
Strong Selective Sweeps on the X Chromosome in the Human-Chimpanzee Ancestor Explain Its Low Divergence
Because the speciation events that led to human, chimpanzee and gorilla were close in time, the genetic relationship of these species varies along the genome. While human and chimpanzee are the closest related species, in 15% of the genome, human and gorilla are more closely related, and in another 15% of the genome the chimpanzee and gorilla are more closely related—a phenomenon called incomplete lineage sorting (ILS). The amount and distribution of ILS can be predicted using population genetics theory and is affected by demography and selection in the ancestral populations. It was previously reported that the X chromosome, in contrast to autosomes, has less than the expected level of ILS. Using a full genome alignment of the X chromosome, we show that this low level of ILS affects only one third of the chromosome. Regions with low level of ILS also show reduced diversity in the extant populations of human and great apes and coincide with regions devoid of Neanderthal introgression. We propose that these regions are targets of selection and that they played a role in the formation of reproductive barriers.
Published in the journal:
. PLoS Genet 11(8): e32767. doi:10.1371/journal.pgen.1005451
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005451
Summary
Because the speciation events that led to human, chimpanzee and gorilla were close in time, the genetic relationship of these species varies along the genome. While human and chimpanzee are the closest related species, in 15% of the genome, human and gorilla are more closely related, and in another 15% of the genome the chimpanzee and gorilla are more closely related—a phenomenon called incomplete lineage sorting (ILS). The amount and distribution of ILS can be predicted using population genetics theory and is affected by demography and selection in the ancestral populations. It was previously reported that the X chromosome, in contrast to autosomes, has less than the expected level of ILS. Using a full genome alignment of the X chromosome, we show that this low level of ILS affects only one third of the chromosome. Regions with low level of ILS also show reduced diversity in the extant populations of human and great apes and coincide with regions devoid of Neanderthal introgression. We propose that these regions are targets of selection and that they played a role in the formation of reproductive barriers.
Introduction
Despite constituting only 5–6% of the human genome, the human X chromosome is important for elucidating evolutionary mechanisms. Because of its particular inheritance pattern and its cosegregation with the very different Y chromosome, evolutionary forces may act upon it in different ways than on the autosomes [1,2]. Thus contrasting the evolution of the X chromosome with that of the autosomes provides clues to the relative importance of different evolutionary forces.
Hemizygosity of males implies that there are fewer X chromosomes than autosomes in a population (3/4 for even sex ratios). Thus, genetic drift is expected to be relatively stronger on the X chromosome. New variants with recessive fitness effects will also be selected for or against more efficiently on the X chromosome, where they are always exposed in males, than on the autosomes, potentially overriding the increased genetic drift.
Empirical studies have shown that nucleotide diversity is more reduced around genes on the X chromosome than on the autosomes [3–5]. This has been interpreted as the result of more efficient selection on coding variants on the X chromosome, which affects linked positions around the genes. However, no distinction is made here between linked effects of positive selection (genetic hitchhiking [6]) and linked effect of selection against deleterious mutations (background selection [7]). For recessive variants, hitchhiking is expected to be more wide ranging for X chromosomes, whereas a different distribution of fitness effects of deleterious variants on the X is needed to cause stronger background selection on the X. Contrasting non-synonymous and synonymous substitutions with non-synonymous and synonymous polymorphisms, several recent studies have reported evidence for more positive selection on protein changes on the X chromosome in both primates and rodents [8–11]. Whether this is due to hemizygosity, different gene content of the X chromosome, antagonistic selection between sexes being more prevalent on the X chromosome, or some fourth reason is not known.
A separate observation is that the X chromosome in most investigated species is disproportionately involved with speciation, as it (i) contributes disproportionately to hybrid incompatibility (the large X effect) and (ii) together with the Y chromosome is responsible for stronger hybrid depression in males than in females (Haldane’s rule). We refer to Laurie (1997) [12] and Schilthuizen, Giesbers and Beukeboom (2011) [13] for several non-exclusive hypotheses for the underlying genetic mechanisms leading to Haldane’s rule.
Recent introgression from Neanderthals into modern humans was recently reported to be far less common on the X chromosome than on the autosomes. This can be interpreted as evidence for emerging incompatibilities between the two species preferentially residing on the X chromosome [14]. It has been suggested that incompatibilities can accrue due to genetic conflicts between the X and the Y [15–19] and some hybrid incompatibility factors in Drosophila do show evidence of causing meiotic drive [20].
We, and others, have previously reported that the X chromosome shows much less divergence between humans and chimpanzees than expected from autosomal divergence [21–23]. This observation is not based on the nucleotide divergence of the X chromosome versus the autosomes—which will be affected by a difference in mutation rate—but on estimating the effective population size of the ancestral species from the proportion of discordant gene trees.
Because the speciation event between human and chimpanzee and the speciation event between the human-chimpanzee ancestor and the gorilla occurred close in time, around 30% of the autosomal genome shows a gene tree different from the species tree—a phenomenon called incomplete lineage sorting (ILS). The expected amount of ILS depends on the difference between the two speciation times and the effective population size in the human-chimpanzee ancestor. For estimates of the two speciation times in question [24], and assuming that the effective population size of the X chromosome is three quarters of that of the autosomes, the X chromosome is expected to show 24% of ILS. The observed mean amount of ILS, however, is around 15%.
We recently reported that certain regions of the X chromosome in different great ape species often experience what looks like very strong selective sweeps [18]. Here we study the amount of incomplete lineage sorting between human, chimpanzee and gorilla along the X chromosome. We observe a striking pattern of mega-base sized regions with extremely low amounts of ILS, interspersed with regions with the amount of ILS expected from the effective population size of the X chromosome (that is, three quarters that of the autosomes). We show that the most plausible explanation is several strong selective sweeps in the ancestral species to humans and chimpanzees. The low-ILS regions overlap strongly with regions devoid of Neanderthal ancestry in the human genome, which suggests that selection in these regions may create reproductive barriers. We propose that the underlying mechanism is meiotic drive resulting from genetic conflict between the sex chromosomes, and that this is caused by testis expressed ampliconic genes found only on sex chromosomes and enriched in the regions where we find signatures of selective sweeps.
Results
Distribution of incomplete lineage sorting along the X chromosome
To explore the pattern of human-chimpanzee divergence across the full X chromosome we performed a detailed analysis of the aligned genomes of human, chimpanzee, gorilla and orangutan [21]. Using the coalescent hidden Markov model (CoalHMM) approach [25], we fitted a model of speciation by isolation, with constant but distinct ancestral effective population sizes for the human-chimpanzee (HC) and the human-chimpanzee-gorilla (HCG) ancestors. The parameters of the model are (i) two speciation times τHC and τHCG for human vs. chimpanzee and for HC vs. gorilla, respectively, (ii) two ancestral population sizes θHC and θHCG for the HC and HCG ancestral populations, respectively, as well as the recombination rate r assumed to be constant along both the alignment and phylogeny. An additional parameter is used to account for the divergence with the outgroup sequence. The speciation time, effective population size and recombination rate parameters are scaled according to 2.Ne.u.g, 2.Ne.u and u, respectively, where u is the mutation rate per generation, g the generation time and Ne the population size of a reference extant species [22,25]. Extant population sizes are not parameters of the model, and only serve for the purpose of scaling parameters. To account for putative variation of parameters along the genome alignment, we estimated demographic parameters in non-overlapping 1 Mb windows. We inferred the proportion of ILS using posterior decoding averaged over each of these 1Mb windows. The expected proportion of ILS in a 3-species alignment is given by the formula:

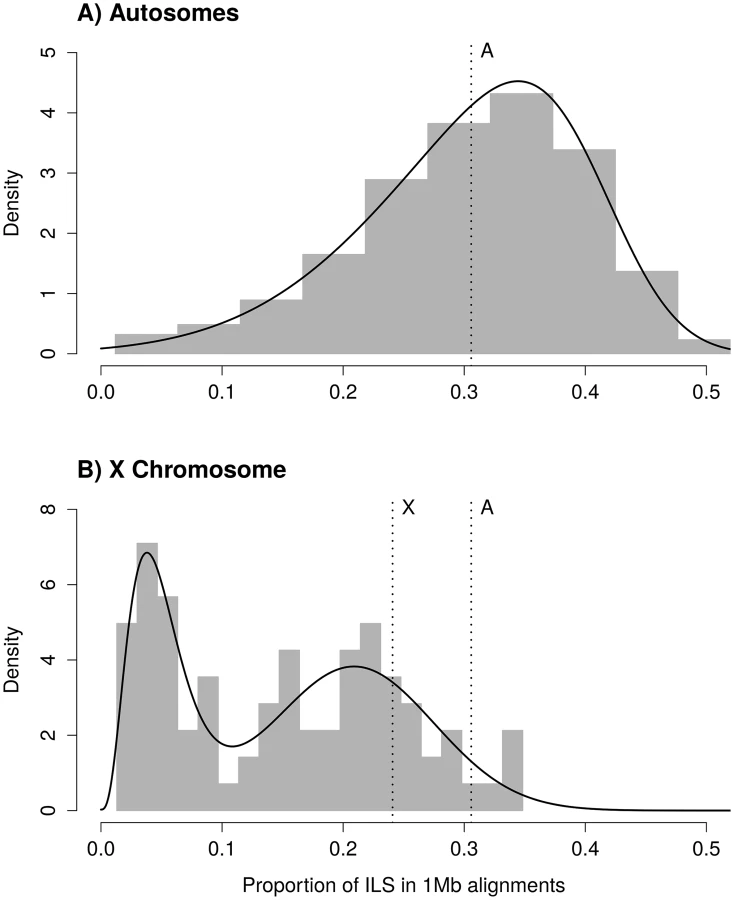
Assuming that the ancestral effective population size of the X chromosome, θX, is three quarters that of the ancestral effective population size of the autosomes, the expected amount of ILS on the X chromosome should be 24.08%. The distribution of ILS proportions on the X chromosome is bimodal (Fig 1B) and in stark contrast to the distribution on the autosomes (see also S1 Fig for a breakdown on individual autosomes). One mode represents 63% of the alignment, with a mean proportion of ILS of 21%, close to the expectation of 24% (the 99% confidence interval of the high ILS mode is [17.6%, 24.5%], estimated using parametric bootstrap). The second mode is estimated to represent 37% of the alignment and shows a mean proportion of ILS below 5%. The regions exhibiting low ILS form 8 major segments spread across the X chromosome (Table 1 and Fig 2A) and cover 29 Mb out of a total alignment length of 84 Mb. Region X5 is split in two by the centromeric region, where alignment data are missing. Regions with comparatively low amount of ILS have a higher frequency of genealogy where the human and chimpanzee coalesce within the HC ancestor, while in ILS genealogies, the human and chimpanzee lineages coalesce further back in time, within the HCG ancestor. As a result, low-ILS regions display a lower divergence compared to the rest of the genome. These results are two-fold: (i) they demonstrate that one third of the X chromosome explains the previously reported low divergence of the chromosome, as the remaining two thirds display a divergence compatible with the expectation under a simple model of divergence with an ancestral effective population size equal to three quarters that of the autosomes and (ii) that unique evolutionary forces have shaped the ancestral diversity in the low-ILS regions.


Robustness of ILS estimation
In Scally et al. [21], we independently estimated parameters in non-overlapping windows of 1 Mb, allowing for parameters to vary across the genome. To test whether inference of very low proportions of ILS could result from incorrect parameter estimation, we compared the inferred amount of ILS under alternative parameterizations with that inferred using fixed parameters (either fixing all parameters or fixing speciation time parameters only) along the genome. These alternative parameterizations result in very similar estimates of ILS (S2 Fig and corresponding UCSC genome browser tracks at http://bioweb.me/HCGILSsupp/UCSCTracks/).
We addressed the possibility that our observation is due to a lower power to detect ILS in the identified regions resulting from reduced mutation rate. We counted the number of informative sites supporting each of the three alternative topologies connecting humans, chimpanzees and gorillas in non-overlapping 100 kb windows along the alignment. If the reduction of ILS is due to a lower mutation rate in these regions, we expect to observe a reduction of the amount of parsimony-informative sites supporting all three topologies. While the total frequency of parsimony-informative sites is significantly lower in the low-ILS regions compared with the rest of the genome (0.00270 vs. 0.00276, Fisher's exact test p-value = 1.34e-05), there is a highly significant excess of sites supporting the species topology (0.00229 vs. 0.00210, Fisher's exact test p-value < 2.2e-16) and deficit of sites in these regions supporting ILS topologies (0.00042 vs. 0.00066, Fisher's exact test p-value < 2.2e-16, Fig 2B and 2C), which suggests that the observed reduction of ILS is not the result of a lower mutation rate.
We computed the ratio of human-chimpanzee divergence to human-gorilla divergence and human-orangutan divergence in 100 kb windows. Assuming a constant mutation rate across the phylogeny and constant ancestral effective population sizes along the genome, these ratios should be on average identical between regions from the genome. In regions with reduced ILS, however, this ratio is expected to be lower because of a more recent human-chimpanzee divergence. In agreement with this latter hypothesis, we observe a significant lower ratio of divergences in low-ILS regions (Fig 2D). A lower mutation rate in these regions would explain this pattern only if the reduction is restricted to the human-chimpanzee lineage.
The effect of background selection on ILS
Deleterious mutations are continuously pruned from the population through purifying selection, reducing the diversity of linked sequences. Such background selection potentially plays an important role in shaping genetic diversity across the genome [28]. The strength of background selection increases with the mutation rate, with density of functional sites, with decreasing selection coefficient against deleterious mutations, and with decreasing recombination rate [29]. Low-ILS regions display both a 0.6-fold lower recombination rate compared to the rest of the chromosome (1.01 cM/Mb versus 1.62 cM/Mb, Wilcoxon test p-value = 2.2e-07) as well as a two-fold higher gene density—a proxy for the proportion of functional sites (3.1% exonic sites versus 1.5% on average, Wilcoxon test p-value < 2.2e-16). Background selection is therefore both expected to be more common (by a factor of ~2.1 due to more functional sites) and to affect larger regions (by a factor of ~1.8 due to less recombination) in the low-ILS regions. To estimate extent to which this may explain our observations, we used standard analytical results that estimate the combined effect of multiple sites under purifying selection (see Material and Methods). Even if we assume that the proportions of functional sites in the candidate regions is two times higher than the observed number of exon base pairs, and that all mutations at these sites are deleterious with a selection coefficient that maximizes the effect of background selection, the expected proportion of ILS should only be reduced by approximately 10% relative to the level found on the remaining X chromosome (19% ILS compared to 21% ILS). To explain the observed reductions in ILS by background selection alone, unrealistic differences of functional site densities are required (e.g. 50% inside identified regions and 10% outside, see Figs 3 and S2). As a further line of evidence, we computed the maximal expected reduction of ILS based on the observed density of exonic sites and average recombination rate (see Methods). We find that only 79 of 252 analyzable windows (31%) could be explained by the action of background selection only, an observation incompatible with the hypothesis that background selection is the sole responsible for the widespread reduction of ILS along the X chromosome.

Finally, recombination rate is lower in males than in females. As X chromosomes spend 2/3 of their time in highly recombining females while autosomes spend only half, background selection is expected to be weaker on the X chromosome than on the autosomes. Consequently, in Drosophila where males do not recombine, X chromosomes display a higher than expected diversity [30]. The fact that we do not observe large regions devoid of ILS on the autosomes further argues against background selection as the major force creating the observed large regions with reduced ILS on the X chromosome.
Selective sweeps and ILS
Adaptive evolution may also remove linked variation during the process of fixing beneficial variants. In the human-chimpanzee ancestor, such selective sweeps will have abolished ILS at the locus under selection and reduced the proportion of ILS in a larger flanking region. Several sweeps in the same region can thus result in a strong reduction of ILS on a mega-base scale. We simulated selective sweeps in the human-chimpanzee ancestor using a rejection sampling method (see Material and Methods). A single sweep is only expected to reduce ILS to less than 5% on a mega-base wide region if selection coefficients are unrealistically high (s > 0.2), suggesting that several sweeps have contributed to the large-scale depletions of ILS (Figs 4 and S4).

If the low-ILS regions are indeed subject to recurrent sweeps, they are expected to also show reduced diversity in human populations. We therefore investigated the patterns of nucleotide diversity in the data of the 1000 Genomes Project [31]. We computed the nucleotide diversity in 100 kb non-overlapping windows along the X chromosome and compared windows within and outside low-ILS regions. Fig 5 summarizes the results for the CEU, JPT and YRI populations (results for all populations are shown in S5 Fig). We find that diversity is significantly reduced in all low-ILS regions compared with the chromosome average (Table 2), and this reduction is on average significantly greater in the Asian and European populations than in the African population (analysis of variance, see Material and Methods). This global difference in magnitude could be explained by phenomena such as sex-biased demography or generation time and population structure during the migration out of Africa [32]. We also compared the eight low-ILS regions separately, and reported differences between regions (Table 3). Plotting population specific diversity across the X chromosome revealed several cases of large-scale depletions of diversity in both Europeans and East Asians. While these depletions affect similar regions, their width differs between populations. This finding suggests that strong sweeps in these regions occurred independently in the European and East Asian population after their divergence less than 100,000 years ago.



Discussion
Using a complete genome alignment of human, chimpanzee, gorilla and orangutan, we report that the human-chimpanzee divergence along the X chromosome is a mosaic of two types of regions: two thirds of the X chromosome display a divergence compatible with the expectation of an ancestral effective population size of the X equal to three quarters that of the autosome, while one third of the X chromosome shows an extremely reduced divergence, and is virtually devoid of incomplete lineage sorting. We have demonstrated that such diversity deserts cannot be accounted for by background selection alone, but must result from recurrent selective sweeps. We recently reported dramatic reductions in X chromosome diversity in other great ape species that almost exclusively affect areas of the low-ILS regions [18] (see S6 Fig).
If the low-ILS regions evolve rapidly through selective sweeps, they could be among the first to accumulate hybrid incompatibility between diverging populations. Recently, the X chromosome was reported to exhibit many more regions devoid of Neanderthal introgression into modern humans than the autosomes. This suggests an association of negative selection driven by hybrid incompatibility with these X-linked regions [14]. We find a striking correspondence between regions of low ILS and the regions devoid of Neanderthal introgression for European populations (p-value = 0.00021, permutation test) and a marginally significant association with the more introgressed Asian populations (p-value = 0.06721, Fig 5). Taken together, these findings show that the regions on the X chromosome that contributed to hybrid incompatibility in the secondary contact between humans and Neanderthals have been affected by recurrent, strong selective sweeps in humans and other great apes.
The occurrence of a secondary contact between initially diverged populations, one of which diverged into modern chimpanzees and the other admixed with the second to form the ancestral human lineage—the complex speciation scenario of Patterson et al. [23]–is also compatible with our observations: if these regions evolved to be incompatible, the lineages within the regions only came from the ancestral population related to chimpanzees while lineages outside the regions come from both ancestral populations, so that we would also expect to see reduced ILS within the regions and not outside the regions. However, such a complex speciation scenario does not explain the observed large-scale reductions of diversity in extant species. Conversely, a scenario consisting only of recurrent sweeps would explain both the divergence patterns along the human and chimpanzee X chromosomes and the reduction of extant diversity, without the need for secondary introgression.
To explain the occurrence of recurrent selective sweeps in the lineage of great apes, we propose a hypothesis that may account for the generality of our findings: Deserts of diversity may arise via meiotic drive, through which fixation of variants that cause preferential transmission of either the X or Y chromosome produces temporary sex ratio distortions [17]. When such distortions are established, mutations conferring a more even sex ratio will be under positive selection. Potential candidates involved in such meiotic drive are ampliconic regions, which contain multiple copies of genes that are specifically expressed in the testis. These genes are postmeiotically expressed in mice, and a recent report suggests that the Y chromosome harbors similar regions [33]. Fourteen of the regions identified in humans [34] are included in our alignment, 11 of which are located in low-ILS regions (Figs 2 and 5), representing a significant enrichment (p-value = 0.01427, permutation test), a result which is even more significant when regions in the centromeric region are included (p-value = 0.00642).
Whatever the underlying mechanism, our observations demonstrate that the evolution of X chromosomes in the human chimpanzee ancestor, and in great apes in general [18], is driven by strong selective forces. The striking overlap between the low-ILS regions we have identified and the Neanderthal introgression deserts identified by Sankararaman et al. [14] further hints that these forces could be driving speciation.
Materials and Methods
Genome alignment and data pre-processing
The Enredo/Pecan/Ortheus genome alignment of the five species human, chimpanzee, gorilla, orangutan and macaque from Scally et al. [21] was used as input. In order to remove badly sequenced and / or ambiguously alignment regions, we filtered the input 5-species alignments using the MafFilter program [35]. We sequentially applied several filters to remove regions with low sequence quality score and high density of gaps. Details on the filters used can be found in the supplementary material of Scally et al. [21]
Inference of incomplete lineage sorting
The divergence of two genomes depends on both the mutation rate and underlying demographic scenario. With a constant mutation rate u and simple demography (constant sized panmictic population evolving neutrally), the time to the most recent common ancestor of two sequences sampled from different species is given by a constant species divergence, τ = T.u, and an ancestral coalescence time following an exponential distribution with mean θ = 2.NeA.u, where T is the number of generations since species divergence and NeA is the ancestral effective population size [22,36]. For species undergoing recombination, a single individual genome is a mosaic of segments with distinct histories, and therefore displays a range of divergence times [22,23,37]. When two speciation events separating three species follow shortly after each other, this variation of genealogy can lead to incomplete lineage sorting (ILS), where the topology of gene trees do not correspond to that of the species tree [22,26]. Reconstructing the distribution of divergence along the genome and the patterns of ILS allows inference of speciation times and ancestral population sizes. We used the CoalHMM framework to infer patterns of ILS along the X chromosome. Model fitting was performed as described in [21]. ILS was estimated using posterior decoding of the hidden Markov model as the proportions of sites in the alignment which supported one of the (HG),C or (CG),H topologies. All parameter estimates can be visualized in the UCSC genome browser using tracks available at http://bioweb.me/HCGILSsupp/.
Distribution of ILS
For the autosomal distribution of ILS, we fitted a skewed normal distribution (R package 'sn' [38]) using the fitdistr function from the MASS package for R. For the X chromosome ILS distribution, we fitted a mixture of gamma and Gaussian distributions. The mixed distribution follows a normal density with probability p, and a gamma density with probability 1-p. In addition to p, the mixed distribution has four parameters: the mean and standard deviation of the Gaussian component, and the shape and rate of the gamma component. The L-BFGS-B optimization method was used to account for parameter constraints. Resulting parameter estimates are 0.209 for the mean of the Gaussian component, 0.066 for the standard deviation of the Gaussian component, 4.139 for the alpha parameter (shape) of the gamma component, 83.369 for the beta parameter (rate) of the gamma component, and p = 0.632. The mean of the gamma component is alpha / beta = 0.0497, that is, less than 5% ILS. We compared the resulting fit with a mixture of skewed normal distributions, which has two extra parameters compared to a Gamma-Gaussian mixture, and found that the skew of the higher mode is very close to zero, while the Gamma distribution offered a better fit of the lower mode. We used a parametric bootstrap approach to estimate the confidence interval of the proportion of ILS for the mean of the normal component of the mixed distribution. We generated a thousand pseudo-replicates by sampling from the estimated distribution, and we re-estimated all parameters from each replicate in order to obtain their distribution. Replicates where optimization failed were discarded (40 out of 1000).
Characterization of low-ILS regions
In order to characterize the patterns of ILS at a finer scale, we computed ILS in 100 kb windows sliding by 20 kb along the posterior decoding of the alignment. To exhibit regions devoid of ILS, we selected contiguous windows with no more than 10% of ILS each. Eight of these regions were greater than 1 Mb in size, and their resulting amount of ILS is less than 5% on average (Table 1). The coordinates of these regions were then translated according to the human hg19 genome sequence. These data are available as a GFF file for visualization in the UCSC genome browser at http://bioweb.me/HCGILSsupp/.
Reduction in ILS by background selection
Background selection reduces diversity by a process in which deleterious mutations are continuously pruned from the population. The strength of background selection in a genomic region is determined by the rate at which deleterious mutations occur, U, the recombination rate of the locus, R, and the strength of negative selection on mutants, s. We consider the diversity measure,π(the pairwise differences between genes) which in a randomly mating population is linearly related to the effective population size. If π0 denotes diversity in the absence of selection and π the diversity in a region subject to background selection, then the expected reduction in diversity is given by

(see Durrett [39] equation (6.24))
The rates U and R are both functions of the locus length (U = uL and R = rL) where r denotes the per-nucleotide-pair recombination rate, u the per-nucleotide deleterious rate, and L the length of the locus. To investigate if background selection can explain the observed reductions in ILS we must compute the expected reduction in diversity in the low-ILS regions relative to the reduction in the remaining chromosome. A larger reduction in low-ILS regions may be caused by weaker negative selection, higher mutation rate, lower recombination rate, and larger proportion of functional sites at which mutation is deleterious. To model the variation of these parameters inside and outside low-ILS regions we simply add a factor to each relevant variable. The relative reduction can thus be expressed as:

The recombination rate, R, and the factor, fR, can be obtained from the deCODE recombination map [40]. We computed the average deCODE recombination rate, as well as the proportion of sites in exons (as a measure of selective constraint) in non-overlapping 100 kb along the human X chromosome.
The recombination rate average outside the low ILS regions is 1.62 cM/Mb and the recombination rate inside the regions is 1.01 cM/Mb which gives us fR = 0.6. For the remaining parameters, s and U, we need to identify realistic values outside the low-ILS regions. Background selection is stronger when selection is weak, but the equation is not valid for very small selection values where selection is nearly neutral. Once s approaches 1/Ne, we do not expect any background selection. Most stimates of effective population sizes, Ne, in great apes are on the order 10,000–100,000 and this puts a lower limit on relevant values of s at 10−4–10−5. To conservatively estimate the largest possible effect of background selection we explore this range of selection coefficients: s = 10−4 and s = 10−5 and allow the selection inside the low ILS regions to be one tenth (fs = 0.1) of that outside. For U values outside low-ILS regions we assume the mean human mutation rate, estimated to be 1.2·10−8 per generation [41]. To obtain the rate of deleterious mutation we must multiply this with the proportion of sites subject to weak negative selection, d. Although this proportion is subject to much controversy it is generally believed to be between 3% and 10% [42]. However, as explained below we explore values up to 100% inside the low-ILS regions.
We assessed the relative diversity for combinations of s and d values (S3 Fig). Each cell represents a combination of parameter values for s, d, fU and fs. The reduction of diversity Δπ translates into reduction of ILS, ΔILS(Fig 3). Assuming the time between speciation events, the generation time and population size reported in Scally et al. [21] (ΔT = 2,250,000 years, g = 20) ILS is given by


For the most extreme parameter values, we see a relative reduction in ILS of nearly 100%. In these cases, however, 100% of the nucleotides within low-ILS regions are under selection. In the cases where 25% of the nucleotides in the low-ILS regions are under selection compared to 5% outside (fU = 5, d = 0.05), the regions retain more than half of the diversity seen outside the regions.
We further computed the expected reduction of ILS due to background selection in 100 kb windows located in low-ILS regions using (eq 4). For each window, we computed the frequency of sites in exons and the average deCODE recombination rate. We further assumed a selection coefficient s = 10−5 and allow the selection inside the low ILS regions to be one tenth (fs = 0.1). Out of 285 windows located in low-ILS regions, we could estimate the maximal reduction of ILS due to background selection in 252 windows for which a deCODE recombination estimate was available. In 79 of these windows only the expected reduction matched the observed one of 0.20.
Simulation of ancient selective sweeps
To assess how hard and soft sweeps in the human-chimpanzee ancestor can have reduced the proportion of ILS we simulated sweeps for different combinations of selection coefficients, s, and frequencies of the selected variant at the onset of selection, f. Frequency trajectories of selected variants are obtained using rejection sampling to obtain trajectories that fix in the population. Trajectories used to simulate hard sweeps begin at one and proceed to fixation at 2N * 3/4 by repeated binomial sampling with probability parameter Nmut/(Nmut + (N − Nmut)(1-s)), where Nmut is the number of selected variants in the previous generation. We use a human-chimpanzee speciation time of 3.7 Myr, a human-gorilla speciation time of 5.95 Myr, a human-chimpanzee effective population size of 73,200 as reported in [21], assuming a mutation rate of 1e-9 and a generation time of 20 years. Trajectories used to simulate soft sweeps are constructed by joining two trajectories. If f is the frequency of the variant at the onset of selection F = f * 2N * 3/4 is the number of variants. We first sample a trajectory that represents the time before the onset of selection. This trajectory is required to reach F at least once before it fixes or is lost, and is truncated randomly at one of the points where it passes the value F. The truncated trajectory is then appended with a trajectory under selection that begins at F and proceeds to fixation.
In each simulation we consider a sample of two sequences that represent 10 cM. As the effect of the sweep is symmetric we only simulate one side of the sweep. We then simulate backwards in the Wright-Fisher process with recombination allowing at most one recombination event per generation per lineage but allowing mergers of multiple lineages expected to occur in strong sweeps. The simulation proceeds until all sequence segments have found a most recent common ancestor (TMRCA). For each combination of parameters s and f we perform 1,000 simulations and the mean TMRCA is computed in bins of 10 kb.
In each simulation individual sequence segments are called as ILS with probability 2/3 if the TMRCA exceeds the time between the speciation events. The width of the region showing less than 5% ILS is then computed for each simulation. In Figs 4 and S3 a recombination rate of 1 cM/Mb is assumed to translate to physical length.
Comparing diversity between human populations
We computed the nucleotide diversity in 100 kb non-overlapping windows along the X chromosome for the 14 populations from the 1,000 genomes project. The windows in each low-ILS region were compared to windows outside the regions using a Wilcoxon test with correction for multiple testing [43] (Table 2). We computed the relative nucleotide diversity in the 1,298 windows located in low-ILS regions by dividing by the average of the rest of the X chromosome. Each population was further categorized according to its origin, Africa, America, Asia or Europe [31]. A linear model was fitted after Box-Cox transformation:

Association with ampliconic regions and Neanderthal introgression-free regions
In order to test the association of low-ILS regions with other genomic features, we developed a Monte-Carlo simulation procedure. In such a test, we wanted to compare a set of "reference" intervals with a set of "query" intervals. The null hypothesis is that the query intervals are independent of the reference intervals. We use the size of the overlap of the two sets of intervals as a statistic. During the randomization procedure, the set of query intervals is shuffled, so that each interval is conserved in length, only the relative order and positions of intervals are changed. Intervals are not allowed to overlap, so that the size of the query set is constant through simulations and identical to the observed one. The distance between two intervals is however allowed to be zero. For each simulation, the size of the overlap with the reference set of intervals is computed. A p-value is calculated by counting the number of simulations with an overlap at least equal to the observed one. In order to randomize intervals, we developed the following procedure: 1) compute the total size S of the chromosome not included in any interval of the query set; 2) draw n breakpoints uniformly between 0 and S, where n in the number of intervals in the query set; 3) insert randomly one query interval at each breakpoint. This procedure has the advantage that it keeps the structure of the reference set, so that the putative auto-correlation of reference intervals along the genome is accounted for. The 'intervals' R package was used for handling intervals and computing their overlap, and 100,000 randomizations were performed for each test.
We applied the randomization test to the two sets of Neanderthal introgression free regions for European and Asian populations, as well as for the ampliconic regions. The coordinates of ampliconic regions tested in [34] were translated to hg19 using the liftOver utility from UCSC. Fourteen regions were included in our alignment. For all tests, the set of low-ILS regions was used as a query set. For ampliconic regions, we performed a second test where ampliconic regions located close to the centromere and not included in our alignment were discarded.
Supporting Information
Zdroje
1. Meisel RP, Connallon T. The faster-X effect: integrating theory and data. Trends Genet TIG. 2013;29 : 537–544.
2. Vicoso B, Charlesworth B. Evolution on the X chromosome: unusual patterns and processes. Nat Rev Genet. 2006;7 : 645–653. 16847464
3. Gottipati S, Arbiza L, Siepel A, Clark AG, Keinan A. Analyses of X-linked and autosomal genetic variation in population-scale whole genome sequencing. Nat Genet. 2011;43 : 741–743. doi: 10.1038/ng.877 21775991
4. Arbiza L, Gottipati S, Siepel A, Keinan A. Contrasting X-linked and autosomal diversity across 14 human populations. Am J Hum Genet. 2014;94 : 827–844. doi: 10.1016/j.ajhg.2014.04.011 24836452
5. Hammer MF, Woerner AE, Mendez FL, Watkins JC, Cox MP, Wall JD. The ratio of human X chromosome to autosome diversity is positively correlated with genetic distance from genes. Nat Genet. 2010;42 : 830–831. doi: 10.1038/ng.651 20802480
6. Maynard Smith J, Haigh J. The hitch-hiking effect of a favourable gene. Genet Res. 1974;23 : 23–35. 4407212
7. Charlesworth B, Morgan MT, Charlesworth D. The effect of deleterious mutations on neutral molecular variation. Genetics. 1993;134 : 1289–1303. 8375663
8. Hvilsom C, Qian Y, Bataillon T, Li Y, Mailund T, Sallé B, et al. Extensive X-linked adaptive evolution in central chimpanzees. Proc Natl Acad Sci U S A. 2012;109 : 2054–2059. doi: 10.1073/pnas.1106877109 22308321
9. Bataillon T, Duan J, Hvilsom C, Jin X, Li Y, Skov L, et al. Inference of purifying and positive selection in three subspecies of chimpanzees (Pan troglodytes) from exome sequencing. Genome Biol Evol. 2015;7 : 1122–1132. doi: 10.1093/gbe/evv058 25829516
10. Veeramah KR, Gutenkunst RN, Woerner AE, Watkins JC, Hammer MF. Evidence for increased levels of positive and negative selection on the X chromosome versus autosomes in humans. Mol Biol Evol. 2014.
11. Kousathanas A, Halligan DL, Keightley PD. Faster-X adaptive protein evolution in house mice. Genetics. 2014;196 : 1131–1143. doi: 10.1534/genetics.113.158246 24361937
12. Laurie CC. The weaker sex is heterogametic: 75 years of Haldane’s rule. Genetics. 1997;147 : 937–951. 9383043
13. Schilthuizen M, Giesbers MCWG, Beukeboom LW. Haldane’s rule in the 21st century. Heredity. 2011;107 : 95–102. doi: 10.1038/hdy.2010.170 21224879
14. Sankararaman S, Mallick S, Dannemann M, Prüfer K, Kelso J, Pääbo S, et al. The genomic landscape of Neanderthal ancestry in present-day humans. Nature. 2014;
15. Hurst LD, Pomiankowski A. Causes of sex ratio bias may account for unisexual sterility in hybrids: a new explanation of Haldane’s rule and related phenomena. Genetics. 1991;128 : 841–858. 1916248
16. Haig D, Grafen A. Genetic scrambling as a defence against meiotic drive. J Theor Biol. 1991;153 : 531–558. 1806752
17. Meiklejohn CD, Tao Y. Genetic conflict and sex chromosome evolution. Trends Ecol Evol. 2010;25 : 215–223. doi: 10.1016/j.tree.2009.10.005 19931208
18. Nam K, Munch K, Hobolth A, Dutheil JY, Veeramah KR, Woerner AE, et al. Extreme selective sweeps independently targeted the X chromosomes of the great apes. Proc Natl Acad Sci U S A. 2015.
19. Frank SA. Divergence of Meiotic Drive-Suppression Systems as an Explanation for Sex - Biased Hybrid Sterility and Inviability. Evolution. 1991;45 : 262–267.
20. McDermott SR, Noor MAF. The role of meiotic drive in hybrid male sterility. Philos Trans R Soc Lond B Biol Sci. 2010;365 : 1265–1272. doi: 10.1098/rstb.2009.0264 20308102
21. Scally A, Dutheil JY, Hillier LW, Jordan GE, Goodhead I, Herrero J, et al. Insights into hominid evolution from the gorilla genome sequence. Nature. 2012;483 : 169–175. doi: 10.1038/nature10842 22398555
22. Hobolth A, Christensen OF, Mailund T, Schierup MH. Genomic relationships and speciation times of human, chimpanzee, and gorilla inferred from a coalescent hidden Markov model. PLoS Genet. 2007;3: e7. 17319744
23. Patterson N, Richter DJ, Gnerre S, Lander ES, Reich D. Genetic evidence for complex speciation of humans and chimpanzees. Nature. 2006;441 : 1103–1108. 16710306
24. Dutheil JY, Ganapathy G, Hobolth A, Mailund T, Uyenoyama MK, Schierup MH. Ancestral population genomics: the coalescent hidden Markov model approach. Genetics. 2009;183 : 259–274. doi: 10.1534/genetics.109.103010 19581452
25. Dutheil JY, Hobolth A. Ancestral population genomics. Methods Mol Biol Clifton NJ. 2012;856 : 293–313.
26. Mailund T, Munch K, Schierup MH. Lineage Sorting in Apes. Annu Rev Genet. 2014;
27. Takahata N. Gene genealogy in three related populations: consistency probability between gene and population trees. Genetics. 1989;122 : 957–966. 2759432
28. McVicker G, Gordon D, Davis C, Green P. Widespread genomic signatures of natural selection in hominid evolution. PLoS Genet. 2009;5: e1000471. doi: 10.1371/journal.pgen.1000471 19424416
29. Nordborg M, Charlesworth B, Charlesworth D. The effect of recombination on background selection. Genet Res. 1996;67 : 159–174. 8801188
30. Charlesworth B. The Role of Background Selection in Shaping Patterns of Molecular Evolution and Variation: Evidence from Variability on the Drosophila X Chromosome. Genetics. 2012;191 : 233–246. doi: 10.1534/genetics.111.138073 22377629
31. Genomes Project Consortium, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491 : 56–65. doi: 10.1038/nature11632 23128226
32. Keinan A, Reich D. Can a sex-biased human demography account for the reduced effective population size of chromosome X in non-Africans? Mol Biol Evol. 2010;27 : 2312–2321. doi: 10.1093/molbev/msq117 20453016
33. Cortez D, Marin R, Toledo-Flores D, Froidevaux L, Liechti A, Waters PD, et al. Origins and functional evolution of Y chromosomes across mammals. Nature. 2014;508 : 488–493. doi: 10.1038/nature13151 24759410
34. Mueller JL, Skaletsky H, Brown LG, Zaghlul S, Rock S, Graves T, et al. Independent specialization of the human and mouse X chromosomes for the male germ line. Nat Genet. 2013;45 : 1083–1087. doi: 10.1038/ng.2705 23872635
35. Dutheil JY, Gaillard S, Stukenbrock EH. MafFilter: a highly flexible and extensible multiple genome alignment files processor. BMC Genomics. 2014;15 : 53. doi: 10.1186/1471-2164-15-53 24447531
36. Hudson RR. Gene genealogies and the coalescent process. 1991. pp. 1–44.
37. Chen FC, Li WH. Genomic divergences between humans and other hominoids and the effective population size of the common ancestor of humans and chimpanzees. Am J Hum Genet. 2001;68 : 444–456. 11170892
38. Azzalini A. A Class of Distributions Which Includes the Normal Ones. Scand J Stat. 1985;12 : 171–178.
39. Durrett R. Probability Models for DNA Sequence Evolution [Internet]. 2nd ed. Springer; 2002. http://www.springer.com/mathematics/probability/book/978-0-387-78168-6
40. Kong A, Gudbjartsson DF, Sainz J, Jonsdottir GM, Gudjonsson SA, Richardsson B, et al. A high-resolution recombination map of the human genome. Nat Genet. 2002;31 : 241–247. 12053178
41. Genomes Project Consortium, Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467 : 1061–1073. doi: 10.1038/nature09534 20981092
42. Rands CM, Meader S, Ponting CP, Lunter G. 8.2% of the Human genome is constrained: variation in rates of turnover across functional element classes in the human lineage. PLoS Genet. 2014;10: e1004525. doi: 10.1371/journal.pgen.1004525 25057982
43. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol. 1995; 289–300.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 8
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Exon 7 Contributes to the Stable Localization of Xist RNA on the Inactive X-Chromosome
- YAP1 Exerts Its Transcriptional Control via TEAD-Mediated Activation of Enhancers
- SmD1 Modulates the miRNA Pathway Independently of Its Pre-mRNA Splicing Function
- TSPO, a Mitochondrial Outer Membrane Protein, Controls Ethanol-Related Behaviors in
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy