
β-Neurexin Is a Ligand for the MSCRAMM SdrC
Gram-positive bacteria contain a family of surface proteins that are covalently anchored to the cell wall of the organism. These cell-wall anchored (CWA) proteins appear to play key roles in the interactions between pathogenic organisms and the host. A subfamily of the CWA has a common structural organization with multiple domains adopting characteristic IgG-like folds. The identified microbial surface components recognizing adhesive matrix molecules (MSCRAMMs) belong to this subfamily, as does SdrC from S. aureus. However, an interactive host ligand for the putative MSCRAMM SdrC was not previously identified. We have screened a phage display peptide library and identified a peptide sequence found in β-neurexin that binds SdrC. A synthetic peptide corresponding to the identified sequence as well as a recombinant form of the β-neurexin 1 exodomain binds SdrC with high affinity and specificity. Furthermore, expression of SdrC on bacteria greatly enhances microbial adherence to cultured mammalian cells expressing β-neurexin on their surface. Taken together, our experimental results demonstrate that β-neurexin is a ligand for SdrC. This interaction involves a specific sequence located in the N-terminal region of the mammalian protein and the N2N3 domain of the MSCRAMM. The fact that these two proteins interact when expressed on the appropriate cells demonstrates the functionality of the interaction. Possible implications of this interaction are discussed.
Published in the journal:
. PLoS Pathog 6(1): e32767. doi:10.1371/journal.ppat.1000726
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1000726
Summary
Gram-positive bacteria contain a family of surface proteins that are covalently anchored to the cell wall of the organism. These cell-wall anchored (CWA) proteins appear to play key roles in the interactions between pathogenic organisms and the host. A subfamily of the CWA has a common structural organization with multiple domains adopting characteristic IgG-like folds. The identified microbial surface components recognizing adhesive matrix molecules (MSCRAMMs) belong to this subfamily, as does SdrC from S. aureus. However, an interactive host ligand for the putative MSCRAMM SdrC was not previously identified. We have screened a phage display peptide library and identified a peptide sequence found in β-neurexin that binds SdrC. A synthetic peptide corresponding to the identified sequence as well as a recombinant form of the β-neurexin 1 exodomain binds SdrC with high affinity and specificity. Furthermore, expression of SdrC on bacteria greatly enhances microbial adherence to cultured mammalian cells expressing β-neurexin on their surface. Taken together, our experimental results demonstrate that β-neurexin is a ligand for SdrC. This interaction involves a specific sequence located in the N-terminal region of the mammalian protein and the N2N3 domain of the MSCRAMM. The fact that these two proteins interact when expressed on the appropriate cells demonstrates the functionality of the interaction. Possible implications of this interaction are discussed.
Introduction
The Gram-positive opportunistic pathogen Staphylococcus aureus can cause a spectrum of infections in humans and animals that differ in severity. Some relatively minor skin infections, such as folliculitis, impetigo and cellulitis, can progress to life threatening diseases like sepsis, endocardatis, osteomylitis and pneumonia [1].
The molecular pathogenic mechanisms of different staphylococcal infections are incompletely understood but studies suggested that a critical factor for the pathogenic success of this organism depends on its ability to adhere effectively to multiple host tissues [2],[3],[4]. The adhesins mediating staphylococcal adherence and colonization often target the extracellular matrix of the host and hence belong to the MSCRAMM family [4]. In Gram-positive bacteria, many MSCRAMMs are cell-wall-anchored (CWA) proteins [5],[6] with a similar structural organization [7]. These proteins contain an amino terminal signal sequence followed by an A-region that often harbors the ligand-binding sites. The A-region is comprised of sub-domains (called N-domains) adopting an immunoglobulin G-like (IgG-like) fold [8]. Sometimes the A-region is followed by a B-region containing repeated β-sandwich modules of unknown function. In the case of the Sdr-subfamily of staphylococcal MSCRAMMs, the B-region is accompanied by a repeat (R) domain composed of multiple Ser-Asp dipeptide repeats (SD-repeat or Sdr) (reviewed in [9]).
The ligand-binding activity of SdrG, a fibrinogen (Fg)-binding MSCRAMM of Staphylococcus epidermidis [10], was shown to proceed via a “dock, lock and latch” mechanism [7],[11]. A crystal structure of a Fg-based peptide in complex with the SdrGN2N3 domain suggested that the peptide “docks” into the groove formed between the N2 and N3 domain. Upon binding the C-terminal extension of the N3 domain is redirected to cover and “lock” the ligand peptide in place and further stabilizes the complex by complementing a β-sheet in the N2 domain, thus functionally serving as a “latch” [11]. The back of the latching trench contains a motif, TYTFTDYVD, conserved in several other staphylococcal MSCRAMMs. Genome-based bioinformatics of five Gram-positive bacterial strains revealed that all organisms contain proteins with predicted IgG-like folded domains, an LPXTG-motif and a TYTFTDYVD-motif. Therefore, the “dock, lock and latch” mechanism was proposed as a common ligand binding strategy among these proteins.
In the current study, we used SdrC as bait for screening a phage library displaying 12-mer linear peptides (New England Biolabs Ph.D.™ -12 Phage Display Peptide Library). The sdrC gene is the first gene in a bi - or tri-partite gene cluster that also contains either sdrD or sdrD and sdrE. The predicted domain organization of SdrC is similar to that of SdrG with the N2 and N3 sub-domains of the A-region adopting an IgG-like fold [12],[13].
The phage display strategy, used in this study, identified beta (β) neurexin as a ligand for the orphan MSCRAMM SdrC. Subsequent biochemical and cell biology experiments confirmed that β-neurexin is a ligand for SdrC.
Neurexins are neuronal cell adhesion molecules that interact with neuroligins and appear to play an important role in synapse function (reviewed in [14]). Previously, the neurexins were discovered as the receptor for the neurotoxin α-latrotoxin of the black-widow spider [15].
Results
Validation of the phage display method to identify MSCRAMM ligands
Analysis of peptides or protein segments displaced on the surface of phage is a useful method to identify partners in protein-protein interaction systems [16],[17],[18],[19]. To determine if this method can be used to identify ligands for MSCRAMMs from Gram-positive bacteria, we performed a validation experiment using SdrGN2N3 as bait in a screen of a commercially available 12-mer linear library (New England Biolabs Ph.D.-12 Phage Display Peptide Library). We carried out three successive rounds of panning against immobilized SdrGN2N3. To confirm that the enrichment of the phage clones between panning rounds was the result of specific interactions, the phage pool before and after each step of amplification was also tested for binding to BSA. We found that the number of phage binding to SdrG increased relative to the number of phage binding to BSA (Table 1) suggesting a specific enrichment for SdrG. We then sequenced the DNA purified from ten phage plaques obtained after the third round of panning. Alignment of the inserted amino acid sequences revealed FSARG as a consensus in the majority of the clones (Table 2). This result was consistent with previous results from our laboratory identifying FFSARG as the binding motif in Fg for SdrG [7]. The successful identification of the SdrG binding sequence suggested that phage display of peptide libraries could be used to find binding partners for orphan MSCRAMMs.


Identification of the putative ligand-binding domain of SdrC
Sequence alignments between the A-region of SdrC (SdrCA or SdrC52-496) and previously identified staphylococcal MSCRAMMs revealed a modest identity (less than 20%). In contrast, a comparison of the predicted structure of SdrCA with the determined structures of the crystallized staphylococcal MSCRAMMs ClfA and SdrG pointed to close structural similarities [7],[20],[21]. A sub-segment of the SdrC A-region corresponding to residues 178–496 was predicted by PHYRE (http://www.sbg.bio.ic.ac.uk/phyre/) to adopt a structure similar to those determined for the N2N3 sub-domain of the Fg-binding MSCRAMMs ClfA (PHYRE e = 3.1×10−32) and SdrG (PHYRE e = 9.6 10−32). Similar to the N2N3 domains of ClfA and SdrG, SdrC178–496 likely contains two IgG-like folded domains and has a TYTFTDYVD-like “latching cleft” motif in the predicted N2 sub-domain. Therefore, we designated SdrC178–496 as the N2N3 domain and SdrC52–177 as the N1 domain.
Both SdrC52–496 (SdrCA) and SdrC178–496 (SdrCN2N3) were expressed with an N-terminal His tag in E. coli (Fig. 1A). SDS-PAGE analysis of purified recombinant proteins indicated apparent molecular weights of 60 KDa (Fig. 1B) and 45 KDa, respectively (Fig. 1C). Matrix-assisted laser desorption ionization mass spectrometry suggested that the masses of the purified recombinant proteins are close to the theoretical molecular masses calculated from the primary amino acid sequences (51,194 KDa compared to 50,924 KDa for SdrCA and 38,358 KDa compared to 38,340 KDa for SdrCN2N3). Thus, the recombinant SdrC proteins migrate aberrantly on SDS-PAGE. Aberrant migration on SDS-PAGE is common for the recombinant A-region of MSCRAMMs and thought to be due to their hydrophilic index [10],[12],[22]. The recombinant A-region was quickly degraded to a single proteolytically stable segment with an apparent molecular mass of 45 KDa. The N-terminal fragment released was apparently further degraded and was rarely detected on SDS-PAGE. N-terminal sequence analysis of the truncated, resistant segment revealed that cleavage had occurred between Ala177 and Ala178. Mass spectrometry indicated a molecular weight of 38,358 KDa compared with 38,340 KDa calculated from the predicted amino acid sequence of the stable fragment suggesting that the cleavage event occurred only at the N-terminus of the A-region. Cleavage of the N-terminal sub-segment (N1) of the A-region has also been observed with other recombinant MSCRAMMs (ClfA, ClfB, SdrG) and may be explained by disordered segments in the N1 sub-domains as well as the presence of minute amounts of contaminating proteases in the MSCRAMM preparations [7],[23],[24]. The proteolysis resistant fragment of SdrCA corresponds to the tandem IgG-like domains (N2N3) as predicted by PHYRE (http://www.sbg.bio.ic.ac.uk/phyre/).

Identification of β-neurexin as a potential binding partner for SdrCN2N3 using phage display
We screened the New England Biolabs Ph.D.-12 Phage Display Peptide Library using immobilized recombinant SdrCN2N3 as bait. After the 3rd round of panning, fifty randomly picked plaques were selected and the corresponding phages were screened again for binding to SdrCN2N3, to SdrGN2N3 and to BSA as controls. All clones bound to SdrC, however, phage from eight clones displayed significantly higher binding to SdrCN2N3 compared to SdrGN2N3 and BSA (Fig. 2A). The clone inserts were sequenced and an alignment of the deduced amino acid sequences is shown in Table 3. From these sequences we could identify a degenerate consensus sequence (P,T,A)HH(I,M)HHFH(G,R,S,Q,T,A) which was then used for a pattern search of the human protein database employing an algorithm allowing for zero, one or two residue mismatches. Zero mismatches returned only neurexin 1β, one mismatch returned neurexin 1β and neurexin 2β and two mismatches returned neurexin 1β, neurexin 2β, neurexin 3β and a T-type voltage-dependent calcium channel (Table S1). All proteins returned by the search were screened for the presence of the consensus sequence within the extracellular domain of the protein. As shown in Fig. 2B, an alignment of the N-terminal extracellular segments of the β-neurexin isoforms revealed variations of the consensus sequence identified by phage display. In addition, the peptide sequence displayed by phage number one confirms a sequence found in neurexin 1β and the sequenced displayed by phage eight is very similar to a sequence found in neurexin 3β [25].

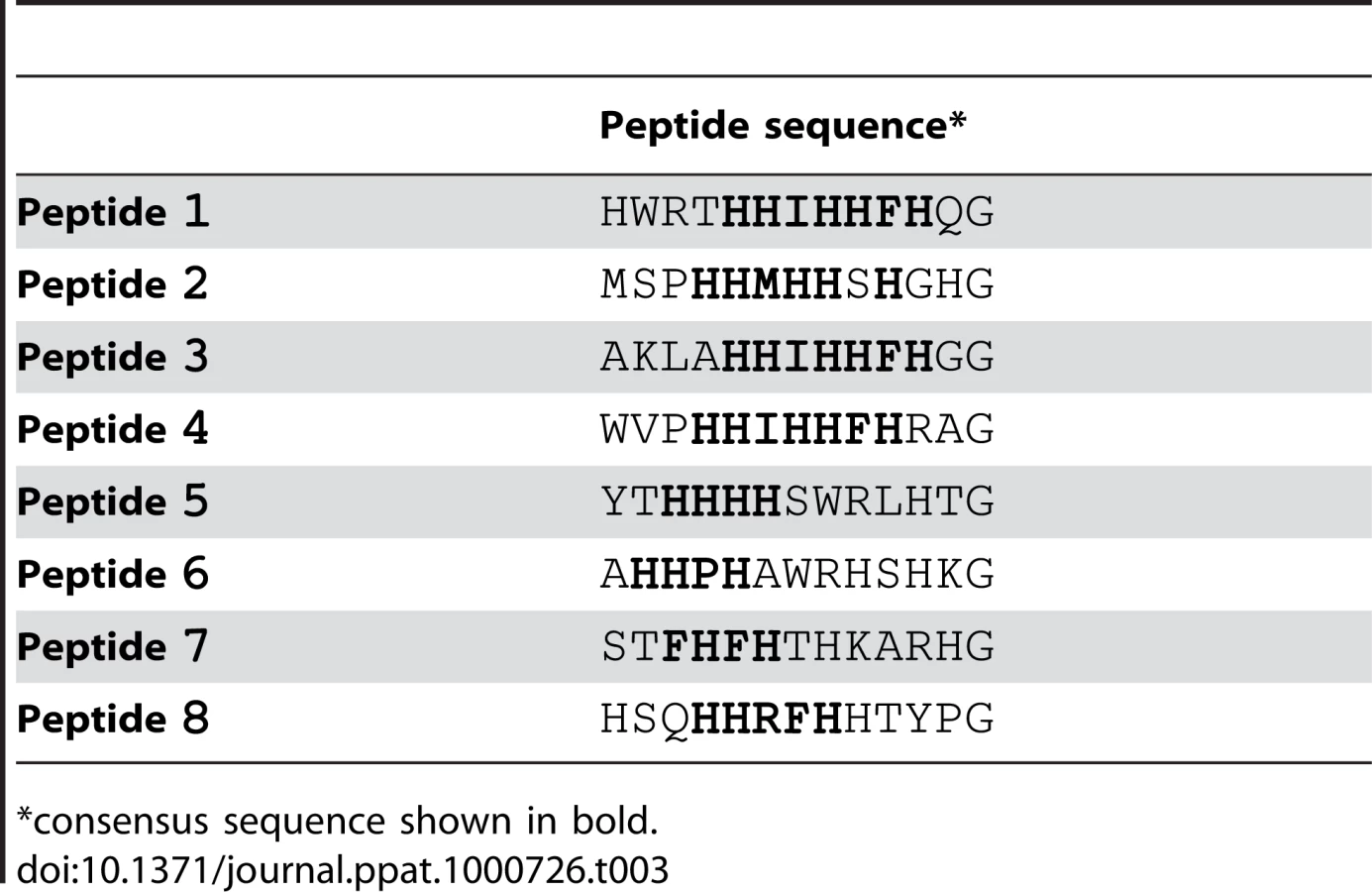
The amino-terminal segments of the β-neurexins contain an unusually long signal sequence (∼50 aa), a neuroligin binding site and a glycosylation site in close proximity to the transmembrane domain. The C-terminal cytoplasmic segment contains a PDZ domain presumably involved in intracellular signaling [25]. The putative SdrC binding site identified by phage display corresponds to amino acid residues 10 to 16 and maps in the neuroligin-binding domain exposed on the cell surface.
Recombinant SdrCN2N3 binds recombinant neurexin 1β (Nrx1β)
The phage display library screening identified beta neurexins as potential binding partners for SdrC. To explore a possible interaction between the recombinant forms of SdrCN2N3 and the exodomain of Nrx1β we expressed in E. coli two recombinant Nrx1β fragments with a C-terminal GST tag (Fig. 3A). The fragment designated Nrx47-255 corresponds to the first 208 amino acids residues of Nrx1β protein which contains the putative SdrC-binding site identified by phage display (amino acid residues 57 through 63). Nrx70-255 is a truncated version of the segment described above in which the putative SdrC-binding site was deleted (Fig. 3B). Both recombinant Nrx1β-GST proteins were immobilized on 96-multiwell plates and used in a solid phase binding assay. SdrCA bound the immobilized Nrx47-255 in a concentration dependent manner but did not bind to either Nrx70-255 or the purified fusion partner GST (Fig. 3C). Similar results were obtained when the intact SdrCN2N3 region (Fig. 3D) or the proteolytically resistant sub-segment released from the recombinant SdrCA (data not shown) were used as probes.

To further demonstrate that HHIHHFH is the specific binding site in Nrx1β for SdrC, we used a synthetic peptide (sp) with a sequence corresponding to the N-terminus of Nrx1β, SLGAHHIHHFHGSSKHHS (spNrxwt), in attempts to inhibit the binding between SdrCN2N3 and Nrx47–255. As a control we used a peptide with the same residues but where the sequence was scrambled to HSHIKLHSHGHSFGHA (spNrxscr). Recombinant SdrCN2N3 (0.5 µM) was incubated with increasing concentration of either peptide prior to being added to the Nrx coated plate. spNrxwt inhibited the interaction between SdrCN2N3 and Nrx47–255 in a concentration dependent manner. We did not observe an inhibition of the MSCRAMM-β-neurexin interaction when spNrxscr was used in the assay (Fig. 3E). The observed biding of recombinant SdrC to Nrx1β and the subsequent synthetic peptide inhibition assay suggests that the interaction between these proteins is specific and that the sequence motif identified in the phage display experiment represents the ligand-binding site in Nrx1β for SdrC.
SdrC binds the Nrxwt peptide with high affinity
We used fluorescence polarization to determine the dissociation constant for the SdrCN2N3-Nrx1β peptide interaction. spNrxwt and spNrxscr were labeled with fluorescein and incubated with increasing concentrations of recombinant MSCRAMM. SdrCN2N3 bound spNrxwt in a concentration dependent, saturable manner with a KD of 2.5±0.5×10-7 M. We did not detect a significant binding of SdrCN2N3 to the fluorescein-labeled spNrxscr. A control MSCRAMM, ClfBN2N3, did not bind to the fluorescein-labeled spNrxwt (Fig. 4A).

To eliminate the possibility that the binding was dependent of the fluorescein label introduced in the wild-type peptide, we tested the ability of unlabeled spNrxwt and spNrxscr to inhibit the binding of SdrCN2N3 to fluorescein-labeled spNrxwt. Increasing concentrations of unlabeled peptides were incubated with SdrCN2N3 for three hours at room temperature before the fluorescent polarization experiment was performed. Fig. 4B shows that the unlabeled spNrxwt but not the spNrxscr inhibited the binding between SdrCN2N3 and the fluorescein-labeled spNrxwt. These results demonstrated that SdrC binds with relatively high affinity to the identified amino acid sequence motif in β-neurexin.
SdrC mediates bacterial attachment to cells expressing Nrx1β
Next we sought to determine if common clinical strains express SdrC and if the SdrC displayed by these isolates are capable of binding Nrx1β. Western blotting analysis of cell wall extractions from USA300, MW2 and MRSA252 display SdrC on their surface during exponential phase of growth in BHI (Fig. 5A1) and RPMI 10% FBS media, and when grown on TSB sheep blood agar (data not shown). SdrC was not found in cell wall extractions from cells in stationary phase of growth (Fig. 5A2). We reasoned that SdrC absence from the cell wall extractions in the late stages of growth might be due to transcription cessation and proteolytical release from the cell wall. Western blotting analysis of concentrated culture supernatants revealed that SdrC was not detected in the supernatant during exponential growth but that all strains tested released SdrC fragments during late stationary growth phase (Fig. 5B1 and Fig. 5B2). SdrC fragments released in the supernatant migrated slower than recombinant SdrCN2N3 on the SDS-PAGE gels suggesting that a larger fragment of the protein is released from the cell wall. Immunoblotting analysis demonstrated that these fragments were recognized by antibodies raised against recombinant N2N3 domains and B-repeats (Fig. 5B3). USA300, MW2 and Newman display and release similar SdrC segments whereas MRSA252 expresses different SdrC species. One possible explanation for this difference may lie in the differences between their amino acid sequences (Fig. S5). These fragments were probed in ligand affinity western blots with recombinant Nrx47–255 protein. Our data indicated that all strains tested release an active form of the SdrC protein capable to interact with Nrx1β (Fig. 5B4). A control membrane not probed with Nrx47–255, did not indicate cross reactivity of anti-GST serum with SdrC fragments released into the supernant (Fig. 5B5).

We next tested whether cell wall anchored SdrC mediates bacterial adherence to Nrx1β-expressing eukaryotic cells. We chose to express both proteins in heterologous systems that do not display any know interaction proteins. The full-length sdrC gene was cloned in pNZ8037 under a nisin inducible promoter. The plasmid was introduced in L. lactis for protein expression. Expression of SdrC on the bacterial cell surface was determined using immunofluorescence microscopy (data not shown).The full length Nrx1β gene was cloned in pCMV5 to obtain the translational fusion Nrx1β mCherry [26] and a mutant FLAG Nrx1β-mCherry where the SdrC binding site was replaced with a FLAG tag. These vectors were transfected into CHOK1 cells which were allowed to grow for 36 hours to ensure protein expression. Transiently transfected cells were incubated with L. lactis transformed with the vector alone, L. lactis expressing SdrC, S. aureus Newman, S. aureus Newman sdrC::Emr (DU 5988), USA300, MW2, or MRSA252 and bacterial attachment to the cultured mammalian cells were followed. Our results indicated that ten to twenty percent of the microorganisms expressing SdrC adhere to CHOK1 cells expressing wild-type Nrx1β. In contrast, we did not detect attachment when bacteria were incubated with untransfected cells or cells transiently transfected with the inactive FLAG Nrx1β-mCherry or when Nrx1-mCherry expressing cells were incubated with the S. aureus mutant DU5988 or L. lactis transformed with an empty vector (Fig. 5C). Fluorescence micrographs revealed that bacteria colocalize with Nrx1β (Fig. S4).
Discussion
The success of S. aureus as a pathogen partly depends on the organism ability to effectively colonize different tissues in the host [9],[27], evade host defense systems [28] and resist antibiotic therapy [29]. The molecular mechanisms involved in the development of different staphylococcal infections are incompletely understood but likely involve a large set of virulence factors. Many of these virulence factors interact with and manipulate specific molecular targets in the host. To interact with host targets most virulence factors are either secreted or exposed on the surface of the bacteria. Gram-positive bacteria express a family of surface proteins that are covalently anchored to the cell wall at their C-terminus. The amino acid sequences of these CWA proteins vary dramatically from one organism to another. Nevertheless, structural predictions identify a subgroup of CWA proteins with a common structural organization. This subgroup includes the known staphylococcal MSCRAMMs but also several proteins with unknown function. We hypothesize that the latter also act as adhesins and interact with molecules in the host [7],[9]. In our search for ligands for these orphan adhesins, we here report that screening a phage display library of linear peptides is a useful strategy to indentify host targets for orphan MSCRAMMs. To demonstrate the validity of this approach we first used SdrGN2N3 to pan a commercially available library of 12 amino acids inserts in the P3 phage protein. The consensus sequence identified from this analysis overlapped with the previously determined SdrG binding site located in the N-terminus of the Fg β chain. We subsequently predicted a N2N3 domain in SdrC and used a recombinant form of this segment as a target in panning the peptide library. A pattern search using the degenerate consensus sequence returned by the phage display experiment against the whole human proteome identified β-neurexin isoforms as the putative binding partners for SdrC. The human proteome contains three β-neurexin proteins, which share approximately 60% identity with each other [14]. Each β-neurexin isoform contains a variant of the peptide sequence identified by phage display located close to the amino-terminal end of the protein, which is exposed on the surface of the eukaryotic cell and therefore is accessible for binding. Moreover, this sequence starts nine residues after the first amino acid of β-neurexin, which is particularly interesting because the SdrGN2N3 binding site in Fg also maps nine residues from the N-terminus of the β chain polypeptide.
Next, we demonstrated that both the SdrCN2N3 and the SdrCA fragments bind with high affinity to the amino-terminal segment of Nrx1β, only when the polypeptide contains the amino acid sequence identified by phage display. The interaction does not promote bacterial internalization (Fig. S1), the Kd of interaction is not influenced by the amount of protein coated on the plates (Fig. S2) and it is not influenced by metal ions (Fig. S3). We further demonstrated that a synthetic peptide corresponding to the binding site in β-neurexin effectively inhibits the binding of SdrC to Nrx1β. These data indicated that recombinant SdrCN2N3 is able to bind recombinant Nrx1β and that the binding site is the amino acid sequence identified by phage display. In this study, we have also shown that the binding of SdrC to Nrxwt peptide exhibits a KD of 2.5×10−7, which is comparable to those recorded for high affinity CWA protein/host protein interactions. Further, we showed that S. aureus Newman, USA300, MW2 and MRSA252 release a fragment of SdrC capable of binding Nrx1β. Heterologous expression of SdrC on the surface of L. lactis mediates bacterial attachment to Nrx1β transiently expressed on CHOK1 cells. Similarly, clinical strains expressing SdrC adhere to wild-type Nrx1β heterologously expressed in CHOK1. This experiment demonstrates that SdrC can interact with Nrx1β when the two proteins are appropriately expressed on bacteria and mammalian cells, respectively.
The biological significance of the SdrC-Nrx1β interaction is unclear. mRNA for β-neurexins is found in many tissues [30] but the protein was only detected in neuronal tissues. It is possible that Nrx1β protein expression occurs in non-neuronal cells or tissues that have not yet been examined and/or that protein expression in non-neuronal tissues is induced under certain conditions. SdrC may serve as an adhesin mediating bacterial attachment to Nrx1β in staphylococcal infections of neuronal tissues or where the target protein is expressed. It is also possible that during the course of the infection biologically active fragments could be released from the bacteria during proteolytic processing of CWA proteins. SdrCN2N3 fragments which are released and resistant to further proteolylic degradation could interact with Nrx1β at tissues distant from the primary infection site. In this scenario it would be interesting to examine if SdrC fragments can induce intracellular signaling through its interaction with Nrx1β or interfere with the neurexin-neuroligin interaction.
A review of the literature reveals that staphylococcal endocarditis and sepsis have been associated with polyneuropathy and in several rare cases even with reversible acute tetraplegia. Studies suggest that polyneuropathy may often follow after sepsis [31],[32],[33]. Before recent advances in medicine, such as life support in the ICUs, septic death likely occurred before the neuromuscular signs could be observed. The most common manifestations of polyneuropathy are difficulty in weaning from the ventilator and limb weakness. Electrophysiologic and histopathologic investigations demonstrated axonal degeneration of motor and sensory fibers that is different from autoimmune syndromes that can develop during infection. The etiology of sepsis-associated polyneuropathy is unclear. It was thought to be caused by neuromuscular blocking agents used to ease mechanical ventilation, antibiotic toxicity, corticosteroids, microanoxia, nutritional deficiency, and high levels of inflammatory mediators. However, the cause of the disease remains unclear and is likely due to a combination of the above-mentioned factors (reviewed in [31]). Moreover, the molecular mechanism of axonal degeneration in sepsis-associated polyneuropathy is unknown. On the other hand, studies investigating neurotransmission, revealed that inhibition of Nrx1β-Nlg interaction led to late-onset neuromuscular deterioration [34]. These observations raise the possibility that SdrC interaction with Nrx1β may contribute to sepsis-associated polyneuropathy. Future studies will investigate in detail the ability of SdrC to interfere with neurexin biology in vitro and in vivo.
Materials and Methods
Media and growth conditions
E. coli was cultured in LB medium (Sigma, St. Louis, MO) containing ampicillin (100 µg/ml) at 37°C with shaking at 250 rpm. S. aureus was cultured in tryptic soy broth at 37°C with shaking at 250 rpm. L. lactis was cultured in GM17 (Oxoid, LTD, Hampshire, UK) containing 0.5% glucose and chloramphenicol (10 µg/ml) (Sigma, St. Louis, MO) at 30°C without shaking. To express SdrC in L. lactis, overnight cultures were diluted 1∶100, grown for another 3 hours and induced with nisin (1.6 ng/ml) overnight, unless otherwise mentioned. CHOK1 cells were grown in CD CHO media (GIBCO, Grand Island, NY) at 37°C in a humidified chamber with 5% CO2.
Bacterial strains used for adhesion
S. aureus strains MW2, USA300 and MRSA were from NARSA or previously described [35]. The sdrC null mutant (sdrC::pG+Host) of S.aureus Newman (DU5988) was described previously [36]. The sdrC gene was cloned into the nisin-inducible expression vector pNZ8037 [37] and transferred into L.lactis NZ9800 by previously described procedures [38].
Molecular modeling
Modeling of SdrC was performed using PHYRE Protein Homology/analogY Recognition Engine Version 2.0 (http://www.sbg.bio.ic.ac.uk/phyre/html/index.html)
Plasmid construction
DNA manipulation was performed using standard methods. DNA modification and restriction enzymes were purchased from New England Biolabs, Inc. (Ipswick, MA) or Promega (Madison, WI) and used according to the manufacturer protocol. Fragments encoding different domains of SdrC or Nrx1β were amplified by PCR from S. aureus genomic DNA, plasmids pCMV5 Nrx1β-Fc or pCMV5 FLAG-Nrx1β-mCherry [26] and oligonucleotide primers listed in Table 4. The PCR products were analyzed by agarose gel electrophoresis and purified using a QIAquick gel extraction kit (Qiagen, Sciences, MD). To construct SdrC expression plasmids, a BamHI-HindIII fragment containing the appropriate gene segment was cloned into pQE30 (Qiagen, Sciences, MD). To construct the Nrx1β-mCherry translational fusion, we first amplified the N-terminal 765 nt of Nrx1β from pCMV5 Nrx1β-Fc to obtain a PCR product corresponding to the first 255 aa of neurexin protein (including the signal sequence). Second, we amplified the Nrx1β 255-mCherry from plasmid pCMV5 FLAG-Nrx1β-mCherry to obtain the translational fusion of the 3′ end fragment of neurexin gene with mCherry from Discosoma sp. The newly synthesized PCR fragments were used as a template for an overlapping PCR to obtain full length neurexin 1β-mCherry as a translational fusion. A BglII-XbaI fragment containing the overlapping PCR was cloned into pCMV5.

To obtain GST-tagged expression proteins we amplified Nrx47–255 (aa) or Nrx70–255 (aa) from pCMV5 Nrx1β-Fc using primers described in Table 1. After purification we cloned these fragments in the E. coli expression system with Gateway Technology (Invitrogen, Inc, Carlsbad, CA) according to the manufacturer's instructions. All plasmid constructs were sequenced to ensure the integrity of the amplified fragments (Baylor College of Medicine DNA Sequencing Core Facility).
Protein expression and purification
Plasmids pQE30-SdrC52–496 and pQE30-SdrC178–496 were transformed into E. coli Topp 3. Overnight starter cultures were diluted 1∶50 in LB containing ampicillin (100 µg/ml) and incubated with shaking until the culture reached OD600 0.6–0.8. Protein expression was induced by adding 0.1 mM IPTG (final concentration) and continuing the incubation for 4 hours. Bacterial cells were harvested by centrifugation, resuspended in PBS and frozen at −80°C. Plasmids pDEST Nrx47–255 and pDEST Nrx70–255 were transformed into BL21-AI (Invitrogen, Inc, Carlsbad, CA). Cultures were induced with 0.1% arabinose for 16 h at room temperature. Bacterial cells were harvested by centrifugation, resuspended in PBS containing EDTA-free Complete Protease Inhibitor (Roche Diagnostics, Mannheim, Germany) and frozen at −80°C.
To purify SdrC52–496 and SdrC178–496, cells containing recombinant protein fragments were passed through a French press (1100 p.s.i.). Cellular debris was removed by centrifugation at 40,000 rpm for 20 minutes followed by filtration through a 0.45 µM membrane. The filtered bacterial lysate were applied at 2 ml/min on a 5 ml nickel-charged HiTrap Chelating column (GE Healthcare, Uppsala, Sweden) equilibrated with 10 mM Tris HCl, 100 mM NaCl pH 7.9. The column was washed with 40 volumes of 10 mM Tris HCl, 100 mM NaCl, 20 mM imidazole. Bound protein was eluted with a linear gradient of imidazole (10 to 200 mM, total volume 200 ml) yielding proteins that were >95% pure.
Fractions containing the SdrC 52–496 recombinant protein were dialyzed overnight in 4 L of 25 mM Tris-Cl pH 7.9 containing 10 mM EDTA and 1 mM 1,10 O-phenantroline. The dialysed sample was applied at 2 ml/minute to a MonoQ anion exchange column equilibrated with 25 mM Tris-Cl pH 7.9 for further purification. Recombinant protein of interest was collected from the flow through and dialyzed against PBS pH 7.4. Sample was reapplied on a nickel-charged HiTrap Chelating column. Pure protein was dialyzed against 50 mM EDTA to remove excess nickel and then in HBS (10 mM HEPES pH 7.4, 150 mM NaCl, 3 mM EDTA) to remove excess EDTA.
Fractions containing the SdrC178-499 recombinant protein were dialyzed overnight in 25 mM Tris-Cl pH 8.7. Dialyzed sample was applied at 2 ml/minute to a MonoQ anion exchange column equilibrated with 25 mM Tris-Cl pH 8.7. Bound protein was eluted with a linear gradient of NaCl (0–1 M NaCl, 160 ml). Fractions containing pure protein were dialyzed against 50 mM EDTA to remove excess nickel and then against HBS to remove excess EDTA.
To purify neurexin GST-tagged fragments, cells were passed through a French press (1100 p.s.i.) in the presence of Complete protease inhibitor tablets (Roche Molecular Biochemicals). Cellular debris was removed as described above. Clear cell lysate was applied to a glutathione-Sepharose column (Sigma, St. Louis, MO). Bound protein was eluted with 10 nM glutathione pH 8. Recombinant protein was concentrated and applied to a S-75 Sephadex (GE Healthcare, Uppsala, Sweden). Proteins were eluted with PBS pH 7.4.
Mass spectrometry analysis and N-terminal sequencing were performed at Tufts Protein Core Facility, Tufts University.
SDS-PAGE and Western
Recombinant proteins were analyzed by SDS-PAGE using standard procedure [39] on 12% acrylamide gels. Gels were stained with Coomassie Blue. CWA proteins from cells grown to exponential (OD600 = 8) or stationary phase (10 h) were harvested, washed 3 times with PBS and concentrated to OD600 = 50. CWA proteins were solubilized with 200 µg/ml lysostaphin in a buffer designed to minimize bacterial lysis composed of 30% sucrose, 50 mM Tris/Base pH 8, 20 mM MgCl2 and 50 µg/ml protease inhibitor (Roche) for 30 min at 37°C. Cell wall fractions were then incubated with 100 µl Sepharore-IgG for 3 h at 4°C to remove protein A. Culture supernatants were concentrated to OD600 = 10 after the addition of 100 µg/ml protease inhibitor (Roche). Protein A was removed by incubating with 100 µl Sepharore-IgG. Extracts or supernatants were separated on 4–15% SDS-PAGE gels. For Western blotting, proteins were transferred electrophoretically to nitrocellulose or polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA) using the semi-dry system (Bio-Rad, Hercules, CA) in Tris (0.02 M)-Glycine (0.15 M)-methanol (20%) buffer. Membranes were blocked in 10% milk and incubated with the appropriate serum. Briefly, membranes A1, A2, B1 and B2 were incubated with rabbit anti-SdrCN2N3 serum (H.T.I. BioProducts, Inc) and the appropriate secondary serum, which is listed below. B2 membrane was stripped (Restore western blot stripping Buffer, Pierce, Rockford, IL) for 10 minutes at room temperature and then re-probed with a rabbit anti-SdrC B-repeats serum (H.T.I. BioProducts, Inc) (B3). The membrane was stripped a second time and incubated for 1 hour at room temperature with Nrx47–255 recombinant protein, followed by goat anti-GST serum. (Invitrogen, Carlsbad, CA) (B4). A control membrane identical with B2 was probed with the same sera but was not incubated with Nrx47–255 recombinant protein (B5). HRP-labeled goat antibody (1∶5000) or HRP-labeled goat anti-mouse or HRP-labeled rabbit anti-goat (BioRad, Hercules, CA) were used to detect bound primary antibody by incubating for 1 hour at room temperature. Membranes were developed with ECL reagent (Pierce, Rockford, IL), exposed to Kodak X-ray film.
Phage display
NEB Ph.D.12-mer random library was incubated with immobilized SdrC N2N3, SdrG N2N3 or BSA at 1 µg/well. The input was 1.5×1011 transducing units with a complexity of 2.7×109 electroporated sequences. The assay was performed according to the manufacturer's instructions. The output phage plaques were 104 transducing units. Wells were washed with 0.5% Tween in TBS buffer, blocked with 10% BSA and incubated with 1×108 transducing units of each amplified phage or insertless phage (fd-tet). To remove unbound phage, wells were washed 10 times with 0.5% Tween in TBS. Bound phage was detected with anti-M13-HRP antibody (GE Healthcare, Buckinghamshire, UK).
Solid phase binding assay
Each well of Immulon 4BH plates was coated with 1 µg of recombinant Nrx47–255-GST, Nrx70–255-GSTor GST overnight at 4°C. Coated wells were blocked for 1 hour at room temperature with 2% BSA in 0.05% Tween-TBS buffer. Increasing concentrations of SdrCN2N3 or A-region were added to the wells and incubated for 1 hour at room temperature. Bound protein was detected with a polyclonal antibody (1∶3000) against SdrCN2N3 (H.T.I. BioProducts) followed by an anti-rabbit HRP-labeled antibody (1∶5000) (Bio-Rad Labs, Hercules, CA). Color development was performed using SigmaFast OPD (Sigma, St. Louis, MO) and the binding was measured using a microtiter plate reader (Molecular Devices) at 450 nm. For inhibition assays, 0.5 µM SdrC178-496 protein was incubated with increasing concentrations of peptides prior to the solid phase binding assay. Data presented represent the mean±SD of three independent experiments performed in triplicate. The binding was analyzed by non-linear regression for one binding site (GraphPad Prism).
Fluorescence polarization
Nrxwt and Nrxscr peptides were labeled with fluorescein as described previously [10]. Increasing concentrations of SdrC178-496 were incubated with 10 nM labeled peptide for 3 hours in the dark at room temperature. Polarization was measured using Luminescence Spectrometer LB50B (Perkin Elmer). The data were analyzed by non-linear regression for one binding site. The equilibrium dissociation constant was calculated using the equation ΔP = ΔPmax x [protein])/(KD + [protein]) (Eq.1) where ΔP is the change in fluorescence polarization, ΔPmax is the maximum change in fluorescence polarization and KD is the equilibrium dissociation constant of the interaction.
For inhibition assays, 0.5 µM SdrC178–496 was first incubated with increasing concentrations of unlabeled Nrxwt or unlabeled Nrxscr for 3 hours at room temperature. The mixture was then incubated for 3 hours with fluorescein-labeled Nrxwt. The results presented represent the mean±SD of three independent experiments.
Adherence assay
CHOK1 cells were grown in 24-well tissue culture plates to approximately 80% confluence. Cells were transfected with either pCMV5 Nrx1β-mCherry or pCMV5 FLAG-Nrx1β-mCherry using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions and grown for 36 hours to allow for protein expression. For attachment assays, bacteria were washed 2 times in PBS, resuspended in CD CHO media and incubated for 1 h with CHOK1 cells at an MOI of 10. To remove unattached bacteria, cells were washed 4 times with PBS. To determine the number of attached bacteria, CHOK1 cells were lysed with sterile water and serial dilutions plated on the appropriate media. The number of attached bacteria was reported as a percent of total bacteria at the end of the incubation period. Each experiment was performed in triplicate wells and repeated three times. Statistical analysis was performed using the Student's t-test.
Protein accession numbers (UniProt) SdrC
S. aureus Newman - O86487; S. aureus USA300 - A8YZQ9; S. aureus MW2 - Q8NXX7; S. aureus MRSA252 - Q6GJA7. Neurexin1β: P58400.
Supporting Information
Zdroje
1. LowyFD
1998 Staphylococcus aureus infections. N Engl J Med 339 520 532
2. ClarkeSR
FosterSJ
2006 Surface adhesins of Staphylococcus aureus. Adv Microb Physiol 51 187 224
3. PattiJM
AllenBL
McGavinMJ
HookM
1994 MSCRAMM-mediated adherence of microorganisms to host tissues. Annu Rev Microbiol 48 585 617
4. PattiJM
HookM
1994 Microbial adhesins recognizing extracellular matrix macromolecules. Curr Opin Cell Biol 6 752 758
5. MarraffiniLA
DedentAC
SchneewindO
2006 Sortases and the art of anchoring proteins to the envelopes of gram-positive bacteria. Microbiol Mol Biol Rev 70 192 221
6. Ton-ThatH
MarraffiniLA
SchneewindO
2004 Protein sorting to the cell wall envelope of Gram-positive bacteria. Biochim Biophys Acta 1694 269 278
7. PonnurajK
BowdenMG
DavisS
GurusiddappaS
MooreD
2003 A “dock, lock, and latch” structural model for a staphylococcal adhesin binding to fibrinogen. Cell 115 217 228
8. DeivanayagamCC
WannER
ChenW
CarsonM
RajashankarKR
2002 A novel variant of the immunoglobulin fold in surface adhesins of Staphylococcus aureus: crystal structure of the fibrinogen-binding MSCRAMM, clumping factor A. EMBO J 21 6660 6672
9. FosterTJ
HookM
1998 Surface protein adhesins of Staphylococcus aureus. Trends Microbiol 6 484 488
10. DavisSL
GurusiddappaS
McCreaKW
PerkinsS
HookM
2001 SdrG, a fibrinogen-binding bacterial adhesin of the microbial surface components recognizing adhesive matrix molecules subfamily from Staphylococcus epidermidis, targets the thrombin cleavage site in the Bbeta chain. J Biol Chem 276 27799 27805
11. BowdenMG
HeuckAP
PonnurajK
KolosovaE
ChoeD
2008 Evidence for the “dock, lock, and latch” ligand binding mechanism of the staphylococcal microbial surface component recognizing adhesive matrix molecules (MSCRAMM) SdrG. J Biol Chem 283 638 647
12. McCreaKW
HartfordO
DavisS
EidhinDN
LinaG
2000 The serine-aspartate repeat (Sdr) protein family in Staphylococcus epidermidis. Microbiology 146(Pt 7) 1535 1546
13. JosefssonE
McCreaKW
Ni EidhinD
O'ConnellD
CoxJ
1998 Three new members of the serine-aspartate repeat protein multigene family of Staphylococcus aureus. Microbiology 144(Pt 12) 3387 3395
14. SudhofTC
2008 Neuroligins and neurexins link synaptic function to cognitive disease. Nature 455 903 911
15. UshkaryovYA
PetrenkoAG
GeppertM
SudhofTC
1992 Neurexins: synaptic cell surface proteins related to the alpha-latrotoxin receptor and laminin. Science 257 50 56
16. MullenLM
NairSP
WardJM
RycroftAN
HendersonB
2006 Phage display in the study of infectious diseases. Trends Microbiol 14 141 147
17. SergeevaA
KoloninMG
MolldremJJ
PasqualiniR
ArapW
2006 Display technologies: application for the discovery of drug and gene delivery agents. Adv Drug Deliv Rev 58 1622 1654
18. KoloninMG
BoverL
SunJ
ZuritaAJ
DoKA
2006 Ligand-directed surface profiling of human cancer cells with combinatorial peptide libraries. Cancer Res 66 34 40
19. SmithGP
PetrenkoVA
1997 Phage Display. Chem Rev 97 391 410
20. DeivanayagamCC
PerkinsS
DanthuluriS
OwensRT
BiceT
1999 Crystallization of ClfA and ClfB fragments: the fibrinogen-binding surface proteins of Staphylococcus aureus. Acta Crystallogr D Biol Crystallogr 55 554 556
21. GaneshVK
RiveraJJ
SmedsE
KoYP
BowdenMG
2008 A structural model of the Staphylococcus aureus ClfA-fibrinogen interaction opens new avenues for the design of anti-staphylococcal therapeutics. PLoS Pathog 4 e1000226 doi:10.1371/journal.ppat.1000226
22. Ni EidhinD
PerkinsS
FrancoisP
VaudauxP
HookM
1998 Clumping factor B (ClfB), a new surface-located fibrinogen-binding adhesin of Staphylococcus aureus. Mol Microbiol 30 245 257
23. HartfordOM
WannER
HookM
FosterTJ
2001 Identification of residues in the Staphylococcus aureus fibrinogen-binding MSCRAMM clumping factor A (ClfA) that are important for ligand binding. J Biol Chem 276 2466 2473
24. McAleeseFM
WalshEJ
SieprawskaM
PotempaJ
FosterTJ
2001 Loss of clumping factor B fibrinogen binding activity by Staphylococcus aureus involves cessation of transcription, shedding and cleavage by metalloprotease. J Biol Chem 276 29969 29978
25. UshkaryovYA
HataY
IchtchenkoK
MoomawC
AfendisS
1994 Conserved domain structure of beta-neurexins. Unusual cleaved signal sequences in receptor-like neuronal cell-surface proteins. J Biol Chem 269 11987 11992
26. BoucardAA
ChubykinAA
ComolettiD
TaylorP
SudhofTC
2005 A splice code for trans-synaptic cell adhesion mediated by binding of neuroligin 1 to alpha - and beta-neurexins. Neuron 48 229 236
27. Bubeck WardenburgJ
PatelRJ
SchneewindO
2007 Surface proteins and exotoxins are required for the pathogenesis of Staphylococcus aureus pneumonia. Infect Immun 75 1040 1044
28. FosterTJ
2005 Immune evasion by staphylococci. Nat Rev Microbiol 3 948 958
29. ChambersHF
2005 Community-associated MRSA–resistance and virulence converge. N Engl J Med 352 1485 1487
30. OcchiG
RampazzoA
BeffagnaG
Antonio DanieliG
2002 Identification and characterization of heart-specific splicing of human neurexin 3 mRNA (NRXN3). Biochem Biophys Res Commun 298 151 155
31. ChenHC
TsaiCS
LeeJT
ChenCA
ChangFY
2005 Acute quadriplegia complicating critical illness polyneuropathy in a patient with infective endocarditis: a case report. J Infect 50 153 157
32. RoderBL
WandallDA
EspersenF
Frimodt-MollerN
SkinhojP
1997 Neurologic manifestations in Staphylococcus aureus endocarditis: a review of 260 bacteremic cases in nondrug addicts. Am J Med 102 379 386
33. CaksenH
UnerA
ArslanS
AnlarO
OdabasD
2004 Severe peripheral polyneuropathy in a child with infective endocarditis caused by Staphylococcus aureus. Acta Neurol Belg 104 114 116
34. AndresC
BeeriR
FriedmanA
Lev-LehmanE
HenisS
1997 Acetylcholinesterase-transgenic mice display embryonic modulations in spinal cord choline acetyltransferase and neurexin Ibeta gene expression followed by late-onset neuromotor deterioration. Proc Natl Acad Sci U S A 94 8173 8178
35. VoyichJM
OttoM
MathemaB
BraughtonKR
WhitneyAR
2006 Is Panton-Valentine leukocidin the major virulence determinant in community-associated methicillin-resistant Staphylococcus aureus disease? J Infect Dis 194(12) 1761 70
36. O'BrienL
KerriganSW
KawG
HoganM
PenadesJ
2002 Multiple mechanisms for the activation of human platelet aggregation by Staphylococcus aureus: roles for the clumping factors ClfA and ClfB, the serine-aspartate repeat protein SdrE and protein A. Mol Microbiol 44 1033 1044
37. de RuyterPG
KuipersOP
de VosWM
1996 Controlled gene expression systems for Lactococcus lactis with the food-grade inducer nisin. Appl Environ Microbiol 62 3662 3667
38. LoughmanA
FitzgeraldJR
BrennanMP
HigginsJ
DownerR
2005 Roles for fibrinogen, immunoglobulin and complement in platelet activation promoted by Staphylococcus aureus clumping factor A. Mol Microbiol 57 804 818
39. LaemmliUK
1970 Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227 680 685
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 1
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Panton-Valentine Leukocidin Is a Very Potent Cytotoxic Factor for Human Neutrophils
- CD8+ T Cell Control of HIV—A Known Unknown
- Polyoma Virus-Induced Osteosarcomas in Inbred Strains of Mice: Host Determinants of Metastasis
- The Deadly Chytrid Fungus: A Story of an Emerging Pathogen
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy