
A Genome Scale Screen for Mutants with Delayed Exit from Mitosis: Ire1-Independent Induction of Autophagy Integrates ER Homeostasis into Mitotic Lifespan
High throughput studies have yielded large collections of genes that together govern post-mitotic longevity in eukaryotic cells. However, it is also clear that mitotic lifespan is subject to regulation via intricate mechanisms that facilitate exit from mitosis. Elucidating these mechanisms has been the subject of intensive research in part because failure to exit mitosis is associated with cell immortalization, a hallmark of neoplastic growth. Yet, to date mechanisms driving mitotic lifespan remain poorly characterized largely due to the absence of a feasible high throughput screening platform. Here we describe a large-scale screen in yeast Saccharomyces cerevisiae for mutants that undergo an atypically high number of cell divisions before exiting mitosis. We report an intricate cross talk between Endoplasmic Reticulum (ER) homeostasis and mitotic longevity. Autophagy, activated in response to ER stress, delays mitotic senescence in part by removing high molecular weight cytoplasmic protein aggregates. This evolutionarily conserved catabolic network similarly extends reproductive lifespan in the nematode Caenorhabditis elegans. Our data highlight that, similar to its role in extending post-mitotic lifespan, catabolism of protein aggregates is among the drivers of mitotic longevity in eukaryotes.
Published in the journal:
. PLoS Genet 11(8): e32767. doi:10.1371/journal.pgen.1005429
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005429
Summary
High throughput studies have yielded large collections of genes that together govern post-mitotic longevity in eukaryotic cells. However, it is also clear that mitotic lifespan is subject to regulation via intricate mechanisms that facilitate exit from mitosis. Elucidating these mechanisms has been the subject of intensive research in part because failure to exit mitosis is associated with cell immortalization, a hallmark of neoplastic growth. Yet, to date mechanisms driving mitotic lifespan remain poorly characterized largely due to the absence of a feasible high throughput screening platform. Here we describe a large-scale screen in yeast Saccharomyces cerevisiae for mutants that undergo an atypically high number of cell divisions before exiting mitosis. We report an intricate cross talk between Endoplasmic Reticulum (ER) homeostasis and mitotic longevity. Autophagy, activated in response to ER stress, delays mitotic senescence in part by removing high molecular weight cytoplasmic protein aggregates. This evolutionarily conserved catabolic network similarly extends reproductive lifespan in the nematode Caenorhabditis elegans. Our data highlight that, similar to its role in extending post-mitotic lifespan, catabolism of protein aggregates is among the drivers of mitotic longevity in eukaryotes.
Introduction
Eukaryotic cells undergo a finite number of cell divisions, both in vitro and in vivo, before irreversibly exiting mitosis [1,2]. Yet single gene mutations can profoundly extend or reduce the number of cell divisions, indicating that mitotic longevity is nevertheless subject to genetic regulation [3–5]. There is now mounting evidence that mitotic senescence is not a default outcome of stochastic cell deterioration, and is instead governed via intricate pathways that in concert limit the number of cell divisions and the onset of complex phenotypes associated with mitotic exit. Elucidating mechanisms that limit the number of cell divisions has been the subject of intensive research in part because failure to exit mitosis is tightly associated with cell immortalization, a hallmark of neoplastic growth [6].
In keeping with the broad conservation of mitotic longevity pathways, the budding yeast S. cerevisiae has proven remarkably well suited to unravelling molecular mechanisms that govern longevity in eukaryotic cells [3,7]. Large-scale screens of yeast mutants designed to map the underlying longevity networks are reported [4,8]. These screens employed a microdissection assay where daughter cells are successively removed and counted until the mother cells stop dividing. However, this assay is highly laborious and requires several weeks to complete, thus limiting its utility as a high throughput screening method. While a valuable genetic resource in dissecting longevity pathways, many of the emerged mutants currently await validation.
Here we report a high throughput, genome scale screen for isolating mutants with delayed mitotic senescence in yeast. We used the age-dependent loss of transcriptional silencing at the HML mating locus [9] to screen a library of 3762 single gene deletions accounting for 2/3 of all yeast annotated ORFs. In parallel to the query library, we similarly screened a control library to search for false positives that display stochastic (not age-dependent) loss of transcriptional silencing. We focused this screen as a positive selection platform for identifying gain of function mutants, i.e., mutants that undergo a higher than wild type number of cell divisions before exiting mitosis. We classified 52 mutants as potentially long-lived and manually validated a randomly selected subset of 20. Many of the isolated genes map to biological functions not previously implicated in mitotic senescence, highlighting that the scope of cell processes that impact mitotic longevity is potentially more extensive than currently anticipated.
In order to demonstrate the utility of the isolated genes as relevant genetic portals towards dissecting longevity networks, we undertook a detailed analysis of an ER-Golgi cluster isolated in this screen. Via investigating RER1, a prototype member of this cluster, we unraveled a proteostatic network that integrates ER homeostasis into mitotic longevity in yeast. Specifically, we provide evidence that the catabolism of high molecular weight protein aggregates, a natural byproduct of high protein synthesis and turn over in dividing cells, is among the principle drivers of mitotic longevity. We extend these findings by demonstrating that this catabolic network is conserved in design and similarly extends the reproductive lifespan in the nematode C. elegans.
Results
A genome scale screen for mutants with delayed exit from mitosis
During late cell divisions, the yeast S. cerevisiae display a marked loss of transcriptional silencing at the mating loci [9]. We exploited this hallmark in a pooled collection of 3762 single deletion mutants to search for mutants that undergo a greater than wild type number of cell divisions before exiting mitosis. A full description of the screen design rationale, the isolated set of potential longevity mutants, along with high-resolution validation of a subset of these mutants are outlined in S1, S2, and S3 Figs. Briefly, we integrated the tractable marker orotidin-5'-phosphate decarboxylase (URA3) at the HML locus in a pool of deletion mutants where non-essential genes were replaced with a kanMX4 cassette [10]. Cells that undergo loss of silencing at the HML locus were selected against using 5-fluoroorotic acid (5-FOA), a cytotoxic uracil analog that inhibits growth of cells expressing URA3 [11]. Long-lived mutants were predicted to be overrepresented in the pool of dividing cells due to the delayed expression of URA3 (S1A, S1B, S1C Fig).
In parallel to the query HML::URA3 library, we also constructed a control library by integrating an identical URA3 reporter at the meiotically induced MEI4 locus (MEI4::URA3). Silencing at both MEI4 and HML loci is mediated via a host of shared gene products [12]. Yet, unlike HML locus, MEI4 remains constitutively silent when cells are maintained in rich growth media. The collective aim of this screen was therefore to isolate mutants that displayed delayed loss of silencing at the HML locus while maintaining transcriptional silence at the control MEI4 locus (Fig 1A).
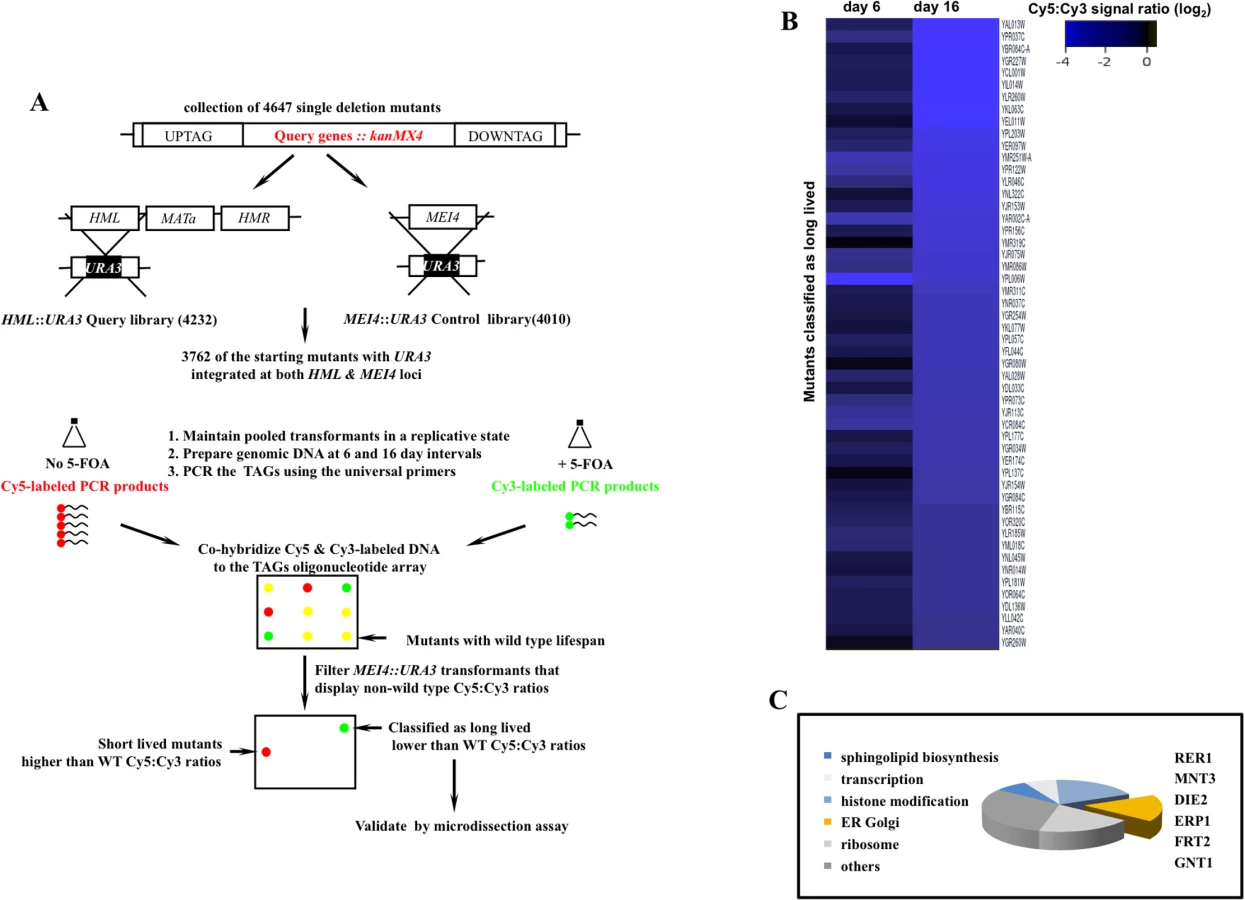
We screened 3762 deletion strains and classified 52 mutants as potentially long-lived (Figs 1B and S3). This rate of recovery is comparable to a previous screen where 13 mutants were identified as long-lived following a query of 564 mutants using mother daughter dissection [4]. The comparatively modest number of long-lived mutants in these screens may be a reflection of low occurrence frequency of long-lived mutations given the complexity of highly interconnected gene networks that converge on cell division.
A subset of the identified genes mapped to broad cellular processes already associated with mitotic lifespan including protein translation [4], sphingolipid biosynthesis [13], and chromatin remodeling [14,15]. Many of the identified genes however had no previous connection to mitotic longevity. Among these, we recovered a cluster that functions in protein modification and transport across the extensive ER-Golgi network in yeast (Fig 1C). We investigated RER1, a prototype member of this gene cluster, in order to understand how the ER-Golgi compartment integrates into longevity networks.
Inactivating RER1 extends mitotic lifespan in yeast and reproductive lifespan in worms
RER1 encodes a receptor that maintains ER compartmentalization by retrieving components of the vesicles that transport cargo from ER to Golgi [16]. Deleting RER1 increased median lifespan by 40% (p-value < 0.001, nonparametric Mann-Whitney U test), while extending the maximal lifespan by 64% relative to the wild type yeast (Fig 2A).

Rer1 displays extensive sequence homology and functional conservation across eukaryotes (S4 Fig) and human Rer1 functionally replaces its yeast counterpart [17]. We therefore examined whether rer-1 loss of function impacts reproductive lifespan in C. elegans by monitoring the ratio of progeny producing animals as a function of time. Hermaphrodite worms exhibit a progressive decline in germ cell proliferation and oocyte fertilization capacity over time that contributes to reproductive cessation. This effect is independent of sperm contribution, brood size, and alterations in reproductive schedule, and instead reflects a physiological deterioration in germ cell quality and/or maintenance that normally occurs as animals age [18]. In rer-1(tm5219) mutants, harboring a 169 bp deletion of exon 1, reproductive decline was slower compared to the wild type N2 (Fig 2B, p-value < 0.0001, Mantel-Cox test). In late reproductive life (days 6 post L4), when 19% of wild type animals remained reproductive, 51% of rer-1 hermaphrodites still generated viable progeny (S1A Fig). The prolonged reproductive lifespan in rer-1(tm5219) worms was eventually tampered by a sharp increase in cases of matricide, when internal hatching of embryos in actively reproducing old rer-1 worms led to death (S1B Fig). A comparatively milder RNAi knockdown of rer-1 with no detectable matricide delayed reproductive senescence in N2 hermaphrodites from day 4 to day 7 of adulthood while increasing the median reproductive lifespan by 66% (Figs 2C and S1D, p-value < 0.0001). Inactivating RER1 thus extends the window for progeny production in yeast and worms.
Loss of RER1 induces ER stress in yeast and worms
Treatment with ER stressors tunicamycin (TM, Fig 3A) or DTT (S2B Fig), which inhibits protein glycosylation or disulfide bond formation in the ER, respectively, resulted in a marked redistribution of GFP-Rer1 to Golgi. Rer1 relocalization to Golgi was reversible (Fig 3A), not due to a stochastic loss of ER structural integrity (S2C Fig), and further displayed a measure of specificity to ER stress since generic stress including amino acid deprivation or extensive heat shock did not alter GFP-Rer1 in situ ER localization (S2D Fig).

In response to ER stress, cells activate an intricate adaptive response, termed the Unfolded Protein Response (UPR). Mediated by a collection of evolutionarily conserved pathways, UPR is aimed at reducing ER protein load in part by signalling a complex program of gene transcription to increase ER protein folding capacity [19]. We hypothesized that the in situ redistribution of Rer1 to Golgi reflected a previously unknown component of UPR evoked to reduce ER load via increased trafficking of misfolded proteins from ER to Golgi for their subsequent packaging into autophagosomes destined for proteolysis in the vacuoles [20]. If so, impaired ER to Golgi trafficking in rer1Δ mutants should result in elevated ER load. Yeast cells upregulate the transcription of Kar2, a member of the Hsp70 family of molecular chaperones, in response to an increase in ER stress [21]. We monitored in situ UPR in rer1Δ cells by measuring lacZ activity expressed from a KAR2 promoter. In unperturbed growth (-TM), lacZ expression was constitutively elevated in rer1Δ mutants relative to the wild type yeast, a reflection of increased ER stress in these cells (Fig 3B). In keeping with its functional conservation, rer-1 inactivation in worms expressing hsp-4::GFP [22] led to a uniform increase in expression of the HSP-4 ER chaperone in the intestine (Fig 3C). Constitutively elevated ER stress in rer1 mutants highlights a requirement for intact ER to Golgi trafficking in maintaining ER homeostasis in yeast and worms.
We next examined whether high basal ER stress drives mitotic longevity. To this end, we monitored lifespan in wild type yeast cells chronically exposed to TM. Growth in 0.02 μg/ml TM, 100x less than the concentration refractory to growth (S3 Fig), extended median lifespan in yeast (Fig 4A). Remarkably, TM also delayed reproductive senescence in wild type C. elegans (Fig 4B). Our data suggest that exposure to chronic sublethal ER stress can autonomously signal reproductive longevity in both yeast and worms.

ER stress extends lifespan in an Ire1-independent manner
The principle sensor of UPR is encoded by IRE1 in yeast and its conserved orthologs in higher eukaryotes [23]. The ER membrane protein Ire1 initiates UPR in response to accumulation of misfolded proteins in the ER lumen. Relative to wild type cells, ire1Δ mutants displayed constitutively impaired basal UPR (Fig 3B), failed to activate UPR after TM treatment (Fig 3B), and as a consequence were exquisitely sensitive to TM (S3 Fig). Our analyses suggest that rer1∆ and ire1∆ genetic interactions are highly complex. First, Ire1 signaled the redistribution of Rer1 to Golgi after TM treatment as GFP-Rer1 maintained an ER localization in ire1Δ mutants treated with TM (Fig 4C). Second, deleting IRE1 entirely abolished the otherwise elevated basal UPR in rer1Δ mutants, indicating a requirement for sensing protein load in the ER lumen by Ire1 (Fig 3B). Finally, deletion of RER1 did not alter ire1∆ TM sensitivity (S3 Fig). Collectively these data suggest that trafficking across the ER-Golgi network is functionally coupled to protein load in the ER lumen. In response to an increase in ER load, Ire1 signals an increase in cargo transport from ER to Golgi, a critical intermediary step in removing misfolded proteins from ER lumen since Golgi, but not ER, is the principle donor of membrane bilayers required for expansion of autophagosomes which ultimately traffic protein cargo imported from ER to vacuoles for degradation [20,24]. In cells with elevated ER load, Rer1 is preferentially distributed to Golgi, presumably because it is required for the retrieval of transport vesicles back to the ER for subsequent rounds of cargo transport [16]. Conversely, deleting RER1 short circuits this pathway, leading to increased protein load in the ER and the ensuing activation of UPR (Fig 4C, lower panel).
Surprisingly, despite its constitutive induction, Ire1-mediated canonical UPR does not contribute to mitotic longevity in yeast; in line with a previous report [4], ire1Δ mutants had a wild type lifespan despite a highly impaired UPR (Fig 4D). Moreover, deleting IRE1 did not impact the otherwise extended lifespan in rer1Δ. While integral to survival under high ER stress, the Ire1-mediated UPR is dispensable in proliferative growth (S3 Fig) and mitotic longevity in yeast (discussed below).
Induction of autophagy in yeast and worm rer1 mutants
In tandem with inducing UPR, ER stress signals the activation of (macro) autophagy in yeast [25] and human cells [26]. Autophagy is a conserved housekeeping mechanism whereby bulk cytoplasm, including proteins and organelles, are sequestered into membrane-bound vesicles (autophagosomes) which subsequently fuse with vacuoles where their content is degraded by a complement of vacuolar proteases [27]. While mechanistically distinct, UPR and autophagy synergistically mitigate ER protein load. We therefore monitored autophagy in yeast rer1Δ cells by using the in situ reporter GFP-Atg8, a functional allele of ATG8 encoding an autophagosome assembly factor [28]. In wild type cells, GFP-Atg8 had a diffuse cytoplasmic distribution with sporadic foci, a reflection of low basal autophagy in unperturbed growth (Fig 5A). In keeping with their elevated ER load, untreated rer1Δ cells or wild type cells treated with a life-extending dose of TM (0.02 μg/ml) routinely had multiple GFP-Atg8 foci (quantified in Fig 5B), denoting increased number of assembled autophagosomes in the cytoplasm.

Exposure to high concentrations of TM induces a selective form of microautophagy, termed ER-phagy, where ER is taken up by vacuoles via invagination of the vacuolar membrane, presumably to reduce the expanded ER volume in these cells [29]. We found that exposure to 1 μg/ml TM led to robust cleavage of Sec63-GFP, a proxy readout for vacuolar internalization of ER [30]. However exposure to 0.02 μg/ml TM, a dose that extends lifespan, did not result in a detectable induction of microautohagy even after prolonged exposure (S4 Fig). Extension of lifespan following chronic treatment with sublethal concentrations of TM therefore does not appear to be accompanied with induction of microautophagy.
We similarly monitored (macro) autophagic flux in the intestine of adult worms expressing an mCherry fusion of LGG-1, the C. elegans Atg8 ortholog (Pnhx-2::mCherry::lgg-1) [31,32]. In wild type worms, mCherry::LGG-1 exhibited a diffuse signal with sporadic punctate foci formation (Fig 5C). In rer-1(tm5219) worms however the number of foci was significantly increased, highlighting a higher abundance of autophagosomes. Elevated ER stress in rer1 mutants therefore signals robust formation of autophagosomes in both yeast and worms.
Protein aggregates are cleared with increased efficiency in yeast and worm rer1 mutants
Autophagy contributes to cell homeostasis in part by removing cytoplasmic protein inclusions [27]. We asked whether the high basal autophagy in rer1Δ mutants culminated in a more efficient clearance of protein aggregates from the cytoplasm. We analyzed the content of the detergent insoluble protein inclusions in yeast rer1Δ and its wild type counterpart by SDS-PAGE [33]. Consistent with their expanded pool of autophagosomes, rer1Δ mutants or wild type cells treated with TM had reduced levels of detergent insoluble protein inclusions relative to the wild type cells (S5 Fig).
To obtain direct evidence for the apparent reduction in protein aggregates in rer1Δ, we monitored the in situ steady state of human α-synuclein-GFP (α-syn-GFP) expressed in yeast [34]. Formation of α-syn aggregates in the brain temporally coincides with the onset of a host of human neurodegenerative disorders broadly associated with aging [35].
When expressed in wild type yeast, human α-syn-GFP forms prominent cytoplasmic foci (Fig 6A). In striking contrast to wild type cells, GFP foci in rer1Δ or wild type cells treated with TM displayed a predominantly vacuolar localization with the cytoplasm essentially devoid of microscopically detectable foci (Fig 6A), a profile consistent with the reduced level of detergent insoluble protein inclusions in these cells (S5 Fig). The apparent clearance of aggregates in yeast rer1Δ was due to the constitutively high basal autophagy in these cells since deleting ATG8 restored α-syn-GFP cytoplasmic foci and concomitantly abolished its intravacuolar proteolysis (Fig 6A and 6B).

Ectopic expression of human α-syn-GFP gives rise to microscopic foci in body wall muscle cells of adult worms stably expressing Punc-54::α-syn::GFP (Fig 6C) [36]. Consistent with their high basal autophagy, rer-1(tm5219) worms or a positive experimental control over - expressing heat shock protein torsinA (tor-2) (Punc-54::tor-2) [37], displayed a marked reduction in the number of the GFP foci (Fig 6C, quantified in 6D). An RNAi knockdown of lgg-1 largely restored the α-syn-GFP foci in muscle cells of rer-1 animals, highlighting a requirement for intact autophagy in removal of protein aggregates in worms. Collectively, these data demonstrate that the expansion in the pool of autophagosomes in rer1 mutants is accompanied with a quantitative reduction in cytoplasmic protein inclusions in yeast and worm.
Autophagy is required for extended lifespan in yeast and worm rer1 mutants
Next we asked whether the constitutively high basal autophagy contributes to mitotic longevity in rer1 mutants. Deletion of genes encoding the autophagosome components Atg8 or Atg5, which mediates Atg8 lipidation required for expansion of autophagosomes [28], abolished the otherwise extended mitotic lifespan in yeast rer1Δ (Fig 7A and 7B). Knockdown of lgg-1 similarly reduced the reproductive lifespan in rer-1 worms (Fig 7C). Enhanced proteostasis thus appears to underlie the extended lifespan in yeast and worm rer1 mutants.

To examine this observation further, we monitored mitotic lifespan in yeast with functionally impaired vacuoles. Vacuoles constitute the biological endpoint for catabolism of protein aggregates. The vacuolar acidic milieu, required for the optimal enzymatic activity of its ensemble of hydrolases, is maintained by a proton-coupled ATPase complex [38]. Deletion of VMA8, encoding a component of the vacuolar ATPase, reduced mitotic lifespan in wild type yeast and concomitantly abolished the extended lifespan in rer1Δ mutants (p-value < 0.01, Fig 7D). The strict requirement for both intact autophagy and vacuolar proteolysis points to enhanced catabolic output as the principle driver of longevity in yeast rer1Δ mutants.
In our screen we also recovered MNT3, encoding a Golgi glycosylase, among the long-lived ER-Golgi cluster of mutants (Fig 1C). As in rer1Δ mutants, deletion of MNT3 led to high basal UPR, elevated autophagy, and extended mitotic lifespan (S10A, S10B, and S10C Fig). Moreover, deleting MNT3 did not further enhance the mitotic lifespan in rer1∆ cells, indicating that both mutants converge on autophagy as the longevity mechanism (S10C Fig).
Discussion
A genome scale screen for identification of mutants that undergo an atypically high number of mitotic divisions before exiting mitosis
Here we report a screen for identification of yeast mutants that undergo a higher than wild type number of cell divisions before exiting mitosis. We classified 52 mutants as potentially long-lived. In a high resolution mother-daughter assay of a randomly selected subset of 20 mutants in this panel, 7 displayed a >15% increase in mean mitotic lifespan relative to the wild type, validating the relevance of this screen as a gene enrichment platform.
Many of the putative longevity genes isolated in this screen map to biological processes with no previous annotation to mitotic lifespan. These include the cAMP-dependent kinase catalytic subunit (TPK2), the regulatory subunits of protein phosphatase 1 (GLC8 and GIP3) and phosphotyrosine LTP1, the polyamine transporter (TPO3), tRNA thiouridylase (SLM3), and the Glutathione-dependent oxidoreductase (GRX4). The potential involvement of these processes in extending mitotic longevity indicates that the regulatory scope of mitotic lifespan may be broader than currently anticipated. Of the 52 potential longevity genes isolated in this screen, 32 have functional orthologs in humans (S3 Fig). This gene set should serve as a starting genetic portal in mapping mitotic longevity pathways in human cells.
Functional coupling of ER homeostasis to mitotic lifespan in yeast and worms
We recovered a gene cluster that functions in ER to Golgi protein transport (RER1, ERP1) and N-linked protein glycosylation (DIE2, MNT3, GNT1). We focused on RER1 to understand how ER integrates into mitotic longevity. Localized to perinuclear ER in unperturbed growth, Rer1 redistributes to Golgi after treatment with ER stressors (Figs 3A and S2B). Redistribution of Rer1 to Golgi is reversible, specific to ER stress, and requires Ire1, the principle inducer of UPR. These observations suggest that Rer1 functions downstream of ER stress and in synergy with canonical UPR where increased ER to Golgi trafficking likely contributes to ER homeostasis by removing misfolded proteins from the ER lumen to Golgi for their subsequent transport to vacuoles by autophagosomes.
Despite its constitutive induction, canonical Ire1-mediated UPR does not contribute to mitotic longevity in yeast rer1∆ mutants; ire1∆ mutants display wild type lifespan despite a complete absence of UPR. Moreover, deletion of IRE1 does not impact the extended mitotic lifespan in rer1Δ mutants. While integral to survival under ER stress, Ire1 is dispensable for growth (S3 Fig) and mitotic longevity in yeast (Fig 4D). To our knowledge a contribution of Ire1-mediated UPR to mitotic lifespan in other eukaryotic models has not been examined.
Enhanced protein catabolism delays mitotic exit in yeast and reproductive senescence in worms
In tandem with UPR, yeast rer1Δ mutants activate autophagy, likely to mitigate an increase in ER load. While complementary in function, we show that induction of autophagy and UPR can be mechanistically uncoupled; despite a strict requirement for Ire1 in initiating canonical UPR (Fig 3B), Ire1 is dispensable for inducing autophagy in yeast (S6 Fig). Our data are in line with a previous report indicating that sensors other than Ire1 signal autophagy in response to increased ER load [29].
We have provided evidence that the elevated basal autophagy in response to high ER stress underlies the extended lifespan in yeast and worm rer1 mutants (Fig 7A, 7B, and 7C). Deletion of MNT3 similarly leads to high basal ER stress, the ensuing increase in the pool of autophagosomes, and extended mitotic longevity (S10 Fig). The induction of autophagy thus appears to be a broad adaptive response evoked to augment UPR in cells with impaired protein modification (mnt3Δ) or trafficking (rer1Δ) across the ER-Golgi network.
The induction of autophagy in rer1Δ mutants is accompanied by a marked clearance of α-syn cytoplasmic inclusions, an observation consistent with the role of autophagy as the principle pathway for removing high molecular weight protein aggregates (Fig 6A and 6C). Protein aggregates form prominent cytological features that segregate with and contribute to senescence in post mitotic models including C.elegans and mice [39,40]. Our data suggest that as in post mitotic aging, formation of protein aggregates also serves as a determinant of reproductive senescence in yeast and worms (Fig 7E).
Finally, we note that despite their extended lifespan, yeast rer1Δ mutants remain sensitive to ER stressors (S3 Fig), display elevated loss of viability after DNA damage [41], and grow poorly under low nutrient condition [42]. While autophagy extends the duration of reproduction, it does not ameliorate other phenotypic defects in rer1 mutants. From an evolutionary viewpoint, the targeted induction of autophagy in rer1 mutants provides a compelling example of a Darwinian adaptation aimed at extending the window for offspring production.
Materials and Methods
Yeast strains and molecular biology
Unless indicated otherwise, experiments were carried out in BY4741 (MATa his3Δ1 Δleu2 Δmet15 Δura3). Respective null mutants were constructed by single step PCR-based gene deletion. Strains were grown in YPD or SC media supplemented with 30 μg/ml of all amino acids. For inducing nutritional stress, cells were grown in SC media without amino acids. Throughout, cells expressing GFP-Atg8 were grown in SC-ura media.
Yeast methods
A CEN based plasmid containing a lacZ gene expressed from a KAR2 promoter [21] provided an in situ readout for UPR. Yeast vacuolar enzymatic activity was measured as described [43]. Total yeast protein lysates were prepared by bead beating as described [44]. Mouse anti-GFP monoclonal antibody (Covance) was used at 1/5000.
Yeast mitotic lifespan analysis
Lifespan analyses were essentially as described [45]. Briefly, cells from logarithmically growing liquid cultures were plated on YPD. After an overnight incubation at 30°C, a minimum starting population of 40 founding daughter cells were removed with a Zeiss Micro-manipulator and buds were successively dissected until all mother cells had ceased dividing. To monitor the effect of ER stress on longevity, tunicamycin (Sigma Aldrich) or DMSO was added to the plate mix. Each experiment was performed at least twice, with most repeated 3 to 5 times. Significance of the differences in mean mitotic lifespan for each experiment was determined using the nonparametric Mann-Whitney U test, as done previously [46]. Eight data points flanking the 50% mitotic lifespan point for each experiment were used in the Mann-Whitney U-value calculator (http://www.socscistatistics.com/tests/mannwhitney/Default.aspx).
Analysis of worm reproductive lifespan
With the exception of RNAi experiments which used an rrf-3(pk1426) temperature sensitive background grown at 16°C, all other experiments were performed at 20°C. Embryos were isolated by hypochlorite treatment and L1 synchronized in diapause. Larvae were allowed to develop to late L4 stage (44 hrs at 20°C or 57 hrs post embryonic development at 16°C) when they were singled out on NGM plates. The reproductive capacity was determined by scoring the number of viable progeny daily. The end of reproductive lifespan was assigned as the last day with progeny followed by two consecutive days without progeny production. Sterile hermaphrodites were excluded from the data. When indicated (Fig 4B), tunicamycin was added to the UV irradiated bacteria (OP50) food supplement. The Mantel-Cox test was used to calculate P-values in the different reproductive lifespan curves.
Monitoring autophagy in C. elegans
Expression of LGG-1 was monitored at 20°C in adult transgenic worms carrying an integrated Pnhx-2::mCherry::lgg-1 transgene. Age-matched wild type and rer-1(tm5219) animals were grown and imaged in parallel.
Analysis of protein aggregates
We used a published protocol [47] to monitor the formation of detergent insoluble materials in yeast. Briefly, spheroplasts were prepared and lysed in a buffer containing 1% sarkosyl and incubated on ice for 5 minutes with occasional vortexing. 2 mg of each lysate was precleared with a 10 minute spin at 1,000g and the detergent insoluble material were pelleted by a 1 hr spin at 20,000g at 4°C. Pellets were resolubilized in 8M urea containing 2% TX-100, resolved by SDS-PAGE, and analyzed by coomassie blue staining. α-syn-GFP expression was induced by growing cells in 0.2% galactose for 4 hours.
Microscopy
Log phase yeast culture cells were spotted on a glass slide and imaged with an Olympus BX-51 microscope equipped with Infinity software v.5.0.3 (Lumenera, Ottawa, Canada) for image acquisition. Alternatively, live cells were imaged using a laser scanning confocal microscope (Zeiss LSM410). MetaMorph v6.1 software (Universal Imaging Corporation; Downington, PA) was used to construct calibrated overlays of z-stacked images. Worms were mounted on 2% agarose pads spotted with 20 μl of 10 mM Na-azide. Confocal microscopy was performed using a Zeiss LSM410 confocal laser scanning microscope. Z-stacks were acquired and processed using the Zeiss LSM software package. Alternatively, imaging was performed using an Applied Precision DeltaVision microscope (GE). Image processing was performed using Softworx software (G.E./Applied Precision).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Hayflick L (1965) The Limited in Vitro Lifetime of Human Diploid Cell Strains. Exp Cell Res 37 : 614–636. 14315085
2. Smith JR, Pereira-Smith OM (1996) Replicative senescence: implications for in vivo aging and tumor suppression. Science 273 : 63–67. 8658197
3. Jazwinski SM, Egilmez NK, Chen JB (1989) Replication control and cellular life span. Exp Gerontol 24 : 423–436. 2698814
4. Kaeberlein M, Powers RW 3rd, Steffen KK, Westman EA, Hu D, et al. (2005) Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science 310 : 1193–1196. 16293764
5. Lesur I, Campbell JL (2004) The transcriptome of prematurely aging yeast cells is similar to that of telomerase-deficient cells. Mol Biol Cell 15 : 1297–1312. 14718559
6. Longo VD, Lieber MR, Vijg J (2008) Turning anti-ageing genes against cancer. Nat Rev Mol Cell Biol 9 : 903–910. doi: 10.1038/nrm2526 18946478
7. Longo VD, Shadel GS, Kaeberlein M, Kennedy B (2012) Replicative and chronological aging in Saccharomyces cerevisiae. Cell Metab 16 : 18–31. doi: 10.1016/j.cmet.2012.06.002 22768836
8. Sutphin GL, Olsen BA, Kennedy BK, Kaeberlein M (2012) Genome-wide analysis of yeast aging. Subcell Biochem 57 : 251–289. doi: 10.1007/978-94-007-2561-4_12 22094426
9. Smeal T, Claus J, Kennedy B, Cole F (1996) Loss of transcriptional silencing causes sterility in old mother cells of S. cerevisiae. Cell 84 : 633–642. 8598049
10. Ooi SL, Shoemaker DD, Boeke JD (2001) A DNA microarray-based genetic screen for nonhomologous end-joining mutants in Saccharomyces cerevisiae. Science 294 : 2552–2556. 11701889
11. Boeke JD, LaCroute F, Fink GR (1984) A positive selection for mutants lacking orotidine-5'-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol Gen Genet 197 : 345–346. 6394957
12. Lustig AJ (1998) Mechanisms of silencing in Saccharomyces cerevisiae. Curr Opin Genet Dev 8 : 233–239. 9610415
13. Schleit J, Johnson SC, Bennett CF, Simko M, Trongtham N, et al. (2013) Molecular mechanisms underlying genotype-dependent responses to dietary restriction. Aging Cell 12 : 1050–1061. doi: 10.1111/acel.12130 23837470
14. Feser J, Truong D, Das C, Carson JJ, Kieft J, et al. (2010) Elevated histone expression promotes life span extension. Mol Cell 39 : 724–735. doi: 10.1016/j.molcel.2010.08.015 20832724
15. Kaeberlein M, Kennedy BK (2005) Large-scale identification in yeast of conserved ageing genes. Mech Ageing Dev 126 : 17–21. 15610758
16. Sato K, Sato M, Nakano A (1997) Rer1p as common machinery for the endoplasmic reticulum localization of membrane proteins. Proc Natl Acad Sci U S A 94 : 9693–9698. 9275186
17. Fullekrug J, Boehm J, Rottger S, Nilsson T, Mieskes G, et al. (1997) Human Rer1 is localized to the Golgi apparatus and complements the deletion of the homologous Rer1 protein of Saccharomyces cerevisiae. Eur J Cell Biol 74 : 31–40. 9309388
18. Luo S, Kleemann GA, Ashraf JM, Shaw WM, Murphy CT (2010) TGF-beta and insulin signaling regulate reproductive aging via oocyte and germline quality maintenance. Cell 143 : 299–312. doi: 10.1016/j.cell.2010.09.013 20946987
19. Ron D, Walter P (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8 : 519–529. 17565364
20. van der Vaart A, Griffith J, Reggiori F (2010) Exit from the Golgi is required for the expansion of the autophagosomal phagophore in yeast Saccharomyces cerevisiae. Mol Biol Cell 21 : 2270–2284. doi: 10.1091/mbc.E09-04-0345 20444982
21. Mori K, Kawahara T, Yoshida H, Yanagi H, Yura T (1996) Signalling from endoplasmic reticulum to nucleus: transcription factor with a basic-leucine zipper motif is required for the unfolded protein-response pathway. Genes Cells 1 : 803–817. 9077435
22. Kuemmerle S, Gutekunst CA, Klein AM, Li XJ, Li SH, et al. (1999) Huntington aggregates may not predict neuronal death in Huntington's disease. Ann Neurol 46 : 842–849. 10589536
23. Sidrauski C, Walter P (1997) The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 90 : 1031–1039. 9323131
24. Ohashi Y, Munro S (2010) Membrane delivery to the yeast autophagosome from the Golgi-endosomal system. Mol Biol Cell 21 : 3998–4008. doi: 10.1091/mbc.E10-05-0457 20861302
25. Yorimitsu T, Nair U, Yang Z, Klionsky DJ (2006) Endoplasmic reticulum stress triggers autophagy. J Biol Chem 281 : 30299–30304. 16901900
26. Ogata M, Hino S, Saito A, Morikawa K, Kondo S, et al. (2006) Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol 26 : 9220–9231. 17030611
27. Cuervo AM, Bergamini E, Brunk UT, Droge W, Ffrench M, et al. (2005) Autophagy and aging: the importance of maintaining "clean" cells. Autophagy 1 : 131–140. 16874025
28. Xie Z, Nair U, Klionsky DJ (2008) Atg8 controls phagophore expansion during autophagosome formation. Mol Biol Cell 19 : 3290–3298. doi: 10.1091/mbc.E07-12-1292 18508918
29. Bernales S, McDonald KL, Walter P (2006) Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol 4: e423. 17132049
30. Schuck S, Gallagher CM, Walter P (2014) ER-phagy mediates selective degradation of endoplasmic reticulum independently of the core autophagy machinery. J Cell Sci 127 : 4078–4088. doi: 10.1242/jcs.154716 25052096
31. Melendez A, Talloczy Z, Seaman M, Eskelinen EL, Hall DH, et al. (2003) Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 301 : 1387–1391. 12958363
32. Jenzer C, Simionato E, Legouis R (2015) Tools and methods to analyze autophagy in C. elegans. Methods 75 : 162–171. doi: 10.1016/j.ymeth.2014.11.019 25484340
33. Fushimi K, Long C, Jayaram N, Chen X, Li L, et al. (2011) Expression of human FUS/TLS in yeast leads to protein aggregation and cytotoxicity, recapitulating key features of FUS proteinopathy. Protein Cell 2 : 141–149. doi: 10.1007/s13238-011-1014-5 21327870
34. Outeiro TF, Lindquist S (2003) Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science 302 : 1772–1775. 14657500
35. Muchowski PJ (2002) Protein misfolding, amyloid formation, and neurodegeneration: a critical role for molecular chaperones? Neuron 35 : 9–12. 12123602
36. Harrington AJ, Knight AL, Caldwell GA, Caldwell KA (2011) Caenorhabditis elegans as a model system for identifying effectors of alpha-synuclein misfolding and dopaminergic cell death associated with Parkinson's disease. Methods 53 : 220–225. doi: 10.1016/j.ymeth.2010.12.036 21195766
37. McLean PJ, Kawamata H, Shariff S, Hewett J, Sharma N, et al. (2002) TorsinA and heat shock proteins act as molecular chaperones: suppression of alpha-synuclein aggregation. J Neurochem 83 : 846–854. 12421356
38. Preston RA, Reinagel PS, Jones EW (1992) Genes required for vacuolar acidity in Saccharomyces cerevisiae. Genetics 131 : 551–558. 1628805
39. David DC, Ollikainen N, Trinidad JC, Cary MP, Burlingame AL, et al. (2010) Widespread protein aggregation as an inherent part of aging in C. elegans. PLoS Biol 8: e1000450. doi: 10.1371/journal.pbio.1000450 20711477
40. Lee JH, Won SM, Suh J, Son SJ, Moon GJ, et al. (2010) Induction of the unfolded protein response and cell death pathway in Alzheimer's disease, but not in aged Tg2576 mice. Exp Mol Med 42 : 386–394. 20368688
41. Kapitzky L, Beltrao P, Berens TJ, Gassner N, Zhou C, et al. (2010) Cross-species chemogenomic profiling reveals evolutionarily conserved drug mode of action. Mol Syst Biol 6 : 451. doi: 10.1038/msb.2010.107 21179023
42. Davey HM, Cross EJ, Davey CL, Gkargkas K, Delneri D, et al. (2012) Genome-wide analysis of longevity in nutrient-deprived Saccharomyces cerevisiae reveals importance of recycling in maintaining cell viability. Environ Microbiol 14 : 1249–1260. doi: 10.1111/j.1462-2920.2012.02705.x 22356628
43. Noda T, Ohsumi Y (1998) Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem 273 : 3963–3966. 9461583
44. Ghavidel A, Kislinger T, Pogoutse O, Sopko R, Jurisica I, et al. (2007) Impaired tRNA nuclear export links DNA damage and cell-cycle checkpoint. Cell 131 : 915–926. 18045534
45. Harkness TA, Shea KA, Legrand C, Brahmania M, Davies GF (2004) A functional analysis reveals dependence on the anaphase-promoting complex for prolonged life span in yeast. Genetics 168 : 759–774. 15514051
46. Menzel J, Malo ME, Chan C, Prusinkiewicz M, Arnason TG, Harkness TA (2014) The anaphase promoting complex regulates yeast lifespan and rDNA stability by targeting Fob1 for degradation. Genetics 196 : 693–709. doi: 10.1534/genetics.113.158949 24361936
47. Fushimi K, Long C, Jayaram N, Chen X, Li L, et al. Expression of human FUS/TLS in yeast leads to protein aggregation and cytotoxicity, recapitulating key features of FUS proteinopathy. Protein Cell 2 : 141–149. doi: 10.1007/s13238-011-1014-5 21327870
48. Soukas AA, Kane EA, Carr CE, Melo JA, Ruvkun G (2009) Rictor/TORC2 regulates fat metabolism, feeding, growth, and life span in Caenorhabditis elegans. Genes Dev 23 : 496–511. doi: 10.1101/gad.1775409 19240135
49. Enomoto S, Berman J (1998) Chromatin assembly factor I contributes to the maintenance, but not the re-establishment, of silencing at the yeast silent mating loci. Genes Dev 12 : 219–232. 9436982
50. Boselli M, Rock J, Unal E, Levine SS, Amon A (2009) Effects of age on meiosis in budding yeast. Dev Cell 16 : 844–855. doi: 10.1016/j.devcel.2009.05.013 19531355
51. Bernstein BE, Tong JK, Schreiber SL (2000) Genomewide studies of histone deacetylase function in yeast. Proc Natl Acad Sci U S A 97 : 13708–13713. 11095743
52. Ooi SL, Shoemaker DD, Boeke JD (2003) DNA helicase gene interaction network defined using synthetic lethality analyzed by microarray. Nat Genet 35 : 277–286. 14566339
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 8
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Exon 7 Contributes to the Stable Localization of Xist RNA on the Inactive X-Chromosome
- YAP1 Exerts Its Transcriptional Control via TEAD-Mediated Activation of Enhancers
- SmD1 Modulates the miRNA Pathway Independently of Its Pre-mRNA Splicing Function
- TSPO, a Mitochondrial Outer Membrane Protein, Controls Ethanol-Related Behaviors in
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy