
The Cardiac Transcription Network Modulated by Gata4, Mef2a, Nkx2.5, Srf, Histone Modifications, and MicroRNAs
The transcriptome, as the pool of all transcribed elements in a given cell, is regulated by the interaction between different molecular levels, involving epigenetic, transcriptional, and post-transcriptional mechanisms. However, many previous studies investigated each of these levels individually, and little is known about their interdependency. We present a systems biology study integrating mRNA profiles with DNA–binding events of key cardiac transcription factors (Gata4, Mef2a, Nkx2.5, and Srf), activating histone modifications (H3ac, H4ac, H3K4me2, and H3K4me3), and microRNA profiles obtained in wild-type and RNAi–mediated knockdown. Finally, we confirmed conclusions primarily obtained in cardiomyocyte cell culture in a time-course of cardiac maturation in mouse around birth. We provide insights into the combinatorial regulation by cardiac transcription factors and show that they can partially compensate each other's function. Genes regulated by multiple transcription factors are less likely differentially expressed in RNAi knockdown of one respective factor. In addition to the analysis of the individual transcription factors, we found that histone 3 acetylation correlates with Srf - and Gata4-dependent gene expression and is complementarily reduced in cardiac Srf knockdown. Further, we found that altered microRNA expression in Srf knockdown potentially explains up to 45% of indirect mRNA targets. Considering all three levels of regulation, we present an Srf-centered transcription network providing on a single-gene level insights into the regulatory circuits establishing respective mRNA profiles. In summary, we show the combinatorial contribution of four DNA–binding transcription factors in regulating the cardiac transcriptome and provide evidence that histone modifications and microRNAs modulate their functional consequence. This opens a new perspective to understand heart development and the complexity cardiovascular disorders.
Published in the journal:
. PLoS Genet 7(2): e32767. doi:10.1371/journal.pgen.1001313
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001313
Summary
The transcriptome, as the pool of all transcribed elements in a given cell, is regulated by the interaction between different molecular levels, involving epigenetic, transcriptional, and post-transcriptional mechanisms. However, many previous studies investigated each of these levels individually, and little is known about their interdependency. We present a systems biology study integrating mRNA profiles with DNA–binding events of key cardiac transcription factors (Gata4, Mef2a, Nkx2.5, and Srf), activating histone modifications (H3ac, H4ac, H3K4me2, and H3K4me3), and microRNA profiles obtained in wild-type and RNAi–mediated knockdown. Finally, we confirmed conclusions primarily obtained in cardiomyocyte cell culture in a time-course of cardiac maturation in mouse around birth. We provide insights into the combinatorial regulation by cardiac transcription factors and show that they can partially compensate each other's function. Genes regulated by multiple transcription factors are less likely differentially expressed in RNAi knockdown of one respective factor. In addition to the analysis of the individual transcription factors, we found that histone 3 acetylation correlates with Srf - and Gata4-dependent gene expression and is complementarily reduced in cardiac Srf knockdown. Further, we found that altered microRNA expression in Srf knockdown potentially explains up to 45% of indirect mRNA targets. Considering all three levels of regulation, we present an Srf-centered transcription network providing on a single-gene level insights into the regulatory circuits establishing respective mRNA profiles. In summary, we show the combinatorial contribution of four DNA–binding transcription factors in regulating the cardiac transcriptome and provide evidence that histone modifications and microRNAs modulate their functional consequence. This opens a new perspective to understand heart development and the complexity cardiovascular disorders.
Introduction
It is long known that an evolutionary conserved orchestra of transcription factors controls cardiac development and function. More recently the contribution of epigenetic and post-transcriptional mechanisms has been identified. Many successful studies have focused on these different aspects independently, but it is still an open question, how these molecular regulatory mechanisms interact. The ability of transcription factor binding to DNA is highly influenced by the chromatin status and epigenetic mechanisms play an important role in establishing and maintaining transcriptional programs. This layer of control comprises posttranslational modification of histones, DNA methylation and chromatin remodeling. To understand networks directing gene expression, the interplay between different transcription and epigenetic factors has to be considered. Furthermore, recent studies have started to unveil powerful roles for microRNAs (miRNAs) in regulating and fine-tuning mRNA profiles via either translational repression or mRNA degradation. It should be noted that gene expression profiles as obtained by standard microarrays or next-generation sequencing reflect mRNA profiles, which depend on the gene transcription as well as the decay of mRNA. In line with this, we present a study integrating mRNA profiles with transcription factor-DNA interaction data, histone modification marks and posttranscriptional regulation by miRNAs.
The DNA-binding transcription factors Gata4, Mef2a, Nkx2.5 and Srf play pivotal roles for the differentiation, maturation and homeostasis of cardiomyocytes. Mice lacking Gata4 die at E8.5 with failure of ventral morphogenesis and heart tube formation [1], [2]. Targeted disruption of Nkx2.5 leads to abnormal heart morphogenesis with lethality at E9.5 [3]. Mef2a knockout mice die within the first postnatal week and exhibit myofibril fragmentation and impaired myocyte differentiation [4]. Srf-null mice show severe defects in the contractile apparatus of cardiomyocytes and die at the gastrulation stage [5], [6]. However, Gata4, Mef2a, Nkx2.5 and Srf not only display independently a central role for cardiac development and function, they also regulate each other's expression [7]–[10]. Despite their impact, we still have limited understanding of the global cardiac transcription networks driven by these factors in a direct and indirect manner.
Moreover, we lack knowledge to which extent epigenetic marks such as histone modifications interfere with the regulation of downstream targets. The N-terminal histone tails serve as targets for a variety of reversible posttranslational modifications including acetylations or methylations. Both have a main impact on chromatin structure and represent binding sites for transcriptional regulators [11]–[13], thus promoting or inhibiting transcription. They are put in place by specific enzyme families and are removed by others [14], [15]. Hence histone acetylation is a dynamic process and for instance, mice lacking histone deacetylases 5 and 9 (HDAC5 and 9) show cardiac defects typical for abnormalities in growth and maturation of cardiomyocytes [16]. A superactivation of Mef2 based on its interaction with HDACs is proposed [17]. However, our understanding of the underlying molecular mechanisms is still premature.
It was reported that only 5–15% of differentially expressed genes in short interference RNA (siRNA) knockdown experiments are also direct transcription factor targets identified by chromatin immunoprecipitation (ChIP) and vice versa [18]–[23]. There is considerable evidence, that cascades of transcriptional regulators form networks. Loss of one factor will directly affect a few genes, but among those genes are other transcriptional regulators whose function is now altered, affecting a further set of downstream genes. In addition, it has to be considered that a significant proportion of downstream effects mediated by DNA-binding transcription factors is promoted via downstream miRNAs or other regulators. For example Srf regulates the transcription of miRNAs such as the smooth muscle relevant miR-143 and miR-145 [24]. Feedback loops between Srf/Mef2 and muscle-specific miR-133/miR-1 have been described, and both miRNAs are expressed throughout heart development and play important roles in muscle proliferation and differentiation [25]–[28]. Furthermore, miR-1 promotes myogenesis by targeting HDAC4 [26], a transcriptional repressor of muscle gene expression, and thus represents an interface to histone acetylation.
Taken the above, we investigated the transcription network driven by Gata4, Mef2a, Nkx2.5 and Srf in cardiomyocytes in a genome-wide approach. First, we focused on the direct downstream targets by evaluating in-vivo DNA-binding sites of the respective factors and correlated these binding events with the expression level of related genes. Second, we investigated the functional consequence of the proposed regulation in knockdown experiments and built respective transcription networks. Third, we analyzed if co-occurrence with activating histone modifications could impact on gene expression levels of direct targets. Fourth, we studied the modulation of mRNA levels by miRNA alterations seen in knockdown experiments. Finally, we integrated the three levels regulating mRNA profiles and generated a comprehensive transcription network centered on Srf. Based on our analysis we argue that transcription networks have a comparable dependency on transcription factor binding, modulation by histone modifications as well as regulation by miRNAs. In addition to the global perspective, our networks provide distinct information on the regulation of individual genes especially with regard to cardiac function.
Results
We used the cardiomyocyte cell line HL-1 to study the global transcription network driven by the DNA-binding transcription factors Gata4, Mef2a, Nkx2.5 and Srf. The mRNA as well as miRNA expression profiles of beating HL-1 cells are highly comparable to the one observed in mouse hearts at P0.5 (Pearson correlation coefficient of 0.95, Figure 1A left) and human right ventricle (Pearson correlation coefficient of 0.90, Figure 1A right; data unpublished). The use of a cell line enabled us to cope with the technical limitations of our different approaches (ChIP-chip/seq, miRNA-seq, siRNA knockdown, microarrays) and supported a single cell-type specific study. We validated the key findings in a time-course of mouse hearts during the cardiac adaptation and maturation period around birth at E18.5, P0.5 and P4.5.
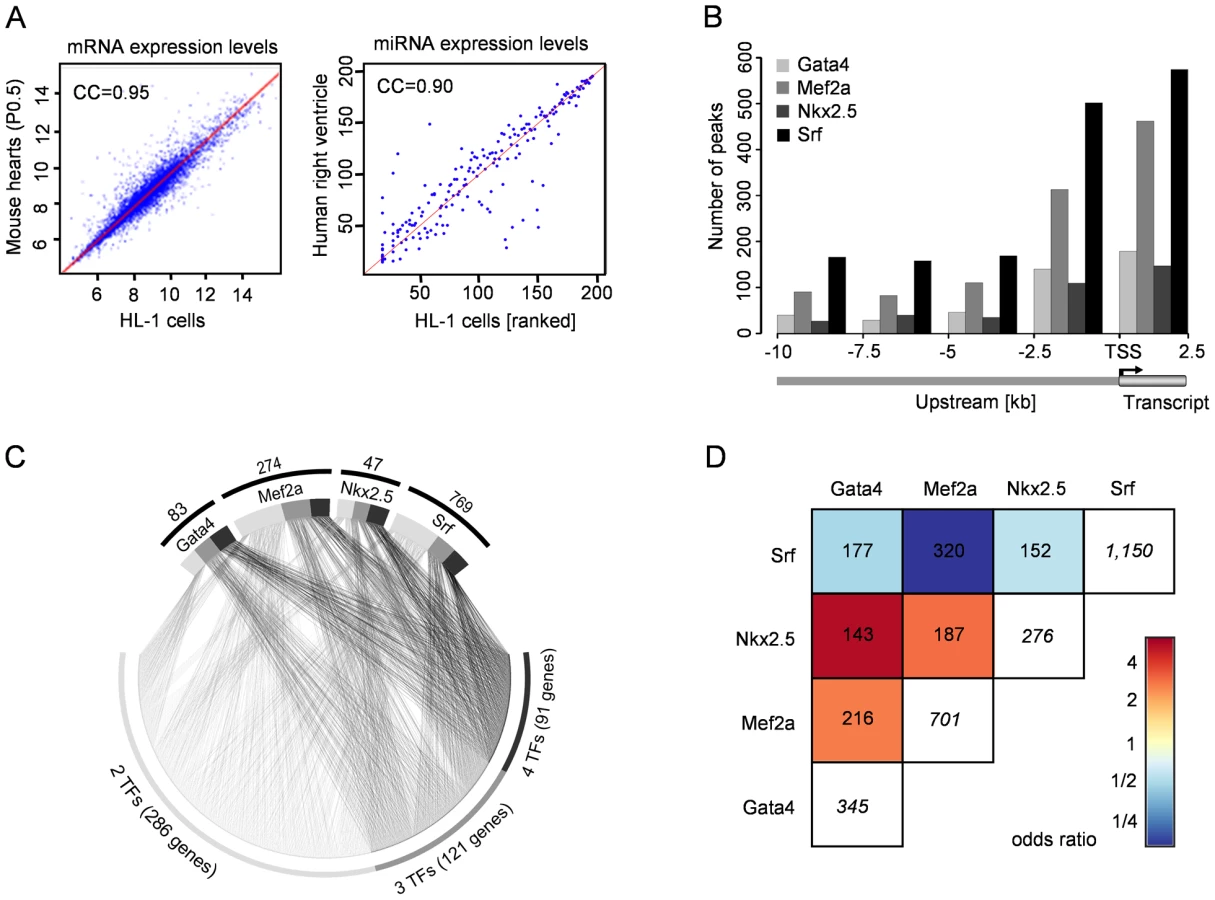
Cardiac Transcription Regulated by Gata4, Mef2a, Nkx2.5, and Srf
We developed a custom two-array set (2×385K) with NimbleGen using a tilling approach for promoter and enhancer regions (10kb upstream), and first exon and intron sequences of 12,625 transcripts (65% of all RefSeq promoters). This enabled the analysis of sequence regions beyond standard promoter arrays. We selected 89Mbp of the mouse genome related to transcripts of 13 data sources (Table S1), which included all known expressed skeletal, smooth and cardiac muscle genes in human and mouse.
Using ChIP-chip analyses we identified several hundreds of transcription factor binding sites (TFBS) for Gata4 (447), Mef2a (999), Nkx2.5 (383) and Srf (1,335) in mouse HL-1 cardiomyocytes, which were related to 345 Gata4, 701 Mef2a, 276 Nkx2.5 and 1,150 Srf target genes (Table S15). Figure 1B shows the distribution of observed binding sites relative to the transcriptional start site (TSS). An average of 24% of TFBS were localized in potential enhancer regions with a distance between 2.5kb to 10kb upstream from any transcriptional start site.
The respective target genes of the studied transcription factors included 42 known targets (Table S2), substantiating the reliability of the system. In addition, we found several genes previously shown to be deregulated in mutants as direct targets of the respective transcription factor. For example, Gata4 and Nkx2.5 levels are decreased in cells depleted of Mef2 [8] and both genes show binding of Mef2a at their promoters in our data.
To gain further insights into the transcription factor functionality, we investigated which Gene Ontology (GO) terms were significantly overrepresented among target genes of each factor when compared to all genes represented on the array. The significant GO terms (p<0.001) show a stronger than expected association with heart development and function and are highly related to the phenotypes reported for the respective transcription factor (Tables S3, S4, S5, S6). For example, the GO terms ‘muscle cell differentiation’ and ‘heart looping’ are significantly overrepresented among Mef2a and Nkx2.5 targets, respectively, and both are key features of corresponding knockout mouse models [3], [4].
We investigated the sequences underlying the transcription factor binding sites in more detail and searched for TRANSFAC [29] motifs within the presumably bound sequences. The TRANSFAC matrices used for motif search are listed in Table S7. For Gata4, Nkx2.5 and Mef2a 84–94% of all ChIP binding events harbored respective binding motifs. For Srf, the fraction of ChIP binding events with predicted motifs was very small (169 out of 1,335 binding sites). However, Srf is well-known to bind the CArG-box CC(A/T)6GG [30], which is only partially represented by TRANSFAC motifs. Using a pattern matching approach we found the CArG-box in 1,063 (approximately 80%) Srf binding events. Furthermore, more than every second binding event of the studied factors occurred at sequence sites containing at least two times the respective transcription factor motif or pattern.
We studied the cross-species conservation of binding motifs and found in ∼10% complete sequence conservation between human and mouse. In 27% the binding sites were localized in regions conserved across 18 vertebrate species based on PhastCons elements [31]. Thus, by focusing only on conserved sequence regions a priori more than two-third of the binding sites would be missed.
Combinatorial Regulation by Multiple Transcription Factors
The investigated transcription factors are known to co-regulate targets and pairwise physical interaction has been described between several of these factors [32], [33]. Nevertheless, it is unknown how frequently this co-binding occurs in-vivo. Consequently, we investigated the assignment of Gata4, Nkx2.5, Mef2a and Srf to the same gene. We observed frequent co-regulation by more than one transcription factor, where Gata4 and Nkx2.5 shared 143 targets (41% and 52%, respectively) and Mef2a and Srf shared 320 target genes (46% and 28%, respectively) (Figure 1C and 1D). For 91 genes co-binding of all four transcription factors was observed and in 85 cases the binding was observed at close proximity within a 500bp window. These data underline the complexity and cooperative regulation of gene regulation shown in our model of four DNA-binding transcription factors.
Functional Consequences of Transcription Factor Binding
We investigated whether the transcription factors act mainly as activators or repressors in a wildtype situation. We carried out genome-wide expression array analysis of the contracting HL-1 cardiomyocytes and classified all transcripts as expressed or non-expressed. We found that for each of the four transcription factors approximately 80% of the target genes were expressed and their expression levels were significantly enhanced compared to non-targets (p<0.005).
Considering the cooperative co-binding of the investigated transcription factors, we were interested in the functional consequence of significantly reducing the quantity of each of the factors. Therefore, we used siRNA technique to reduce the protein levels of investigated transcription factors by more than 70% and studied its consequence for gene transcription. The reduction at mRNA and protein level was monitored by quantitative PCR and Western Blot analysis (Figure S1 and Figure 2A) and the genome-wide effects on transcript levels were measured by expression array analysis (Table S17). All data were based on a total of 4 replicate experiments using duplicates of two different siRNAs per transcription factor. The majority of deregulated transcripts were downregulated in the siRNA treated samples, confirming a primarily activating function of the transcription factors. Performing Annexin assays and Tryptophan Blue staining, we observed an increased apoptosis and cell death in particular when Gata4 or Srf were knocked down (Figure S2), which is in line with previous data [34]–[36].

Analogous to our analyses of common downstream targets based on transcription factor binding events, we studied common differentially expressed genes. The different factors share a comparable proportion of differentially expressed genes when knocked down (Figure 2B). An opposing effect for targets regulated by several transcription factors was only observed in two cases: Myocd and Tpm1. Figure 2C shows the combinatorial regulation of a selection of heart and muscle relevant, directly bound and differentially expressed genes. This includes genes coding for structural proteins like Actc1, Actn2, Tnnt2, Mybpc3 or Myh6; growth factors like Igf1 or apoptosis factors like Casp3. The transcription factor Tbx20 represents an example for a gene that is bound and regulated by all four factors. A broad panel of differentially expressed genes was further confirmed by quantitative real-time PCR (Table S8).
Finally, we compared the differentially expressed genes in siRNA knockdown experiments to the direct target genes identified by ChIP. Analyzing the overlap focusing on the functional role of the respective genes, we found that both datasets share the Gene Ontology terms reflecting heart and muscle development and function. For example, muscle cell differentiation, muscle contraction and heart development are the main functional roles for direct targets of Srf as well as the respective differentially expressed genes. However, only a small fraction of direct target genes (∼10%) was also differentially expressed, pointing to the combinatorial nature of gene regulation. In accordance, we found that genes bound by multiple transcription factors were significantly less likely differentially expressed (χ2-test p<0.001). Likewise transcription factors having a high number of common binding targets share only a small number of co-regulated genes in RNAi knockdown (correlation shown in Figure 2B is inverse to the correlation in Figure 1D). In addition, binding in a poised state or buffering by epigenetic mechanisms such as histone modifications which interfere with the accessibility of the DNA should be considered. It has to be kept in mind that transcription factor binding depends on binding affinity and accessibility of binding sites. The regulatory potential of several factors has been reported to be strongly dosage dependent (e.g. Tbx5 [37] and Gata4 [38]). Furthermore, a significant proportion of differentially expressed genes in RNAi are likely to be regulated in an indirect manner. Recent studies show the powerful roles for miRNAs in controlling mRNA profiles largely by silencing target genes, via either translational repression or mRNA degradation.
Histone 3 Acetylation Correlates with the Activating Potential of Transcription Factors
To explore the influence of histone modifications as an epigenetic mechanism to modulate gene expression, we analyzed our transcription factor binding data in the context of co-occurring histone marks. In a previous study we investigated the localization of four histone modifications, which are known to promote an open chromatin state (H3K9K14ac, H4K5K8K12K16ac, H3K4me2 and H3K4me3) [39]. We found that ∼80% of the respective transcription factor binding events are marked by one or more of these histone modifications, whereas in a randomized simulation only 23% are expected to co-occur (Figure S3). We consequently investigated whether the presence of any of these marks correlates with higher expression levels of direct target genes and found a significant impact for histone 3 acetylation (H3ac) only (Figure 3A and Figure S4). For Nkx2.5 and Mef2a the expression levels of direct targets were significantly higher than the reference group, independent of whether H3ac was present or not. Genes showing neither transcription factor binding nor H3ac were used as a reference. In case of Gata4 and Srf the expression levels of direct targets were only significantly increased when binding sites were additionally marked by H3ac. The enhanced expression levels depending on H3ac co-occurrence is further depicted in Figure 3B, which shows confirmation experiments of nine genes using quantitative PCR. In conclusion, our data provide evidence that acetylation of histone 3 supports the activating function of Gata4 and Srf, which might be mediated via p300. The histone acetyl transferase p300 not only acetylates lysine residues on histone 3 but also on Gata4, thereby enhancing the DNA-binding and activating potential of this transcription factor [40]. The Srf cofactor Myocardin has been reported to recruit p300 to Srf binding sites whereby histone 3 acetylation is induced and gene expression enhanced [41]. Finally, we studied the change of H3ac marks as a consequence of Srf knockdown using ChIP followed by qPCR. Strikingly, we found complementary alterations of H3ac in a panel of relevant promoter regions (Figure 3C and 3D).

Histone 3 Acetylation Correlates with Srf Target Gene Activation
To validate and further investigate the correlation of H3ac with Srf target gene expression, we performed genome-wide ChIP-seq experiments in HL-1 cardiomyocytes (Table S16). We found a synergistic effect of H3ac and Srf binding when compared to non-bound genes or genes solely bound by either of both (Figure 3E). The influence of H3 acetylation marks was further substantiated by RNAi knockdown of Srf in HL-1 cells (Figure 3F). In accordance to its mainly activating function, we found a significant decrease in expression levels of genes bound by Srf. However, this decrease was significantly reduced in genes additionally marked by H3ac in the wild-type.
In a further attempt to confirm our results gathered in cell culture, we studied Srf and H3ac binding and their influence on gene expression in mouse hearts in a time-series during cardiac maturation at three developmental stages E18.5, P0.5 and P4.5 around birth. From the fetal to the postnatal stage, the heart adapts to the body circulation and cardiomyocytes mature. During this process the heart increases in size (Figure 4A), the cells elongate, myofibrils align and cell-cell contacts become bipolar. Immunostaining of α-Actinin-1 and Connexin 43 illustrates hypertrophy of cardiomyocytes, assembly of the sarcomeric z-discs and development of gap junction [42]. Based on ChIP-chip/seq results for Srf and H3ac in HL-1 cells, we analyzed promoter binding regions of genes and miRNAs relevant for this process using ChIP followed by quantitative real-time PCR. The selection comprises (Figure 4B): Dmpk (kinase of myogenin), Slmap (sarcomeric protein), Picalm (clathrin assembly protein), miR-133a (cardiac and muscle-specific miRNA), the growth factor Igf1 and its receptor Igf1r, Pitx2c (cardiac transcription factor), and Nrp2 (interactor of Vegf). We found a high correlation between the changes of Srf and H3ac binding and the gene expression levels over time. In case of Pitx2c and Nrp2 we identified multiple binding events in HL-1 cells by ChIP-chip/seq of which their functionality could be confirmed by common changes over time in the mouse model (Figure 4B). Taken together, these data support an important role for the co-occurrence of Srf and H3ac in the regulation of the cardiac maturation process and underline the influence of histone modifications.

Studying the Impact of miRNAs on the Srf-Driven Transcription Network
Considering that only a small proportion of differentially expressed genes in loss-of-function experiments are direct targets of the respective transcription factors, we were interested in studying the impact of miRNAs as secondary effectors (Figure 5A, 5B). Again we focused on the transcription factor Srf, which is known to regulate cardiac relevant miRNAs like miR-1 and miR-133 [26], [43]. We investigated Srf binding using ChIP-seq technology to map Srf binding sites potentially regulating miRNAs. We found 22 miRNAs from the miRNA database miRBase with Srf binding within a region of 10kb. This includes the previously described miR-208 Srf binding site, as well as other well-known muscle relevant miRNAs like miR-1, miR-125b, miR-133, miR-143 and miR145 (Table S9). Second, we performed Srf knockdown using two different siRNAs and quantified the miRNA expression levels by miRNA-seq (Table S10 and Table S18). We observed 42 miRNAs (49 loci) to be differentially expressed in both siRNA experiments, including miR-208, miR-125b and miR-21. The analysis revealed that most of the miRNAs were downregulated (78%) supporting the role of Srf as an miRNA activator (Figure 5A).

To explore the potential effect of the differentially expressed miRNAs on the Srf network, we assigned confirmed and predicted targets to each miRNA. We found 192 miRNA targets to be also differentially expressed in Srf knockdown, with a higher fraction of upregulated genes (57% of all upregulated genes) compared to downregulated genes (44% of all downregulated genes, Figure 5A). The majority of these dysregulated target genes had 3′UTR target sequences for a panel of our differentially expressed miRNAs (median of 3). The differential expression of miRNAs potentially impacts up to 45% of all differentially expressed genes by Srf knockdown, a higher proportion of genes than expected (Fisher's exact test, p = 1.77e-5), and provides a feasible explanation for the observed consequences on the transcriptional portrait (Figure 5B). A representative example is shown in Figure 5C. It comprises the three genes Igfbp5 (insulin-like growth factor binding protein 5), Nfic (nuclear factor I/C) and Ctnnal1 (catenin alpha-like 1). None of these factors has a direct Srf binding site in ChIP-chip/seq but all are found to be upregulated in the Srf siRNA knockdown experiment. Using miRNA target prediction a number of downregulated miRNAs were found that provide a possible explanation for this indirect regulation (see Table S11).
Confirmation of Novel Transcription Factor Binding Sites
We confirmed a panel of observed transcription factor binding sites by qPCR (Figure S5). Using luciferase reporter gene assays, we validated an Srf binding site in the regulatory region of mouse miR-125b-1 as well as an Nkx2.5 binding element in the core promoter region of human DPF3. Mmu-miR-125b-1 is known to be deregulated in heart diseases [44] and was found to be differentially expressed in Srf siRNA knockdown. Figure 6A shows the Srf binding motif and respective Srf ChIP-seq peak within the regulatory region of miR-125b-1. Luciferase reporter gene assays with wildtype and mutated fusion constructs confirm its functionality. Mutation of the potential Srf binding sequence (CAGCCAAC→CATAGTAC) significantly reduced the transcriptional activity of the reporter gene. DPF3 is a novel epigenetic regulator of heart and skeletal muscle development [13]. Within the 1.2kbp promoter region we found three Mef2 matrices and one Nkx2.5 matrix using TRANSFAC MATCH [45]. In case of Mef2a, all three potential binding sites can drive reporter gene expression as reported [13]. Figure 6B shows the binding of Nkx2.5 to the human DPF3 core promoter. Subsequently, co-transfection of reporter construct and increasing amounts of Nkx2.5 expression vector revealed a dose-dependent transcriptional activation by Nkx2.5. In line with this, deletion of the potential Nkx2.5 binding element (TCCACTTTCC) showed that transcriptional activity was indeed mediated through this motif, as activation was lost in the mutated construct.

Srf-Centered Transcription Network Integrating Srf-Binding Events, H3ac, miRNAs, and Differential Expression in Srf Knockdown
In addition to a genome-wide perspective, our analysis also provides useful information on the level of individual genes. We conducted an extensive literature search and built an Srf centered cardiac transcription network, where we subsequently integrated our findings from the Srf and histone 3 acetylation ChIP and Srf siRNA-mediated knockdown experiments (Figure 7). Thus our data add regulatory content to the nodes, which are connected by referenced interactions. The network depicts common regulation by Srf and H3ac as well as the impact of the posttranscriptional modulation of expression levels by miRNAs. Target genes important in the cardiovascular context are arranged to their biological roles like regulation in muscle contractility or cardiac growth and conduction. As an example the apoptotic machinery is regulated at all three levels (Srf, H3ac and miRNAs) through several pathways involving pro-apoptotic (Casp3, miR-320, Hsp20/a8/a5, Bax) as well as anti-apoptotic (miR-21, Bcl2, Mcl1) genes.

Discussion
We present a systematic in-vivo analysis of three levels regulating cardiac mRNA profiles, namely regulation of gene transcription by epigenetic and genetic factors and posttranscriptional regulation by short noncoding RNAs. We performed genome-wide profiling of the DNA occupancy of four key cardiac transcription factors (Gata4, Nkx2.5, Mef2a and Srf) and studied their co-occurrence with four activating histone modifications (H3ac, H4ac, H3K4me2 and H3K4me3) as well as the potential regulatory impact of miRNAs. We combined these data with mRNA expression profiles in wildtype and RNAi mediated knockdown cells and finally confirmed key conclusions in a time-course of cardiac maturation in mouse around birth.
In human and mouse ∼2,000 transcription factors, more than 100 different modifications of histone residues and ∼700 miRNAs modulate the mRNA profiles corresponding to ∼23,000 genes. Major insights have been gained into the regulation of the transcription process by DNA-binding transcription factors [46]–[48]. The role of histone modifications in establishing and maintaining the chromatin status and their function as protein interaction partners has been discovered [12], [13], [49]. More recently, the high impact of miRNAs on mRNA profiles and their function as inhibitors of the translation process has emerged [43], [50]–[52]. However, we lack data showing the interaction between these three levels of regulation. The initial insights were obtained by focusing on the different levels independently, and it was long thought that transcription factors are the main driving force. We feel that it is a fine-tuned balance and our data favor a comparable impact for all three levels with a high degree of interdependency. Our data indicate that histone 3 acetylation is involved in the regulation of Srf as well as Gata4 dependent cardiac genes and moreover potentially compensates the loss of transcriptional activation in Srf knockdown. Vice versa histone modifying enzymes represent an important group of direct downstream targets of Srf (e.g. histone demethylases containing a Jumonji domain such as Jmjd1c, Jmjd2b, Jmjd3, Jmjd4 and Jmjd5, see Figure 7). A similar picture emerges for the relationship of miRNAs and Srf such that the Argonaute proteins Eif2c2 (Ago2) and Eif2c3 (Ago3), which are direct Srf targets, play a key role for miRNA mediated-mRNA cleavage via the RISC complex [53]. In line with this, we found a panel of miRNAs deregulated in Srf knockdown, explaining three times more differentially expressed genes than Srf binding events alone could do. We are convinced that these data reflect the high degree of interdependency between the different levels. In addition, our data underline the high potency of compensatory regulation between DNA-binding transcription factors. We show that genes regulated by multiple transcription factors were significantly less likely differentially expressed in RNAi knockdown of one respective factor. So far, it had been postulated that members of a gene family (e.g. Mef transcription factors [4], [54], [55]) or factors with redundant paralogs could buffer each others dysfunction [18]. Our data extend these findings to primarily unrelated transcription factors, which share common targets.
The observed correlation of histone 3 acetylation with Srf and Gata4 target gene activation underlines the beneficial effects seen for HDAC inhibitors for a variety of disease states [17]. Further, we favor the view that modulation of the histone modification status might be a plausible explanation for incomplete penetrance or phenotypic diversity as frequently observed in mouse models with identical genetic background or in human disease such as congenital heart disease. Here, a distinct gene mutation can lead to a broad portfolio of phenotypes, such as mutations in Cited-2 [56], [57]. Environmental factors are potentially causative for these observation and recent reports show a link between environment and alterations of histone modifications. Thus, the change of the phosphorylation status and thereof the activity of histone modifying enzymes mediated for example via the calcium/calmodulin-dependent protein kinase II (CaMKII) could represent a mechanistic explanation [58]–[60].
In accordance with others, we found that the overwhelming proportion of differentially expressed genes in our RNAi experiments were indirect targets of the respective transcription factor. Computational studies suggest that up to 30% of all human genes are regulated by miRNAs, while each miRNA may control hundreds of gene targets [61], [62]. Our in-vivo data highlight the global impact of miRNAs on expression profile alterations seen in transcription factor loss-of-function studies. Differentially expressed miRNAs in Srf knockdown potentially explain up to 45% of the altered mRNA profile in our study.
In summary, our data indicate that the different levels regulating mRNA profiles have a high degree of interdependency. The different nodes of the regulatory network have the potential to modulate each other and should therefore be viewed in context. Further functional tests will be required to evaluate regulatory circuits on a single gene basis. It will be of interest to study how the interdependency of the different factors stabilizes the overall function of given networks, and how it contributes to the resistance to external disturbances as well as to the impact of novel therapeutic tools such as HDAC inhibitors or antagomirs.
Materials and Methods
All methods are abbreviated and additional information is provided in the online supplement.
Ethics Statement
Human cardiac tissue was obtained from the German Heart Center with ethical approval by the responsible institutional review committee (Charité 129/2000) and informed consent of patients [63].
Cell Culture and Cardiac Samples
HL-1 cells were provided by Prof. William C. Claycomb (Departments of Biochemistry and Molecular Biology and Cell Biology and Anatomy, Louisiana State University Medical Center, New Orleans, LA 70112) and cultured as described [64]. The cells were used for experiments at their maximum contraction. HEK293T cells were cultivated according to standard protocols. Mouse hearts at the indicated stages of CD1 and C57/Bl6 strain were dissected in cold PBS from the rest of the body. For subsequent RNA isolation heart samples were directly snap frozen in liquid nitrogen and stored at −80°C. For ChIP or histology experiments heart samples were fixed with formaldehyde or paraformaldehyde, respectively.
siRNA and Cell Transfection
For RNAi knockdown HL-1 cells were transfected with two different siRNAs (Qiagen) per transcription factor (Table S12) using two biological replicates each (4 replicates in total). As a control, the cells were transfected with an unspecific siRNA (siNon). Cells were grown to 70–80% confluence for at least two days without addition of antibiotics. 3×105 cells were seeded into 6-well plates with 2ml media resulting in 70–80% confluence after 4h. The mixture of 9µl (20µM) siRNA in 270µl of DMEM media and 16µl Lipofectamine 2000 (Invitrogen) in 470µl DMEM media was incubated for 20min at room temperature and added drop wise to the cells. The cell culture media was changed after 24h and cells were harvested for protein extraction or RNA preparation after 48h. For reporter gene assays HEK293T cells were transfected with Transfast (Promega) according to manufacturer's instruction.
mRNA Expression Analysis in Wild-Type and siRNA–Treated Cells
Total RNA of cultured cells and heart tissues was isolated using TRIzol reagent (Invitrogen) followed by DNase digest (Promega) and ethanol precipitation according to standard protocols. Reverse transcription reactions were carried out via AMV-RT (Promega) with random hexamers (Amersham Pharmacia Biotech). Illumina array analysis was performed by Integragen (France). For each set of experiments two biological and two technical replicates were analyzed using Illumina Mouse-6 v1.1 genome-wide microarrays.
To verify transcript expression levels of HL-1 cells and mouse hearts, quantitative real-time PCR measurements were performed using SYBR Green PCR Master Mix (ABgene) and the ABI PRISM 7900HT Sequence Detection System. Gene expression was calculated using the ΔCT method with normalization to the housekeeping gene Hprt. Primer sequences and additional results are given in Tables S8 and S13 and Figure S1.
Analysis of mRNA Expression Data in Wild-Type and siRNA–Treated Cells
The raw and transformed data of the Illumina expression microarrays (Mouse-6 v1.1 genome wide arrays) were deposited in the ArrayExpress database at the EBI (accession code E-TABM-376). Probe intensities were obtained from Integragen (France). Probes were filtered according to the detection score given by the Illumina array analysis software BeadStudio. Only probes with a detection score greater or equal to 0.95 in at least one experiment were retained. Probe intensities were qspline normalized and probes assigned to one transcript (Ensembl v46, mm8) were normalized using the median polish procedure. Differential expression was determined using the limma package [65] of Bioconductor 2.0 [66] and p-values were corrected for multiple testing according to Benjamin and Yekutieli [67]. Only transcripts with p-value smaller or equal to 0.05 in both siRNA-mediated knockdowns when compared to siNon-treated cells were considered to be significantly differentially expressed.
MicroRNA Expression Analysis
Small RNAs were isolated from total RNA of HL-1 cells and prepared for miRNA sequencing using Illumina Kit FG-102-1009 according to manufacturer's protocol. For quantification of miR-133a-1 in mouse hearts stem-loop qPCR (primer sequence: GTTGGCTCTGGTGCAGGGTCCGAGGTATTCGCACCAGAGCCAACCAGCTG) and TaqMan qPCR (forward primer: 5′ATTAATTTGGTC CCCTTCAAC, reverse primer:5′GTGCAGGGTCCGAGGT, TaqMan probe 21 (Roche)) were performed as described elsewhere [68], [69].
MicroRNA-seq Data Analysis
Small RNAs were sequenced by Illumina/Solexa next-generation (single-end) sequencing technology. The small RNA-seq data were deposited in the Gene Expression Omnibus (GEO) database at the NCBI (accession code GSE26397). In two independent siRNA-mediated knockdowns of Srf (Srf-si1 & Srf-si2) and an unspecific siRNA we retrieved 14,911,499 (Srf-si1), 14,518,157 (Srf-si2) and 14,742,382 (siNon) unfiltered 36bp reads, which yielded 5,634,650 (Srf-si1), 5,503,661 (Srf-si2) and 5,674,429 (siNon) unique (i.e. non-redundant) read sequences. These reads were mapped to the mouse reference genome (NCBI v37, mm9) using MicroRazerS [70] allowing at most 20 equally-best hits for each read and using a seed length of 16 bases with at most one mismatch. For Srf-si1 96.7%, for Srf-si2 96.2% and for siNon 96.5% of all unique sequences could be mapped to the mouse genome. In total 402 miRNAs were identified, corresponding to 450 different loci. To annotate the aligned sequence reads with miRNAs, we checked for overlaps with miRNA positions (http://www.miRBase.org/, release 14.0). We tested for differential expression between the Srf-si1/2 and siNon libraries using Fisher's exact test with FDR correction for multiple testing (p≤0.05). For all miRNAs identified as significantly differentially expressed in at least one siRNA knockdown of Srf (but both either up - or downregulated) compared to negative control we did target gene predictions using the miRanda v3.0 algorithm [71]. Finally, the target prediction revealed 192 of 429 differentially expressed genes. Using a fisher exact test we found the number to be statistical significant when compared to a prediction based on all versus differentially expressed genes (p = 1.77e-5).
miRNA Expression Levels in HL-1 Cells Compared to Human Right Ventricle
The number of unfiltered 36bp reads for wildtype HL-1 was 14,440,535, while the human heart samples produced 14,475,968 (NH-1, normal heart (NH)), 16,270,049 (NH-2), 12,940,172 (NH-3) and 14,890,970 (NH-4) unfiltered reads, respectively, yielding 5,541,954 (HL-1), 5,176,852 (NH-1), 7,189,852 (NH-2), 3,397,365 (NH-3) and 5,075,129 (NH-4) unique reads. Mapping to mouse and human reference genomes (mm9, NCBI v37 and hg18, NCBI v36) was performed by MicroRazerS [70] allowing at most one mismatch and 20 equally-best hits per read with a seed length of 16 (mouse) and 18 (human), respectively. 97% (HL-1) and 87–91% (NH-1-4) of all unique sequences could be mapped to the corresponding reference genome. In total 196 common, 107 mouse-specific and 180 human-specific miRNA families were found (http://www.miRBase.org, release 14.0). To account for cross-species differences in the specific-expression levels, we used rank-transformed miRNA expression levels for comparison.
Chromatin Immunoprecipitation
ChIP experiments with HL-1 cells and mouse hearts were carried out as previously described [72] with minor modifications. The antibodies used are given in the Table S14. ChIP-chip experiments of HL-1 cells were performed on NimbleGen custom made microarrays with two biological duplicates (containing two pooled technical replicates each). Samples were labeled and hybridized according to NimbleGen standard procedure. Sample preparation for ChIP-seq of HL-1 cells was performed according the Illumina library preparation procedure. Two pooled biological replicates for Srf and H3ac were sequenced using Illumina/Solexa next-generation (single-end) sequencing technology. ChIP-chip and ChIP-seq data were confirmed by quantitative real-time PCR using the SyberGreen I PCR Master Mix (Abgene) and the ABI PRISM 7900HT Sequence Detection System or using the RealTime ready DNA Probes Master with the Universal ProbeLibrary and the LightCycler 1536 (Roche). Results of ChIP-qPCR experiments are given in Figure S5 and primer sequences for verifications in Table S13. ChIP after siRNA knockdown of Srf in Hl-1 cells was performed using the LowCell ChIP Protein A Kit from Diagenode according to the manufacture's instructions.
ChIP-chip Data Analysis
The raw and transformed data of the ChIP-chip experiments and the array design were deposited in the ArrayExpress database (accession code E-TABM-378 and A-MEXP-893). We designed a set of two 385k NimbleGen arrays to represent enhancer and promoter regions of 12,625 transcriptional start sites based on a broad panel of muscle relevant data source (Table S1) [13]. The arrays represented 89Mbp of the mouse genome build mm8 and contained 740,000 probes with a tiling of 110bp (50–60bp gap between probes). This included conserved regions (based on PhastCons [31] score thresholds of 0.2) within 10kb upstream, the full sequence within 2kb upstream and the first exon and intron of the corresponding transcript.
The array intensities of each channel were normalized and log-transformed using VSN [73]. Log-ratio enrichment levels for each probe were calculated by subtraction of log Cy3 (Input) from log Cy5 (ChIP sample). The signal of transcription factors were smoothed by calculating a median over the probes inside a sliding window of 600bp. To distinguish enriched probes a z-score and empirical p-value for each probe on the null hypothesis that these z-scores have a symmetric distribution with mean zero was calculated. Significant probe positions (corrected for multiple testing [74], FDR<0.1), with a distance less than 210bp were combined into transcription factor binding sites. The histone binding sites were identified as described previously [39].
ChIP-seq Data Analysis
The ChIP-seq data were deposited in the GEO database (accession code GSE26397). Of the initial 6,967,318 and 8,364,328 sequence reads obtained in the Srf and H3ac ChIP-seq experiment, respectively, 4,543,634 (65.2%) for Srf and 6,141,144 (73.4%) for H3ac could be mapped to the mouse reference genome (NCBI v37, mm9) using the read mapping tool RazerS [75]. Only uniquely mapped 36nt reads with at most two mismatches were retained. To identify enriched regions we used the CisGenome software [76]. For Srf we used a window size of 100bp, a step size of 25bp and a read count level of 10 (FDR = 1.6%). For H3ac we applied a window size of 250bp, a step size of 50bp and a read count level of 10 (FDR = 4.7%). After peak localization the found Srf and H3ac peaks were filtered (see Text S1) identifying 2,190 Srf and 10,486 H3ac ChIP-seq peaks.
TFBS Conservation Analysis
The occurrence of transcription factor binding motifs within observed peaks was analyzed with TRANSFAC MATCH program [45] by using ±250bp sequence surrounding the peak center and position weight matrices corresponding to the studied factors obtained from TRANSFAC [29]. The TRANSFAC matrices used for motif search are listed in Table S7. Presence of the CArG-box was determined by searching the pattern CC(A/T)6GG with two errors at most. The degree of conservation of respective motifs was studied using PhastCons conserved elements [31]. In addition 100% conservation between human and mouse was defined using 100bp windows.
Occurrence and Co-Occurrence of TFBS
Identified ChIP-chip or ChIP-seq binding sites were assigned to transcriptional start sites if located within 10kb upstream or in the transcribed region. General co-regulation of a gene by two or more transcription factors was defined irrespective of the distance between the binding sites. In addition, co-occurrence between transcription factors or histone modifications was defined if the centers of the peaks had a distance below ±500bp.
Gene Ontology Associations to Gene Groups
The association of gene groups to Gene Ontology (GO) terms [77] was assessed as described previously [78] (conditional hypergeometric test, p<0.001). To analyze the association of differentially expressed transcripts with GO categories, transcripts were mapped to genes. Overrepresentation was tested against all genes represented on the ChIP and siRNA array, respectively.
Data Analysis
Standard bioinformatic analysis was carried out using R and Bioconductor packages [66] as well as Perl and its BioPerl modules. If not mentioned otherwise, p-values given are based on Student's t-test.
Protein Extraction and Western Blot
Specific or non-specific siRNA treated HL-1 cardiomyocytes were used for Western Blot analysis to monitor the knockdown efficiency at protein level. HL-1 cells were treated with lysis buffer (20mM Tris-HCl pH 7.4, 150nM NaCl, 1mM EDTA, 1% Triton, 1mM DTT, 0.1mM PMSF, 1× Protease Inhibitor Cocktail, 1mM NaVO4) for protein extraction. Western Blot was performed according to standard protocols. All antibodies with their respective dilution are given in Table S14.
Reporter Gene Assays and Site-Directed Mutagenesis
Reporter constructs were made by cloning the 385bp long human DPF3 minimal promoter (chr14 : 72.430.563–72.430.943, NCBI36/hg18) and the 485bp long regulatory region downstream of mmu-miR-125b-1 (chr9 : 41.390.238–41.390.700, NCBI37/mm9) into the pGL3 basic vector (Promega). Transient co-transfections were carried out in triplicates in 96-well plates in HEK293T cells by transfecting 50ng of reporter vector, 5ng of Firefly luciferase vector for internal normalization of transfection efficiency and 50–150ng of the respective expression vectors. Activity was measured by Dual-Luciferase assay (Promega) after 48 hours. Site-directed mutagenesis of DNA was carried out using the QuikChange site-directed mutagenesis kit (Stratagene) according to manufacturer's instructions. Oligonucleotides for mutagenesis were designed to introduce deletions or mutations of the potential Nkx2.5 or Srf binding sites. Mutagenesis was confirmed by plasmid sequencing carried out at MWG Biotech.
Immunocytochemistry
For immunofluorescence analyses, the heart tissues were fixed over night with 4% paraformaldehyde, dehydrated and embedded in paraffin. Subsequently sections of 8µm were de-paraffinized, rehydrated and antigen retrieval was performed in 10mM citric acid buffer (pH 6). Blocking was carried out in 5% normal goat serum in PBS for 1h at room temperature. Primary and secondary antibodies were applied in the same buffer for 2h at room temperature, each followed by three washes in PBS and a DAPI counterstaining. Antibodies with their respective dilution are listed in Table S14. The sections were mounted in Flouromount G (Electron Microscopy Science) and examined on a Zeiss LSM 510 META confocal microscope (Carl Zeiss).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. MolkentinJD
LinQ
DuncanSA
OlsonEN
1997 Requirement of the transcription factor GATA4 for heart tube formation and ventral morphogenesis. Genes Dev 11 1061 1072
2. KuoCT
MorriseyEE
AnandappaR
SigristK
LuMM
1997 GATA4 transcription factor is required for ventral morphogenesis and heart tube formation. Genes Dev 11 1048 1060
3. LyonsI
ParsonsLM
HartleyL
LiR
AndrewsJE
1995 Myogenic and morphogenetic defects in the heart tubes of murine embryos lacking the homeo box gene Nkx2-5. Genes Dev 9 1654 1666
4. NayaF
BlackB
WuH
Bassel-DubyR
RichardsonJ
2002 Mitochondrial deficiency and cardiac sudden death in mice lacking the MEF2A transcription factor. Nat Med 8 1303 1309
5. NiuZ
YuW
ZhangSX
BarronM
BelaguliNS
2005 Conditional mutagenesis of the murine serum response factor gene blocks cardiogenesis and the transcription of downstream gene targets. J Biol Chem 280 32531 32538
6. MianoJM
RamananN
GeorgerMA
de Mesy BentleyKL
EmersonRL
2004 Restricted inactivation of serum response factor to the cardiovascular system. Proc Natl Acad Sci USA 101 17132 17137
7. BalzaROJr
MisraRP
2006 Role of the serum response factor in regulating contractile apparatus gene expression and sarcomeric integrity in cardiomyocytes. J Biol Chem 281 6498 6510
8. KaramboulasC
DakuboGD
LiuJ
De RepentignyY
YutzeyK
2006 Disruption of MEF2 activity in cardiomyoblasts inhibits cardiomyogenesis. J Cell Sci 119 4315 4321
9. SearcyRD
VincentEB
LiberatoreCM
YutzeyKE
1998 A GATA-dependent nkx-2.5 regulatory element activates early cardiac gene expression in transgenic mice. Development 125 4461 4470
10. SpencerJA
MisraRP
1996 Expression of the serum response factor gene is regulated by serum response factor binding sites. J Biol Chem 271 16535 16543
11. KouzaridesT
2007 Chromatin modifications and their function. Cell 128 693 705
12. RuthenburgAJ
LiH
PatelDJ
AllisCD
2007 Multivalent engagement of chromatin modifications by linked binding modules. Nat Rev Mol Cell Biol 8 983 994
13. LangeM
KaynakB
ForsterUB
TonjesM
FischerJJ
2008 Regulation of muscle development by DPF3, a novel histone acetylation and methylation reader of the BAF chromatin remodeling complex. Genes Dev 22 2370 2384
14. ThorneJL
CampbellMJ
TurnerBM
2009 Transcription factors, chromatin and cancer. Int J Biochem Cell Biol 41 164 175
15. WangZ
ZangC
CuiK
SchonesDE
BarskiA
2009 Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 138 1019 1031
16. ChangS
McKinseyTA
ZhangCL
RichardsonJA
HillJA
2004 Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol Cell Biol 24 8467 8476
17. HaberlandM
MontgomeryRL
OlsonEN
2009 The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet 10 32 42
18. GitterA
SiegfriedZ
KlutsteinM
FornesO
OlivaB
2009 Backup in gene regulatory networks explains differences between binding and knockout results. Mol Syst Biol 5 276
19. YuM
RivaL
XieH
SchindlerY
MoranT
2009 Insights into GATA-1-Mediated Gene Activation versus Repression via Genome-wide Chromatin Occupancy Analysis. Molecular Cell 36 682 695
20. Phuc LeP
FriedmanJR
SchugJ
BrestelliJE
ParkerJB
2005 Glucocorticoid receptor-dependent gene regulatory networks. PLoS Genet 1 e16 doi:10.1371/journal.pgen.0010016
21. KwonY-S
Garcia-BassetsI
HuttKR
ChengCS
JinM
2007 Sensitive ChIP-DSL technology reveals an extensive estrogen receptor alpha-binding program on human gene promoters. Proc Natl Acad Sci USA 104 4852 4857
22. HuZ
KillionPJ
IyerVR
2007 Genetic reconstruction of a functional transcriptional regulatory network. Nat Genet 39 683 687
23. HarbisonC
GordonD
LeeT
RinaldiN
MacisaacK
2004 Transcriptional regulatory code of a eukaryotic genome. Nature 431 99 104
24. CordesKR
SheehyNT
WhiteMP
BerryEC
MortonSU
2009 miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 460 705 710
25. KwonC
HanZ
OlsonEN
SrivastavaD
2005 MicroRNA1 influences cardiac differentiation in Drosophila and regulates Notch signaling. Proc Natl Acad Sci U S A 102 18986 18991
26. ChenJF
MandelEM
ThomsonJM
WuQ
CallisTE
2006 The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet 38 228 233
27. NiuZ
LiA
ZhangSX
SchwartzRJ
2007 Serum response factor micromanaging cardiogenesis. Curr Opin Cell Biol 19 618 627
28. ZhaoY
RansomJF
LiA
VedanthamV
von DrehleM
2007 Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell 129 303 317
29. KnuppelR
DietzeP
LehnbergW
FrechK
WingenderE
1994 TRANSFAC retrieval program: a network model database of eukaryotic transcription regulating sequences and proteins. J Comput Biol 1 191 198
30. MianoJM
LongX
FujiwaraK
2007 Serum response factor: master regulator of the actin cytoskeleton and contractile apparatus. Am J Physiol Cell Physiol 292 C70 81
31. SiepelA
BejeranoG
PedersenJ
HinrichsA
HouM
2005 Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15 1034 1050
32. AkazawaH
KomuroI
2005 Cardiac transcription factor Csx/Nkx2-5: Its role in cardiac development and diseases. Pharmacol Ther 107 252 268
33. ClarkKL
YutzeyKE
BensonDW
2006 Transcription factors and congenital heart defects. Annual Review of Physiology 68 97 121
34. KobayashiS
LackeyT
HuangY
BispingE
PuWT
2006 Transcription factor gata4 regulates cardiac BCL2 gene expression in vitro and in vivo. Faseb J 20 800 802
35. VickersER
KaszaA
KurnazIA
SeifertA
ZeefLAH
2004 Ternary complex factor-serum response factor complex-regulated gene activity is required for cellular proliferation and inhibition of apoptotic cell death. Mol Cell Biol 24 10340 10351
36. SuzukiYJ
EvansT
2004 Regulation of cardiac myocyte apoptosis by the GATA-4 transcription factor. Life Sci 74 1829 1838
37. BruneauBG
NemerG
SchmittJP
CharronF
RobitailleL
2001 A murine model of Holt-Oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell 106 709 721
38. PuWT
IshiwataT
JuraszekAL
MaQ
IzumoS
2004 GATA4 is a dosage-sensitive regulator of cardiac morphogenesis. Developmental Biology 275 235 244
39. FischerJJ
ToedlingJ
KruegerT
SchuelerM
HuberW
2008 Combinatorial effects of four histone modifications in transcription and differentiation. Genomics 91 41 51
40. TakayaT
KawamuraT
MorimotoT
OnoK
KitaT
2008 Identification of p300-targeted acetylated residues in GATA4 during hypertrophic responses in cardiac myocytes. J Biol Chem 283 9828 9835
41. CaoD
WangZ
ZhangCL
OhJ
XingW
2005 Modulation of smooth muscle gene expression by association of histone acetyltransferases and deacetylases with myocardin. Mol Cell Biol 25 364 376
42. HirschyA
SchatzmannF
EhlerE
PerriardJC
2006 Establishment of cardiac cytoarchitecture in the developing mouse heart. Dev Biol 289 430 441
43. van RooijE
LiuN
OlsonEN
2008 MicroRNAs flex their muscles. Trends Genet 24 159 166
44. LatronicoMV
CatalucciD
CondorelliG
2008 MicroRNA and cardiac pathologies. Physiol Genomics 34 239 242
45. KelAE
GosslingE
ReuterI
CheremushkinE
Kel-MargoulisOV
2003 MATCH: A tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res 31 3576 3579
46. ENCODE 2007 Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447 799 816
47. FarnhamPJ
2009 Insights from genomic profiling of transcription factors. Nat Rev Genet 10 605 616
48. VaquerizasJM
KummerfeldSK
TeichmannSA
LuscombeNM
2009 A census of human transcription factors: function, expression and evolution. Nat Rev Genet 10 252 263
49. KochCM
AndrewsRM
FlicekP
DillonSC
KaraözU
2007 The landscape of histone modifications across 1% of the human genome in five human cell lines. Genome Res 17 691 707
50. WangY
LiangY
LuQ
2008 MicroRNA epigenetic alterations: predicting biomarkers and therapeutic targets in human diseases. Clin Genet 74 307 315
51. CordesKR
SrivastavaD
2009 MicroRNA regulation of cardiovascular development. Circ Res 104 724 732
52. BonauerA
CarmonaG
IwasakiM
MioneM
KoyanagiM
2009 MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science 324 1710 1713
53. KimVN
HanJ
SiomiMC
2009 Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol 10 126 139
54. LinQ
SchwarzJ
BucanaC
OlsonEN
1997 Control of mouse cardiac morphogenesis and myogenesis by transcription factor MEF2C. Science 276 1404 1407
55. VongLH
RagusaMJ
SchwarzJJ
2005 Generation of conditional Mef2cloxP/loxP mice for temporal - and tissue-specific analyses. Genesis 43 43 48
56. SperlingS
GrimmCH
DunkelI
MebusS
SperlingHP
2005 Identification and functional analysis of CITED2 mutations in patients with congenital heart defects. Hum Mutat 26 575 582
57. MacDonaldST
BamforthSD
ChenC-M
FarthingCR
FranklynA
2008 Epiblastic Cited2 deficiency results in cardiac phenotypic heterogeneity and provides a mechanism for haploinsufficiency. Cardiovasc Res 79 448 457
58. McGeeS
FairlieE
GarnhamA
HargreavesM
2009 Exercise-induced histone modifications in human skeletal muscle. J Physiol (Lond)
59. SaccaniS
PantanoS
NatoliG
2002 p38-Dependent marking of inflammatory genes for increased NF-kappa B recruitment. Nat Immunol 3 69 75
60. SripichaiO
KieferCM
BhanuNV
TannoT
NohS-J
2009 Cytokine-mediated increases in fetal hemoglobin are associated with globin gene histone modification and transcription factor reprogramming. Blood 114 2299 2306
61. ShalgiR
LieberD
OrenM
PilpelY
2007 Global and local architecture of the mammalian microRNA-transcription factor regulatory network. PLoS Comput Biol 3 e131 doi:10.1371/journal.pcbi.0030131
62. SiomiH
SiomiMC
2009 On the road to reading the RNA-interference code. Nature 457 396 404
63. ToenjesM
SchuelerM
HammerS
PapeUJ
FischerJJ
2008 Prediction of cardiac transcription networks based on molecular data and complex clinical phenotypes. Mol Biosyst 4 589 598
64. ClaycombWC
LansonNAJr
StallworthBS
EgelandDB
DelcarpioJB
1998 HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci U S A 95 2979 2984
65. SmythGK
2004 Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3 Article3
66. GentlemanRC
CareyVJ
BatesDM
BolstadB
DettlingM
2004 Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5 R80
67. BenjaminiY
YekutieliD
2001 The control of the false discovery rate in multiple testing under dependency. Annals of Statistics 29 1165 1188
68. ChenC
RidzonDA
BroomerAJ
ZhouZ
LeeDH
2005 Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res 33 e179
69. WuRM
WoodM
ThrushA
WaltonEF
Varkonyi-GasicE
2007 Real-Time PCR Quantification of Plant miRNAs Using Universal ProbeLibrary Technology. Biochemica 2
70. EmdeAK
GrunertM
WeeseD
ReinertK
SperlingSR
2009 MicroRazerS: rapid alignment of small RNA reads. Bioinformatics 26 123 124
71. JohnB
EnrightAJ
AravinA
TuschlT
SanderC
2004 Human MicroRNA targets. PLoS Biol 2 e363 doi:10.1371/journal.pbio.0020363
72. HorakCE
MahajanMC
LuscombeNM
GersteinM
WeissmanSM
2002 GATA-1 binding sites mapped in the beta-globin locus by using mammalian chIp-chip analysis. Proc Natl Acad Sci U S A 99 2924 2929
73. HuberW
von HeydebreckA
SultmannH
PoustkaA
VingronM
2002 Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics 18 Suppl 1 S96 104
74. StoreyJD
TibshiraniR
2003 Statistical significance for genomewide studies. Proc Natl Acad Sci U S A 100 9440 9445
75. WeeseD
EmdeAK
RauschT
DoringA
ReinertK
2009 RazerS–fast read mapping with sensitivity control. Genome Res 19 1646 1654
76. JiH
JiangH
MaW
JohnsonDS
MyersRM
2008 An integrated software system for analyzing ChIP-chip and ChIP-seq data. Nat Biotechnol 26 1293 1300
77. AshburnerM
BallCA
BlakeJA
BotsteinD
ButlerH
2000 Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25 25 29
78. AlexaA
RahnenfuhrerJ
LengauerT
2006 Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 22 1600 1607
79. AndersonC
CatoeH
WernerR
2006 MIR-206 regulates connexin43 expression during skeletal muscle development. Nucleic Acids Res 34 5863 5871
80. BoutzPL
ChawlaG
StoilovP
BlackDL
2007 MicroRNAs regulate the expression of the alternative splicing factor nPTB during muscle development. Genes Dev 21 71 84
81. CallisTE
DengZ
ChenJF
WangDZ
2008 Muscling through the microRNA world. Exp Biol Med (Maywood) 233 131 138
82. CareA
CatalucciD
FelicettiF
BonciD
AddarioA
2007 MicroRNA-133 controls cardiac hypertrophy. Nat Med 13 613 618
83. ChenCZ
LiL
LodishHF
BartelDP
2004 MicroRNAs modulate hematopoietic lineage differentiation. Science 303 83 86
84. ChengAM
ByromMW
SheltonJ
FordLP
2005 Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res 33 1290 1297
85. CimminoA
CalinGA
FabbriM
IorioMV
FerracinM
2005 miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A 102 13944 13949
86. ClopA
MarcqF
TakedaH
PirottinD
TordoirX
2006 A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat Genet 38 813 818
87. FanGC
ChuG
KraniasEG
2005 Hsp20 and its cardioprotection. Trends Cardiovasc Med 15 138 141
88. FelliN
FontanaL
PelosiE
BottaR
BonciD
2005 MicroRNAs 221 and 222 inhibit normal erythropoiesis and erythroleukemic cell growth via kit receptor down-modulation. Proc Natl Acad Sci U S A 102 18081 18086
89. KimHK
LeeYS
SivaprasadU
MalhotraA
DuttaA
2006 Muscle-specific microRNA miR-206 promotes muscle differentiation. J Cell Biol 174 677 687
90. KutayH
BaiS
DattaJ
MotiwalaT
PogribnyI
2006 Downregulation of miR-122 in the rodent and human hepatocellular carcinomas. J Cell Biochem 99 671 678
91. Lagos-QuintanaM
RauhutR
MeyerJ
BorkhardtA
TuschlT
2003 New microRNAs from mouse and human. Rna 9 175 179
92. LalA
NavarroF
MaherCA
MaliszewskiLE
YanN
2009 miR-24 Inhibits cell proliferation by targeting E2F2, MYC, and other cell-cycle genes via binding to “seedless” 3′UTR microRNA recognition elements. Mol Cell 35 610 625
93. LatronicoMV
CatalucciD
CondorelliG
2007 Emerging role of microRNAs in cardiovascular biology. Circ Res 101 1225 1236
94. LuoX
LinH
PanZ
XiaoJ
ZhangY
2008 Down-regulation of miR-1/miR-133 contributes to re-expression of pacemaker channel genes HCN2 and HCN4 in hypertrophic heart. J Biol Chem 283 20045 20052
95. MarsonA
LevineSS
ColeMF
FramptonGM
BrambrinkT
2008 Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell 134 521 533
96. McCarthyJJ
EsserKA
2007 MicroRNA-1 and microRNA-133a expression are decreased during skeletal muscle hypertrophy. J Appl Physiol 102 306 313
97. MengF
HensonR
Wehbe-JanekH
GhoshalK
JacobST
2007 MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer Gastroenterology 647 658
98. MottJL
KobayashiS
BronkSF
GoresGJ
2007 mir-29 regulates Mcl-1 protein expression and apoptosis. Oncogene 26 6133 6140
99. NaguibnevaI
Ameyar-ZazouaM
PolesskayaA
Ait-Si-AliS
GroismanR
2006 The microRNA miR-181 targets the homeobox protein Hox-A11 during mammalian myoblast differentiation. Nat Cell Biol 8 278 284
100. ParkSY
LeeJH
HaM
NamJW
KimVN
2009 miR-29 miRNAs activate p53 by targeting p85 alpha and CDC42. Nat Struct Mol Biol 16 23 29
101. PetroccaF
VisoneR
OnelliMR
ShahMH
NicolosoMS
2008 E2F1-regulated microRNAs impair TGFbeta-dependent cell-cycle arrest and apoptosis in gastric cancer. Cancer Cell 13 272 286
102. RaoPK
KumarRM
FarkhondehM
BaskervilleS
LodishHF
2006 Myogenic factors that regulate expression of muscle-specific microRNAs. Proc Natl Acad Sci U S A 103 8721 8726
103. RenXP
WuJ
WangX
SartorMA
QianJ
2009 MicroRNA-320 is involved in the regulation of cardiac ischemia/reperfusion injury by targeting heat-shock protein 20. Circulation 119 2357 2366
104. RosenbergMI
GeorgesSA
AsawachaicharnA
AnalauE
TapscottSJ
2006 MyoD inhibits Fstl1 and Utrn expression by inducing transcription of miR-206. J Cell Biol 175 77 85
105. SmirnovaL
GrafeA
SeilerA
SchumacherS
NitschR
2005 Regulation of miRNA expression during neural cell specification. Eur J Neurosci 21 1469 1477
106. TangY
ZhengJ
SunY
WuZ
LiuZ
2009 MicroRNA-1 regulates cardiomyocyte apoptosis by targeting Bcl-2. Int Heart J 50 377 387
107. ThumT
CatalucciD
BauersachsJ
2008 MicroRNAs: novel regulators in cardiac development and disease. Cardiovasc Res 79 562 570
108. ThumT
GaluppoP
WolfC
FiedlerJ
KneitzS
2007 MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation 116 258 267
109. ThumT
GrossC
FiedlerJ
FischerT
KisslerS
2008 MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 456 980 984
110. TiliE
MichailleJJ
CiminoA
CostineanS
DumitruCD
2007 Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J Immunol 179 5082 5089
111. TuddenhamL
WheelerG
Ntounia-FousaraS
WatersJ
HajihosseiniMK
2006 The cartilage specific microRNA-140 targets histone deacetylase 4 in mouse cells. FEBS Lett 580 4214 4217
112. UrbichC
KuehbacherA
DimmelerS
2008 Role of microRNAs in vascular diseases, inflammation, and angiogenesis. Cardiovasc Res 79 581 588
113. van RooijE
SutherlandLB
QiX
RichardsonJA
HillJ
2007 Control of stress-dependent cardiac growth and gene expression by a microRNA. Science 316 575 579
114. XiaoJ
LuoX
LinH
ZhangY
LuY
2007 MicroRNA miR-133 represses HERG K+ channel expression contributing to QT prolongation in diabetic hearts. J Biol Chem 282 12363 12367
115. XuC
LuY
PanZ
ChuW
LuoX
2007 The muscle-specific microRNAs miR-1 and miR-133 produce opposing effects on apoptosis by targeting HSP60, HSP70 and caspase-9 in cardiomyocytes. J Cell Sci 120 3045 3052
116. YangB
LinH
XiaoJ
LuY
LuoX
2007 The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat Med 13 486 491
117. YuasaK
HagiwaraY
AndoM
NakamuraA
TakedaS
2008 MicroRNA-206 is highly expressed in newly formed muscle fibers: implications regarding potential for muscle regeneration and maturation in muscular dystrophy. Cell Struct Funct 33 163 169
118. ZhaoY
SamalE
SrivastavaD
2005 Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature 436 214 220
119. ZhuS
SiML
WuH
MoYY
2007 MicroRNA-21 targets the tumor suppressor gene tropomyosin 1 (TPM1). J Biol Chem 282 14328 14336
120. ZhuS
WuH
WuF
NieD
ShengS
2008 MicroRNA-21 targets tumor suppressor genes in invasion and metastasis. Cell Res 18 350 359
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 2
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Meta-Analysis of Genome-Wide Association Studies in Celiac Disease and Rheumatoid Arthritis Identifies Fourteen Non-HLA Shared Loci
- MiRNA Control of Vegetative Phase Change in Trees
- Risk Alleles for Systemic Lupus Erythematosus in a Large Case-Control Collection and Associations with Clinical Subphenotypes
- The Cardiac Transcription Network Modulated by Gata4, Mef2a, Nkx2.5, Srf, Histone Modifications, and MicroRNAs
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy