
Genome-Wide Association and Trans-ethnic Meta-Analysis for Advanced Diabetic Kidney Disease: Family Investigation of Nephropathy and Diabetes (FIND)
Type 2 diabetes is the most common cause of severe kidney disease worldwide and diabetic kidney disease (DKD) associates with premature death. Individuals of non-European ancestry have the highest burden of type 2 DKD; hence understanding the causes of DKD remains critical to reducing health disparities. Family studies demonstrate that genes regulate the onset and progression of DKD; however, identifying these genes has proven to be challenging. The Family Investigation of Diabetes and Nephropathy consortium (FIND) recruited a large multi-ethnic collection of individuals with type 2 diabetes with and without kidney disease in order to detect genes associated with DKD. FIND discovered and replicated a DKD-associated genetic locus on human chromosome 6q25.2 (rs955333) between the SCAF8 and CNKSR genes. Findings were supported by significantly different expression of genes in this region from kidney tissue of subjects with, versus without DKD. The present findings identify a novel kidney disease susceptibility locus in individuals with type 2 diabetes which is consistent across subjects of differing ancestries. In addition, FIND results provide a rich catalogue of genetic variation in DKD patients for future research on the genetic architecture regulating this common and devastating disease.
Published in the journal:
. PLoS Genet 11(8): e32767. doi:10.1371/journal.pgen.1005352
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005352
Summary
Type 2 diabetes is the most common cause of severe kidney disease worldwide and diabetic kidney disease (DKD) associates with premature death. Individuals of non-European ancestry have the highest burden of type 2 DKD; hence understanding the causes of DKD remains critical to reducing health disparities. Family studies demonstrate that genes regulate the onset and progression of DKD; however, identifying these genes has proven to be challenging. The Family Investigation of Diabetes and Nephropathy consortium (FIND) recruited a large multi-ethnic collection of individuals with type 2 diabetes with and without kidney disease in order to detect genes associated with DKD. FIND discovered and replicated a DKD-associated genetic locus on human chromosome 6q25.2 (rs955333) between the SCAF8 and CNKSR genes. Findings were supported by significantly different expression of genes in this region from kidney tissue of subjects with, versus without DKD. The present findings identify a novel kidney disease susceptibility locus in individuals with type 2 diabetes which is consistent across subjects of differing ancestries. In addition, FIND results provide a rich catalogue of genetic variation in DKD patients for future research on the genetic architecture regulating this common and devastating disease.
Introduction
Diabetic kidney disease (DKD) is a devastating complication in patients with diabetes mellitus (DM) and is associated with high risk for cardiovascular disease and death.[1,2] DKD is the leading cause of end-stage kidney disease (ESKD) requiring renal replacement therapy in developed nations; these procedures incur high healthcare costs with great personal, family and societal burden.[3] The prevalence of DKD continues to rise in the United States in proportion to the growing prevalence of DM. Unfortunately, intensification of glycemic, lipid and blood pressure control have not dramatically impacted the prevalence of DKD.[3,4] Hyperglycemia alone is insufficient to cause DKD. Genetic factors appear critical in its pathogenesis based upon variable incidence rates of DKD between population groups, aggregation of DKD-associated ESKD in families, and the highly heritable nature of diabetic renal histologic changes, estimated glomerular filtration rate (eGFR) and proteinuria.[5]
Genome-wide association studies (GWAS) have identified multiple loci for kidney function and chronic kidney disease (CKD) in population - and community-based cohorts, primarily of European ancestry.[6–10] However, CKD phenotypes in many studies included minimally to moderately reduced eGFR, not fully reflective of the progressive forms of CKD seen in kidney disease clinics. In early reports, published GWAS signals for DKD were equivocal, confounded by small sample sizes and failure to consistently replicate. Recently, the GEnetics of Nephropathy: an International Effort (GENIE) consortium identified genome-wide significant, replicated signals in a meta-analysis of over 12,000 type 1 (T1) DM patients with DKD of European ancestry.[9] Type 2 (T2) DM is far more prevalent than T1DM, accounting for 90% of cases worldwide and for the majority of prevalent cases of DKD. Relative to European Americans (EAs) with T2DM, African American (AA), American Indian (AI), and Mexican American (MA) patients with T2DM are disproportionately affected by severe DKD,[3] yet under-represented in genetic analyses. Defining the underlying genetic architecture responsible for advanced T2DM-associated kidney disease in multiple populations could provide critical insights into pathogenesis and identify new molecular targets for therapy. We report the results of a GWAS in AA, EA, MA, and AI patients with DKD enrolled in the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK)-sponsored “Family Investigation of Nephropathy and Diabetes” (FIND) [11] and the corresponding large replication study and trans-ethnic meta-analysis.
Results and Discussion
Demographic characteristics of the Discovery and Find Large Replication study (FILR) samples that met FIND phenotype qualifications and genotype quality control (QC) are summarized in Table 1 and S1 Table. The proportion of females, age at ESKD or enrollment, hemoglobin A1c and proportion with diabetic retinopathy (DR) varied by ancestry but was generally comparable between the Discovery and FILR samples within a specific ethnic population. Phenotypic differences among these populations and their genetic and DKD prevalence differences motivated the meta-analysis approach.

The principal component (PC) analysis identified PCs that genetically partitioned the Discovery sample into ancestry groups consistent with self-report. S1 Fig displays the two-dimensional partitioning via PC analysis with the boundaries for inclusion into the GWAS analysis. The logistic regression model that included the PCs as covariates reduced the inflation factor to nominal levels and combined with the P-P plot show no evidence of a systematic inflation (S2 Fig). In the replication, the inflation factor was λ = 1.05 using 278 AIMs. If we scale to 1,000 cases and 1,000 controls this would be λ1000 = 1.017, an appropriate inflation factor for the replication study. S3 Fig provides a summary of the statistical power analyses for the race-specific discovery, replication analysis, and meta-analyses. These calculations show that for risk-predisposing variants shared across ancestries the study has power >0.50 and 0.80 to detect odds ratios (OR) on the order of 1.06 to 1.15 for a minor allele frequency (MAF) = 0.45 and 1.30 to 1.36 for a MAF = 0.05, respectively. Further, leveraging the differences in linkage disequilibrium (LD) among the four ancestries, the study is powered to potentially reduce the size of the associated region via trans-ethnic mapping.
Trans-ethnic Meta-Analysis Associations
The only locus that reached genome wide significance for DKD in the trans-ethnic meta-analysis encompassing all FIND Discovery and Replication samples was rs955333 on chromosome 6 (minimum p-value 1.31x10-8 [additive]; minimum p-value 9.02x10-11 [dominant]) (Table 2). Fig 1 contains the Manhattan plot for the meta-analysis across all ancestries included in the Discovery and Replication samples. Consistent directions of association were present in three ethnic groups (only AA samples did not pass QC) and several supporting single nucleotide polymorphisms (SNPs) were detected in the region (regional plot in Fig 2). This SNP lies between the SR-like carboxyl-terminal domain associated factor 8 gene (SCAF8) and the connector enhancer of KSR family of scaffold proteins gene (CNKSR3), suggesting a possible role in transcription regulation. CNKSR3 is a direct mineralocorticoid receptor target gene involved in regulation of the epithelial sodium channel (ENaC) on the apical membrane of cells in the distal nephron.[12] CNKSR3 is highly expressed in the renal cortical collecting duct and upregulated in response to physiologic aldosterone concentrations. ENaC precisely regulates renal sodium absorption and plays important roles in maintenance of plasma volume and blood pressure. Ziera et al. [12] suggested that CNKSR3, a PSD-95/DLG-1/ZO-1 (PDZ) domain containing protein, inhibits the RAS/ERK signaling pathway, stimulating ENaC activity with enhanced renal sodium absorption. More recently, CNKSR3 was shown to function as an aldosterone-induced scaffolding platform that orchestrated assembly of ENaC and its regulators Nedd4-2, Raf-1 and SGK-1 and was essential for stimulation of ENaC function by aldosterone.[13] Clinically, renin-angiotensin-aldosterone system (RAAS) blockade serves as a mainstay of therapy for patients with DKD and other proteinuric kidney diseases.[14,15] Inhibition of aldosterone may further limit renal fibrosis, independent of natriuretic effects.[16,17] Hence, significant association between DKD and markers near CNKSR3 is consistent with clinical trial data demonstrating that blockade of the renin angiotensin system or the aldosterone receptor slows DKD progression. However, further experiments are needed to demonstrate that the associated SNP regulates the pathogenesis of progressive DKD. Further studies will be necessary to assess if the CNKSR3 regulates DKD pathogenesis indirectly by its effects on ENaC activity or directly by promoting aldosterone-dependent fibrosis.

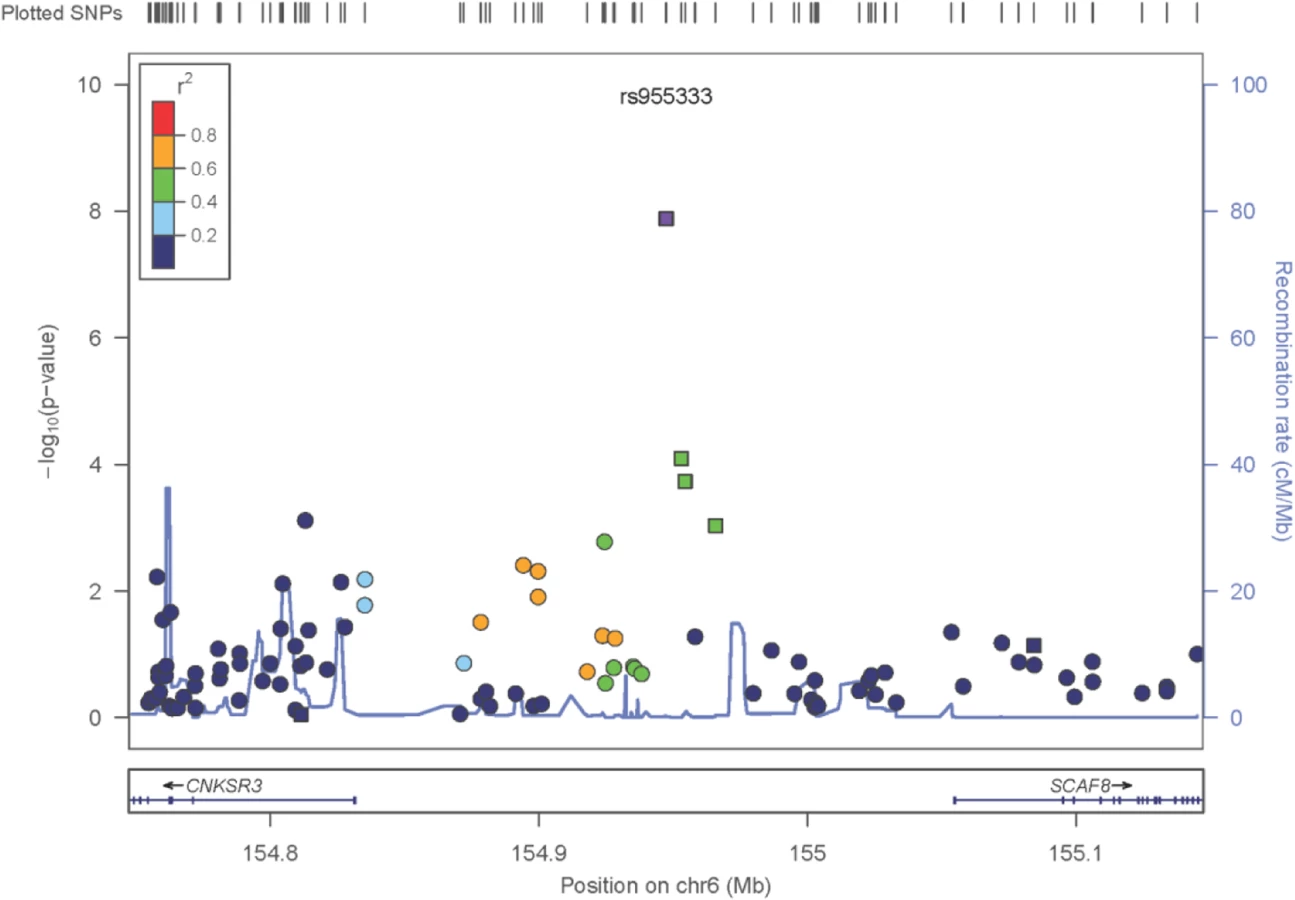

Less is known about the function of SCAF8, also known as RBM16. SCAF8 is a RNA maturation factor recruited to the carboxy-terminal domain of RNA polymerase II in a phosphorylation-dependent manner.[18] It also is a target for ataxia telangiectasia mutated (ATM) kinase, a crucial component of the DNA damage response required for DNA repair and cell cycle control.[19] ATM kinase is associated with responsiveness of patients with DM to the insulin sensitizer metformin in some but not all studies.[20,21] Thus, genes in the region of rs955333 are suggestive of DKD-related pathogenesis.
GWAS loci identify elements that may regulate gene expression, and recent data indicate GWAS associations are located in regions bounded by recombination hot spots near non-coding causal variants, which regulate transcription.[22,23] We next contrasted transcript abundance of the genes within the megabase region centered on rs955333, TIAM2, SCAF8, CNKSR3, IPCEF1 and OPRM1, in DKD and living donor kidney biopsies. DKD biopsies were obtained from European and AI cohorts and were analyzed separately. All five genes show statistically significant differential expression in at least one kidney tissue compartment of one population. SCAF8 steady state mRNA levels show increased expression in DKD compared to living donor biopsies in glomerular and tubulo-interstitial compartments of both populations (S2B Table); TIAM2 and OPRM1 show glomerular-specific differential expression; IPCEF1 is repressed in both tissue compartments of AI subjects; and CNKSR3 is increased in the tubulo-interstitial compartment of AIs (S2B Table). Normalized tubulo-interstitial expression of CNKSR3 correlated with urine albumin (r = 0.78, q = 0.0056) and urine albumin:creatinine ratio (UACR) (r = 0.74, q = 0.0107). In addition, IPCEF1, located downstream of CNKSR3, has been reported to be translated with CNKSR3 as one protein,[24] and has a tubulo-interstitial expression quantitative trait locus (eQTL) (NM_001130699, rs249964, P = 2.34E-04) (S2A Table). LD between this SNP and the sentinel variants in the region significantly associated with DKD in the trans-ethnic (rs955333) and AI association analysis (rs12523822; see below) is negligible (D’ = 0.43, r2 = 0.01 in AI). However, tubulo-interstitial expression of IPCEF1 in kidney tissue from AIs was significantly correlated with the DKD phenotype UACR (r = -0.54, q = 0.031). These studies were limited by the small number of available biopsies the narrow criteria used to define the region of interest (see Methods). As proxies, disease-dependent differential gene expression and the rs249964 eQTL demonstrate DKD regulatory activity in the locus. Significant results of eQTL and differential gene expression analyses for other loci in Table 2 are also sown in S2A Table and S2B Table, respectively.
African American Associations
No SNP reached genome-wide significance (P<5x10-8) in the AA GWAS; however, a number provided suggestive evidence for association with DKD (Table 3; S5A Table and S6A Table summarize the top 200 SNP associations in the discovery GWAS and replication study, respectively). The strongest associations were found within the apolipoprotein L1 (APOL1) and non-muscle heavy chain 9 gene (MYH9) region on 22q (Table 2, Discovery + FILR meta-analysis: rs5750250, P = 7.7x10-8; rs136161, P = 5.23x10-7). Since G1 and G2 variants of APOL1 are strongly associated with non-diabetic nephropathy in AA patients,[25–27] the G1/G2 compound risk was modeled under a recessive genetic model and these variants accounted for the associations on 22q in Table 2 (rs5750250 P = 7.70x10-8, OR = 1.27; rs136161 P = 5.23x10-7, OR = 1.36). Association with G1/G2 within APOL1 likely exists due to inclusion of non-FIND AA cases with coincident DM and unrecognized non-diabetic kidney disease.[28] APOL1 was not associated with T2D-ESKD in a logistic regression analysis adjusting for age, gender and global ancestry restricted to FIND MALD and CHOICE (Choices for Healthy Outcomes In Caring for End-stage renal disease) study cases meeting the original FIND DKD case definition (rs73885319 P = 0.1098; rs71785313 P = 0.1182).[29]
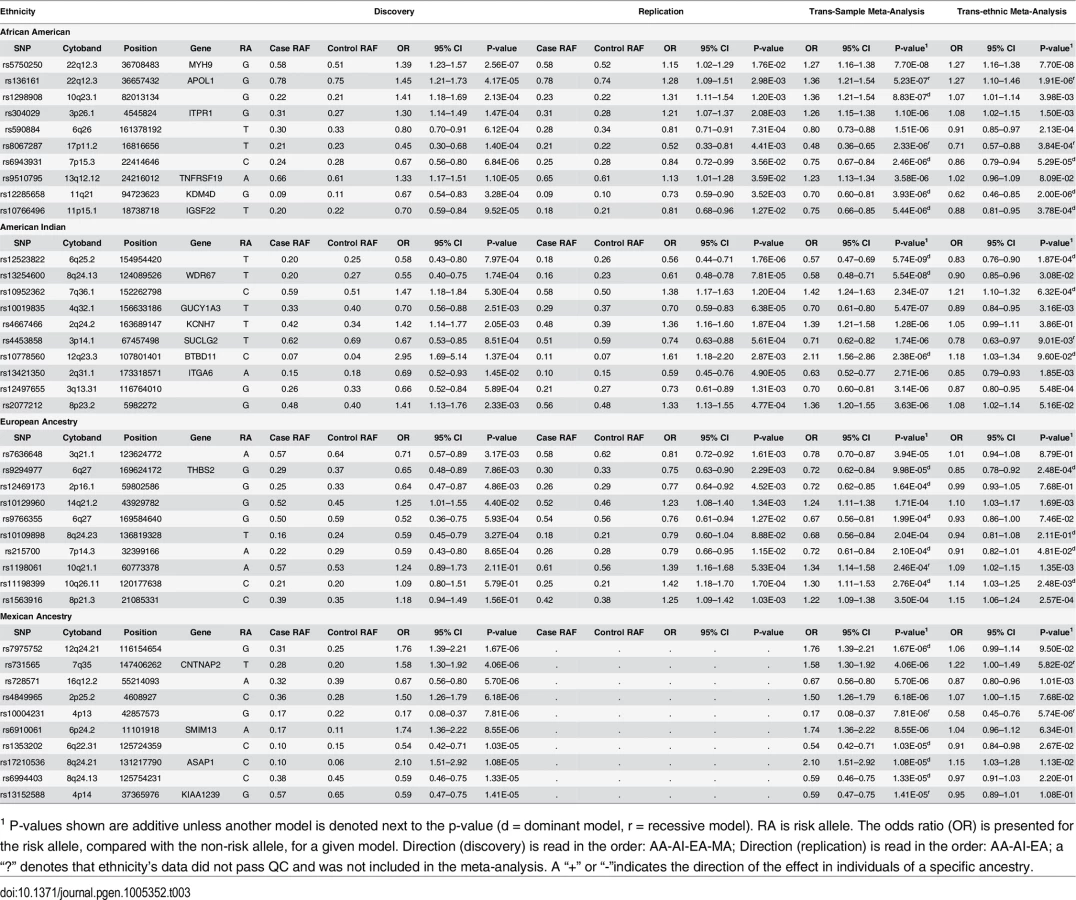
Regions beyond 22q provided suggestive evidence of association in the AA Discovery + FILR meta-analysis including rs1298908 on 10q22 (OR = 1.36, P = 8.83x10-7) between MAT1A and ANXA11, in a region dense with regulatory elements and transcription factors. There was also an association on 3p26 (rs304029, OR = 1.26 P = 1.10x10-6) within inositol 1,4,5-trisphosphate receptor, type 1 (ITPR1), a gene involved in cerebellar and autoimmune disorders but not renal involvement.[30] The genes in these other candidate regions (ANXA11, MAT1A and ITPR1) also show statistically significant differential expression in at least one population and compartment; as do IGSF22 near candidate rs11766496 on chromosome 11, and TNFRSF19 near rs95107795 on chromosome 13. Other top AA associated regions in Table 2 do not have clear connections to kidney disease. Since APOL1 association likely reflected inclusion of non-FIND cases with non-diabetic nephropathy, a GWAS was re-computed within AAs in the discovery sample, which only included subjects lacking two APOL1 risk variants. The top 200 associations from this GWAS are summarized in S7 Table. The correlation between the–log10 (p-value) for GWAS with and with AA subjects with and without two APOL1 risk variants is r = 0.82 (S4 Fig). The top association in this subset GWAS was rs2780902 on 1p31 (OR = 0.52, P = 2.98x10-7) within Janus kinase 1 (JAK1), a member of the protein-tyrosine kinases.[31] The ENCODE data shows that this SNP resides within a region with numerous transcription factors and DNase I hypersensitivity sites. JAK1 is a widely expressed membrane associated phosphoprotein and is involved in interferon transduction pathway. This kinase links cytokine ligand binding to tyrosine phosphorylation of various known signaling proteins and the signal transducers and activators of transcription (STATs). Another interesting association among the top 10 associations is rs2596230 on 15q14 (OR = 1.56, P = 9.36x10-6) within ryanodine receptor 3 (RYR3).[32] The protein encoded by RYR3 functions to release calcium from intercellular storage in many cellular processes and the gene is expressed in the kidney. The closely related gene, RYR2, is associated with albuminuria.[33] Our prior analyses of transcript expression in DKD biopsies provide additional support for the associations. Both JAK1 and RYR3 (and RYR2) show differential expression that is restricted to the European subjects with Stage III and Stage IV CKD. JAK1 expression is increased in DKD in both compartments, while RYR3 and RYR2 are depressed in the glomerulus.[34] We also recomputed the genome wide discovery and trans-ethnic meta-analysis removing AA subjects with APOL1. The top 200 associations are summarized in S8 Table.
American Indian Associations
Several regions provided evidence of association with DKD in AIs (Table 3; S5B Table and S6B Table summarize the top 200 SNP associations in the discovery GWAS and replication study, respectively). The strongest association was with rs12523822 on 6q.25 in the SCAF8-CNKSR3 gene region (OR = 0.57, P = 5.74x10-9). This SNP is in strong LD with rs955333, the top hit in the trans-ethnic meta-analysis (r2 = 0.96 in AI unrelated controls); S5 Fig graphically illustrates the extended linkage disequilibrium in this region in all but the AA samples. The A allele at rs955333 is the ancestral allele and confers susceptibility to DKD (as the G allele has OR<1 in Table 2); the A allele has a frequency of 0.76 in the American Indian samples and 0.85 in European American samples, but is nearly monomorphic in African American samples. The allele frequencies are very similar in population-based samples: 0.85 in HapMap CEU, 1.00 in YRI, 0.77 in MEX and 0.76 in full-heritage American Indians from the southwestern United States (R Hanson, personal communication). Thus, the high risk allele at this locus does not appear to be Amerindian specific. The p-value for association in European Americans is 0.0013 and 1.3x10-6 in American Indian, suggesting that the signal does not come entirely from American Indians samples. Further fine-mapping or sequencing will be necessary to fully characterize the association signal within and across ethnic groups. Another association that approached genome-wide significance was rs13254600 (OR = 0.58, P = 5.54x10-8) on 8q24 within WD repeat domain 67 (WDR67). This gene is expressed in a wide variety of tissues, including kidney, and may affect cellular membrane functions by regulating Rab GTPase activity.[35] TBC1D31 (WDR67) mRNA is increased in both compartments of kidney tissue from AIs, but only in the glomerulus for European subjects with more advanced DKD.
Another SNP of interest is rs10019835 (OR = 0.70, 5.47x10-7) on 4q32 within guanylate cyclase 1, soluble, alpha 3 (GUCY1A3); the protein encoded by GUCY1A3 serves as a receptor for nitric oxide,[36] which through its role in endothelial function may be a mediator of DKD.[37] GUCY1A3 is differentially expressed in both tissue compartments and both DKD biopsy cohorts, and shows one of the strongest differences of all genes in candidate regions (especially among the European subjects who have more advanced DKD) (S3A Table and S3B Table; S6 Fig). In addition, the candidate SNP rs10019835 has a tubulo-interstitial specific eQTL with the full-length isoform of GUCY1A3 (NM_000856, P = 4.97x10-4). The shortest isoform of the gene (NM_001130687) has a glomerular eQTL with rs12504357 (P = 2.63x10-5), an intronic SNP that is 5kb upstream of the associated variant. These two eQTL SNPs have D’ = 1 in some populations, likely reflecting low allele frequencies in the reference populations. Integrin alpha 6 (ITGA6, rs13421350, 2q31, OR = 0.58, P = 5.54x10-8) is involved in cell adhesion and is expressed in the kidney. The gene shows negative differential expression in Europeans with DKD, and it has both glomerular and tubulo-interstitial eQTL. The glomerular eQTL is with the SNP rs6758468 (P = 5.41x10-4), which is 143kb from the candidate; while the tubulo-interstitial eQTL is with rs12469788 (P = 3.26x10-4), which is 5kb from the candidate with D’ = 1, but negligible r2. Finally, rs10952362 on 7q36 near XRCC2 (rs10952362, OR = 1.91, P = 7.99x10-8), a gene involved in DNA repair was strongly associated with DKD.[38] We find that XRCC2 is repressed in the tubulo-interstitial kidney tissue from AIs.
European American Associations
EA subjects comprised the smallest group within FIND and power to detect variants associated with DKD was limited (S3 Fig). None of the associations in the EA Discovery + FILR meta-analysis had a p-value <10−5 (Table 3; S5C Table and S6C Table summarize the top 200 SNP associations in the discovery GWAS and replication study, respectively).
Mexican American Associations
Several suggestive associations were identified in the MA Discovery GWAS (Table 3; S5D Table summarizes the top 200 SNP associations in the GWAS). No replication cohort was available to be genotyped in FILR, so only the Discovery GWAS and trans-ethnic meta-analysis are reported (Tables 2 and 3). The strongest association was on 12q24 for rs7975752, located ~242 kb downstream of the mediator complex subunit 13-like (MED13L) gene (OR = 1.76, P = 1.67 x 10−6). MED13L functions as a transcriptional coactivator for RNA polymerase II-transcribed genes. While its functional significance in DKD is unclear, gene variants 4 Mb downstream (rs614226) and upstream (rs653178) on 12q24 show genome-wide significant association with ESKD [9] and CKD [39] in Europeans. We see that MED13L is repressed in both compartments in kidney tissue from AIs but only in the glomerular transcriptome in the European subjects. Association was observed between DKD and rs731565 (P = 4.06 x 10−6) residing within an intronic region of the contactin-associated protein-like 2 (CNTNAP2) gene on 7q36. SNP rs7805747, approximately 4 Mb downstream from rs731565 has been associated with CKD in European populations [39] Finally, rs4849965, 1.2 Mb upstream of the SRY-related HMG-box 11 (SOX11) gene on 2p25.2 trended toward association with DKD (OR 1.50, 95% CI 1.26–1.79; P = 6.18x10-6) and has previously been associated with CKD in Europeans.[39] We find that absolute tubulo-interstitial expression of SOX11 in AIs is correlated with ACR (r = 0.66, q = 0.029).
Conclusions
The current FIND GWAS comprises the largest genetic analysis for severe DKD based upon risk for progression to ESKD in EA and high-risk non-European ethnic groups including AAs, AIs, and MAs. As in other GWAS, results support a role for multiple DKD susceptibility genes, each with weak effects. A number of the SNPs most strongly associated with DKD had additional support from compartment-specific gene expression measures and eQTL analysis obtained in European and American Indian populations. A novel chromosome 6q25.2 DKD locus was identified in AI samples; SNPs in this region had genome-wide significant association and consistent directions of effect in the meta-analysis across all ethnic groups. Independent support for this region comes from an association with serum creatinine/eGFR in a GWAS in East Asian populations (P = 2.6 x 10−5 at rs4870304) [40]. Strengths of the FIND GWAS were the severe phenotype in cases, focus on DKD in T2D, and inclusion of non-European populations. The 6q25.2 locus requires fine mapping and additional replication in independent sample sets of diabetic subjects with and without DKD that has sufficient power to detect associated, common variants with moderate effect size. Once localized and replicated, functional studies in animal and cell culture models will be necessary to discover the biological mechanisms responsible for the association of DKD with the underlying genetic architecture.
As in other GWAS for complex disease, many previously identified DKD loci were not replicated in the FIND analyses. The inconsistency between our data and published DKD GWAS could reflect that FIND limited the DKD case group to subjects with ESKD and DKD with heavy proteinuria felt to be at high risk for progression to ESKD. FIND did not include microalbuminuric participants as “cases” in the Discovery cohort, choosing instead to focus on advanced nephropathy. However, some microalbuminuric participants with ACR<100 mg/g were included in the replication analysis. Prior GWAS focused on European and Asian DKD populations, often enriched for T1D-associated DKD. Genetic associations may not replicate across other populations; for example, association of APOL1 variants with non-diabetic kidney disease is limited to populations with recent African ancestry. Another possible interpretation is the variants, which regulate DKD pathogenesis, are distinct for T1D and T2D, although a meta-analysis including both T1D and T2D subjects may identify shared loci. Finally, the DKD phenotype in the FIND GWAS relied on standard, stringent clinical criteria for advanced DKD. This approach limited phenotypic heterogeneity but potentially minimized the utility of cross-study comparisons. Although heavy proteinuria is a hallmark of DKD, recent analyses suggest approximately one third of patients with diabetes and an eGFR <60 ml/min per 1.73 m2 had normal urinary protein excretion.[4] This would justify the focus of FIND on advanced DKD. Although not the only DKD phenotype with a genetic component, several investigators recently proposed using ESKD as the optimal DKD phenotype in genetic association studies.[41,42] The availability of bio-samples from patients with advanced DKD is limited. Therefore, entry criteria in the present replication cohorts were loosened to increase sample size; this likely included a small number of participants with non-diabetic CKD (or DKD less likely to progress to ESKD). The AA non-FIND cases used in our replication cohort appear to have included individuals with DM and coincident focal segmental glomerulosclerosis (FSGS), an effect addressed via partitioning based on APOL1 G1 and G2.[28] As in all GWAS, some non-nephropathy controls may develop DKD. This effect would bias results toward the null making it less likely to detect significant association.
FIND was well-powered to detect common risk variants with moderate effect sizes shared across ethnic groups. It was also well powered to use differences in effect sizes to help localize the region of association via transracial mapping. However, it was not powered to detect modest ethnic-specific effects that are not shared with another ethnicity or gene-gene interactions. Thus, these ethnic-specific scans provide important hypothesis generating results for subsequent meta-analyses, pathway enrichment analyses and hypothesis generation.
Materials and Methods
Ethics Statement
The FIND was completed in accordance with the principles of the Declaration of Helsinki. Written informed consent was obtained from all participants. The Institutional Review Board at each participating center (Case Western Reserve University, Cleveland, OH, Harbor-University of California Los Angeles Medical Center, Johns Hopkins University, Baltimore, National Institute of Diabetes and Digestive and Kidney Diseases, Phoenix, AZ, University of California, Los Angeles, CA, University of New Mexico, Albuquerque, NM, University of Texas Health Science Center at San Antonio, San Antonio, TX, Wake Forest School of Medicine, Winston-Salem, NC) approved all procedures, and all study subjects provided written informed consent. A certificate of confidentiality was filed at the National Institutes of Health.
Samples
Discovery cohorts
FIND is a multi-ancestry family study of severe DKD.[11] Index cases had advanced DKD, likely to progress to ESKD based on clinical criteria, and at least one informative sibling with either DKD or long-standing DM without nephropathy. Detailed phenotype criteria for enrollment have been reported.[26,43,44] Index cases of AA, EA, MA and AI ethnicity were included in the Discovery GWAS; all had DM duration >5 years and/or DR, with UACR >1 g/g or ESKD. Unrelated controls had DM duration ≥9 years, UACR <30 mg/g (equating to overnight albumin excretion <20 mcg/min), and serum creatinine <1.6 mg/dl ([122 μmol/L] men) or <1.4 mg/dl ([107 μmol/L] women). In AA, EA and MA only unrelated cases and controls were included; since AI participants were largely recruited from relatively small communities all available cases and controls meeting criteria were included, regardless of relationships.
Additional non-FIND-study DKD cases and controls (with and without DM) were genotyped to increase power (S1 Table). Non-FIND samples included unrelated DKD cases and controls of self-reported African American ethnicity recruited at Wake Forest,[45] Case Western Reserve [46] and Howard Universities;[47] unrelated cases and controls of EA ethnicity recruited at Wake Forest [48] and Case Western Reserve;[49,50] cases and controls of AI ethnicity recruited at NIDDK-Phoenix; [51] and cases and controls of MA ethnicity recruited in San Antonio and Los Angeles.[52]
Replication cohorts
The FIND Large Replication (FILR) Study was comprised of samples independent from the Discovery cohorts. AA and EA replication cohorts were unrelated individuals recruited at Wake Forest, Johns Hopkins, Case Western Reserve and Harbor UCLA Universities and out-of-study control data from the Genetic Association Information Network (GAIN) consortium;[53] AI replication cohorts consisted of pedigree data from NIDDK-Phoenix [51] and from the Dakota and Oklahoma centers of the Strong Heart Family Study.[54] Replication cases had DM duration >5 years and/or DR, UACR ≥0.3 g/g (equating to overnight albumin excretion >200 mcg/min) and/or proteinuria >500 mg/day or ESKD. DM controls had an eGFR >60 ml/min/1.73 m2, UACR <30 mg/g after 10 year DM duration or UACR <100 mg/g after 15 year DM duration. GAIN study subjects with and without DM were used as controls; no kidney function data were available for these individuals. GAIN samples were excluded for specific SNPs, if MAFs were inconsistent with those in FIND controls. Additional MA subjects were not available for inclusion in FILR.
Samples analyzed
Based on ancestry, the FIND discovery GWAS samples included: (i) AA: 1564 DKD cases (633 in FIND, 931 out of study), 369 controls with DM lacking nephropathy (277 in FIND, 92 out of study) and 1,288 non-diabetic non-nephropathy controls (all out of study); (ii) AI: 538 DKD cases, 319 controls with DM lacking nephropathy; (iii) EA: 342 DKD cases, 404 controls with DM lacking nephropathy; and (iv) MA: 779 DKD cases and 594 controls with DM lacking nephropathy. The FILR replication study included: (i) AA: 950 DKD cases, 50 controls with DM lacking nephropathy and 1,887 non-diabetic non-nephropathy controls; (ii) AI: 471 DKD cases, 340 controls with DM lacking nephropathy and 486 non-diabetic non-nephropathy controls and (iii) EA: 582 DKD cases, 205 controls with DM lacking nephropathy and 2,568 non-diabetic non-nephropathy controls. FILR samples were genotyped at loci including the top associated SNPs from the Discovery GWAS, eQTL associations, literature-based candidate gene polymorphisms and ancestry informative markers (AIMs). S1 Table delineates the sample sources in the Discovery GWAS and FILR, stratified by ancestry.
Genotyping and Statistical Methods
See Supplementary Methods (S1 Text).
SNP Selection for the Discovery and Replication Study
The DNA samples that comprise the Discovery cohorts, plus an additional 244 blind duplicates were genotyped on the Affymetrix Genome-Wide Human 6.0 SNP array (see S1 Text Supplemental Methods for details). The FILR replication samples were genotyped for 3,937 SNPs selected based on the strength of the statistical association from the Discovery GWAS. Additional SNPs were included based on the FIND eQTL association and candidate gene SNPs previously reported to be associated with DKD (see S1 Text Supplemental Methods for details). Specifically, within each ancestry group, the SNPs with the strongest statistical evidence of association were identified; a few additional SNPs from each region with supportive but weaker evidence of association were also identified (i.e., associations due to LD but r2<0.95 with the primary associated SNP). This redundancy was designed to limit the number of regions not represented in the replication study due to genotyping failure. In total, 3,019 SNPs (821 AA, 790 AI, 608 EA, and 800 MA) were genotyped for FILR based solely on statistical association with DKD within an ethnicity. The trans-ethnic meta-analysis of the discovery cohort identified another 436 SNPs nominally associated with DKD (p<0.0003). In addition, 482 SNPs (121 AA, 133 AI, 122 EA, 14 MA, meta-analysis 92) were chosen with the smallest L2-norm (i.e., Euclidean distance) of the–log10 (p-values) from GWAS and eQTL association analyses, provided that p <0.01 from GWAS. Here, the L2-norm was defined relative to the maximum of the–log10 (p-values) from the GWAS and eQTL and provides an ordering of the combined evidence for eQTL and association with DKD. SNP associations in FILR were considered “replicated” if both the association reached statistical significance and direction of the association was consistent with the Discovery analysis. Finally, 278 AIMs were genotyped to allow for adjustment of potential population substructure. Thus, FILR was designed as a replication study and not a large-scale trans-ethnic fine-mapping study. Subsequent studies will complete fine-mapping to localize associations.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Ninomiya T, Perkovic V, de Galan BE, Zoungas S, Pillai A et al. (2009) Albuminuria and kidney function independently predict cardiovascular and renal outcomes in diabetes. J Am Soc Nephrol 20 : 1813–1821. doi: 10.1681/ASN.2008121270 19443635
2. Berhane AM, Weil EJ, Knowler WC, Nelson RG, Hanson RL (2011) Albuminuria and estimated glomerular filtration rate as predictors of diabetic end-stage renal disease and death. Clin J Am Soc Nephrol 6 : 2444–2451. doi: 10.2215/CJN.00580111 21852671
3. [Anonymous] (2012) U.S. Renal Data System, USRDS 2012 Annual Data Report, Vol 1: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States, National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases.
4. de Boer I, Rue TC, Hall YN, Heagerty PJ, Weiss NS et al. (2011) Temporal trends in the prevalence of diabetic kidney disease in the United States. JAMA 305 : 2532–2539. doi: 10.1001/jama.2011.861 21693741
5. Freedman BI, Bostrom M, Daeihagh P, Bowden DW (2007) Genetic factors in diabetic nephropathy. Clin J Am Soc Nephrol 2 : 1306–1316. 17942768
6. Pezzolesi MG, Poznik GD, Mychaleckyj JC, Paterson AD, Barati MT et al. (2009) Genome-wide association scan for diabetic nephropathy susceptibility genes in type 1 diabetes. Diabetes 58(6): 1403–1410. doi: 10.2337/db08-1514 19252134
7. Maeda S, Kobayashi MA, Araki S, Babazono T, Freedman BI et al. (2010) A single nucleotide polymorphism within the acetyl-coenzyme A carboxylase beta gene is associated with proteinuria in patients with type 2 diabetes. PLoS Genet 6: e1000842. doi: 10.1371/journal.pgen.1000842 20168990
8. Pezzolesi MG, Poznik GD, Skupien J, Smiles AM, Mychaleckyj JC et al. (2011) An intergenic region on chromosome 13q33.3 is associated with the susceptibility to kidney disease in type 1 and 2 diabetes. Kidney Int 80 : 105–111. doi: 10.1038/ki.2011.64 21412220
9. Sandholm N, Salem RM, McKnight AJ, Brennan EP, Forsblom C et al. (2012) New susceptibility loci associated with kidney disease in type 1 diabetes. PLoS Genet 8: e1002921. doi: 10.1371/journal.pgen.1002921 23028342
10. Sandholm N, McKnight AJ, Salem RM, Brennan EP, Forsblom C et al. (2013) Chromosome 2q31.1 associates with ESRD in women with type 1 diabetes. J Am Soc Nephrol 24 : 1537–1543. doi: 10.1681/ASN.2012111122 24029427
11. Knowler WC, Coresh J, Elston RC, Freedman BI, Iyengar SK et al. (2005) The Family Investigation of Nephropathy and Diabetes (FIND): design and methods. J Diabetes Complications 19 : 1–9. 15642484
12. Ziera T, Irlbacher H, Fromm A, Latouche C, Krug SM et al. (2009) Cnksr3 is a direct mineralocorticoid receptor target gene and plays a key role in the regulation of the epithelial sodium channel. FASEB J 23 : 3936–3946. doi: 10.1096/fj.09-134759 19567370
13. Soundararajan R, Ziera T, Koo E, Ling K, Wang J et al. (2012) Scaffold protein connector enhancer of kinase suppressor of Ras isoform 3 (CNK3) coordinates assembly of a multiprotein epithelial sodium channel (ENaC)-regulatory complex. J Biol Chem 287 : 33014–33025. 22851176
14. Lewis EJ, Hunsicker LG, Bain RP, Rohde RD (1993) The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med 329(20): 1456–1462. 8413456
15. Brenner BM, Cooper ME, de ZD, Keane WF, Mitch WE et al. (2001) Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 20;345(12): 861–869. 11565518
16. Bertocchio JP, Warnock DG, Jaisser F (2011) Mineralocorticoid receptor activation and blockade: an emerging paradigm in chronic kidney disease. Kidney Int 79 : 1051–1060. doi: 10.1038/ki.2011.48 21412221
17. Rubin MF, Townsend RR (2009) Aldosterone blockade in diabetic nephropathy: relative risks and potential promise. J Am Soc Nephrol 20 : 2487–2489. doi: 10.1681/ASN.2009101036 19875814
18. Becker R, Loll B, Meinhart A (2008) Snapshots of the RNA processing factor SCAF8 bound to different phosphorylated forms of the carboxyl-terminal domain of RNA polymerase II. J Biol Chem 283 : 22659–22669. doi: 10.1074/jbc.M803540200 18550522
19. Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER III, Hurov KE et al. (2007) ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316 : 1160–1166. 17525332
20. Zhou K, Bellenguez C, Spencer CC, Bennett AJ, Coleman RL et al. (2011) Common variants near ATM are associated with glycemic response to metformin in type 2 diabetes. Nat Genet 43 : 117–120. doi: 10.1038/ng.735 21186350
21. Florez JC, Jablonski KA, Taylor A, Mather K, Horton E et al. (2012) The C allele of ATM rs11212617 does not associate with metformin response in the Diabetes Prevention Program. Diabetes Care 35 : 1864–1867. 22751958
22. Green ED, Guyer MS (2011) Charting a course for genomic medicine from base pairs to bedside. Nature 470 : 204–213. doi: 10.1038/nature09764 21307933
23. Lango AH, Estrada K, Lettre G, Berndt SI, Weedon MN et al. (2010) Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature 467 : 832–838. doi: 10.1038/nature09410 20881960
24. Attar MA, Salem JC, Pursel HS, Santy LC (2012) CNK3 and IPCEF1 produce a single protein that is required for HGF dependent Arf6 activation and migration. Exp Cell Res 318 : 228–237. doi: 10.1016/j.yexcr.2011.10.018 22085542
25. Kopp JB, Smith MW, Nelson GW, Johnson RC, Freedman BI et al. (2008) MYH9 is a major-effect risk gene for focal segmental glomerulosclerosis. Nat Genet 40 : 1175–1184. doi: 10.1038/ng.226 18794856
26. Kao WH, Klag MJ, Meoni LA, Reich D, Berthier-Schaad Y et al. (2008) MYH9 is associated with nondiabetic end-stage renal disease in African Americans. Nat Genet 40 : 1185–1192. doi: 10.1038/ng.232 18794854
27. Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P et al. (2010) Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 329 : 841–845. doi: 10.1126/science.1193032 20647424
28. Freedman BI, Langefeld CD, Lu L, Divers J, Comeau ME et al. (2011) Differential Effects of MYH9 and APOL1 Risk Variants on FRMD3 Association with Diabetic ESRD in African Americans. PLoS Genet 7: e1002150. doi: 10.1371/journal.pgen.1002150 21698141
29. Kao WH (2012) Diabetic Nephropathy Fails to Associate with the APOL1/MYH9 Locus or Type 2 Diabetes Mellitus Susceptibility Genes: The Family Investigation of Nephropathy and Diabetes (FIND) Consortium. J Am Soc Nephol 23 : 249A.
30. Yamada N, Makino Y, Clark RA, Pearson DW, Mattei MG et al. (1994) Human inositol 1,4,5-trisphosphate type-1 receptor, InsP3R1: structure, function, regulation of expression and chromosomal localization. Biochem J 302 (Pt 3): 781–790. 7945203
31. Wilks AF, Harpur AG, Kurban RR, Ralph SJ, Zurcher G et al. (1991) Two novel protein-tyrosine kinases, each with a second phosphotransferase-related catalytic domain, define a new class of protein kinase. Mol Cell Biol 11 : 2057–2065. 1848670
32. Marks AR, Tempst P, Hwang KS, Taubman MB, Inui M et al. (1989) Molecular cloning and characterization of the ryanodine receptor/junctional channel complex cDNA from skeletal muscle sarcoplasmic reticulum. Proc Natl Acad Sci U S A 86 : 8683–8687. 2813419
33. Hwang SJ, Yang Q, Meigs JB, Pearce EN, Fox CS (2007) A genome-wide association for kidney function and endocrine-related traits in the NHLBI's Framingham Heart Study. BMC Med Genet 19;8 Suppl 1: S10. 17903292
34. Berthier CC, Zhang H, Schin M, Henger A, Nelson RG et al. (2009) Enhanced expression of Janus kinase-signal transducer and activator of transcription pathway members in human diabetic nephropathy. Diabetes 58 : 469–477. doi: 10.2337/db08-1328 19017763
35. Stenmark H, Olkkonen VM (2001) The Rab GTPase family. Genome Biol 2: REVIEWS3007.
36. Zabel U, Weeger M, La M, Schmidt HH (1998) Human soluble guanylate cyclase: functional expression and revised isoenzyme family. Biochem J 335 (Pt 1): 51–57. 9742212
37. Prabhakar SS (2004) Role of nitric oxide in diabetic nephropathy. Semin Nephrol 24 : 333–344. 15252773
38. Thacker J, Tambini CE, Simpson PJ, Tsui LC, Scherer SW (1995) Localization to chromosome 7q36.1 of the human XRCC2 gene, determining sensitivity to DNA-damaging agents. Hum Mol Genet 4 : 113–120. 7711722
39. Kottgen A, Pattaro C, Boger CA, Fuchsberger C, Olden M et al. (2010) New loci associated with kidney function and chronic kidney disease. Nat Genet 42 : 376–384. doi: 10.1038/ng.568 20383146
40. Okada Y, Sim X, Go MJ, Wu JY, Gu D et al. (2012) Meta-analysis identifies multiple loci associated with kidney function-related traits in east Asian populations. Nat Genet 44 : 904–909. doi: 10.1038/ng.2352 22797727
41. Bowden DW, Freedman BI (2012) The challenging search for diabetic nephropathy genes. Diabetes 61 : 1923–1924. doi: 10.2337/db12-0596 22826311
42. Pezzolesi MG, Krolewski AS (2013) Diabetic nephropathy: is ESRD its only heritable phenotype? J Am Soc Nephrol 24 : 1505–1507. doi: 10.1681/ASN.2013070769 24029425
43. Iyengar SK, Abboud HE, Goddard KA, Saad MF, Adler SG et al. (2007) Genome-wide scans for diabetic nephropathy and albuminuria in multiethnic populations: the family investigation of nephropathy and diabetes (FIND). Diabetes 56 : 1577–1585. 17363742
44. Schelling JR, Abboud HE, Nicholas SB, Pahl MV, Sedor JR et al. (2008) Genome-wide scan for estimated glomerular filtration rate in multi-ethnic diabetic populations: the Family Investigation of Nephropathy and Diabetes (FIND). Diabetes 57(1): 235–243. 18003762
45. McDonough CW, Palmer ND, Hicks PJ, Roh BH, An SS et al. (2011) A genome-wide association study for diabetic nephropathy genes in African Americans. Kidney Int 79 : 563–572. doi: 10.1038/ki.2010.467 21150874
46. Iyengar SK, Fox KA, Schachere M, Manzoor F, Slaughter ME et al. (2003) Linkage analysis of candidate loci in end-stage renal disease due to diabetic nephropathy. J Am Soc Nephol 14: S195–S201.
47. Ramos E, Chen G, Shriner D, Doumatey A, Gerry NP et al. (2011) Replication of genome-wide association studies (GWAS) loci for fasting plasma glucose in African-Americans. Diabetologia 54 : 783–788. doi: 10.1007/s00125-010-2002-7 21188353
48. Cooke JN, Bostrom MA, Hicks PJ, Ng MC, Hellwege JN et al. (2012) Polymorphisms in MYH9 are associated with diabetic nephropathy in European Americans. Nephrol Dial Transplant 27 : 1505–1511. doi: 10.1093/ndt/gfr522 21968013
49. Iyengar SK, Fox KA, Schachere M, Manzoor F, Slaughter ME et al. (2003) Linkage analysis of candidate loci for end-stage renal disease due to diabetic nephropathy. J Am Soc Nephrol 14: S195–S201. 12819328
50. Gunzler D, Bleyer AJ, Thomas RL, Brien O, Russell GB et al. (2013) Diabetic nephropathy in a sibling and albuminuria predict early GFR decline: a prospective cohort study. BMC Nephrol 14 : 124. doi: 10.1186/1471-2369-14-124 23773264
51. Pavkov ME, Knowler WC, Hanson RL, Williams DE, Lemley KV et al. (2013) Comparison of serum cystatin C, serum creatinine, measured GFR, and estimated GFR to assess the risk of kidney failure in American Indians with diabetic nephropathy. Am J Kidney Dis 62 : 33–41. doi: 10.1053/j.ajkd.2012.11.044 23347458
52. Thameem F, Puppala S, Lehman DM, Stern MP, Blangero J et al. (2010) The Ser(326)Cys Polymorphism of 8-Oxoguanine Glycosylase 1 (OGG1) Is Associated with Type 2 Diabetes in Mexican Americans. Hum Hered 70 : 97–101. doi: 10.1159/000291964 20606456
53. Manolio TA, Rodriguez LL, Brooks L, Abecasis G, Ballinger D et al. (2007) New models of collaboration in genome-wide association studies: the Genetic Association Information Network. Nat Genet 39 : 1045–1051. 17728769
54. Franceschini N, Haack K, Almasy L, Laston S, Lee ET et al. (2014) Generalization of associations of kidney-related genetic loci to American Indians. Clin J Am Soc Nephrol 9 : 150–158. doi: 10.2215/CJN.02300213 24311711
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 8
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Exon 7 Contributes to the Stable Localization of Xist RNA on the Inactive X-Chromosome
- YAP1 Exerts Its Transcriptional Control via TEAD-Mediated Activation of Enhancers
- SmD1 Modulates the miRNA Pathway Independently of Its Pre-mRNA Splicing Function
- TSPO, a Mitochondrial Outer Membrane Protein, Controls Ethanol-Related Behaviors in
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy