unfolded protein response in C. elegans. PLoS Genet 1 e37 doi:10.1371/journal.pgen.0010037
34. HollienJ
WeissmanJS
2006 Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 313 104 107
35. MurrayJI
WhitfieldML
TrinkleinND
MyersRM
BrownPO
2004 Diverse and specific gene expression responses to stresses in cultured human cells. Mol Biol Cell 15 2361 2374
36. ColasanteC
RoblesA
LiCH
SchwedeA
BenzC
2007 Regulated expression of glycosomal phosphoglycerate kinase in Trypanosoma brucei. Mol Biochem Parasitol 151 193 204
37. EllgaardL
MolinariM
HeleniusA
1999 Setting the standards: quality control in the secretory pathway. Science 286 1882 1888
38. RutkowskiDT
ArnoldSM
MillerCN
WuJ
LiJ
2006 Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol 4 e374 doi:10.1371/journal.pbio.0040374
39. LeeAS
2005 The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods 35 373 381
40. DatemaR
LezicaRP
RobbinsPW
SchwarzRT
1981 Deoxyglucose inhibition of protein glycosylation: effects of nucleotide deoxysugars on the formation of glucosylated lipid intermediates. Arch Biochem Biophys 206 65 71
41. BackSH
SchroderM
LeeK
ZhangK
KaufmanRJ
2005 ER stress signaling by regulated splicing: IRE1/HAC1/XBP1. Methods 35 395 416
42. BernalesS
McDonaldKL
WalterP
2006 Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol 4 e423 doi:10.1371/journal.pbio.0040423
43. BommiasamyH
BackSH
FagoneP
LeeK
MeshinchiS
2009 ATF6alpha induces XBP1-independent expansion of the endoplasmic reticulum. J Cell Sci 122 1626 1636
44. TkaczID
LustigY
SternMZ
BitonM
Salmon-DivonM
2007 Identification of novel snRNA-specific Sm proteins that bind selectively to U2 and U4 snRNAs in Trypanosoma brucei. RNA 13 30 43
45. HuryA
GoldshmidtH
TkaczID
MichaeliS
2009 Trypanosome spliced-leader-associated RNA (SLA1) localization and implications for spliced-leader RNA biogenesis. Eukaryot Cell 8 56 68
46. GoldshmidtH
SheinerL
ButikoferP
RoditiI
UlielS
2008 Role of protein translocation pathways across the endoplasmic reticulum in Trypanosoma brucei. J Biol Chem 283 32085 32098
47. DarzynkiewiczZ
BednerE
SmolewskiP
2001 Flow cytometry in analysis of cell cycle and apoptosis. Semin Hematol 38 179 193
48. van EngelandM
NielandLJ
RamaekersFC
SchutteB
ReutelingspergerCP
1998 Annexin V-affinity assay: a review on an apoptosis detection system based on phosphatidylserine exposure. Cytometry 31 1 9
49. MandelboimM
BarthS
BitonM
LiangXH
MichaeliS
2003 Silencing of Sm proteins in Trypanosoma brucei by RNA interference captured a novel cytoplasmic intermediate in spliced leader RNA biogenesis. J Biol Chem 278 51469 51478
50. SternMZ
GuptaSK
Salmon-DivonM
HahamT
BardaO
2009 Multiple roles for polypyrimidine tract binding (PTB) proteins in trypanosome RNA metabolism. RNA 15 648 665
51. PintonP
GiorgiC
SivieroR
ZecchiniE
RizzutoR
2008 Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 27 6407 6418
52. LuHG
ZhongL
ChangKP
DocampoR
1997 Intracellular Ca2+ pool content and signaling and expression of a calcium pump are linked to virulence in Leishmania mexicana amazonesis amastigotes. J Biol Chem 272 9464 9473
53. BenaimG
Lopez-EstranoC
DocampoR
MorenoSN
1993 A calmodulin-stimulated Ca2+ pump in plasma-membrane vesicles from Trypanosoma brucei; selective inhibition by pentamidine. Biochem J 296 (Pt3) 759 763
54. NolanDP
ReverlardP
PaysE
1994 Overexpression and characterization of a gene for a Ca(2+)-ATPase of the endoplasmic reticulum in Trypanosoma brucei. J Biol Chem 269 26045 26051
55. MorenoSN
DocampoR
VercesiAE
1992 Calcium homeostasis in procyclic and bloodstream forms of Trypanosoma brucei. Lack of inositol 1,4,5-trisphosphate-sensitive Ca2+ release. J Biol Chem 267 6020 6026
56. DocampoR
MorenoSN
1999 Acidocalcisome: A novel Ca2+ storage compartment in trypanosomatids and apicomplexan parasites. Parasitol Today 15 443 448
57. DocampoR
ScottDA
VercesiAE
MorenoSN
1995 Intracellular Ca2+ storage in acidocalcisomes of Trypanosoma cruzi. Biochem J 310 ( Pt3) 1005 1012
58. RidgleyEL
XiongZH
RubenL
1999 Reactive oxygen species activate a Ca2+-dependent cell death pathway in the unicellular organism Trypanosoma brucei brucei. Biochem J 340 ( Pt1) 33 40
59. GeeKR
BrownKA
ChenWN
Bishop-StewartJ
GrayD
2000 Chemical and physiological characterization of fluo-4 Ca(2+)-indicator dyes. Cell Calcium 27 97 106
60. FlorykD
HoustekJ
1999 Tetramethyl rhodamine methyl ester (TMRM) is suitable for cytofluorometric measurements of mitochondrial membrane potential in cells treated with digitonin. Biosci Rep 19 27 34
61. BassDA
ParceJW
DechateletLR
SzejdaP
SeedsMC
1983 Flow cytometric studies of oxidative product formation by neutrophils: a graded response to membrane stimulation. J Immunol 130 1910 1917
62. LiuX
KimCN
YangJ
JemmersonR
WangX
1996 Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86 147 157
63. GannavaramS
VedvyasC
DebrabantA
2008 Conservation of the pro-apoptotic nuclease activity of endonuclease G in unicellular trypanosomatid parasites. J Cell Sci 121 99 109
64. LevineB
2005 Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell 120 159 162
65. YorimitsuT
NairU
YangZ
KlionskyDJ
2006 Endoplasmic reticulum stress triggers autophagy. Journal of Biological Chemistry 281 30299 30304
66. GozuacikD
KimchiA
2007 Autophagy and cell death. Curr Top Dev Biol 78 217 245
67. IzquierdoL
AtrihA
RodriguesJA
JonesDC
FergusonMA
2009 Trypanosoma brucei UDP-glucose:glycoprotein glucosyltransferase has unusual substrate specificity and protects the parasite from stress. Eukaryot Cell 8 230 240
68. ConteI
LabriolaC
CazzuloJJ
DocampoR
ParodiAJ
2003 The interplay between folding-facilitating mechanisms in Trypanosoma cruzi endoplasmic reticulum. Mol Biol Cell 14 3529 3540
69. KramerS
QueirozR
EllisL
WebbH
HoheiselJD
2008 Heat shock causes a decrease in polysomes and the appearance of stress granules in trypanosomes independently of eIF2{alpha} phosphorylation at Thr169. J Cell Sci 121 3002 3014
70. MoraesMC
JesusTC
HashimotoNN
DeyM
SchwartzKJ
2007 Novel membrane-bound eIF2alpha kinase in the flagellar pocket of Trypanosoma brucei. Eukaryot Cell 6 1979 1991
71. FigarellaK
RawerM
UzcateguiNL
KubataBK
LauberK
2005 Prostaglandin D2 induces programmed cell death in Trypanosoma brucei bloodstream form. Cell Death Differ 12 335 346
72. VassellaE
ReunerB
YutzyB
BoshartM
1997 Differentiation of African trypanosomes is controlled by a density sensing mechanism which signals cell cycle arrest via the cAMP pathway. J Cell Sci 110 ( Pt21) 2661 2671
73. FigarellaK
UzcateguiNL
BeckA
SchoenfeldC
KubataBK
2006 Prostaglandin-induced programmed cell death in Trypanosoma brucei involves oxidative stress. Cell Death Differ 13 1802 1814
74. DocampoR
MorenoSN
2001 The acidocalcisome. Mol Biochem Parasitol 114 151 159
75. MorenoSN
DocampoR
2009 The role of acidocalcisomes in parasitic protists. J Eukaryot Microbiol 56 208 213
76. ShimizuS
NaritaM
TsujimotoY
1999 Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 399 483 487
77. JeongSY
SeolDW
2008 The role of mitochondria in apoptosis. BMB Rep 41 11 22
78. LiLY
LuoX
WangX
2001 Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 412 95 99
79. FeissnerRF
SkalskaJ
GaumWE
SheuSS
2009 Crosstalk signaling between mitochondrial Ca2+ and ROS. Front Biosci 14 1197 1218
80. RaoRV
EllerbyHM
BredesenDE
2004 Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ 11 372 380
81. LinJH
LiH
ZhangY
RonD
WalterP
2009 Divergent effects of PERK and IRE1 signaling on cell viability. PLoS ONE 4 e4170 doi:10.1371/journal.pone.0004170
82. WangZ
MorrisJC
DrewME
EnglundPT
2000 Inhibition of Trypanosoma brucei gene expression by RNA interference using an integratable vector with opposing T7 promoters. J Biol Chem 275 40174 40179
83. BrunR
Schonenberger
1979 Cultivation and in vitro cloning or procyclic culture forms of Trypanosoma brucei in a semi-defined medium. Short communication. Acta Trop 36 289 292
84. HaileS
EstevezAM
ClaytonC
2003 A role for the exosome in the in vivo degradation of unstable mRNAs. RNA 9 1491 1501
85. HirumiH
HirumiK
1989 Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J Parasitol 75 985 989
86. LiuL
LiangXH
UlielS
UngerR
UlluE
2002 RNA interference of signal peptide-binding protein SRP54 elicits deleterious effects and protein sorting defects in trypanosomes. Journal of Biological Chemistry 277 47348 47357
87. CominiMA
GuerreroSA
HaileS
MengeU
LunsdorfH
2004 Validation of Trypanosoma brucei trypanothione synthetase as drug target. Free Radic Biol Med 36 1289 1302
88. KellyS
ReedJ
KramerS
EllisL
WebbH
2007 Functional genomics in Trypanosoma brucei: a collection of vectors for the expression of tagged proteins from endogenous and ectopic gene loci. Mol Biochem Parasitol 154 103 109
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 1
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Panton-Valentine Leukocidin Is a Very Potent Cytotoxic Factor for Human Neutrophils
- CD8+ T Cell Control of HIV—A Known Unknown
- Polyoma Virus-Induced Osteosarcomas in Inbred Strains of Mice: Host Determinants of Metastasis
- The Deadly Chytrid Fungus: A Story of an Emerging Pathogen
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy