
Functional Activation of the Flagellar Type III Secretion Export Apparatus
Bacteria build needle-like injectsomes to secrete toxins into host cells and build propeller-like flagella to swim through their environment using a molecular machine called the type III secretion system (T3SS). Both the injectisome and the flagellum are large self-assembling complexes and regulation of the T3SS ensures that proteins are secreted sequentially for proper structure and function. Here we report genetic and cytological data that the SwrB protein of Bacillus subtilis helps the base of the flagellum adopt a completed conformation which in turn activates the enclosed T3SS to export proteins for the next stage of flagellar assembly. Thus SwrB presents a novel mechanism to supervise an early structural checkpoint regulating machine assembly. Targeting functional regulators like SwrB could inhibit T3SS-based strategies of pathogens.
Published in the journal:
. PLoS Genet 11(8): e32767. doi:10.1371/journal.pgen.1005443
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1005443
Summary
Bacteria build needle-like injectsomes to secrete toxins into host cells and build propeller-like flagella to swim through their environment using a molecular machine called the type III secretion system (T3SS). Both the injectisome and the flagellum are large self-assembling complexes and regulation of the T3SS ensures that proteins are secreted sequentially for proper structure and function. Here we report genetic and cytological data that the SwrB protein of Bacillus subtilis helps the base of the flagellum adopt a completed conformation which in turn activates the enclosed T3SS to export proteins for the next stage of flagellar assembly. Thus SwrB presents a novel mechanism to supervise an early structural checkpoint regulating machine assembly. Targeting functional regulators like SwrB could inhibit T3SS-based strategies of pathogens.
Introduction
Some bacteria swim through liquid and swarm over surfaces by synthesizing trans-envelope nanomachines called flagella. Flagella spontaneously self-assemble from over twenty separate proteins thought to be organized into three structural domains called the basal body, the hook and the filament [1–3]. The basal body is composed of a ring of a transmembrane protein called FliF that surrounds a membrane-embedded, dedicated type III secretion export apparatus [4–7]. Beneath the basal body sits the FliG rotor that interacts with the proton-conducting stators to generate torque, as well as the C-ring proteins FliM and FliN that control the direction of flagellar rotation [8,9]. The export apparatus secretes subunits of the drive-shaft rod that transits the peptidoglycan, the universal joint hook, and the long helical filament. When the basal body rotates, rotational energy is transmitted through the rod and the hook to turn the filament that generates propulsion like a propeller. Central to flagellar assembly is the control of the flagellar type III secretion export apparatus.
Type III secretion export apparati are housed within both the flagellar basal body and the evolutionarily-related “injectisome” used by various pathogenic bacteria to secrete toxins directly into eukaryotic host cells [10,11]. Recent cytological evidence suggests that the export apparatus is likely the first substructure to form and serves as the nucleation point for subsequent flagellar/injectisome basal body assembly [7,12,13]. The export apparatus requires a conserved set of 5 transmembrane proteins: FliP, FliQ, FliR, FlhA, and FlhB [6,14]. FlhA is important for the recognition of secretion substrates and FlhB controls the substrate specificity of the export apparatus [15–18]. The roles of FliP, FliQ, and FliR are poorly understood, but presumably these proteins either transduce the proton motive force that powers protein export or serve as the export channel [19,20]. Flagellar export systems differ from injectisome export systems in that flagella require a sixth transmembrane protein FliO that appears to function as a regulator [21,22].
The flagellar export apparatus secretes two classes of proteins considered “early” and “late” depending on the temporal order in which they are secreted. Early class flagellar structural components like the rod and the hook subunits are secreted first, and are recognized by information encoded within the N-terminus of the secreted protein [23–25]. Late class subunits, which include the hook-associated proteins and flagellin, are ushered by chaperones and are secreted only after a substrate specificity switch occurs within the export apparatus in response to hook completion [17,26–28]. Furthermore, the expression of the genes encoding the late class secretion substrates is inhibited by the anti-sigma factor FlgM prior to hook completion. Once the hook is complete and the substrate specificity switch occurs, FlgM is exported, its cognate sigma factor is liberated, and late class flagellar genes are expressed [29,30]. Thus, the activity of the export apparatus is morphogenetically coupled to flagellar structure so as to govern subsequent flagellar gene expression and assembly. Finally, there is evidence for another morphogenetic coupling event that precedes hook completion as mutants defective in the cytoplasmic flagellar rotor FliG and C-ring components FliM and FliN have flagellar type III secretion defects [31–37].
Here we provide further evidence for an early morphogenetic checkpoint in flagellar assembly whereby completion of the flagellar basal body and the single pass transmembrane protein SwrB of Bacillus subtilis functionally activate the flagellar type III export apparatus to become secretion proficient. Mutants defective for SwrB assemble a reduced number of flagella due to a reduced frequency of hook assembly, and a secretion defect was implicated as many genetic suppressors increased the abundance of the secretion apparatus component FliP. We show that the proficiency of the export apparatus for hook subunit secretion was coupled to the conformation of the basal body as C-ring mutants phenocopied the absence of SwrB. Furthermore, overexpression of SwrB rescued hook assembly to cells defective for FliG, and FliG gain-of-function alleles suppressed the absence of SwrB. We conclude that SwrB chaperones the formation of a completed basal body conformation thereby activating the flagellar type III export apparatus for the secretion of the hook, and likely rod, structural subunits.
Results
SwrB is required for hook, but not basal body (C-ring) assembly
SwrB is a single-pass transmembrane protein of unknown function that is required for swarming motility in B. subtilis [38,39]. Swarming motility over a solid surface requires an increase in the number of flagella relative to swimming in liquid, and mutants defective in the master regulator of flagellar biosynthesis, SwrA, have reduced flagellar number and are unable to swarm [40–42]. To determine whether cells mutated for SwrB have a defect in flagellar filament number, a variant of the flagellar filament protein Hag that could be labeled with a fluorescent dye (HagT209C) was introduced to the wild type, swrB, and swrA mutant backgrounds [43]. Cells mutated for SwrB appeared to have fewer filaments than wild type and resembled cells mutated for SwrA (Fig 1A). We conclude that SwrB is required for the assembly of wild type numbers of flagellar filaments and we infer that the reduction in filament number accounts for the swarming defect of a swrB mutant.

Consistent with a reduction in flagellar filament assembly, cells lacking either SwrB or SwrA showed a reduced level of the flagellar filament protein, Hag, by Western blot analysis (Fig 2A). The reduction in Hag protein was likely due to reduced transcription of the hag gene as cells mutated for either SwrB or SwrA showed lower expression from a reporter in which the hag promoter (Phag) was fused to the lacZ gene encoding β-galactosidase (Phag-lacZ) (Fig 2B, white bars), and a reduced frequency of cells expressing a reporter in which the Phag promoter was fused to green fluorescent protein (GFP) (Phag-gfp) (Fig 2C, white bars, and S1 Fig) [39,41]. The Phag promoter is transcribed by RNA polymerase and the alternative sigma factor, σD [44]. Whereas cells mutated for SwrA exhibited a reduced level of σD protein, cells mutated for SwrB exhibited a level of σD protein comparable to the wild type (Fig 2A). We conclude that the absence of SwrB resulted in reduced expression of the hag gene but unlike the absence of SwrA, the defect occurred downstream of σD protein levels.
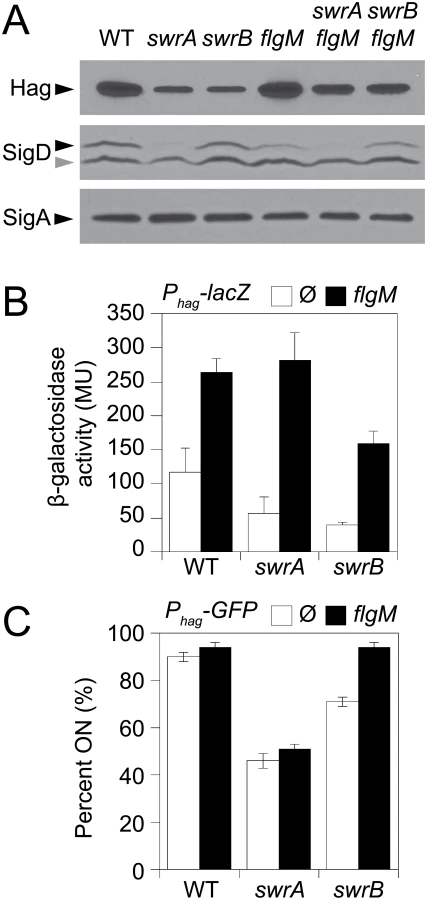
One reason that hag expression might be reduced in the SwrB mutant despite wild type levels of σD is due to enhanced inhibition of σD by its cognate anti-sigma factor FlgM [45–48]. Consistent with enhanced inhibition by FlgM, mutation of FlgM increased the magnitude of expression of the Phag-lacZ reporter (Fig 2B, black bars), and increased the frequency of expression of the Phag-GFP reporter in the wild type and swrB mutant backgrounds (Fig 2C, black bars, and S1 Fig). The SwrA protein controls the frequency of cells that have σD protein levels above a critical threshold upstream of FlgM regulation [49,50] and thus simultaneous mutation of both FlgM and SwrA increased expression magnitude in a subpopulation (Fig 2B) but did not increase the frequency of σD activity (Fig 2C). Finally, mutation of FlgM, did not appear to increase flagellar filament number in the absence of either SwrB or SwrA (Fig 1B) despite a modest increase in flagellin protein levels (Fig 2A). We conclude that enhanced inhibition of σD by FlgM was a consequence and not a cause of the reduction of flagellar number in the swrB mutant.
FlgM inhibition of σD activity is enhanced when cells are defective in flagellar hook synthesis, and a defect in flagellar hook synthesis would also account for the reduction in flagellar filaments observed in the SwrB mutant [51,52]. To measure flagellar hook numbers, a variant of the flagellar hook protein FlgE that could be labeled with a fluorescent dye (FlgET123C) was introduced to the wild type, swrB, and swrA mutant backgrounds [51]. Cells mutated for swrB appeared to have fewer hooks than the wild type and instead resembled the reduced numbers of hooks in cells mutated for swrA (Fig 1C). To count flagellar hooks, 3D structured illumination microscopy (3D-SIM) was conducted on hook-stained cells of each genetic background and each individual cell was expressed as a point on a scatter plot representing the number of hooks versus cell length (Fig 3, green symbols). Whereas wild type cells had an average of 15 hooks, swrB and swrA mutants had an average of 5 and 4 hooks per cell, respectively. We conclude that the swrB mutant was defective in hook assembly and as a result exhibited enhanced inhibition of σD by FlgM.

Because hook assembly depends on flagellar basal body synthesis, the reduction in hook numbers observed in a swrB mutant could be due to a commensurate reduction in basal body number. To measure flagellar basal body synthesis, a translational fusion of GFP to the C-ring protein FliM (amyE::Pfla/che-fliM-GFP) was introduced to the wild type, swrB, and swrA mutant backgrounds [53]. Cells mutated for swrB appeared to have wild type numbers of flagellar C-rings, whereas cells mutated for swrA appeared to have a reduction in flagellar basal body number, as previously reported (Fig 1D) [53]. To count flagellar C-rings 3D-SIM was conducted on cells of each genetic background and each individual cell was expressed as a point on a scatter plot representing the number of basal bodies versus cell length (Fig 3, red symbols). On average, wild type had 24 C-rings, the swrB mutant had 23 C-rings, and the swrA mutant had 9 C-rings per cell. We conclude that SwrB and SwrA increased flagellar numbers at two different steps in flagellar assembly by promoting hook and C-ring (basal body) assembly respectively. We further conclude that SwrB somehow promoted hook assembly at a step downstream of the incorporation of FliM into the basal body.
SwrB activates the flagellar export apparatus
To determine how SwrB regulates hook assembly, spontaneous suppressor mutations were isolated that restored swarming motility to a swrB mutant. When a swrB mutant was inoculated in the center of a swarm agar plate, cells initially grew as a tight central colony and, unlike the wild type, failed to spread from the inoculum origin (Fig 4A). However, after 24 hours of incubation, flares of cells that had regained the ability to swarm emerged from the central colony and cells from these flares were clonally isolated. Twenty-four spontaneous sob (suppressor of swrB) mutants were independently isolated. Each suppressor was validated by PCR length polymorphism to confirm the presence of the swrB mutant allele. A combination of candidate gene sequencing, phage transduction linkage mapping, and Illumina whole-genome sequencing was used to identify the location of each of the sob suppressor mutations. Based on the swarm behavior and location of the suppressor mutations, the sob alleles were divided into six classes and analyzed separately (Table 1 and Figs 4B–4G and 5A).



Class I–Improved Pfla/che consensus
The sob class I alleles were identified by PCR amplification and directed sequencing of the Pfla/che promoter region, as the Pfla/che promoter has been found to be a common site for suppressors of motility defects [41,54]. Five independently-isolated sob alleles contained the same single base pair point mutation that changed the TACAAT -10 element of Pfla/che promoter to a consensus TATAAT sequence (Table 1 and Fig 5B). We note that the same kinds of mutations increased Pfla/che promoter activity in previously-reported suppressors of swrA (soa) [41], in 8 out of 23 newly-isolated soa alleles (S2 Fig and Table 2), and in suppressors of a dominant degU32(Hy) allele (dhs) [54]. We infer that one way to suppress a defect in SwrB is by increasing Pfla/che promoter activity.
To directly test the consequence of a sob class I allele in a swrB mutant background, the Pfla/che promoter region was cloned either from wild type or a class I sob21 allele upstream of a promoterless lacZ gene and inserted at an ectopic locus (amyE::Pfla/cheWT-lacZ and amyE::Pfla/chesobclassI-lacZ). Consistent with previous reports, the improved -10 promoter element of the sob class I allele increased expression of Pfla/che in wild type, swrB, and swrA mutant backgrounds relative to the wild type promoter (Fig 6, compare white and gray bars within strains). Unlike mutation of SwrA, however, mutation of SwrB did not reduce the expression of the Pfla/che wild type reporter in an otherwise wild type genetic background (Fig 6, compare white bars between strains). We conclude that although SwrB does not normally act to increase the expression of the Pfla/che promoter, increased transcriptional activity from the Pfla/che promoter nonetheless compensates for the absence of SwrB.

Expression from the Pfla/che promoter is thought to control transcript abundance of the enitre fla/che operon [49,50,55,56]. To determine fla/che transcript levels in the absence of SwrB, QRT-PCR was conducted at various positions along the length of operon. Consistent with results observed with the Pfla/che-lacZ reporter assay, mutation of SwrA resulted in a reduction in fla/che operon transcript abundance but mutation of SwrB did not (Fig 7A). When the sob21-improved Pfla/che -10 element class I allele was present at the native site in the chromosome, transcript levels increased from 2-to-8 fold depending on the genetic position in the fla/che operon (Fig 7B). We conclude that although SwrB does not normally act to increase fla/che operon transcript levels, increased fla/che transcript levels nonetheless compensates for the absence of SwrB.

Class II–Deletions of the codY terminator
The sob class II alleles were identified by PCR amplification and directed sequencing of the Pfla/che promoter region. The PCR of some amplicons, however, were shorter than the wild type and sequencing indicated nested deletions such that the smallest common mutation deleted one arm of a Rho-independent terminator of the codY gene located immediately upstream of the fla/che operon (Fig 5B and Table 1). We note that similar deletions are thought to cause read-through transcription from the codY gene into the fla/che operon in previously identified suppressors of swrA (soa) [41], in 13 out of 23 newly-isolated of soa alleles (S2 Fig and Table 2), and also in suppressors of a dominant degU32(Hy) allele (dhs) [54]. When the codY terminator was deleted in the sob37 genetic background, fla/che transcript levels increased from 4-to-14 fold depending on the genetic position in the fla/che operon (Fig 7C). We conclude that the class II codY terminator deletions compensated for the absence of SwrB in a manner similar to the class I alleles by increased expression of the fla/che operon transcript.
Class III–Mutations upstream of Pfla/che
The sob class III alleles were identified by PCR amplification and directed sequencing of the Pfla/che promoter region. Four independently isolated sob alleles contained a single base pair mutation at position -110 and one sob allele contained a single base pair point mutation at position -104 relative to the fla/che transcriptional start site, upstream of the Pfla/che promoter (Fig 5B). Like the sob class I and class II mutations, the class III sob6 allele elevated fla/che transcript levels, but to a lesser extent: only 1-to-4 fold depending on the position in the operon (Fig 7D). To test the role of the class III mutations on Pfla/che promoter activity, the sob6 allele was cloned as part of the Pfla/che promoter region upstream of a promoter-less lacZ gene and inserted at an ectopic locus (amyE::Pfla/chesobclassIII-lacZ). Unlike the class I alleles, the class III allele did not increase Pfla/che reporter expression in the wild type, swrB, or swrA mutant backgrounds (Fig 6, compare white and black bars within strains). Thus, the class III alleles appeared to bypass the absence of SwrB without increasing transcription initiation from the Pfla/che promoter.
We considered the possibility that the sob class III alleles functioned in trans by mutating an as-yet undiscovered diffusible gene product encoded within the Pfla/che promoter region. The sob6 allele did not appear to cause a loss-of-function phenotype to a diffusible gene product, however, because the swrB swarming motility defect was not restored to the swrB sob6 background when the wild type Pfla/che promoter region was introduced at an ectopic site (S3A Fig). Furthermore, the sob6 allele did not appear to cause a gain-of-function phenotype in a diffusible gene product because swarming motility was not rescued to a swrB mutant when the Pfla/chesob6 promoter region was ectopically integrated (S3B Fig). We conclude that the sob6 allele does not function in trans, but rather functions in cis in a manner seemingly independent of activating transcription initiation from the Pfla/che promoter. Similar mutations in the -110/-114 region were not in any of 23 newly-isolated suppressors of swrA (soa) and thus the region could potentially represent an enhanced SwrA binding site (S2 Fig and Table 2). Mutations in this region, however, do not act like SwrA to increase expression of the Pfla/che promoter and fall outside of the putative binding site of the response regulator DegU with which SwrA has been reported to interact [54,57–59]. What recognizes the -110/-114 element is unknown.
Class IV–Mutation of the intergenic region upstream of cspB
The four sob class IV alleles were not within the Pfla/che promoter region, and classical SPP1 transduction-based genetic linkage analysis failed due to an inability to generate linked transposon insertions. Instead, the sob class IV alleles were identified by Illumina whole-genome sequencing. Three of the class IV sob alleles were large deletions that overlapped with one another and all had in common the deletion of the gene cspB, encoding the RNA binding cold shock protein CspB, and the upstream intergenic region [60,61] (Fig 5C). Likewise, the fourth allele was a single base pair mutation in the intergenic region upstream of cspB and within the overlapping region common to the other three alleles. One way in which sob class IV alleles could restore motility to the swrB mutant is by increasing the expression of the fla/che operon in a manner similar to sob classes I, II, and III. The swrB sob24 background containing a deletion spanning the intergenic region and cspB increased fla/che operon transcript abundance 2-to-7 fold but did not increase expression of the Pfla/che-lacZ reporter over wild type (Fig 7E and S4 Fig). We conclude that the class IV alleles rescued the swrB swarming defect by mutating a trans factor that directly or indirectly reduced fla/che operon transcript levels.
To explore the identity of the putative trans factor, four nested complementation constructs encoding combinations of cspB, the intergenic region, and the adjacent yhcJ gene were integrated at the ectopic amyE locus in the chromosome (Fig 5C). Only those constructs that contained both the intergenic region and the cspB open reading frame were able to restore swarming inhibition to the swrB sob24 mutant (Fig 4H). Thus, one potentially relevant diffusible gene product disrupted by the sob class IV alleles was the CspB protein. To test the role of cspB in the inhibition of swarming motility in the swrB mutant, an in-frame markerless deletion was generated in the cspB open reading frame that left the upstream intergenic region intact. Deletion of cspB in an otherwise wild type background resulted in wild type swarming behavior but did not rescue swarming in the absence of SwrB (Fig 4I). We conclude that the sob class IV mutants rescued swrB swarming by mutation of a gene product emanating from the intergenic region upstream of cspB. The molecular nature of the diffusible gene product encoded upstream of the cspB gene is unknown but its mutation compensates for the absence of SwrB by increasing fla/che transcript levels like the sob class I, II, and III alleles.
Class V–Enhanced translation of FliP
The single sob class V allele (sob22) was found to be genetically linked to the swrB::tet parental allele by classical SPP1-transduction based linkage analysis. Sequencing of fla/che operon DNA upstream of swrB revealed a missense point mutation near the 3’ end of the fliO gene (FliOK212E) (Fig 5D and Table 1). One way in which the sob22 mutation might restore swarming to the swrB mutant is by the generation of a gain-of-function substitution in FliO. To determine the consequence of the sob22 allele on the function of FliO, the native fliO gene was first mutated by an in-frame marker-less deletion and then complemented with either the wild type fliO allele or the fliOsob22 allele cloned downstream of the Pfla/che promoter region and inserted at the ectopic amyE locus (amyE::Pfla/che-fliO and amyE::Pfla/che-fliOsob22). Deletion of fliO abolished swarming motility and the fliO deletion was partially complemented when either the wild type or sob22 allele of fliO was ectopically expressed (Fig 4J). Neither the fliO wild type allele nor the fliOsob22 allele, however, rescued swarming motility to a swrB fliO double mutant (Fig 4K). We conclude that the fliOsob22 allele resulted in a neutral FliOK212E substitution that did not suppress the swrB phenotype.
Another way in which the sob22 mutation might restore swarming to the swrB mutant is by enhancing translation of the fliP gene (Fig 5D). The open reading frame of fliP is immediately downstream of, and overlaps with, the open reading frame of fliO. The sob22 allele is an A to G transition fifteen base pairs upstream of the fliP translational start and results in a sequence with increased similarity to the consensus for a Shine-Dalgarno ribosome binding site (AGAAGA > AGGAGA) [62] (Fig 5D). To determine the consequence of the sob22 allele on FliP, the native fliP gene was first mutated by an in-frame markerless deletion and then complemented with either the wild type fliP allele or the fliPsob22 allele cloned downstream of the Pfla/che promoter region and inserted at the ectopic amyE locus (amyE::Pfla/che-fliP and amyE::Pfla/che-fliPsob22). Deletion of fliP abolished swarming motility and the fliP deletion was partially complemented with the wild type fliP allele and fully complemented by the fliPsob22 allele (Fig 4L). Whereas the wild type allele of fliP did not restore swarming motility to a swrB fliP double mutant, the fliPsob22 allele rescued swarming to a level comparable to that observed in the original sob22 background (Fig 4M). We conclude that sob22 suppresses the swrB non-swarming phenotype by improving the translation of FliP.
To determine whether increasing expression of FliP by another mechanism was sufficient to restore swarming motility to the swrB mutant, the wild type fliP allele was cloned downstream of the IPTG-inducible Physpank promoter and inserted at the amyE locus (amyE::Phsypank-fliP). As specificity controls, the fliO and fliQ genes encoded immediately upstream and downstream of fliP respectively were separately cloned under the Physpank promoter and inserted at the amyE locus. Whereas IPTG induction of fliP restored swarming motility to the swrB mutant, IPTG induction of fliO (Fig 4N) and fliQ did not (S5 Fig). FliO, FliP and FliQ are core transmembrane components of the flagellar type III secretion export apparatus, and we conclude that increased synthesis of FliP specifically is sufficient to bypass the requirement of SwrB in swarming motility. We infer that increased expression of FliP likely accounts for the Class I, II, III, and IV mutations that act to increase the expression of the fla/che operon, of which fliP is a member (Fig 5A).
Class VI–Gain-of-function allele in FliG
The two sob class VI alleles were independently-isolated and identified using Illumina whole genome sequencing. Both sob class VI alleles were identical missense mutations in the fliG gene encoding the flagellar rotor protein FliG (FliGQ132R) (Fig 5E and Table 1). To determine the consequences of FliGQ132R, the native fliG gene was first mutated by an in-frame markerless deletion and complemented with the either the wild type fliG or the fliGQ132R allele cloned downstream of the Pfla/che promoter region and inserted at the ectopic amyE locus (amyE::Pfla/che-fliG and amyE::Pfla/che-fliGQ132R). Deletion of fliG abolished swarming motility and the fliG deletion was partially complemented by the wild type FliG protein (Fig 4O). In contrast, the FliGQ132R protein fully complemented the swarming defect of the fliG mutant and thus appeared to have enhanced activity (Fig 4O). Furthermore, the swarming defect of a swrB fliG double mutant was rescued in the presence of FliGQ132R but not wild type FliG (Fig 4P). We conclude that the FliGQ132R substitution confers a gain-of-function phenotype sufficient to restore swarming in the absence of SwrB.
The FliGQ132R substitution is located in the central armadillo (ARM) motif associated with polymerization of the rotor and assembly of the FliM C-ring component [32,63,64]. In E. coli and S. enterica, loss-of-function mutations in either the FliG ARM motif or FliM result in assembly defects of extracellular flagellar components [31,34–37,63]. Consistent with a role for FliM in flagellar assembly, mutation of fliM in B. subtilis reduced the flagellar hook frequency similar to that observed in the absence of SwrB (Fig 8). Introduction of the gain-of-function fliGQ132R allele to the fliM mutant background, however, increased hook frequency similar to that found in the isogenic fliGQ132R allele alone (Fig 8). We conclude that the gain-of-function fliGQ132R allele bypasses the absence of FliM and increases the frequency of hook assembly. We infer that FliGQ132R alters the conformation of the flagellar rotor and that this conformation is normally adopted only after complete assembly of FliM into the C-ring.

SwrB activates hook secretion in a manner dependent on FliF and FliP
Mutations in FliP and FliG that suppressed the absence of SwrB suggested that the mechanism by which SwrB enhanced hook assembly was related to flagellar secretion by way of basal body structure. Therefore, we further explored the relationship of SwrB to components of the basal body and C-ring for enhancing hook polymerization. To do so, strains were generated that contained the FlgET123C allele for fluorescent labeling of the flagellar hook in backgrounds mutated for SwrB, FliM, FliG, and FliF. Cells mutated for either FliM or FliG displayed a low frequency of flagellar hooks and resembled cells mutated for SwrB (Fig 9). By contrast, no hooks were detected in cells mutated for FliF (Fig 9). Next, the swrB gene was cloned downstream of an IPTG inducible Physpank promoter and integrated at an ectopic site in each of the strains tested (amyE::Physpank-swrB). In the presence of IPTG, overexpression of SwrB increased the frequency of hooks for the swrB, fliM and fliG mutants but did not restore hook formation to the fliF mutant (Fig 9). We conclude that the activation of hook assembly by SwrB does not require FliG or FliM, but that FliGQ132R nonetheless compensates for the absence of SwrB. We further conclude that the basal body structural component FliF is required for SwrB to activate hook assembly.

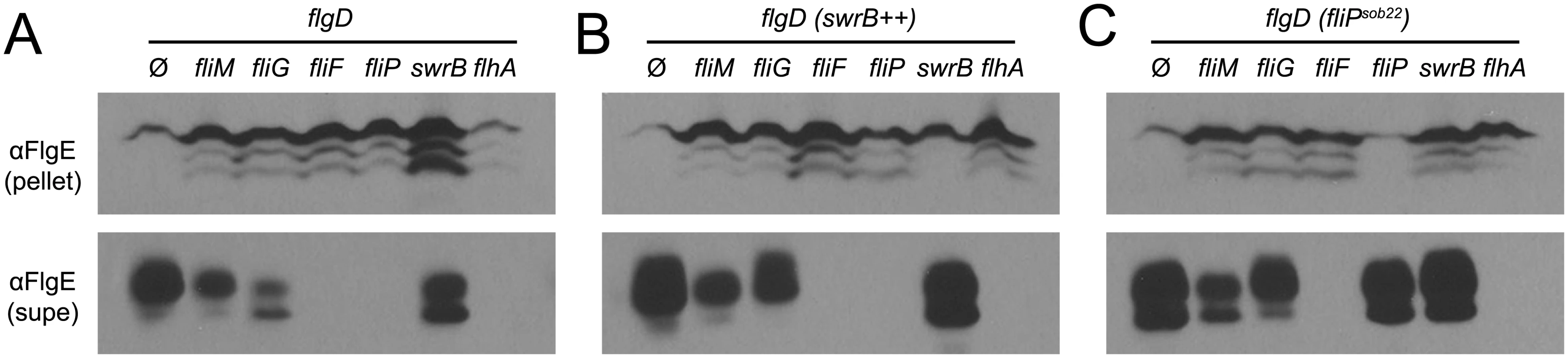
FliF could be required for SwrB activation of hook assembly because FliF also serves as the polymerization platform for the flagellar rod and hook. Thus, SwrB could stimulate flagellar secretion in the absence of FliF but the hook subunits would accumulate in the supernatant due to an inability to polymerize. To determine the effect SwrB has on hook protein secretion, strains were generated that were mutated for the extracellular hook chaperone protein FlgD such that all strains would fail to polymerize FlgE, and FlgE would thus be secreted into the supernatant [51,65]. Cells mutated for FliM and FliG reduced the amount of secreted hook protein whereas cells mutated for FliF, FliP, and FlhA abolished hook secretion (Fig 10A). Next, the IPTG-inducible Physpank-swrB construct was added to each strain to determine the effect that over-expression of SwrB would have on hook secretion. When SwrB was over-expressed, the amount of hook protein in the supernatant appeared to increase in the fliM, fliG, and swrB mutants, but not in the fliF, fliP, or flhA mutants (Fig 10B). Finally, addition of the Pfla/che-fliPsob22 complementation construct provided in trans appeared to increase FlgE secretion in fliM, fliG, swrB, and fliP mutants but not in the fliF or flhA mutant (Fig 10C). We conclude FliF, FliP, and FlhA are downstream of SwrB for hook protein secretion. In sum, we conclude that SwrB activates an early flagellar morphogenetic checkpoint by catalyzing a FliF-basal body conformation that activates the type III secretion export apparatus for hook subunit secretion.
Discussion
Flagellar regulation is morphogenetically coupled to flagellar structural intermediates to ensure that the structural subunits are assembled in the proper order. A classic example of morphogenetic coupling is illustrated by how the completion of the flagellar hook induces a change in the substrate specificity of the export apparatus and governs the expression of the flagellar filament protein via secretion of the anti-sigma factor FlgM [2,27]. Here we find evidence of an earlier morphogenetic coupling event in which SwrB and the completion of the flagellar basal body govern the secretion of the hook (and likely rod) structural subunits. Given that the type III secretion export apparatus appears to be the first flagellar subdomain assembled within the membrane, we suggest that if the export apparatus was immediately active upon assembly, structural components would be secreted in the absence of the basal body upon which they are polymerized. Instead, it appears that the export apparatus becomes functional only after the basal body is complete; an event indicated by a conformational change adopted by the flagellar base plate protein FliF. Thus, basal body completion is a discrete flagellar morphogenetic checkpoint and we argue that the B. subtilis membrane protein SwrB potentiates the ability of the basal body to adopt a “completed” conformation.
SwrB (YlxL) was originally discovered as the product encoded by the last gene in the 32 gene fla/che operon and was shown to be required for the activation of σD-dependent gene expression and swarming motility [38,39,41]. Here we account for both previously reported phenotypes of the swrB mutant. We found that cells defective for SwrB were unable to swarm because they failed to synthesize wild type numbers of flagellar filaments and resembled the hypoflagellated state of cells defective for another swarming regulator, SwrA [53]. The SwrB defect in flagellar number was not due to a reduction in the number of flagellar basal bodies as seen in cells defective for SwrA however, but rather due to a reduction in the number of flagellar hooks. The swrB mutant defect in hook synthesis is consistent with the observed reduction in σD-dependent gene expression as hook completion is needed to antagonize the σD anti-sigma factor FlgM [39,51]. Thus, SwrB increased the probability that basal bodies became proficient for hook assembly. Spontaneous suppressors that restored swarming motility to swrB mutants indicated that SwrB increased the frequency of hook assembly by activating hook subunit secretion via the type III secretion component FliP.
FliP is a transmembrane protein that is incorporated early in the nascent basal body and required for nucleation of other components of the flagellar type III secretion export apparatus FliO, FliQ, FliR, FlhA and FlhB [5,66,67]. Mutation of FliP abolishes secretion, and while the precise function of FliP is unknown, it appears to be regulated by at least two mechanisms. First, the FliP N-terminal signal sequence appears to be inhibitory in S. enterica and is processed presumably after assembly of the export apparatus [5,68]. B. subtilis FliP however lacks the N-terminal signal sequence altogether, perhaps an indication of the need for additional regulatory mechanisms [68,69]. Second, the levels of FliP appear to be important in S. enterica as the accessory protein FliO protects FliP from proteolytic degradation [21,22]. Here we further support the importance of FliP protein levels in B. subtilis as 22 out of 24 spontaneous suppressor-of-swrB (sob) mutations restored swarming to a swrB mutant by increasing FliP expression.
FliP was directly implicated by a single suppressor-of-swrB (sob) mutation that mutated the FliP Shine-Dalgarno sequence closer to consensus, thus enhancing translation. Furthermore, enhanced transcription of the native fliP gene within the fla/che operon by another 21 sob alleles or an ectopically integrated fliP gene expressed from artificial IPTG-inducible promoter was sufficient to rescue swarming to a swrB mutant. FliP is hypothesized to sit within the confines of a ring of the basal body protein FliF, the stoichiometry of which along with the rest of the type III secretion export apparatus components is thought to be definite and precise [5]. Thus, how extra copies of wild type FliP would improve swarming and/or become incorporated into basal bodies is unclear. Perhaps extra FliP protein titrates an inhibitor of the type III secretion export apparatus. Alternatively, FliP is thought to be one of the earliest proteins assembled in the flagellum and overexpression may increase the population of FliP molecules in a secretion-active conformation that preferentially promotes basal body nucleation. A potential candidate for either the inhibitor and/or conformational regulator of the type III secretion system is found in the final class of SwrB suppressors that fall within the rotor protein FliG.
FliG forms the gear-like rotor that docks to the cytoplasmic surface of the FliF basal body protein, interacts with the MotA/MotB proton channel stator, and serves as a scaffold for the assembly of the FliM/FliN(FliY) cytoplasmic C-ring [33,70–75]. Although FliG, FliM, and FliN are found in the cytoplasm, it has long been known that mutations in each protein cause extracytoplasmic flagellar assembly defects [31–37]. Here we show that a gain-of-function mutation found in the ARM motif that controls FliG polymerization bypasses the C-ring requirement for flagellar hook (but not flagellar filament) assembly [64] (Fig 8 and S6 Fig). We posit that proper conformation of the FliG rotor in vivo, normally promoted by the completion of the C-ring and the presence of SwrB, activates the type III secretion export apparatus. Since FliG and the export apparatus are not known to directly interact, we infer that a regulatory conformational change is propagated from the rotor through basal body protein FliF [74].
FliF is a critical transmembrane structural protein that defines the flagellar basal body as it docks to the FliG rotor, surrounds the type III export apparatus, and forms a polymerization platform for the rod [4,76–78]. FliF has been hypothesized to exist in active and inactive conformations. In S. enterica, FliF has a large periplasmic domain that may regulate the export apparatus as electron microscopy of FliF rings show two conformations, one with a lumen that appears to be open and one in which the lumen appears to be closed [4,79–81]. Furthermore, a FliF closed conformation was hypothesized to be adopted when the poorly-understood periplasmic protein FliE was mutated [6,82,83]. In B. subtilis, FliF is required for secretion as cells defective in FliF fail to secrete both the early class substrate FlgE and the late class substrate FlgM (Fig 10) [52]. Thus, FliF could be required for secretion by surrounding the type III export apparatus and regulating its function.
Based on cytological and genetic suppressor data, we conclude that SwrB functions as an assembly chaperone to enhance the probability that the flagellar basal body adopts a conformation proficient for secretion (Fig 11). We propose that the flagellar type III secretion apparatus and FliF form first in the membrane as a “proto-basal body” that is inactive for export (Fig 11A) [7]. The proto-basal body is able to spontaneously mature to become proficient for hook secretion at a low frequency (Fig 11B). Under normal conditions, the frequency of proto-basal body maturation is increased by both SwrB and assembly of the C-ring as the absence of either share a low hook-to-basal body ratio (Fig 11C). Indeed, SwrB and FliG appear to be required at the same step as FliG gain-of-function alleles that enhance rotor stability bypass the requirement of SwrB for hook assembly, and artificial overexpression of SwrB bypasses the need for FliG. The most likely convergence point for SwrB and FliG is the membrane-basal body protein FliF, as FliG is a known interactor and SwrB, itself a membrane protein, could be adjacent. We hypothesize that the previously documented changes in FliF conformation are responsible for activating the export apparatus and do so by activating the membrane protein FliP. In sum, maturation of the flagellar basal body to a secretion proficient state is a morphogenetic checkpoint that must be passed prior to proceeding to the export and assembly of more distal components.

SwrB is conserved only in members of the genus Bacillus but the notion that flagellar basal body completion acts as a conformational liscencing event to permit subsequent flagellar secretion is consistent with genetic and biochemical observations in a variety of systems. In Campylobacter jejuni, the conformation adopted by the completed basal body activates expression of the rod and hook genes as a separate regulatory “tier”, and similar “4-tier” flagellar regulatory hierarchies have been demonstrated in Caulobacter crescentus and Pseudomonas aeruginosa [84–91]. Thus instead of regulating the type III export apparatus functionally, completion of the basal body transcriptionally controls the availability of the rod and hook cargo. Between our results in B. subtilis and the “4-tier” flagellar systems, we suggest that the rod should be considered separate from the basal body and therefore the structural domains of the flagellum should be considered the “basal body”, the “rod-hook”, and the “filament”, at least for these organisms. Finally, our conformation control model is consistent with the notion that a conformational change propagates from the needle tip to the export apparatus at the base of the type III secretion injectosomes of pathogens to activate the secretion of effectors in response to direct contact with host cells [92–94]. Thus, we conclude that the activation of secretion is a regulated morphogenetic checkpoint, and disrupting functional regulators could be a strategy to attenuate both flagella and injectisome virulence factors.
Materials and Methods
Strains and growth conditions
B. subtilis strains were grown in Luria-Bertani (LB) (10 g tryptone, 5 g yeast extract, 5 g NaCl per L) broth or on LB plates fortified with 1.5% Bacto agar at 37°C. When appropriate, antibiotics were included at the following concentrations: 10 μg/ml tetracycline, 100 μg/ml spectinomycin, 5 μg/ml chloramphenicol, 5 μg/ml kanamycin, and 1 μg/ml erythromycin plus 25 μg/ml lincomycin (mls). Isopropyl β-D-thiogalactopyranoside (IPTG, Sigma) was added to the medium at the indicated concentration when appropriate. For the swarm expansion assay, swarm agar plates containing 25 ml LB fortified with 0.7% Bacto agar were prepared fresh and the following day were dried for a total of 20 minutes in a laminar flow hood (see below).
Strain construction
All constructs were first introduced into the competent ancestral strain DS2569 by natural competence and then transferred to the 3610 background using SPP1-mediated generalized phage transduction or by transformation in the competent ancestral strain DK1042 (as indicated by the presence of the comIQ12L allele in the genotype) [95,96]. All strains used in this study are listed in Table 3. All plasmids used in this study are listed in S1 Table. All primers used in this study are listed in S2 Table.

LacZ reporter constructs
To generate the β-galactosidase (lacZ) reporter construct pAP50, a PCR product containing the Pflache promoter including the sob6 point mutation was amplified from B. subtilis strain DS7063 using primer pair 321/322. The PCR product was digested with EcoRI and BamHI and cloned into the EcoRI and BamHI sites of plasmid pDG1728, which carries a spectromycin-resistance marker and a polylinker upstream of the lacZ gene between two arms of the amyE gene [97].
In-frame deletions
To generate the ΔfliO in frame marker-less deletion construct, the region upstream of fliO was PCR amplified using the primer pair 1692/1693 and digested with EcoRI and XhoI, and the region downstream of fliO was PCR amplified using the primer pair 1694/1695 and digested with XhoI and BamHI. The two fragments were then simultaneously ligated into the EcoRI and BamHI sites of pMiniMAD which carries a temperature sensitive origin of replication and an erythromycin resistance cassette to generate pDP332 [98]. The plasmid pDP332 was introduced to DS2569 by single cross-over integration by transformation at the restrictive temperature for plasmid replication (37°C) using mls resistance as a selection. The integrated plasmid was then transduced into 3610. To evict the plasmid, the strain was incubated in 3ml LB broth at a permissive temperature for plasmid replication (22°C) for 14 hours, diluted 30-fold in fresh LB broth, and incubated at 22°C for another 8 hours. Dilution and outgrowth was repeated 2 more times. Cells were then serially diluted and plated on LB agar at 37°C. Individual colonies were patched on LB plates and LB plates containing mls to identify mls sensitive colonies that had evicted the plasmid. Chromosomal DNA from colonies that had excised the plasmid was purified and screened by PCR using primers 1692/1695 to determine which isolate had retained the ΔfliO allele.
To generate the ΔfliP in frame marker-less deletion construct, the region upstream of fliP was PCR amplified using the primer pair 2290/2291 and digested with EcoRI and XhoI, and the region downstream of fliP was PCR amplified using the primer pair 2292/2293 and digested with XhoI and BamHI. The two fragments were then simultaneously ligated into the EcoRI and BamHI sites of pMiniMAD which carries a temperature sensitive origin of replication and an erythromycin resistance cassette to generate pDP346. The plasmid pDP346 was introduced to DS2569 by single cross-over integration by transformation at the restrictive temperature for plasmid replication (37°C) using mls resistance as a selection. The integrated plasmid was then transduced into 3610 and evicted as described above. Chromosomal DNA from colonies that had excised the plasmid was purified and screened by PCR using primers 2292/2293 to determine which isolate had retained the ΔfliP allele.
To generate the ΔcspB in frame marker-less deletion construct, the region upstream of cspB was PCR amplified using the primer pair 3944/3945 and digested with EcoRI and BamH1, and the region downstream of cspB was PCR amplified using the primer pair 3946/3947 and digested with BamHI and XhoI. The two fragments were then simultaneously ligated into the EcoRI and SalI sites of pMiniMAD which carries a temperature sensitive origin of replication and an erythromycin resistance cassette to generate pAP94. The plasmid pAP94 was introduced to DK1042 by single cross-over integration by transformation at the restrictive temperature for plasmid replication (37°C) using mls resistance as a selection. The plasmid was evicted as described above and chromosomal DNA from colonies that had excised the plasmid was purified and screened by PCR using primers 3944/3947 to determine which isolate had retained the ΔcspB allele.
fliGQ132R ΔfliM
To generate the fliGsob28 allelic exchange construct, the region spanning the fliGsob28 point mutation was PCR amplified from DS9155 using the primer pair 1229/3632 and digested with EcoRI and SalI. The fragment was then ligated into the EcoRI and SalI sites of pMiniMAD which carries a temperature sensitive origin of replication and an erythromycin resistance cassette to generate pAP80. The plasmid pAP80 was introduced to DK1042. Plasmid eviction was conducted as described above and chromosomal DNA from colonies that had excised the plasmid was purified and screened by PCR using primers 1229/3632 and sequencing to determine which isolate had retained the fliGsob28 point mutation to generate DK1285. The plasmid pSG32 was then transformed into DK1285 and evicted (as described in 53) and screened by PCR using primers 1569/1572 to generate strain DK1412.
Pflachesob6
To generate the Pflachesob6 allelic exchange construct, the region spanning the Pflachesob6 point mutation was PCR amplified from DS7063 using the primer pair 1648/1482 and digested with EcoRI and BamHI. The fragment was then ligated into the EcoRI and BamHI sites of pMiniMAD which carries a temperature sensitive origin of replication and an erythromycin resistance cassette to generate pAP46. The plasmid pAP46 was introduced to DS2569 by transformation and transduced to 3610. Plasmid eviction was conducted in was conducted as described above and chromosomal DNA from colonies that had excised the plasmid was purified and screened by PCR using primers 1648/1482 and sequencing to determine which isolate had retained the Pflachesob6 point mutation.
Pflachesob21
To generate the Pflachesob21 allelic exchange construct, the region spanning the Pflachesob21 point mutation was PCR amplified from DS9148 using the primer pair 1648/1482 and digested with EcoRI and BamHI. The fragment was then ligated into the EcoRI and BamHI sites of pMiniMAD which carries a temperature sensitive origin of replication and an erythromycin resistance cassette to generate pAP44. The plasmid pAP44 was introduced to DS2569 by transformation and transduced to 3610. Plasmid eviction was conducted in was conducted as described above and chromosomal DNA from colonies that had excised the plasmid was purified and screened by PCR using primers 1648/1482 and sequencing to determine which isolate had retained the Pflachesob21 point mutation.
Complementation constructs
To generate the Pflache-fliO complementation construct pAP60 and the Pflache-fliOsob22 complementation construct pAP61, PCR using primer pair 3217/3218 amplified fliO from B. subtilis 3610 and DS9149 chromosomal DNA respectively and the resulting products were digested with XhoI and BamHI. Concurrently, a PCR product containing the flache promoter was amplified from B. subtilis 3610 chromosomal DNA using the primer pair 2460/2461 and digested with EcoRI and XhoI. The two fragments were then simultaneously ligated into the EcoRI and BamHI sites of pAH25 containing a spectinomycin resistance cassette between two arms of the amyE gene (gift from Amy Camp, Mount Holyoke University).
To generate the Pflache-fliP complementation construct pAP69 and the Pflache-fliPsob22 complementation construct pAP71, PCR using primer pair 3449/3450 containing amplified fliP from B. subtilis 3610 and DS9149 chromosomal DNA respectively and the resulting products were digested with XhoI and BamHI. Concurrently, a PCR product containing the flache promoter was amplified from B. subtilis 3610 chromosomal DNA using the primer pair 2460/2461 and digested with EcoRI and XhoI. The two fragments were then simultaneously ligated into the EcoRI and BamHI sites of pAH25 containing a spectinomycin resistance cassette between two arms of the amyE gene.
Gibson isothermal assembly was used to generate the series of constructs to complement the region mutated by sob24 as restriction enzyme based cloning strategies failed when DNA fragments containing the 5’ end of the yhcJ gene was introduced to E. coli. To generate arms for homologous recombination, one arm containing the amyE gene and upstream region was amplified with primers 3733/3180, and one arm containing the amyE gene, the spectinomycin resistance cassette, and the downstream region was amplified with primers 3177/4086 both using chromosomal DNA purified from DK1118 as a template. Next, the complementation fragments were amplified from 3610 template DNA as follows: “comp1” region was amplified with primers 4087/4088, “comp2” region was amplified with 4090/4088, “comp3” region was amplified using primers 4087/4089, and “comp4” region was amplified using primers 4090/4089. Each complementation region was combined with the arms from the amyE locus, assembled using Gibson assembly, and transformed into DK1042 selecting for spectinomycin resistance [99].
Inducible constructs
To generate the inducible amyE::Physpank-fliP spec construct pAP70, a PCR product containing fliP was amplified from B. subtilis 3610 chromosomal DNA using the primer pair 3451/3473, digested with NheI and SphI, and cloned into the NheI and SphI sites of pDR111 containing a spectinomycin resistance cassette, a polylinker downstream of the Physpank promoter, and the gene encoding the LacI repressor between the two arms of the amyE gene [100].
To generate the inducible amyE::Physpank-fliO spec construct pAP85, a PCR product containing fliP was amplified from B. subtilis 3610 chromosomal DNA using the primer pair 3741/3742, digested with NheI and SphI, and cloned into the NheI and SphI sites of pDR111.
To generate the inducible amyE::Physpank-fliQ spec construct pKRH44, a PCR product containing fliQ was amplified from B. subtilis 3610 chromosomal DNA using the primer pair 4560/4561, digested with NheI and SphI, and cloned into the NheI and SphI sites of pDR111.
SPP1 phage transduction
To 0.2 ml of dense culture grown in TY broth (LB broth supplemented after autoclaving with 10 mM MgSO4 and 100 μM MnSO4), serial dilutions of SPP1 phage stock were added and statically incubated for 15 minutes at 37°C. To each mixture, 3 ml TYSA (molten TY supplemented with 0.5% agar) was added, poured atop fresh TY plates, and incubated at 30°C overnight. Top agar from the plate containing near confluent plaques was harvested by scraping into a 15 ml conical tube, vortexed, and centrifuged at 5,000 x g for 5 minutes. The supernatant was treated with 25 μg/ml DNase before being passed through a 0.45 μm syringe filter and stored at 4°C.
Recipient cells were grown to stationary phase in 3 ml TY broth at 37°C. 1 ml cells were mixed with 25 μl of SPP1 donor phage stock. 9 ml of TY broth was added to the mixture and allowed to stand at 37°C for 30 minutes. The transduction mixture was then centrifuged at 5,000 x g for 5 minutes, the supernatant was discarded and the pellet was resuspended in the remaining volume. 100 μl of cell suspension was then plated on LB fortified with 1.5% agar, the appropriate antibiotic, and 10 mM sodium citrate.
Swarm expansion assay
Cells were grown to mid-log phase at 37°C in LB broth and resuspended to 10 OD600 in pH 8.0 PBS buffer (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 2 mM KH2PO4) containing 0.5% India ink (Higgins). Freshly prepared LB containing 0.7% Bacto agar (25 ml/plate) was dried for 10 minutes in a laminar flow hood, centrally inoculated with 10 μl of the cell suspension, dried for another 10 minutes, and incubated at 37°C [101]. The India ink demarks the origin of the colony and the swarm radius was measured relative to the origin. For consistency, an axis was drawn on the back of the plate and swarm radii measurements were taken along this transect. For experiments including IPTG, cells were propagated in broth in the presence of IPTG, and IPTG was included in the swarm agar plates.
Western blotting
B. subtilis strains were grown in LB broth to OD600 ~0.5, 1 ml was harvested by centrifugation, and resuspended to 10 OD600 in Lysis buffer (20 mM Tris pH 7.0, 10 mM EDTA, 1 mg/ml lysozyme, 10 g/ml DNAse I, 100 g/ml RNAse I, 1 mM PMSF) and incubated 30 minutes at 37°C. Each lysate was then mixed with the appropriate amount of 6x SDS loading dye to dilute the loading dye to 1x concentration. Samples were separated by 12% Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The proteins were electroblotted onto nitrocellulose and developed with a 1 : 20,000 dilution of (anti-FliG, anti-FlgE), 1 : 10,000 dilution of (anti-SigD), or 1 : 80,000 dilution of (anti-Hag, anti-SigA) of primary antibody and a 1 : 10,000 dilution secondary antibody (horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin G). Immunoblot was developed using the Immun-Star HRP developer kit (Bio-Rad).
FlgE secretion assay
For the pellet fraction (cytoplasmic and cell-associated proteins), B. subtilis strains were grown in 20 ml LB broth in the presence 1 mM IPTG when appropriate to an OD600 of 1.2–1.7, and 10 ml samples of broth culture were harvested by centrifugation, resuspended to 10 OD600 units in lysis buffer (20 mM Tris [pH 7.0], 10 mM EDTA, 1 mg/ml lysozyme, 10 μg/ml RNase I, 1 mM PMSF), and incubated 30 minutes at 37°C. For the supernatant fraction (secreted extracellular proteins), 10 ml of supernatant was collected from the same cultures as those used to generate the pellet fractions. The supernatant was clarified by centrifugation at 5,000 × g for 30 min and treated with 1 ml of freshly prepared 0.015% sodium deoxycholate for 10 min at room temperature. Proteins from the supernatant were precipitated by adding 500 μl chilled trichloroacetic acid (TCA) and incubating the mixture for >2 h on ice at 4°C. Precipitated proteins were pelleted at 9,447 × g for 10 min at 4°C, washed twice with 1 ml ice-cold acetone, and resuspended to 10 OD600 units in 0.1 N sodium hydroxide. Ten microliters of cell pellet or supernatant sample was mixed with 2 μl 6× SDS loading buffer. Samples were separated in parallel by 15% SDS-PAGE. Proteins were electroblotted onto nitrocellulose for 1 hour at 400 mA and probed with a 1 : 20,000 dilution of anti-FlgE primary antibody and with a 1 : 10,000 dilution of secondary antibody (horseradish peroxidase [HRP]-conjugated goat anti-rabbit immunoglobulin G). Immunoblots were developed using the Pierce ECL Western blotting substrate kit (Thermo Scientific).
Microscopy
Fluorescence microscopy was performed with a Nikon 80i microscope along with a phase contrast objective Nikon Plan Apo 100X and an Excite 120 metal halide lamp. FM4-64 was visualized with a C-FL HYQ Texas Red Filter Cube (excitation filter 532–587 nm, barrier filter >590 nm). GFP was visualized using a C-FL HYQ FITC Filter Cube (FITC, excitation filter 460–500 nm, barrier filter 515–550 nm). Images were captured with a Photometrics Coolsnap HQ2 camera in black and white, false colored and superimposed using Metamorph image software.
For Phag-GFP microscopy, cells were grown at 37°C in LB broth to OD600 0.6–1.0, resuspended in 30 μl PBS buffer containing 5 μg/ml FM 4–64 and incubated for 5 min at room temperature. The cells were pelleted, resuspeneded in 30μl PBS buffer, and were observed by spotting 4 μl of suspension on a cleaned microscope slide and immobilized with a poly-L-lysine-treated glass coverslip.
For fluorescent microscopy of flagellar filaments, 1.0 ml of broth culture was harvested at 0.6–1.0 OD600, resuspended in 50 μl of PBS buffer containing 5μg/ml Alexa Fluor 488 C5 maleimide (Molecular Probes), incubated for 3 min at room temperature, and washed once in 1.0 ml of PBS buffer. The suspension was pelleted, resuspended in 30 μl of PBS buffer containing 5 μg/ml FM 4–64, and incubated for 5 min at room temperature. The cells were pelleted, resuspeneded in 30 μl PBS buffer, and were observed by spotting 4 μl of suspension on a cleaned microscope slide and immobilized with a poly-L-lysine-treated glass coverslip.
For fluorescent microscopy of flagellar hooks, 1.0 ml of broth culture was harvested at 0.6–1.0 OD600, resuspended in 50 μl of PBS buffer containing 5μg/ml Alexa Fluor 488 C5 maleimide (Molecular Probes), incubated for 3 min at room temperature, and washed once in 1.0 ml of PBS buffer. The suspension was pelleted, resuspended in 30 μl of PBS buffer containing 5 μg/ml FM 4–64, and incubated for 5 min at room temperature. The cells were pelleted, resuspeneded in 30 μl PBS buffer, and were observed by spotting 4 μl of suspension on a cleaned microscope slide and immobilized with a poly-L-lysine-treated glass coverslip.
For PfliM-fliM-GFP microscopy, cells were grown at 37°C in LB broth to OD600 0.6–1.0, resuspended in 30 μl PBS buffer containing 5 μg/ml FM 4–64 and incubated for 5 min at room temperature. The cells were pelleted, resuspeneded in 30μl PBS buffer, and were observed by spotting 4 μl of suspension on a cleaned microscope slide and immobilized with a poly-L-lysine-treated glass coverslip.
For super-resolution microscopy, the OMX 3D-SIM Super-Resolution system was used. Supper-resolution microscopy was performed by using a 1.4-numerical-aperture (NA) Olympus 100X oil objective. FM4-64 was observed using laser line 561 and emission filter 609 nm to 654 nm, and GFP (along with Alexa Fluor 488 nm) was observed using laser line 488 nm and emission filter 500 nm to 550 nm. Images were captured using a Photometrics Cascade II electron-multiplying charge-coupled-device camera, processed using SoftWorx imaging software, and analyzed using Imaris software.
OMX 3D-SIM counts
Images that were processed as described above were used in Imaris (Bitplane) to determine the number and location of FliM-GFP or FlgE labeled with a fluorescent dye (FlgET123C). The spots feature within the software labelled each puncta by the search parameter of identifying spots of 1 μM in the 488 nm wavelength. Cell pole positions were determined by using the 561 nm wavelength using the slice feature. The x, y, and z coordinates of each puncta in each cell was exported from Imars. Scripts were developed in MATLAB (The Mathworks) for importing two-dimensional Cartesian coordinates for basal body, hook, and cell lengths from Imaris.
β-Galactosidase assays
Cells were harvested from cultures growing at 37°C in LB broth. Cells were collected in 1.0 ml aliquots and suspended in an equal volume of Z buffer (40 mM NaH2PO4, 60 mM NaHPO4, 1.0 mM MgSO4, 10 mM KCl, and 38 mM 2-mercaptoethanol). Lysozyme was added to each sample to a final concentration of 0.2 mg/ml and incubated at 37°C for 30 min. Each sample was diluted in Z buffer to a final volume of 500 μl, and the reaction was started with 100 μl of 4 mg/ml 2-nitrophenyl β-galactopyranoside in Z buffer and stopped with 250 μl of 1M Na2CO3. The OD420 of the reaction mixture was measured, and the β-galactosidase-specific activity was calculated according the equation [OD420/(time X OD600)] X dilution factor X 1000.
Direct Sanger sequencing
For sob and soa mutations contained within the flache promoter, a PCR product containing the flache promoter was amplified from B. subtilis chromosomal DNA (either from strain 3610 or the appropriate suppressor strain) using the primer set 1921/1922. The Pflache PCR product was then sequenced using primer 1921 and 1922 individually.
Library construction and genome sequencing
Genomic samples were fragmented using the Corvaris S220 ultrasonicator and then assayed using the Agilent TapeStatin using D1K HS tapes. The fagmenented samples were processed into Illumina DNA-Seq libraries using Bechman SWHT chemistry in conjunction with BioScientific NextFlex Adaptors on the BIomekFx automated workstation. The large (350bp-750bp) size selection option was selected, and 12 μl of preamplified library was used as template in a 10 cycle amplification reaction. Following amplification, the libraries were cleaned using a 1X AmpureXP ratio and eluted in EB buffer.
After Illumina sequencing, MiSeq reads were trimmed using a quality cutoff of 20 and remaining sequencing adapters were removed using Cutadapt 1.2.1. FLASH 1.2.2 was then used to merge read pairs in which the forward and reverse read overlapped. The assemblies were performed using Newbler 2.7 using the merged reads as singletons and the unmerged reads as paired end reads. For SNP prediction, reads were mapped against both the NCIB 3610 reference and the DS234 assembly. Mapping was performed with bowtie 2.0.2 using the default parameters. Samtools was used to convert the mapping data to a pileup format. VarScan 2.3.2 was used to call SNPs, using parameters to require a minimum read depth of 20 and a minimum variant frequency of 90%.
To verify the point mutation in sob28, A PCR product containing fliG was amplified from B. subtilis chromosomal DNA strain DS9155 using the primer set 1229/3632. The fliG PCR product was then sequenced using primer 788 to verify the sob mutation. To verify the deletion boundaries within sob24 and sob25, A PCR product containing the deletion region was amplified from B. subtilis chromosomal DNA strain DS9151 and DS9152 using the primer set 3628/3629. The PCR product was then sequenced using primer 3476 to verify the deletion boundaries.
SPP1-mediated phage co-transduction linkage mapping
To determine the frequency of co-transduction of the sob22 mutation to the swrB::tet allele, a lysate was generated on the suppressor strain (DS9149) and the swrB::tet allele was transduced to the wildtype 3610 strain. Three hundred of the resulting colonies were then picked onto 0.7% LB swarm agar plates to enumerate the number of swarm proficient colonies. The percentage of colonies that were swarm proficient was inversely proportional to the distance between the swrB::tet and the suppressor mutation. The primer set 1692/2293 was used to generate a PCR product 13kb upstream of the swrB::tet allele within the flache operon. The flache PCR product was then sequenced using primer 1601.
RNA purification
25 ml of cells were grown in triplicate in LB broth at 37°C until the cultures reached 1.0 OD600. 10 ml of each culture was then harvested into 15 ml conical tubes containing 1.25 ml of stop solution (-20°C—5% phenol diluted in 100% ethanol) and centrifuged at 5,800 rpm for 10 min at 4°C. The resulting pellets were then transferred to 1.5 ml microfuge tubes containing 500 μl of -80°C methanol, centrifuged at 14,000 rpm for 1 min at 4°C, and then stored at -80°C. The pellets where then resuspended in 850 μl TE buffer and then transferred to microfuge tubes containing 10 mg/ml lysozyme, inverted 4–6 times and placed at 37°C for 45 min. After this incubation, 50 μl of 10% SDS (Sodium dodecyl sulfate) was as added to the samples, the samples were inverted 4–6 times, and then 50 μl of 3M sodium acetate, pH 5.2 was added and the tubes were again inverted 4–6 times. Each sample was then split into to 500 μl volumes into separate microfuge tubes. 500 μl of phenol was added to each sample, the samples were inverted 10 times, and then placed in a 64°C water bath for 6 min. The tubes were inverted every minute during this water bath incubation. After the water bath incubation, the samples were centrifuged at 14,000 rpm for 10 min at 4°C. The resulting aqueous layer was then transferred to a fresh 1.5 ml microfuge tube where an equal volume of chloroform was added. The tubes were inverted 6–10 times, and then centrifuged at 14,000 rpm for 5 min at 4°C. The resulting aqueous layer was then transferred to a fresh 1.5 ml microfuge tubes where 1/10 the volume of 3M sodium acetate, pH 5.2 and 2 volumes of -20°C 100% ethanol were added to the tubes. The resulting mixtures were incubated at -80°C for 30 min and then centrifuged at 14,000 rpm for 40 min at 4°C. The ethanol layer was then removed, and the resulting white pellet was washed with 1 ml of -20°C 75% ethanol, centrifuged at 14,000 rpm for 5 min at 4°C. The ethanol layer was removed and the pellet was resuspended in 50 μl of RNase free water, and each split pool was rejoined together an incubated at 50°C to encourage resuspension.
Quantitative PCR primer optimization
Each primer pair was diluted to both 1 μM and 5 μM stocks and subsequently mixed into 25 separate reactions in duplicate. Each of the 25 reactions was a permutation of one forward primer at either 50 nM, 100 nM, 300 nM, 600 nM, or 900 nM concentration with its corresponding reverse primer at either 50 nM, 100 nM, 300 nM, 600 nM, or 900 nM concentration along with SYBR Green SuperMix reagent (Quanta Biosciences), and 106 copies of template. Each primer optimization assay also contained two additional control reactions containing forward and reverse primers each at either 50nM or 900 nM, Green SuperMix reagent (Quanta Biosciences) and no template. Quantitative PCR was conducted on each reaction set for each primer used in downstream quantitative PCR assays on the Stratagene MX3500 Pro thermocycler. Data were analyzed using the MXPro Stratagene software package.
Quantitiative reverse transcriptase PCR
Total RNA was isolated as described above. Isolated RNA was DNase digested used TURBO DNA free ket (Ambion). cDNA was reverse transcribed from each DNase-digested RNA sample using random dT primers of the qScript cDNA superMix (Quanta Biosciences). Quantitave PCR was performed with specific primer pairs whose concentrations were optimized as described above and either diluted cDNA templeate, DNase digested RNA, or no template using SYBR Green SuperMix (Quanta Biosciences) on the Stratagene MX3500 Pro thermocycler. Data were analyzed using the MXPro Stratagene software package. The following primers where used: 1448/1449 (sigD), 1450/1451 (sigA), 1560/1561 (flgB), 1562/1563 (cheD), 1564/1565 (hag), 1596/1597 (fliF), 1598/1599 (flgE), 1602/1603 (flhA), 1604/1605 (cheB), 1652/1653 (fliI), 1654/1655 (fliK), 1656/1657 (fliY), 1658/1659 (fliR), 1662/1663 (cheW), and 3758/3759 (fliP)
Supporting Information
Zdroje
1. Macnab RM (2003) How bacteria assemble flagella. Annu Rev Microbiol 57 : 77–100. 12730325
2. Chevance FFV, Hughes KT (2008) Coordinating assembly of a bacterial macromolecular machine. Nat Rev Microbiol 6 : 455–465. doi: 10.1038/nrmicro1887 18483484
3. Mukherjee S, Kearns DB (2015) The structure and regulation of flagella in Bacillus subtilis. Annu. Rev. Genet. 48 : 319–340.
4. Ueno T, Oosawa K, Aizawa S-I (1992) M ring, S ring and proximal rod of the flagellar basal body of Salmonella typhimurium are composed of subunits of a single protein, FliF. J Mol Biol 227 : 672–677. 1404383
5. Fan F, Ohnishi K, Francis NR, Macnab RM. (1997) The FliP and FliR proteins of Salmonella typhimurium, putative components of the type III flagellar export apparatus, are located in the flagellar basal body. Mol. Microbiol. 26 : 1035–1046. 9426140
6. Minamino T, Macnab RM (1999) Components of the Salmonella flagellar export apparatus and classification of export substrates. J Bacteriol 181 : 1388–1394. 10049367
7. Li H, Sourjik V (2011) Assembly and stability of flagellar motor in Escherichia coli. Mol Microbiol 80 : 886–899. doi: 10.1111/j.1365-2958.2011.07557.x 21244534
8. Blair DF (2003) Flagellar movement driven by proton translocation. FEBS Lett 545 : 86–95. 12788496
9. Paul K, Brunstetter D, Titen S, Blair DF (2011) A molecular mechanism of direction switching in the flagellar motor of Escherichia coli. Proc Natl Acad Sci USA 108 : 17171–17176. doi: 10.1073/pnas.1110111108 21969567
10. Abby SS, Rocha EPC (2012) The non-flagellar type III secretion system evolved from the bacterial flagellum and diversified into host-cell adapted systems. PLoS Genet 8:e1002983. doi: 10.1371/journal.pgen.1002983 23028376
11. Erhardt M, Namba K, Hughes KT (2010) Bacterial nanomachines: the flagellum and type III injectisome. Cold Spring Harb Perspect Biol 2:a000299. doi: 10.1101/cshperspect.a000299 20926516
12. Wagner S, Königsmaier L, Lara-Tejero M, Lefebre M, Marlovits TC, Galán JE (2010) Organization and coordinated assembly of the type III secretion export apparatus. Proc Natl Acad Sci USA 107 : 17745–17750. doi: 10.1073/pnas.1008053107 20876096
13. Diepold A, Amstutz M, Abel S, Sorg I, Jenal U, Cornelis GR (2010) Deciphering the assembly of the Yersinia type III secretion injectisome. EMBO J 29 : 1928–1940. doi: 10.1038/emboj.2010.84 20453832
14. Pallen MJ, Penn CW, Chaudhuri RR (2005) Bacterial flagellar diversity in the post-genomic era. Trends Microbiol 13 : 143–149. 15817382
15. Minamino T, Macnab RM (2000) Domain structure of Salmonella FlhB, a flagellar export component responsible for substrate specificity switching. J Bacteriol 182 : 4906–4914. 10940035
16. Fraser GM, Hirano T, Ferris HU, Devgan LL, Kihara M, Macnab RM (2003) Substrate specificity of type III flagellar protein is Salmonella is controlled by subdomain interactions in FlhB. Mol Microbiol 48 : 1043–1057. 12753195
17. Bange G, Kümmerer N, Engel C, Bozkurt G, Wild K, Sinning I (2010) FlhA provides the adaptor for coordinated delivery of late flagella building blocks to the type III secretion system. Proc Natl Acad Sci USA 107 : 11295–11300. doi: 10.1073/pnas.1001383107 20534509
18. Kinoshita M, Hara N, Imada K, Namba K, Minamino T (2013) Interactions of bacterial flagellar chaperone-substrate complexes with FlhA contribute to co-ordinating assembly of the flagellar filament. Mol Microbiol 90 : 1249–1261. doi: 10.1111/mmi.12430 24325251
19. Minamino T, Namba K (2008) Distinct roles of the FliI ATPase and proton motive force in bacterial flagellar protein export. Nature 451 : 485–488. doi: 10.1038/nature06449 18216858
20. Paul K, Erhardt M, Hirano T, Blair DF, Hughes KT (2008) Energy source of flagellar type III secretion. Nature 451 : 489–492. doi: 10.1038/nature06497 18216859
21. Barker CS, Meshcheryakova IV, Kostyukova AS, Samatey FA (2010) FliO regulation of FliP in the formation of the Salmonella enterica flagellum. PLoS Genet 6:e1001143. doi: 10.1371/journal.pgen.1001143 20941389
22. Barker CS, Meshcheryakova IV, Inoue T, Samatey FA. (2014) Assembling flagella in Salmonella mutant strains producing a type III export apparatus without FliO. J. Bacteriol. 196 : 4001–4011. doi: 10.1128/JB.02184-14 25201947
23. Iyoda S, Kutsukake K (1995) Molecular dissection of the flagellum-specific anti-sigma factor, FlgM, of Salmonella typhimurium. Mol Gene Genet 249 : 417–424.
24. Chilcott GS, Hughes KT (1998) The type III secretion determinants of the flagellar anti-transcription factor, FlgM, extend from the amino terminus in the anti-σ28 domain. Mol Microbiol 30 : 1029–1040. 9988479
25. Hirano T, Minamino T, Namba K, Macnab RM (2003) Substrate specificity classes and the recognition signal for Salmonella type III flagellar export. J Bacteriol 185 : 2485–2492. 12670972
26. Auvray F, Thomas J, Fraser GM, Hughes C (2001) Flagellin polymerization control by a cytoplasmic export chaperone. J Mol Biol 308 : 221–229. 11327763
27. Ferris HU, Minamino T (2006) Flipping the switch: bringing order to flagellar assembly. Trends Microbiol 14 : 519–526. 17067800
28. Minamino T, Kinoshita M, Hara N, Takeuchi S, Hida A, Koya S, Glenwright H, Imada K, Aldridge PD, Namba K (2012) Interaction of a bacterial flagellar chaperone FlgN with FlhA is required for efficient export of its cognate substrates. Mol Microbiol 83 : 775–788. doi: 10.1111/j.1365-2958.2011.07964.x 22233518
29. Hughes KT, Gillen KL, Semon MJ, Karlinsey JE (1993) Sensing structural intermediates in bacterial flagellar assembly by export of a negative regulator. Science 262 : 1277–1280. 8235660
30. Kutsukake K (1994) Excretion of the anti-sigma factor through a flagellar substructure couples flagellar gene expression with flagellar assembly in Salmonella typhimurium. Mol Gen Genet 243 : 605–612. 8028576
31. Sockett H, Yamaguchi S, Kihara M, Irikura VM, Macnab RM (1992) Molecular analysis of the flagellar switch protein FliM of Salmonella typhimurium. J Bacteriol 174 : 793–806. 1732214
32. Irikura VM, Kihara M, Yamaguchi S, Sockett H, Macnab RM (1993) Salmonella typhimurium fliG and fliN mutations causing defects in assembly, rotation, and switching of the flagellar motor. J Bacteriol 175 : 802–810. 8423152
33. Lloyd SA, Tang H, Wang X, Billing S, Blair DF (1996) Torque generation in the flagellar motor of Escherichia coli: evidence of a direct role for FliG but not FliM or FliN. J Bacteriol 178 : 223–331. 8550421
34. González-Pedrajo B, Minamino T, Kihara M, Namba K (2006) Interactions between C-ring proteins and export apparatus components: a possible mechanism for facilitating type III protein export. Mol. Microbiol. 60 : 984–998. 16677309
35. McMurray JL, Murphy JW, González-Pedrajo B. (2006) The FliN-FliH interaction mediates localization of flagellar export ATPase FliI to the C ring complex. Biochemistry 45 : 11790–11798. 17002279
36. Paul K, Harmon JG, Blair DF. (2006) Mutational analysis of the flagellar rotor protein FliN: identification of surfaces important for flagellar assembly and switching. J. Bacteriol. 188 : 5240–5248. 16816196
37. Minamino T, Yoshimura SD, Morimoto YV, González-Pedrajo B, Kami-Ike N, Namba K. (2009) Roles of the extreme N-terminal region of FliH for efficient localization of the FliH-FliI complex to the bacterial flagellar type III export apparatus. Mol. Microbiol. 74 : 1471–1483. doi: 10.1111/j.1365-2958.2009.06946.x 19889085
38. Kearns DB, Chu F, Rudner R, Losick R (2004) Genes governing swarming in Bacillus subtilis and evidence for a phase variation mechanism controlling surface motility. Mol Microbiol 52 : 357–369. 15066026
39. Werhane H, Lopez P, Mendel M, Zimmer M, Ordal GW, Márquez-Magaña LM (2004) The last gene of the fla/che operon in Bacillus subtilis, ylxL, is required for maximal σD function. J Bacteriol 186 : 4025–4029. 15175317
40. Calvio C, Celandroni F, Ghelardi E, Amati G, Salvetti S, Ceciliani F, Galizzi A, Senesi S (2005) Swarming differentiation and swimming motility in Bacillus subtilis are controlled by swrA, a newly identified dicistronic operon. J Bacteriol 187 : 5356–5366. 16030230
41. Kearns DB, Losick R (2005) Cell population heterogeneity during growth of Bacillus subtilis. Genes Dev 19 : 3083–3094. 16357223
42. Mukherjee S, Bree AC, Liu J, Patrick JE, Chien P, and Kearns DB. (2015) Adaptor-mediated Lon proteolysis restricts Bacillus subtilis hyperflagellation. Proc. Natl. Acad. Sci. USA. 112 : 250–255. doi: 10.1073/pnas.1417419112 25538299
43. Blair KM, Turner L, Winkelman JT, Berg HC, Kearns DB (2008) A molecular clutch disables flagella in the Bacillus subtilis biofilm. Science 320 : 1636–1638. doi: 10.1126/science.1157877 18566286
44. Mirel DB, Chamberlin MJ (1989) The Bacillus subtilis flagellin gene (hag) is transcribed by the σ28 form of RNA polymerase. J Bacteriol 171 : 3095–3101. 2498284
45. Ohnishi K, Kutsukake K, Suzuki H, Iino T (1992) A novel transcriptional regulation mechanism in the flagellar regulon of Salmonella typhimurium: an anti-sigma factor inhibits the activity of the flagellum-specific sigma factor, σF. Mol Microbiol 6 : 3149–3157. 1453955
46. Caramori T, Barillà D, Nessi C, Sacchi L, Galizzi A (1996) Role of FlgM in σD-dependent gene expression in Bacillus subtilis. J Bacteriol 178 : 311–3118.
47. Fredrick K, Helmann JD (1996) FlgM is a primary regulator of σD activity, and its absence restores motility to a sinR mutant. J Bacteriol 178 : 7010–7013. 8955328
48. Bertero MG, Gonzales B, Tarricone C, Ceciliani F, Galizzi A (1999) Overproduction and characterization of the Bacillus subtilis anti-sigma factor FlgM. J Biol Chem 274 : 12103–12107. 10207036
49. Cozy LM, Kearns DB (2010) Gene position in a long operon governs motility development in Bacillus subtilis. Mol Microbiol 76 : 273–285. doi: 10.1111/j.1365-2958.2010.07112.x 20233303
50. Cozy LM, Phillips AM, Calvo RA, Bate AR, Hsueh Y-H, Bonneau R, Eichenberger P, Kearns DB (2012) SlrA/SinR/SlrR inhibits motility gene expression upstream of a hypersensitive and hysteretic switch at the level of σD in Bacillus subtilis. Mol Microbiol 83 : 1210–1228. doi: 10.1111/j.1365-2958.2012.08003.x 22329926
51. Courtney CR, Cozy LM, Kearns DB (2012) Molecular characterization of the flagellar hook in Bacillus subtilis. J Bacteriol 194 : 4619–4629. doi: 10.1128/JB.00444-12 22730131
52. Calvo R, and Kearns DB. (2015) FlgM is secreted by the flagellar export apparatus in Bacillus subtilis. J. Bacteriol. 197 : 81–91. doi: 10.1128/JB.02324-14 25313396
53. Guttenplan SB, Shaw S, Kearns DB (2013) The cell biology of peritrichous flagella in Bacillus subtilis. Mol Microbiol 87 : 211–229. doi: 10.1111/mmi.12103 23190039
54. Amati G, Bisicchia P, Galizzi A (2004) DegU-P represses expression of the motility fla-che operon in Bacillus subtilis. J Bacteriol 186 : 6003–6014. 15342569
55. Márquez-Magaña LM, Chamberlin MJ (1994) Characterization of the sigD transcriptional unit of Bacillus subtilis. J Bacteriol 176 : 2427–2434. 8157612
56. West JT, Estacio W, Marquez-Magana L (2000) Relative roles of the fla/che PA, PD-3, and PsigD promoters in regulating motility and sigD expression in Bacillus subtilis. J Bacteriol 182 : 4841–4848. 10940026
57. Tsukahara K, Ogura M. (2008) Promoter selectivity of the Bacillus subtilis response regulator DegU, a positive regulator of the fla/che operon and sacB. BMC Microbiol. 8 : 8. doi: 10.1186/1471-2180-8-8 18197985
58. Ogura M, Tsukahara K (2012) SwrA regulates assembly of Bacillus subtilis DegU via its interaction with N-terminal domain of DegU. J Biochem 6 : 643–655.
59. Mordini S, Osera C, Marini S, Scavone F, Bellazzi R, Galizzi A, Calvio C (2013) The role of SwrA, DegU and PD3 in fla/che expression in B. subtilis. PLoS One 8:e85065/ doi: 10.1371/journal.pone.0085065 24386445
60. Willimsky G, Bang H, Fischer G, Marahiel MA (1992) Characterization of cspB, a Bacillus subtilis inducible cold shock gene affecting cell viability at low temperatures. J Bacteriol 174 : 6326–6335. 1400185
61. Graumann P, Marahiel MA (1994) The major cold shock protein of Bacillus subtilis CspB binds with high affinity to the ATTGG - and CCAAT sequences in single stranded oligonucleotides. FEBS Lett 338 : 157–160. 8307174
62. Vellanoweth RL, Rabinowitz JC (1992) The influence of ribosome-binding-site elements on translational efficiency in Bacillus subtilis and Escherichia coli in vivo. Mol Microbiol 6 : 1105–1114. 1375309
63. Brown PN, Terrazas M, Paul K, Blair DF (2007) Mutational analysis of the flagellar protein FliG: sites of interaction with FliM and implications for organization of the switch complex. J Bacteriol 189 : 305–312. 17085573
64. Lee LK, Ginsburg MA, Crovace C, Donohoe M, Stock D (2010) Structure of the torque ring of the flagellar motor and the molecular basis for rotational switching. Nature 996–1000.
65. Ohnishi K, Ohto Y, Aizawa S, Macnab RM, Iino T. (1994) FlgD is a scaffolding protein needed for flagellar hook assembly in Salmonella typhimurium. J Bacteriol 176 : 2272–2281. 8157595
66. Jones CJ, and Macnab RM. (1990) Flagellar assembly in Salmonella typhimurium: analysis with temperature-senstive mutants. J. Bacteriol. 172 : 1327–1339. 2407720
67. Morimoto YV, Ito M, Kiraoka KD, Che Y-S, Bai F, Kami-ike N, Namba K, Minamino T. (2014) Assembly and stoichiometry of FliF and FlhA in Salmonella flagellar basal body. Mol. Microbiol. 91 : 1214–1226. doi: 10.1111/mmi.12529 24450479
68. Ohnishi K, Fan F, Schoenhals GJ, Kihara M, Macnab RM. (1997) The FliO, FliP, FliQ, and FliR proteins of Salmonella: putative components for flagellar assembly. J. Bacteriol. 179 : 6092–6099. 9324257
69. Bischoff DS, Weinreich MD, Ordal GW (1992) Nucleotide sequences of Bacillus subtilis flagellar biosynthetic genes fliP and fliQ and identification of a novel flagellar gene, fliZ. J Bacteriol 174 : 4017–4025. 1597417
70. Francis NR, Irikura VM, Yamaguchi S, DeRosier DJ, Macnab RM (1992) Localization of the Salmonella typhimurium flagellar switch protein FliG to the cytoplasmic M-ring face of the basal body. Proc Natl Acad Sci USA 89 : 6304–6308. 1631122
71. Garza AG, Harris-Haller LW, Stoebner RA, Manson MD (1995) Motility protein interactions in the bacterial flagellar motor. Proc Natl Acad Sci USA 92 : 1970–1974. 7892209
72. Marykwas DL, Schmidt SA, Berg HC (1996) Interacting components of the flagellar motor of Escherichia coli revealed by the two-hybrid system in yeast. J Mol Biol 256 : 564–576. 8604139
73. Thomas D, Morgan DG, DeRosier DJ (2001) Structures of bacterial flagellar motors from two FliF-FliG gene fusion mutants. J Bacteriol 183 : 6404–6412. 11591685
74. Levenson R, Zhou H, Dahlquist FW (2012) Structural insights in the interaction between the bacterial flagellar motor proteins FliF and FliG. Biochem 51 : 5052–5060.
75. Tang H, Braun TF, Blair DF (1996) Motility protein complexes in the bacterial flagellar motor. J Mol Biol 261 : 209–221. 8757288
76. Kubori T, Shimamoto N, Yamaguchi S, Namba K, Aizawa S-I (1992) Morphological pathway of flagellar assembly in Salmonella typhimurium. J Mol Biol 226 : 433–446. 1640458
77. Kubori T, Yamaguchi S, Aizawa S-I (1997) Assembly of the switch complex onto the MS ring complex of Salmonella typhimurium does not require any other flagellar proteins. J Bacteriol 179 : 813–817. 9006037
78. Suzuki T, Iino T, Horiguchi T, Yamaguchi S (1978) Incomplete flagellar structures in nonflagellate mutants of Salmonella typhimurium. J. Bacteriol. 33 : 904–915.
79. Ueno T, Oosawa K, Aizawa S-I (1994) Domain structures of the MS ring component protein (FliF) of the flagellar basal body of Salmonella typhimurium. J Mol Biol 236 : 546–555. 8107139
80. Katayama E, Shiraishi T, Ooswaw K, Baba N, Aizawa S-I (1996) Geometry of the flagellar motor in the cytoplasmic membrane of Salmonella typhimurium as determined by stereo-photogrammetry of quick-freeze deep-etch replica images. J. Mol. Biol. 255 : 458–475. 8568890
81. Suzuki H, Yonejura K, Murata K, Hirai T, Oosawa K, Namba K (1998) A structural feature in the central channel of the bacterial flagellar FliF ring complex is implicated in type III protein export. J Struct Biol 124 : 104–114. 10049798
82. Minamino T, Yamaguchi S, Macnab RM (2000) Interaction between FliE and FlgB, a proximal rod component of the flagellar basal body of Salmonella. J Bacteriol 182 : 3029–3036. 10809679
83. Zhao X, Zhang K, Boquoi T, Hu B, Motaleb MA, Miller KA, James ME, Charon NW, Manson MD, Norris SJ, Li C, Liu J (2013) Cryoelectron tomography reveals the sequential assembly of bacterial flagella in Borrelia burgdorferi. Proc Natl Acad Sci USA 110 : 14390–14395. doi: 10.1073/pnas.1308306110 23940315
84. Hendrixson DR, DiRita VJ (2003) Transcription of σ54-dependent but not σ28-dependent flagellar genes in Campylobacter jejuni is associated with formation of the flagellar secretory apparatus. Mol Microbiol 50 : 687–702. 14617189
85. Joslin SN, Hendrixson DR (2009) Activation of the Campylobacter jejuni FlgSR two-component system is linked to the flagellar export apparatus. J. Bacteriol. 191 : 2656–2667. doi: 10.1128/JB.01689-08 19201799
86. Boll JM, Hendrixson DR (2013) A regulatory checkpoint during flagellar biogenesis in Campylobacter jejuni initiates signal transduction to activate transcription of flagellar genes. mBio:e00432–13. doi: 10.1128/mBio.00432-13 24003178
87. Ramakrishnan G, Zhao J-L, Newton AN. 1994. Multiple structural proteins are required for both transcriptional activation and negative autoregulation of Caulobacter crescentus flagellar genes. J. Bacteriol. 176 : 7587–7600. 8002583
88. Mohr CD, MacKichan JK, Shapiro L. 1998. A membrane-associated protein, FliX, is required for an early step in Caulobacter flagellar assembly. J. Bacteriol. 180 : 2175–2185. 9555902
89. Boyd CH, Gober JW. 2001. Temporal regulation of genes encoding the flagellar proximal rod in Caulobacter crescentus. J. Bacteriol. 183 : 725–735. 11133968
90. Muir RE, O’Brien TM, Gober JW. 2001. The Caulobacter crescentus flagellar gene, fliX, encodes a novel trans-acting factor that couples flagellar assembly to transcription. Mol. Microbiol. 39 : 1623–1637. 11260478
91. Dasgupta N, Wolfgang MC, Goodman AL, Arora SK, Jyot J, Lory S, Ramphal R. 2003. A four-tiered transcriptional regulatory circuit controls flagellar biogenesis in Pseudomonas aeruginosa. Mol. Microbiol. 50 : 809–824. 14617143
92. Kenjale R, Wilson J, Zenk SF, Saurya S, Picking WL, Picking WD, Blocker A (2005) The needle component of the type III secretion of Shigella regulates the activity of the secretion apparatus. J Biol Chem 280 : 42929–42937. 16227202
93. Deane JE, Roversi P, Cordes FS, Johnson S, Kenjale R, Daniell S, Booy F, Picking WD, Picking WL, Blocker AJ, Lea SM (2006) Molecular model of a type III secretion system needle: implications for host-cell sensing. Proc Natl Acad Sci USA 103 : 12529–12533. 16888041
94. Diepold A, Wiesand U, Amstutz M, Cornelis GR (2012) Assembly of the Yersinia injectisome: the missing pieces. Mol Microbiol 85 : 878–892. doi: 10.1111/j.1365-2958.2012.08146.x 22788867
95. Yasbin RE, Young FE (1974) Transduction in Bacillus subtilis by bacteriophage SPP1. J Virol 14 : 1343–1348. 4214946
96. Konkol MA, Blair KM, Kearns DB (2013) Plasmid-encoded ComI inhibits competence in the ancestral 3610 strain of Bacillus subtilis. J Bacteriol 195 : 4085–4093. doi: 10.1128/JB.00696-13 23836866
97. Guérout-Fleury A-M, Frandsen N, Stragier P (1996) Plasmids for ectopic integration in Bacillus subtilis. Gene 180 : 57–61. 8973347
98. Patrick JE, Kearns DB (2008) MinJ (YvjD) is a topological determinant of cell division in Bacillus subtilis. Mol Microbiol 70 : 1166–1179. doi: 10.1111/j.1365-2958.2008.06469.x 18976281
99. Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchinson CA III, Smith HO (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Meth 6 : 343–345.
100. Ben-Yehuda S, Rudner DZ, Losick R (2003) RacA, a bacterial protein that anchors chromosomes to the cell poles. Science 299 : 532–536. 12493822
101. Kearns DB, Losick R (2003) Swarming motility in undomesticated Bacillus subtilis. Mol. Microbiol. 49 : 581–590. 12864845
102. Chan JM, Guttenplan SB, Kearns DB (2014) Defects in the flagellar motor increase synthesis of poly-γ-glutamate in Bacillus subtilis. J. Bacteriol. 196 : 740–753. doi: 10.1128/JB.01217-13 24296669
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 8
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Exon 7 Contributes to the Stable Localization of Xist RNA on the Inactive X-Chromosome
- YAP1 Exerts Its Transcriptional Control via TEAD-Mediated Activation of Enhancers
- SmD1 Modulates the miRNA Pathway Independently of Its Pre-mRNA Splicing Function
- TSPO, a Mitochondrial Outer Membrane Protein, Controls Ethanol-Related Behaviors in
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy