
Mouse Genome-Wide Association and Systems Genetics Identify As a Regulator of Bone Mineral Density and Osteoclastogenesis
Significant advances have been made in the discovery of genes affecting bone mineral density (BMD); however, our understanding of its genetic basis remains incomplete. In the current study, genome-wide association (GWA) and co-expression network analysis were used in the recently described Hybrid Mouse Diversity Panel (HMDP) to identify and functionally characterize novel BMD genes. In the HMDP, a GWA of total body, spinal, and femoral BMD revealed four significant associations (−log10P>5.39) affecting at least one BMD trait on chromosomes (Chrs.) 7, 11, 12, and 17. The associations implicated a total of 163 genes with each association harboring between 14 and 112 genes. This list was reduced to 26 functional candidates by identifying those genes that were regulated by local eQTL in bone or harbored potentially functional non-synonymous (NS) SNPs. This analysis revealed that the most significant BMD SNP on Chr. 12 was a NS SNP in the additional sex combs like-2 (Asxl2) gene that was predicted to be functional. The involvement of Asxl2 in the regulation of bone mass was confirmed by the observation that Asxl2 knockout mice had reduced BMD. To begin to unravel the mechanism through which Asxl2 influenced BMD, a gene co-expression network was created using cortical bone gene expression microarray data from the HMDP strains. Asxl2 was identified as a member of a co-expression module enriched for genes involved in the differentiation of myeloid cells. In bone, osteoclasts are bone-resorbing cells of myeloid origin, suggesting that Asxl2 may play a role in osteoclast differentiation. In agreement, the knockdown of Asxl2 in bone marrow macrophages impaired their ability to form osteoclasts. This study identifies a new regulator of BMD and osteoclastogenesis and highlights the power of GWA and systems genetics in the mouse for dissecting complex genetic traits.
Published in the journal:
. PLoS Genet 7(4): e32767. doi:10.1371/journal.pgen.1002038
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002038
Summary
Significant advances have been made in the discovery of genes affecting bone mineral density (BMD); however, our understanding of its genetic basis remains incomplete. In the current study, genome-wide association (GWA) and co-expression network analysis were used in the recently described Hybrid Mouse Diversity Panel (HMDP) to identify and functionally characterize novel BMD genes. In the HMDP, a GWA of total body, spinal, and femoral BMD revealed four significant associations (−log10P>5.39) affecting at least one BMD trait on chromosomes (Chrs.) 7, 11, 12, and 17. The associations implicated a total of 163 genes with each association harboring between 14 and 112 genes. This list was reduced to 26 functional candidates by identifying those genes that were regulated by local eQTL in bone or harbored potentially functional non-synonymous (NS) SNPs. This analysis revealed that the most significant BMD SNP on Chr. 12 was a NS SNP in the additional sex combs like-2 (Asxl2) gene that was predicted to be functional. The involvement of Asxl2 in the regulation of bone mass was confirmed by the observation that Asxl2 knockout mice had reduced BMD. To begin to unravel the mechanism through which Asxl2 influenced BMD, a gene co-expression network was created using cortical bone gene expression microarray data from the HMDP strains. Asxl2 was identified as a member of a co-expression module enriched for genes involved in the differentiation of myeloid cells. In bone, osteoclasts are bone-resorbing cells of myeloid origin, suggesting that Asxl2 may play a role in osteoclast differentiation. In agreement, the knockdown of Asxl2 in bone marrow macrophages impaired their ability to form osteoclasts. This study identifies a new regulator of BMD and osteoclastogenesis and highlights the power of GWA and systems genetics in the mouse for dissecting complex genetic traits.
Introduction
Osteoporosis is a common disease characterized by bone fragility and an increased risk of fracture [1]. One of strongest predictors of fracture is low bone mineral density (BMD) [2] and while BMD is influenced by both genetic and environmental factors, most (between 60% and 80%) of its variance is heritable [3]. Thus, the identification of novel BMD genes is critical for the discovery of new pathways and gene networks that will advance our understanding of basic bone biology and identify new therapeutic targets with the potential to combat osteoporosis.
Due in large part to its many advantages, such as the ability to experimentally cross genetically defined strains and perturb candidate genes, the mouse has played an instrumental role in the genetic analysis of BMD [4]. However, progress has been limited by low-resolution linkage-based quantitative trait loci (QTL) mapping approaches and the difficulties inherent to QTL cloning [5]. As a result, mouse linkage approaches have lead to the identification of only three BMD quantitative trait genes, Alox12 [6], Sfrp4 [7] and Darc [8], even though hundreds of QTL have been mapped [9]. In part due to the success of genome-wide association (GWA) in humans, several groups have investigated the prospects of high-resolution association mapping approaches in the mouse. One of the main challenges facing GWA in the mouse has been determining the most ideal population for such analyses. To date, mouse GWA studies have been performed with varying success using small sets of classical laboratory strains [10], advanced intercross lines [11], heterogeneous stocks [12], outbred mice [13], [14] and the Hybrid Mouse Diversity Panel (HMDP) [15]. One of the most promising is the HMDP, a collection of ∼100 classical laboratory and recombinant inbred (RI) strains that have been genotyped at ∼135,000 SNPs [15]. The primary advantage of the HMDP is that mapping resolution is over an order of magnitude higher than with linkage. We have recently demonstrated through simulations that associations explaining 5% of the phenotypic variance in a trait have 95% confidence intervals (CIs) of ∼2.6 megabases (Mb) [15]. This is in comparison to CIs for mouse linkage studies that are typically in the range of 40–60 Mb [13]. Additionally, statistical power in the HDMP has been found to be adequate to map variants affecting complex traits [15]. Moreover, phenotypes can be mapped in the HMDP without the need for costly genotyping and one can collect multiple specimens (e.g. tissues, individual cell-types, etc.) from strains for molecular profiling that are difficult or impossible to collect from a single mouse [15].
A drawback of both mouse and human GWA studies (and all “genotype to phenotype” mapping approaches) is their inability to provide information on how associated genes actually influence disease [16]. In many cases, it takes years to decipher the underlying biology of novel gene discoveries. One way to begin to provide functional information is through the use of systems genetics [17]. Systems genetics is an approach that incorporates molecular phenotypes, most commonly gene expression microarray profiles, into the genetic analysis of clinical phenotypes. One way that systems genetics can be used to functionally annotate genes of unknown function is through the generation of gene co-expression networks. Co-expression networks are created by clustering genes based on patterns of co-expression across a series of perturbations, such as the differing genetic backgrounds in the HMDP [18]. Co-expressed gene clusters or “modules” have been shown to be enriched for genes involved in the same general function [19], [20], [21], allowing one to annotate genes through “guilt-by-association” [22]. For example, if an uncharacterized gene is co-expressed with genes known to be involved in a biological process such as “apoptosis”, then it is more likely than not the unknown gene is also involved in “apoptosis”. The use of network analysis of systems genetic data can help to inform GWA discoveries by providing clues as to a gene's function in a physiologically-relevant context.
The goal of the current study was to identify and functionally characterize novel BMD genes using GWA and systems genetics in the HMDP. This approach implicated additional sex combs like-2 (Asxl2) as the gene responsible for a BMD association on chromosome (Chr.) 12. This was further strengthened by the observation that Asxl2 knockout mice had reduced BMD. Furthermore, gene co-expression analysis of bone transcriptomic data predicted that Asxl2 was involved in the differentiation of bone-resorbing osteoclasts. In support of this prediction, osteoclastogenesis was impaired in bone marrow macrophages in which Asxl2 expression was reduced by RNA interference. Together, these data are consistent with the hypothesis that Asxl2 is a novel regulator of BMD and osteoclastogenesis.
Results
Genome-wide association analysis of BMD in the HMDP
A detailed description of the HMDP, including strain selection and evaluation of statistical power and mapping resolution, is provided in [15]. Towards identifying genomic regions associated with BMD, we phenotyped 16-week old male mice (N = 879) from 97 HMDP strains (N = 9.1 mice/strain) for total body (TBMD), lumbar spine (SBMD) and femur (FBMD) areal BMD (Table S1). A wide range of BMD values were observed across the HMDP with differences of 1.4, 1.6 and 1.6-fold between the lowest and highest strains for TBMD, SBMD and FBMD, respectively (Figure 1).

To identify associations for the three BMD phenotypes we used the Efficient Mixed-Model Association (EMMA) algorithm [23]. Adjusted association P-values were calculated for 108,064 SNPs with minor allele frequencies >5%. We have previously demonstrated that the P<0.05 genome-wide equivalent for GWA using EMMA in the HMDP is P = 4.1×10−6 (−log10P = 5.39) [23]. At this threshold, associations on chromosomes (Chrs.) 7, 12 and 17 were identified influencing TBMD (Figure 2A). A fourth unique association on Chr. 11 was identified for SBMD (Figure 2B) and the only significant locus for FBMD was the Chr. 7 association also affecting TBMD (Figure 2C). The details of each association are provided in Table 1. Importantly, each of these regions overlap with the location of QTL for areal BMD measures previously identified by linkage in F2 crosses [9].


Characterization of BMD associations
We have previously shown that the 95% confidence interval (CI) for the distribution of distances between the most significant and true causal SNPs, for simulated associations that explain 5% of the variance in the HMDP, is ∼2.6 Mb [24]. Therefore, we used this interval to conservatively define the boundaries of the four associations (Table 1). Within each association there were a total of 112 (Chr. 7), 14 (Chr. 11), 18 (Chr. 12) and 19 (Chr. 17) unique RefSeq genes (a full list of genes is provided in Table S2). We next identified those genes possessing functional alterations that might underlie the associations. Genes were selected based on whether they were regulated by a local expression QTL (eQTL) in the HMDP or if they harbored a non-synonymous (NS) SNP that was predicted to have functional consequences. For the eQTL analysis, we generated gene expression microarray profiles using RNA isolated from cortical bone in 95 of the 97 HMDP strains (N = 1–3/arrays per strain). EMMA was then used to perform an association analysis between all SNPs and array probes mapping within each region. A total of 74 genes were represented by at least one probe, after excluding probes that overlapped SNPs present among the classical inbred strains used in the HMDP (see Methods). Of these, 11 genes (8 within the Chr. 7 association and 3 within the Chr. 17 association) were identified with at least one probe whose expression was regulated by a significant (P≤5.1×10−4; Bonferroni corrected for the number of probes tested) local eQTL (Table 2 and data for all genes is provided in Table S2). In addition, we identified a total of 19 NS SNPs in 14 genes that were predicted to be either “Possibly Damaging” or “Probably Damaging” by PolyPhen [25], [26] (Table 3 and a list of all NS SNPs is provided in Table S3). A nonsense SNP was also identified in the meprin A, alpha (Mep1a) gene (Table 3). Therefore, of the 163 positional candidate genes, 26 were found to be regulated by a local eQTL in bone or harbored potentially functional NS SNPs. The number of functional candidate genes within each association was 12 (Chr. 7), 2 (Chr. 11), 3 (Chr. 12) and 9 (Chr. 17).


We also determined if any of the 163 genes implicated by GWA have been previously implicated in bone development. The associations on Chrs. 11 and 12 did not harbor known bone genes. In contrast, three (Fosb [27], RelB [28] and Apoe [29]) of the Chr. 7 genes have been linked to the regulation of bone mass. All three were located in close proximity to the association peak. Fosb was 124 kilobases (Kb) upstream, Relb was 180 Kb downstream and Apoe was 270 Kb downstream of rs32149600, the most significantly associated SNP on Chr. 7. Additionally, of the 19 Chr. 17 genes, Runx2, the “master regulator” of osteoblast differentiation [30], was located 1.0 Mbp downstream of rs33294019, the most significant SNP. None of the known bone genes were regulated by local bone eQTL or harbored potentially functional NS SNPs.
Detailed analysis of the Chr. 12 association implicates Asxl2 as a regulator of BMD
All 26 of the genes identified above are candidates for the BMD associations and warrant further investigation. However, the goal of this analysis was to identify a gene(s) that was the most likely causal gene for an association. Due to Chr. 7 and Chr. 17 possessing multiple functional candidates (12 and 9, respectively), we could not identify the most likely candidate based on the existing data for either of the associations; therefore, we focused on the Chr. 11 and Chr. 12 associations due to the presence of only 2 and 3 functional candidates, respectively. All five of these genes harbored potentially functional NS SNPs. The expression of these genes was not regulated by local eQTL. Of the five, the additional sex-combs like 2 (Asxl2) was the most compelling candidate due to the fact that rs29131970, a NS SNP in Asxl2 that was predicted to be functional, was also the peak SNP for Chr. 12 BMD association (Table 3 and Figure 3). Rs29131970 results in a phenylalanine (F) to serine (S) substitution at amino acid 1191 of the mouse ASXL2 protein. The mouse reference genome (the C57BL/6J strain) harbors the “C” allele, which codes for S; whereas, the rat, human, orangutan, dog, horse, opossum and chicken reference genomes all have the “T” allele, which codes for F. HMDP strains homozygous for the “C” allele had lower BMD relative to strains with the “T” allele (data not shown). A role for the other four candidates possessing NS SNPs on Chr. 11 and Chr. 12 appear to be less likely because of the modest linkage disequilibrium (LD) (r2 between 0.09 and 0.36) among the HMDP classical inbred strains between these SNPs and the SNPs most significantly associated with BMD for each region (Table 3). Therefore, based on the existing data we hypothesized that Asxl2 was the causal gene for the Chr. 12 association.

Asxl2 knockout mice have reduced BMD
To directly test the hypothesis that Asxl2 was involved in the regulation of BMD we characterized TBMD, SBMD and FBMD in Asxl2 knockout mice (Asxl2−/−). The mice used for this experiment were between the ages of 2–4 months and as previously reported [31] a significant (P<0.05) reduction in body weight in Asxl2−/− mice was observed (data not shown). To evaluate bone mass in the absence of these confounding effects we evaluated the BMD residuals across genotype after adjusting for age and body weight within each sex. This analysis revealed significant (P<0.05) reductions in TBMD, SBMD and FBMD residuals in male Asxl2−/− mice as compared to wild-type (Asxl2+/+) controls (Figure 4A–4C). In addition, male heterozygous (Asxl2+/−) mice demonstrated an intermediate phenotype for all BMD measures, although the differences were not statistically different from either homozygous genotype (Figure 4A–4C). We also observed similar decreases in TBMD, SBMD and FBMD residuals in female Asxl2−/− mice as compared to Asxl2+/+ controls (Figure 4D–4F). These data confirm that Asxl2 is a regulator of BMD and are consistent with the hypothesis that Asxl2 is responsible for the genetic association on Chr. 12 in the HMDP.

Transcriptional network analysis predicts that Asxl2 is involved in osteoclastogenesis
We next used systems genetics to begin to identify a potential function for Asxl2 in bone. For this analysis we utilized the cortical bone gene expression data from the HMDP strains. An analysis of Asxl2 expression revealed that while its expression was not under regulation by detectable local (described above) or distant eQTL (data not shown), Asxl2 was expressed in cortical bone (in the top 10% of probes based on average expression) and most importantly, its expression differed by 1.5-fold between the lowest and highest expressing HMDP strains (Figure S1). We reasoned that its high level of expression and variation in expression among strains would allow us to identify biologically meaningful co-expression relationships between Asxl2 and genes sharing similar functions, even though a difference in its expression does not underlie the Chr. 12 association. To identify the genes that were co-expressed with Asxl2 in bone, genes were grouped into “co-expression modules” using Weighted Gene Co-expression Network Analysis (WGCNA) [20]. We used WGCNA to generate a bone co-expression network comprised of the 3600 most variable and highly connected genes (see Methods). The 3600 genes were subsequently partitioned into eight gene modules (Figure 5A). Of the eight, we focused our attention on the blue module that contained Asxl2 along with 1334 other genes (full list is provided in Table S4). The DAVID knowledge base (http://david.abcc.ncifcrf.gov/) was used to determine if the blue module was enriched for specific gene ontology (GO) categories. We were most interested in identifying enrichments in specific gene functions; thus, we restricted the analysis to the GO biological process and molecular function categories and excluded the cellular component category. DAVID's functional annotation clustering tool was used to identify 14 significant (enrichment score (ES) >3.0; see Methods) gene clusters containing highly related GO terms (Table S5). The top four clusters contained genes involved in: 1) the cell cycle/chromosome/DNA replication/cell division, 2) hematopoiesis/myeloid cell differentiation, 3) ATP binding and 4) chromosome organization/chromatin organization (Table 4). Asxl2 is thought to regulate the function of Polycomb (PcG) and Trithorax (trxG) protein complexes, which are involved in the establishment and maintenance of chromatin [31]. Its membership in a module enriched for genes involved in the GO terms “chromosome” (Bonferroni corrected enrichment P = 3.2×10−7) and “chromatin organization” (Bonferroni corrected enrichment P = 9.2×10−3) is consistent with its known function and suggests that this module is comprised of biologically meaningful co-expression relationships.

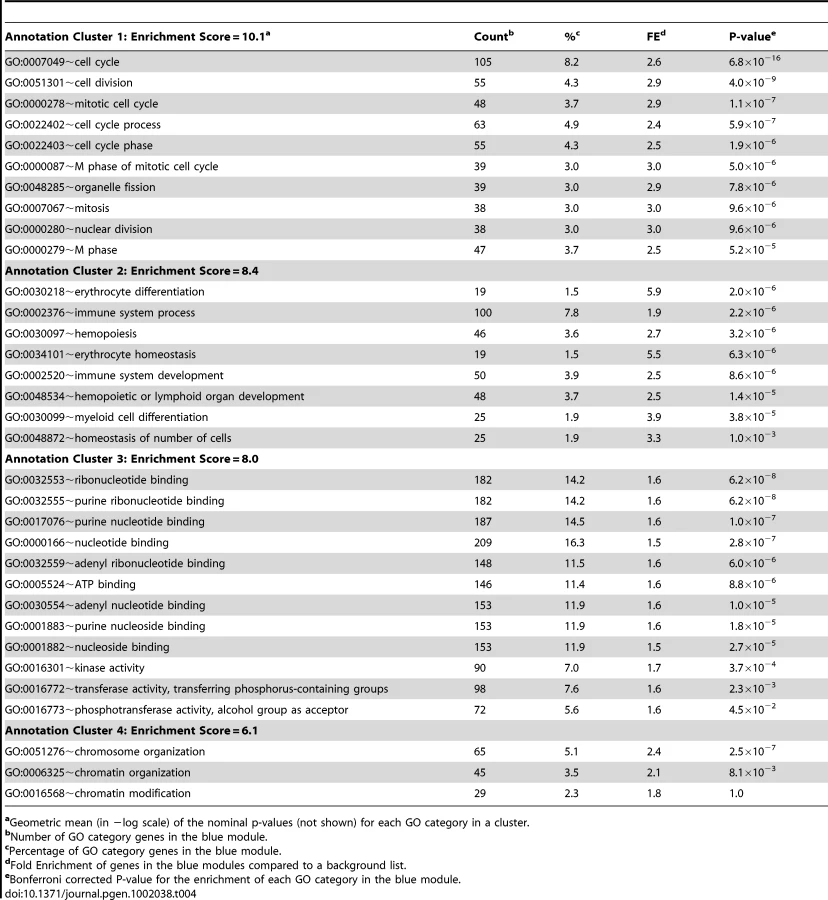
We next characterized the nature of the genes most closely connected to Asxl2 in the blue module. For this purpose, a network view was generated for the 34,690 strongest connections among 1256 (94%) of the 1335 blue module genes (Figure 5B). In this view of the blue module, Asxl2 was connected to a single node, the apoptotic peptidase-activating factor 1 (Apaf1) gene. Apaf1, in turn, was connected to 149 genes; all located within the cluster of genes on the left side of the network depiction Figure 5B. To examine the gene composition of the cluster connected to Asxl2, genes were colored based on their membership in three of the four top enrichments described above, including the “cell cycle”, “myeloid cell differentiation” and “chromatin organization” (Table 4). We excluded the “ATP-binding category” since genes in this category are involved in a wide-range of biological processes. The blue module as a whole was enriched for all three categories (Table 4); however, the cluster of genes most closely connected to Asxl2 contained most (75%; enrichment P = 2.0×10−3) of the genes involved in myeloid cell differentiation (red nodes in Figure 5B) and very few cell cycle (green nodes) or chromatin organization (yellow nodes) genes. These data indicate that in our bone network, Asxl2 is most closely connected with genes involved in myeloid cell differentiation and may play a role in this process.
Asxl2 is a regulator of osteoclastogenesis
The GO category “myeloid cell differentiation” is comprised of genes that are involved in the general process of myeloid precursors acquiring characteristics of downstream cell lineages. With respect to bone cells, osteoclasts are bone-resorbing cells of myeloid origin; thus, we hypothesized that Asxl2 played a role in the differentiation of pre-osteoclasts. Additionally, many of the blue module genes in this category have been implicated in osteoclastogenesis, such as Inpp5d (aka SHIP) [32], Smad5 [33], Id2 [34], Plcg2 [35], Twsg1 [36], among others. To test the hypothesis that Asxl2 was involved in osteoclastogenesis, we infected bone marrow macrophages (BMMs) with lentiviral constructs expressing short-hairpin RNAs (shRNA) targeting Asxl2. BMMs were infected with a vector only control (NC) or one of five lentiviral constructs (A1–A5) expressing distinct shRNAs targeting Asxl2. After five days of culture, Asxl2 expression was particularly lower in cells infected with the A3 and A5 lentiviral constructs (Figure 6A). Osteoclastogenesis was induced in infected BMMs by culturing in the presence of M-CSF and RANKL. After five days of culture the number of TRAP+ (tartrate-resistant acid phosphatase, a marker of osteoclasts) multinuclear cells (MNCs) was significantly (P<0.05) reduced in cultures infected with lentiviral constructs A3 and A5 compared to NC treated cells (Figure 6B and 6C). We observed a strong positive correlation (r = 0.74, P = 0.04) between the relative expression of Asxl2 and TRAP+ MNCs across the six treatments (Figure 6D). These data are consistent with our network inference and confirm that Asxl2 is a regulator of osteoclastogenesis and strengthen the hypothesis that Asxl2 is a regulator of BMD.

Discussion
The mouse has numerous advantages for the genetic analysis of BMD; however, historically mapping approaches in the mouse have been plagued by the lack of resolution. Additionally, GWA approaches provide no information on the function of associated genes. We have addressed both limitations through the use of high-resolution GWA in the HMDP to identify associations confined to narrow genomic intervals and gene co-expression analysis of bone microarray data to provide insight on gene function. This novel analytical paradigm resulted in the discovery of Asxl2 as a regulator of BMD and osteoclastogenesis. This study identified a new gene and possibly an entire network of genes that play an important role in BMD and osteoclast function.
Polycomb (PcG) and trithorax (trxG) are highly conserved protein complexes that are involved in the repression and activation of gene expression, respectively, through the establishment and maintenance of chromatin modifications at specific target genes [37]. With respect to bone cells, PcG and trxG have been implicated in cell proliferation [38], myeloid precursor maturation [39] and osteoblast differentiation [40]. In Drosophila, Additional sex combs (Asx) belongs to a group of proteins known as the Enhancers of trxG and PcG (ETP) [41]. Although its specific mechanism is unknown, Asx is thought to promote both PcG-mediated silencing and trxG-mediated activation of gene expression. In humans and mice there are three Asx homologues, Asx-like 1, 2 and 3 [42], [43], [44]. Mutations in ASXL1 result in myleoproliferative neoplasms [45] and little is known regarding the function of ASXL3. Recently described Asxl2−/− knockout mice display a global reduction in the PcG-associated histone modification trimethylation of histone H3 lysine 27. This is consistent with a conserved function as an ETP in mammals [31]. Additionally, Asxl2−/− mice develop anterior and posterior transformations of the axial skeleton [31], which further supports our observation that Asxl2 is involved in bone development.
An important open question is whether Asxl2 impacts bone development through its expression in other cell-types, such as osteoblasts. If Asxl2 functioned exclusively in osteoclasts, we would expect based on the in vitro osteoclastogenesis data, that loss of Asxl2 function would impair osteoclast function and bone resorption in vivo, resulting in increased BMD. In contrast, we observed a decrease in BMD in Asxl2−/− mice. It has been shown in a number of mouse models that loss of specific genes can lead to an impairment of both osteoblast and osteoclast function. This results in a condition referred to as low-turnover osteopenia in which bone formation by osteoblasts and resorption by osteoclasts are impaired with a net loss of bone. As examples, osteoblasts and osteoclasts from mice deficient in klotho [46], JunB [47] and Akt1 [48] have impaired in vitro differentiation. These mice also have reduced BMD due to low-turnover osteopenia. In addition, Synaptotagmin VII (Syt11) has been shown to alter protein secretion in osteoblasts and osteoclasts, resulting in decreased bone mass in Syt−/− mice [49]. In data not presented we observed (using publically available data from the BioGPS browser; http://biogps.gnf.org, probes 1460597_at and 1439063_at) high and ubiquitous expression of Asxl2 in 96 different mouse tissues and cell-lines, including primary osteoclasts and osteoblasts. In addition, while the blue module was highly enriched for genes involved in myeloid differentiation there were also a number of genes such as Bmp4, Chrd, Hdac5 and Igf2 that play a role in osteoblast differentiation (Table S3). These data suggest that the decreases in BMD seen in Asxl2−/− mice may be due to deficiencies in both osteoclast and osteoblast function. Further work is needed to clarify the precise role of Asxl2 in bone.
We have previously characterized the HMDP as a novel population for GWA in the mouse [15]. This study extends our original observations by demonstrating the feasibility of identifying associations affecting additional complex phenotypes. In contrast to more traditional mouse linkage mapping strategies, we used association in the HMDP to identify four associations containing a relatively small number of genes. Based on prior work, the boundaries of the associations were defined as the 2.6 Mb region surrounding the most significant SNP. We expect that these intervals are conservative and would likely be smaller if based on region specific LD patterns, as shown for Chr. 12 (Figure 3). However, defining the associations in this way allowed us to be confident that the regions contained the causal gene(s). This study also highlights the other key advantage of the HMDP; the ability to collect and molecularly profile tissues, such as bone, that are difficult or impossible to collect from a single mouse or in human populations. In the future, the accumulation of many bone-related clinical and molecular phenotypes in the HMDP will enable the large-scale systems-level analyses that should provide significant insight on bone physiology and genetics. Additionally, the HMDP will provide the opportunity to address the genetic basis of extremely important phenotypes, such as bone loss, nanostrucutal properties of bone and bone cell aging that are difficult to address in humans. Moreover, as we have used systems genetic to gain functional insight for a gene identified by mouse GWA, it is also possible that the HMDP could be used to dissect the function of genes identified in human GWA studies.
However, the HMDP is not without limitations. First, the statistical power to detect effects of subtle variants is modest. We have previously estimated that for highly heritable phenotypes, such as BMD, the power to detect variants explaining 10% of the trait variance is 50% [15]. The power drops precipitously for variants explaining less than 10% of the variance. Thus, this version of the HMDP (with ∼100 strains) is unable to identify the many more variants with subtle effects that are undoubtedly affecting BMD in this population. This problem will be less of an issue for future HMDP panels containing a larger number of strains. Additionally, one of the side effects of the breeding history of inbred mouse strains is the presence of LD between markers on separate chromosomes (i.e. non-syntenic LD). It is thought that this is due to selection for allelic combinations that confer increased fitness during the inbreeding process [50]. False-positive associations can arise if a region associated with a phenotype is in LD with other regions of the genome. Although this is always a potential pitfall when using the HMDP, it is easily identifiable and we did not observe LD (r2<0.4) between any of the four BMD associations (data not shown).
Most GWA studies stop at gene discovery. However, without physiologically relevant functional information it is often difficult to be confident that the true causal gene has been identified and to begin unraveling the mechanistic underpinnings of significant associations. Recently, some GWA studies have begun to incorporate gene expression information to determine if a significant variant regulates expression. A positive finding in such an analysis suggests that the genotype dependent differential gene expression is the basis of the association. This approach has recently been used to discover that variants in the promoter of the serine racemase (SRR) gene regulate its expression and BMD [51]. We have taken that application one step further and developed a bone transcriptional network to aid in the functional annotation of genes of unknown function. Using this network, we were able to determine that Asxl2 was closely connected to genes with links to osteoclast differentiation. This simple connection allowed us to test the hypothesis that Asxl2 was involved in a bone specific function. Another important point is that, as we demonstrated for Asxl2, a gene's expression does not have to be under genetic regulation for this approach to work.
In conclusion, we have used mouse GWA and gene co-expression network analysis to identify Asxl2 as a novel regulator of BMD and osteoclastogenesis. Our analysis has revealed a new gene and pathway that play an important role in bone development. Additionally, this study demonstrates the feasibility of using the HMDP for the dissection of complex genetic traits.
Materials and Methods
Ethics statement
The animal protocol for the HMDP mice was approved by the Institutional Care and Use Committee (IACUC) at the University of California, Los Angeles. The animal protocol for the Asxl2−/− mice was approved by the Animal Care Committee (ACC) at the University of Illinois at Chicago.
The Hybrid Mouse Diversity Panel
Approximately nine male mice for each HMDP strain (Table S1) were purchased from the Jackson Labs (Bar Harbor, ME). Mice were between 6 and 10 weeks of age and to ensure adequate acclimatization to a common environment the mice were aged until 16 weeks before sacrifice. All mice were maintained on a chow diet (Ralston-Purina Co, St. Louis, Mo) until sacrifice.
Genotyping
Inbred strains were previously genotyped by the Broad Institute (available via www.mousehapmap.org). Genotypes of recombinant inbred strains were imputed as previously described [15]. Of the 140,000 SNPs available at the Broad Institute, 108,064 were informative with an allele frequency ≥5% and less than 20% missing data, and were used for the association analysis.
BMD determination
All carcasses were stored at −20°C after sacrifice and then thawed overnight at 4°C prior to BMD scans. The entire thawed carcass was scanned. BMD scans were performed using a Lunar PIXImus II Densitometer (GE Healthcare, Piscataway, NJ). The PIXImus II was calibrated daily using a phantom of known BMD. BMD was calculated for the entire carcass minus the skull, the lumbar spine and the left femur.
RNA and microarray processing
At sacrifice the diaphysis of the right femur was excised and cleaned free of soft tissue. Bone marrow was removed by flushing with PBS using a 22-guage needle and 3 ml syringe. The bone was then flash frozen in LN2 and stored at −80C. Total RNA was isolated using the Trizol Plus RNA Purification Kit (Invitrogen, Carlsbad, CA) following homogenization of the whole bone sample. RNA integrity was confirmed using the Agilent 2100 Bioanalyzer (Agilent, Palo Alto, CA). Microarray expression profiles were generated (N = 1–3 per strain) using the Illumina MouseWG-6 v1.1 BeadChips (Illumina, San Diego, CA) by the Southern Genotyping Consortium at UCLA. Biotin-labeled cRNA was synthesized by the total prep RNA amplification kit from Ambion (Austin, TX). cRNA was quantified and normalized to 77 ng/µl, and then 850 ng was hybridized to Beadchips.
Microarray data processing
The expression values were transformed using the Variance Stabilizing Transformation (VST) [52], and normalized with the Robust Spline Normalization (RSN) algorithm using the LumiR R package [53]. After normalization, the ComBat software was used to adjust for batch effects using an empirical Bayes methods [54]. Microarray data has been submitted the the NCBI Gene Expression Omnibus (GEO) database (GSE27483).
EMMA
Population structure is a major confounding for genome-wide association analyses in the HMDP. This is due to the fact that many phenotypes correlate with the phylogeny of HMDP strains (i.e. genetically similar strains have similar phenotypes) and any SNP that correlates with these strain relationships will be falsely associated with the phenotype. The Efficient Mixed-Model Association (EMMA) algorithm has been shown to effectively reduce this confounding [23]. We applied the following linear mixed model to perform association mapping between BMD or a gene's expression and a marker under the confounding effect from population structure [23], [55], [56]; , where is a phenotypes of each mouse, is their genotype, is an incidence matrix mapping each mouse to corresponding strain, is a random effect accounting for population structure effect with , and is the uncorrelated random effect with . The Efficient Mixed Model Association (EMMA) [23] is used for efficient and reliable estimation of restricted maximum likelihood (REML) parameters and hypothesis testing under the linear mixed model. After estimating REML parameters, a standard F test is used to test the statistical significance of the marker-phenotype association.
Candidate gene characterization
To characterize the genes located in each BMD association, we downloaded all RefSeq genes in the four regions from the USCS genome browser (http://genome.ucsc.edu/cgi-bin/hgGateway) using the NCBI Build37 genome assembly. From the Illumina MouseWG-6 microarray we identified all probes corresponding to the 163 RefSeq genes. Probes were excluded if they overlapped with SNPs (dbSNP 128) to avoid the hybridization artifacts that can arise due [57], [58]. EMMA was used to calculate association P-values for all probes corresponding to the 163 RefSeq genes. Only SNPs mapping to each associated region were used in this analysis. Known non-synonymous SNPs within each region were downloaded from the Mouse Phenome Database (http://phenome.jax.org/) using a set of over 7 million genotyped and imputed SNPs. We only selected SNPs that were variant in at least one of the classical inbred strains represented in the HMDP. Prediction of the functional effect of these SNPs was performed using the PolyPhen tool (http://genetics.bwh.harvard.edu/pph/). R2 was calculated for each non-synonymous SNP and the peak BMD SNP within the 30 classical inbred strains in the HMDP.
Characterizing BMD in Asxl2−/− mice
The generation and initial characterization of Asxl2−/− gene trap mice has been previously described [31]. These mice harbor a gene trap cassette downstream of exon 1. Homozygotes for the gene trap allele show little (∼3%) Asxl2 expression whereas heterozygotes expressed Asxl2 at ∼50% of wild type levels. BMD in littermate male and female mice (2–4 months of age) of varying Asxl2 genotype was measured as described above. Age and weight at sacrifice were recorded. To assess the effects of Asxl2 deficiency on BMD independent of age and weight we generated BMD residuals using a simple linear regression. A Student's t-test was used to test the significance of the differences in BMD residuals in the different genotypes.
Weighted Gene Co-Expression Network Analysis
Network analysis was performed using the WGCNA R package [59]. An extensive overview of WGCNA, including numerous tutorials, can be found at http://www.genetics.ucla.edu/labs/horvath/CoexpressionNetwork/. To begin, we filtered the array data to remove lowly and non-expressed genes by selecting probes based on a detection P-value of <0.05 in 95% of the samples. Next, we selected the 8000 most varying genes based on variance across the 95 samples and then selected the most connected (based on k.total described below) 3600 genes for network analysis. Our group and others have used this number of genes previously (as examples [19], [60]), mainly because most other genes have very low k.total values. If multiple probes existed for a given gene only the most connected probe per gene was included in the list of 3600. To generate a co-expression network for the selected probes, we first calculated Pearson correlation coefficients for all gene-gene comparisons across the 95 microarray samples. The matrix of correlations was then converted to an adjacency matrix of connection strengths. The adjacencies were defined as where and are the and gene expression traits. The power was selected using the scale-free topology criterion previously outlined by Zhang and Horvath [18]. Network connectivity (k.total) of the gene was calculated as the sum of the connection strengths with all other network genes, . This summation performed over all genes in a particular module was defined as the intramodular connectivity (k.in). Modules were defined as sets of genes with high topological overlap [18]. The topological overlap measure (TOM) between the and gene expression traits was taken as , where denotes the number of nodes to which both and are connected, and indexes the nodes of the network. A TOM-based dissimilarity measure was used for hierarchical clustering. Gene modules corresponded to the branches of the resulting dendrogram and were precisely defined using the “Dynamic Hybrid” branch cutting algorithm [61]. Highly similar modules were identified by clustering and merged together. In order to distinguish modules each was assigned a unique color.
Osteoclast culture and lentiviral transduction
Whole bone marrow was extracted from femora of mice with α-MEM and cultured overnight in α-MEM (Sigma-Aldrich, St Louis, MO) containing 10% heat-inactivated FBS, 100 IU/ml penicillin, and 100 µg/ml streptomycin (α10 medium). The nonadherent cells were collected by centrifugation and re-plated in a new 10-cm petri dish in α10 medium. To generate osteoclasts, 100 ng/ml RANKL and 1/100 vol CMG 14-12 culture supernatant were added to α10 medium for 4–5 days. Osteoclasts were stained for TRAP as described by the manufacturer's instructions (Sigma-Aldrich). The SHCLNG-NM_172421 MISSION lentiviral shRNA (Sigma-Aldrich) clone set (containing five separate lentiviral shRNA clones each expressing a distinct Asxl2 targeting sequence) was used to transduce BMMs according to manufacturer's specifications. The MISSION pLKO.1-puro control vector was used as a negative control.
Semi-quantitative RT-PCR
Total RNA was isolated from cultures using the RNeasy Mini Kit (Qiagen). Purified RNA was DNase treated using the DNA-free kit (Ambion). The High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) was used to synthesize cDNA in a volume of 20 µl. The reaction mixture was adjusted to 200 µl with dH2O and 5 µl of the dilute cDNA was used for PCR. PCR was performed for 22 cycles for Asxl2 and Actb using the following primers (Asxl2-F, 5′-ACCCACCATTCCAGCAAGTA-3′, Asxl2-R, 5′-TGGCTGCTTTGACAGTCTTG-3′, Actb-F, 5′-CCAACCGTGAAAAGATGACC-3′, Actb-R, 5′-ACCAGAGGCATACAGGGACA-3′). PCR products were separated on a 1.5% agarose gel containing 0.5 mg/ml ethidium bromide. Band densitometry was performed using the Image J software (NIH). Normalized Asxl2 expression levels were determined by subtracting lane specific backgrounds for each sample and dividing by Actb intensities.
Supporting Information
Zdroje
1. 2001 Osteoporosis prevention, diagnosis, and therapy. JAMA 785 795 2001/02/15 ed
2. JohnellOKanisJAOdenAJohanssonHDe LaetC 2005 Predictive value of BMD for hip and other fractures. J Bone Miner Res 20 1185 1194
3. RalstonSH 2007 Genetics of osteoporosis. Proc Nutr Soc 66 158 165
4. XiongQJiaoYHastyKACanaleSTStuartJM 2009 Quantitative trait loci, genes, and polymorphisms that regulate bone mineral density in mouse. Genomics 93 401 414
5. FlintJValdarWShifmanSMottR 2005 Strategies for mapping and cloning quantitative trait genes in rodents. Nat Rev Genet 6 271 286
6. KleinRFAllardJAvnurZNikolchevaTRotsteinD 2004 Regulation of bone mass in mice by the lipoxygenase gene Alox15. Science 303 229 232
7. NakanishiRShimizuMMoriMAkiyamaHOkudairaS 2006 Secreted frizzled-related protein 4 is a negative regulator of peak BMD in SAMP6 mice. J Bone Miner Res 21 1713 1721
8. EdderkaouiBBaylinkDJBeamerWGWergedalJEPorteR 2007 Identification of mouse Duffy antigen receptor for chemokines (Darc) as a BMD QTL gene. Genome Res 17 577 585
9. Ackert-BicknellCLKarasikDLiQSmithRVHsuYH 2010 Mouse BMD quantitative trait loci show improved concordance with human genome-wide association loci when recalculated on a new, common mouse genetic map. J Bone Miner Res 25 1808 1820
10. LiuPWangYVikisHMaciagAWangD 2006 Candidate lung tumor susceptibility genes identified through whole-genome association analyses in inbred mice. Nat Genet 38 888 895
11. ChengRLimJESamochaKESokoloffGAbneyM 2010 Genome-wide Association Studies and the Problem of Relatedness Among Advanced Intercross Lines and Other Highly Recombinant Populations. Genetics
12. ValdarWSolbergLCGauguierDBurnettSKlenermanP 2006 Genome-wide genetic association of complex traits in heterogeneous stock mice. Nat Genet 38 879 887
13. FarberCRvan NasAGhazalpourAAtenJEDossS 2009 An integrative genetics approach to identify candidate genes regulating BMD: combining linkage, gene expression, and association. J Bone Miner Res 24 105 116
14. GhazalpourADossSKangHFarberCWenPZ 2008 High-resolution mapping of gene expression using association in an outbred mouse stock. PLoS Genet 4 e1000149 doi:10.1371/journal.pgen.1000149
15. BennettBJFarberCROrozcoLKangHMGhazalpourA 2010 A high-resolution association mapping panel for the dissection of complex traits in mice. Genome Res 20 281 290
16. SchadtEE 2009 Molecular networks as sensors and drivers of common human diseases. Nature 461 218 223
17. FarberCRLusisAJ 2009 Future of osteoporosis genetics: enhancing genome-wide association studies. J Bone Miner Res 24 1937 1942
18. ZhangBHorvathS 2005 A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol 4 Article17
19. GhazalpourADossSZhangBWangSPlaisierC 2006 Integrating genetic and network analysis to characterize genes related to mouse weight. PLoS Genet 2 e130 doi:10.1371/journal.pgen.0020130
20. HorvathSZhangBCarlsonMLuKVZhuS 2006 Analysis of oncogenic signaling networks in glioblastoma identifies ASPM as a molecular target. Proc Natl Acad Sci U S A 103 17402 17407
21. OldhamMCKonopkaGIwamotoKLangfelderPKatoT 2008 Functional organization of the transcriptome in human brain. Nat Neurosci 11 1271 1282
22. WolfeCJKohaneISButteAJ 2005 Systematic survey reveals general applicability of “guilt-by-association” within gene coexpression networks. BMC Bioinformatics 6 227
23. KangHMZaitlenNAWadeCMKirbyAHeckermanD 2008 Efficient control of population structure in model organism association mapping. Genetics 178 1709 1723
24. BennettBJFarberCROrozcoLKangHMGhazalpourA A high-resolution association mapping panel for the dissection of complex traits in mice. Genome Res 20 281 290
25. RamenskyVBorkPSunyaevS 2002 Human non-synonymous SNPs: server and survey. Nucleic Acids Res 30 3894 3900
26. SunyaevSRamenskyVKochILatheW3rdKondrashovAS 2001 Prediction of deleterious human alleles. Hum Mol Genet 10 591 597
27. SabatakosGSimsNAChenJAokiKKelzMB 2000 Overexpression of DeltaFosB transcription factor(s) increases bone formation and inhibits adipogenesis. Nat Med 6 985 990
28. SoysaNSAllesNWeihDLovasAMianAH 2009 The Pivotal Role of the Alternative NF-kappaB Pathway in Maintenance of Basal Bone Homeostasis and Osteoclastogenesis. J Bone Miner Res
29. SchillingAFSchinkeTMunchCGebauerMNiemeierA 2005 Increased bone formation in mice lacking apolipoprotein E. J Bone Miner Res 20 274 282
30. DucyPZhangRGeoffroyVRidallALKarsentyG 1997 Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell 89 747 754
31. BaskindHANaLMaQPatelMPGeenenDL 2009 Functional conservation of asxl2, a murine homolog for the Drosophila enhancer of trithorax and polycomb group gene asx. PLoS ONE 4 e4750 doi:10.1371/journal.pone.0004750
32. ZhouPKitauraHTeitelbaumSLKrystalGRossFP 2006 SHIP1 negatively regulates proliferation of osteoclast precursors via Akt-dependent alterations in D-type cyclins and p27. J Immunol 177 8777 8784
33. KanekoHArakawaTManoHKanedaTOgasawaraA 2000 Direct stimulation of osteoclastic bone resorption by bone morphogenetic protein (BMP)-2 and expression of BMP receptors in mature osteoclasts. Bone 27 479 486
34. LeeJKimKKimJHJinHMChoiHK 2006 Id helix-loop-helix proteins negatively regulate TRANCE-mediated osteoclast differentiation. Blood 107 2686 2693
35. MaoDEppleHUthgenanntBNovackDVFaccioR 2006 PLCgamma2 regulates osteoclastogenesis via its interaction with ITAM proteins and GAB2. J Clin Invest 116 2869 2879
36. Sotillo RodriguezJEManskyKCJensenEDCarlsonAESchwarzT 2009 Enhanced osteoclastogenesis causes osteopenia in twisted gastrulation-deficient mice through increased BMP signaling. J Bone Miner Res 24 1917 1926
37. SchuettengruberBChourroutDVervoortMLeblancBCavalliG 2007 Genome regulation by polycomb and trithorax proteins. Cell 128 735 745
38. MartinezAMCavalliG 2006 The role of polycomb group proteins in cell cycle regulation during development. Cell Cycle 5 1189 1197
39. AraiSMiyazakiT 2005 Impaired maturation of myeloid progenitors in mice lacking novel Polycomb group protein MBT-1. Embo J 24 1863 1873
40. ZhangHWDingJJinJLGuoJLiuJN 2010 Defects in mesenchymal stem cell self-renewal and cell fate determination lead to an osteopenic phenotype in Bmi-1 null mice. J Bone Miner Res 25 640 652
41. GildeaJJLopezRShearnA 2000 A screen for new trithorax group genes identified little imaginal discs, the Drosophila melanogaster homologue of human retinoblastoma binding protein 2. Genetics 156 645 663
42. FisherCLRandazzoFHumphriesRKBrockHW 2006 Characterization of Asxl1, a murine homolog of Additional sex combs, and analysis of the Asx-like gene family. Gene 369 109 118
43. KatohMKatohM 2003 Identification and characterization of ASXL2 gene in silico. Int J Oncol 23 845 850
44. KatohMKatohM 2004 Identification and characterization of ASXL3 gene in silico. Int J Oncol 24 1617 1622
45. CarbucciaNMuratiATrouplinVBrecquevilleMAdelaideJ 2009 Mutations of ASXL1 gene in myeloproliferative neoplasms. Leukemia 23 2183 2186
46. KawaguchiHManabeNMiyauraCChikudaHNakamuraK 1999 Independent impairment of osteoblast and osteoclast differentiation in klotho mouse exhibiting low-turnover osteopenia. J Clin Invest 104 229 237
47. KennerLHoebertzABeilTKeonNKarrethF 2004 Mice lacking JunB are osteopenic due to cell-autonomous osteoblast and osteoclast defects. J Cell Biol 164 613 623
48. KawamuraNKugimiyaFOshimaYOhbaSIkedaT 2007 Akt1 in osteoblasts and osteoclasts controls bone remodeling. PLoS ONE 2 e1058 doi:10.1371/journal.pone.0001058
49. ZhaoHItoYChappelJAndrewsNWTeitelbaumSL 2008 Synaptotagmin VII regulates bone remodeling by modulating osteoclast and osteoblast secretion. Dev Cell 14 914 925
50. PetkovPMGraberJHChurchillGADiPetrilloKKingBL 2005 Evidence of a large-scale functional organization of mammalian chromosomes. PLoS Genet 1 e33 doi:10.1371/journal.pgen.0010033
51. GrundbergEKwanTGeBLamKCKokaV 2009 Population genomics in a disease targeted primary cell model. Genome Res 19 1942 1952
52. LinSMDuPHuberWKibbeWA 2008 Model-based variance-stabilizing transformation for Illumina microarray data. Nucleic Acids Res 36 e11
53. DuPKibbeWALinSM 2008 lumi: a pipeline for processing Illumina microarray. Bioinformatics 24 1547 1548
54. JohnsonWELiCRabinovicA 2007 Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8 118 127
55. YuJPressoirGBriggsWHVroh BiIYamasakiM 2006 A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38 203 208
56. ZhaoKAranzanaMJKimSListerCShindoC 2007 An Arabidopsis example of association mapping in structured samples. PLoS Genet 3 e4 doi:10.1371/journal.pgen.0030004
57. ChenLPageGPMehtaTFengRCuiX 2009 Single nucleotide polymorphisms affect both cis - and trans-eQTLs. Genomics 93 501 508
58. AlbertsRTerpstraPLiYBreitlingRNapJP 2007 Sequence polymorphisms cause many false cis eQTLs. PLoS ONE 2 e622 doi:10.1371/journal.pone.0000622
59. LangfelderPHorvathS 2008 WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9 559
60. FarberCR 2010 Identification of a gene module associated with BMD through the integration of network analysis and genome-wide association data. J Bone Miner Res 25 2359 2367
61. LangfelderPZhangBHorvathS 2008 Defining clusters from a hierarchical cluster tree: the Dynamic Tree Cut package for R. Bioinformatics 24 719 720
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 4
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Survival Motor Neuron Protein Regulates Stem Cell Division, Proliferation, and Differentiation in
- PTG Depletion Removes Lafora Bodies and Rescues the Fatal Epilepsy of Lafora Disease
- Evolution of Vertebrate Transient Receptor Potential Vanilloid 3 Channels: Opposite Temperature Sensitivity between Mammals and Western Clawed Frogs
- An Evolutionary Genomic Approach to Identify Genes Involved in Human Birth Timing
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy