
Srf1 Is a Novel Regulator of Phospholipase D Activity and Is Essential to Buffer the Toxic Effects of C16:0 Platelet Activating Factor
During Alzheimer's Disease, sustained exposure to amyloid-β42 oligomers perturbs metabolism of ether-linked glycerophospholipids defined by a saturated 16 carbon chain at the sn-1 position. The intraneuronal accumulation of 1-O-hexadecyl-2-acetyl-sn-glycerophosphocholine (C16 : 0 PAF), but not its immediate precursor 1-O-hexadecyl-sn-glycerophosphocholine (C16 : 0 lyso-PAF), participates in signaling tau hyperphosphorylation and compromises neuronal viability. As C16 : 0 PAF is a naturally occurring lipid involved in cellular signaling, it is likely that mechanisms exist to protect cells against its toxic effects. Here, we utilized a chemical genomic approach to identify key processes specific for regulating the sensitivity of Saccharomyces cerevisiae to alkyacylglycerophosphocholines elevated in Alzheimer's Disease. We identified ten deletion mutants that were hypersensitive to C16 : 0 PAF and five deletion mutants that were hypersensitive to C16 : 0 lyso-PAF. Deletion of YDL133w, a previously uncharacterized gene which we have renamed SRF1 (Spo14 Regulatory Factor 1), resulted in the greatest differential sensitivity to C16 : 0 PAF over C16 : 0 lyso-PAF. We demonstrate that Srf1 physically interacts with Spo14, yeast phospholipase D (PLD), and is essential for PLD catalytic activity in mitotic cells. Though C16 : 0 PAF treatment does not impact hydrolysis of phosphatidylcholine in yeast, C16 : 0 PAF does promote delocalization of GFP-Spo14 and phosphatidic acid from the cell periphery. Furthermore, we demonstrate that, similar to yeast cells, PLD activity is required to protect mammalian neural cells from C16 : 0 PAF. Together, these findings implicate PLD as a potential neuroprotective target capable of ameliorating disruptions in lipid metabolism in response to accumulating oligomeric amyloid-β42.
Published in the journal:
. PLoS Genet 7(2): e32767. doi:10.1371/journal.pgen.1001299
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001299
Summary
During Alzheimer's Disease, sustained exposure to amyloid-β42 oligomers perturbs metabolism of ether-linked glycerophospholipids defined by a saturated 16 carbon chain at the sn-1 position. The intraneuronal accumulation of 1-O-hexadecyl-2-acetyl-sn-glycerophosphocholine (C16 : 0 PAF), but not its immediate precursor 1-O-hexadecyl-sn-glycerophosphocholine (C16 : 0 lyso-PAF), participates in signaling tau hyperphosphorylation and compromises neuronal viability. As C16 : 0 PAF is a naturally occurring lipid involved in cellular signaling, it is likely that mechanisms exist to protect cells against its toxic effects. Here, we utilized a chemical genomic approach to identify key processes specific for regulating the sensitivity of Saccharomyces cerevisiae to alkyacylglycerophosphocholines elevated in Alzheimer's Disease. We identified ten deletion mutants that were hypersensitive to C16 : 0 PAF and five deletion mutants that were hypersensitive to C16 : 0 lyso-PAF. Deletion of YDL133w, a previously uncharacterized gene which we have renamed SRF1 (Spo14 Regulatory Factor 1), resulted in the greatest differential sensitivity to C16 : 0 PAF over C16 : 0 lyso-PAF. We demonstrate that Srf1 physically interacts with Spo14, yeast phospholipase D (PLD), and is essential for PLD catalytic activity in mitotic cells. Though C16 : 0 PAF treatment does not impact hydrolysis of phosphatidylcholine in yeast, C16 : 0 PAF does promote delocalization of GFP-Spo14 and phosphatidic acid from the cell periphery. Furthermore, we demonstrate that, similar to yeast cells, PLD activity is required to protect mammalian neural cells from C16 : 0 PAF. Together, these findings implicate PLD as a potential neuroprotective target capable of ameliorating disruptions in lipid metabolism in response to accumulating oligomeric amyloid-β42.
Introduction
Perturbations in glycerophosphocholine (GPC) metabolism are linked to the pathogenesis of Alzheimer's Disease (AD) with the accumulation of choline-containing lipids in AD patients associated with accelerated cognitive decline [1]–[4]. Soluble amyloid-β42 (Aβ42) oligomers can increase hydrolysis of structural membrane lipids by activating cytosolic phospholipase A2 (cPLA2), a Group IVa PLA2 that preferentially hydrolyzes arachidonic acid from the sn-2 position of 1-O-alkyl-2-arachidonoyl - and 1-O-acyl-2-arachidonoyl - glycerophospholipids [2], [5], [6]. Sustained activation results in arachidonic acid signaling cascades, as well as the intraneuronal accumulation of choline-containing second messengers [1], [2], [5].
The fate and functions of these GPC second messengers are of particular interest. Recent evidence points to the accumulation of specific choline-containing metabolites [1], [2], [5]. Underlying mechanisms have yet to be fully elucidated. PC(O-16 : 0/2 : 0) platelet activating factor (PAF) species are elevated in AD brain and human neurons exposed to Aβ42 [1]. Of these elevated species, 1-O-hexadecyl-2-acetyl-sn-glycerophosphocholine (C16 : 0 PAF), but not its immediate precursor 1-O-hexadecyl-sn-glycerophosphocholine (C16 : 0 lyso-PAF), is implicated in Aβ42 toxicity [1]. Rising intraneuronal concentrations of C16 : 0 PAF activate an endoplasmic reticulum (ER)-stress signaling cascade leading to the hyperphosphorylation of tau that ultimately compromises neuronal viability [1]. Molecular and pharmacological approaches designed to promote the hydrolysis of C16 : 0 PAF to C16 : 0 lyso-PAF and/or block downstream signaling are sufficient to inhibit Aβ-mediated neurotoxicity [7]–[10]. These findings underscore the importance of this lipid species in Alzheimer's disease and emphasize the rationale for identifying key targets involved in shielding its toxic effects and/or promoting its hydrolysis and inactivation.
Unbiased approaches exploiting the cross-species conservation of biochemical pathways between man and the budding yeast Saccharomyces cerevisiae have proven to be successful in elucidating the mode-of-action of various compounds [11]. Further, S. cerevisiae has also been used as a model organism to delineate key aspects of eukaryotic lipid metabolism and to investigate various neurodegenerative diseases [12], [13]. Despite conservation of the PAF metabolic pathway genes and detection of PAF species in yeast [14], the function of the different species in yeast is currently unclear, although PAF species are suggested to play a role in cell cycle progression [15], [16]. Similar to the effects of PAF on mammalian cells, yeast cells treated with PAFs and structurally related alkylacylglycerophosphocholine analogs induce disruptions in lipid metabolism and reduce viability [17], [18]. Interestingly, S. cerevisiae strains harboring deletions for enzymes involved in phosphatidic acid (PA) metabolism, including phospholipase D (SPO14) and glycerol 3-phosphate acyltransferase (SCT1), exhibit increased susceptibility to PAF species and other choline-containing lipids suggesting an essential role for PA in mediating the toxic effects of PAFs [17]. Whether the toxic effects of alkylacylglycerolipids are solely dependent upon SPO14 and similar pathways impinging upon PA metabolism or whether other aspects of cellular metabolism are involved has not been systematically assessed at a genome-wide level. Moreover, it remains unclear why PAF second messengers accumulate in AD tissue without compensatory metabolism.
To identify additional requisite proteins and/or pathways which serve to regulate the cytotoxic affects of C16 : 0 PAF, we performed genome-wide yeast chemical genomic screens with both C16 : 0 PAF (pathogenic in AD) and C16 : 0 lyso-PAF (non-pathogenic in AD). We found that the two PAF species identify largely distinct chemical genetic interactions and that the deletion mutant that exhibited the greatest differential sensitivity to the pathogenic C16 : 0 PAF species was YDL133w, a previously uncharacterized ORF encoding a putative transmembrane protein with unknown function. Upon subsequent investigation we determined that Ydl133w physically interacts with Spo14 and is required for PLD catalytic activity in S. cerevisiae. Importantly, Ydl133w, here in referred to as Srf1 (Spo14 Regulatory Factor 1), represents the only reported regulator of Spo14 required for PLD catalytic activity in mitotic cells. We report that though C16 : 0 PAF does not impact global PLD activity, it does cause the delocalization of Spo14 and PA from the periphery. Importantly our observations can be extended from the yeast model system as PLD activity in mammalian cells was found to confer protection against the toxic effects of C16 : 0 PAF.
Results
Sensitivity of yeast to C16 : 0 PAF and C16 : 0 lyso-PAF
Previous studies have determined that, similar to neuronal cells [1], S. cerevisiae are sensitive to C16 : 0 PAF [17]. To determine whether C16 : 0 PAF and C16 : 0 lyso-PAF differentially impact the growth of S. cerevisiae and to identify an appropriate working concentration range for these lipids in subsequent studies we performed liquid growth curve analysis using wild type haploid yeast cultured with increasing concentrations of C16 : 0 PAF, C16 : 0 lyso-PAF or ethanol (carrier control). As expected, C16 : 0 PAF inhibited wild type haploid yeast growth in liquid culture in a concentration-dependent manner whereas C16 : 0 lyso-PAF was found to be comparatively less toxic at similar concentrations (Figure 1). Although both lipids impact viability at higher concentrations, the distinct effects of these two lipids at lower concentrations have not previously been appreciated and parallels their toxicity to neuronal cells [1], [19].

C16 : 0 PAF and C16 : 0 lyso-PAF chemicogenomic screen
To identify critical proteins and/or pathways which are involved in regulating the cytotoxic effects of C16 : 0 PAF, the yeast haploid deletion mutant array (DMA) was robotically pinned onto agar plates containing either ethanol or a sublethal concentration of C16 : 0 PAF (120 µM). To facilitate the identification of potentially AD-relevant pathways we also screened with equimolar concentrations of C16 : 0 lyso-PAF (120 µM) to distinguish between pathways that regulate AD-associated (C16 : 0 PAF) and non-associated (C16 : 0 lyso-PAF) phenotypes. The screen was performed in triplicate and 90 strains displaying putative increased sensitivity to C16 : 0 PAF or C16 : 0 lyso-PAF were further subjected to quantitative liquid growth curve measurements in the presence of C16 : 0 PAF, lyso-PAF, or ethanol (Table S1). A reduced concentration of 40 µM was employed in liquid cultures as this was found to cause moderate growth inhibition in wild type cells, thereby permitting quantitative analysis of the sensitivity of the individual strains to C16 : 0 PAF and C16 : 0 lyso-PAF relative to ethanol treatment using logistic growth curve analysis (LGCA, see Material and Methods and Text S1 for details). This rigorous methodology accounts for the repeated measurements of individual liquid cultures and also exploits full, sigmoidal growth curves, since it does not assume exponential growth. Furthermore, we also normalize for plate and plate-treatment effects. LGCA revealed 13 deletion mutants that were hypersensitive to at least one PAF species, compared to the wild type response: ten strains were hypersensitive to C16 : 0 PAF, five strains were hypersensitive to C16 : 0 lyso-PAF, and two overlapping strains were hypersensitive to both (Bonferroni corrected p-value <0.04; Figure 2A, Table S1). As expected, the spo14Δ mutant was hypersensitive to C16 : 0 PAF as has previously been described [17]. sct1Δ mutants have also been reported to be hypersensitive to C16 : 0 PAF [17], and though it did not make our stringent cut-off, the sct1Δ mutant was ranked 13th in sensitivity to C16 : 0 PAF (Table S1).

Using LGCA we also assessed the differential sensitivity of each deletion mutant to C16 : 0 PAF versus C16 : 0 lyso-PAF. At the 40 µM treatment level, wild type cells exhibited approximately the same growth inhibition when exposed to either PAF species and we defined differential sensitivity as a departure from the status quo. Eleven deletion mutants exhibited differential sensitivity to one of the PAF species (Bonferroni-corrected p-value <0.04, Figure 2B and Table S1). Of the two mutants that were identified in both screens, agp2Δ cells were equally sensitive to both lipids whereas taf14Δ displayed greater sensitivity to C16 : 0 PAF. Though LGCA identified ccs1Δ as being sensitive to C16 : 0 PAF its differential sensitivity was not significant. The differential sensitivity of four strains was striking. Namely, nup84Δ cells displayed the greatest differential sensitivity to C16 : 0 lyso-PAF, whereas srf1Δ, snf6Δ and spo14Δ were significantly more sensitive to C16 : 0 PAF than C16 : 0 lyso-PAF. The largely distinct chemical genetic profiles for C16 : 0 PAF and C16 : 0 lyso-PAF indicates that these related alkylacylglycerophospholipids impact upon distinct cellular pathways in yeast.
Srf1 interacts with Spo14
Our results indicate that at the 40 µM treatment level, Srf1 is pivotal for buffering the effects of C16 : 0 PAF. The biological function(s) of Srf1 is unknown but it is predicted to be a transmembrane protein. Therefore we sought to decipher its cellular function by identifying proteins that interact with Srf1. As traditional tandem affinity purification (TAP) protocols were not successful in purifying Srf1-TAP [data not shown and ref. 20], [21], we utilized a less stringent single step affinity purification approach based on the modified chromatin immunopurification (mChIP) technique [22]. Though this technique was developed for improving the purification of insoluble chromatin associated proteins, it is also applicable to other subclasses, including membrane associated proteins [23]. Using mChIP we successfully purified Srf1-TAP and identified five co-purifying proteins by mass spectrometry, of which the largest number of peptides correspond to Spo14 (Figure 3). The physical interaction between Srf1 and Spo14, combined with the sensitivity of the corresponding deletion mutants to C16 : 0 PAF [Figures 2, 4 and ref. 17] suggest Srf1 may work in a complex with Spo14 to regulate PA metabolism.


Srf1 and Spo14 buffer the toxic effects of C16 : 0 PAF
To determine if Srf1 and Spo14 function together or work in parallel pathways, we compared the sensitivity of the srf1Δspo14Δ double mutant to C16 : 0 PAF with that of the single mutants (Figure 4A). In agreement with our chemical genomic screen, srf1Δ cells display greater sensitivity to C16 : 0 PAF than spo14Δ cells. Interestingly, deletion of both SRF1 and SPO14 did not result in an additive increase in C16 : 0 PAF sensitivity but rather, the double mutant and spo14Δ exhibited similar sensitivity to C16 : 0 PAF. This indicates that spo14Δ is epistatic to srf1Δ and is in agreement with the hypothesis that Spo14 and Srf1 are in a complex and not in parallel pathways. In light of the physical interaction between Srf1 and Spo14, one interpretation of this result is that Spo14 activity is misregulated in the absence of Srf1. To further examine this possibility, we overexpressed SPO14 in wild type, srf1Δ and spo14Δ strains (Figure 4B). Overexpression of catalytically active (SPO14 and GFP-SPO14) [24], but not catalytically inactive (GFP-SPO14K–H) [24], SPO14 was sufficient to rescue the spo14Δ strain from C16 : 0 PAF-mediated toxicity, whereas overexpression of SPO14 was not found to have any effect in srf1Δ cells. Our findings suggest that Srf1 functions either downstream of Spo14 in mediating an aspect of PA-dependent signaling or directly upon the regulation of Spo14 function. In consideration of the physical interaction between Spo14 and Srf1 (Figure 3), it is more likely that Spo14 and Srf1 act in concert to mediate choline hydrolysis and PA production.
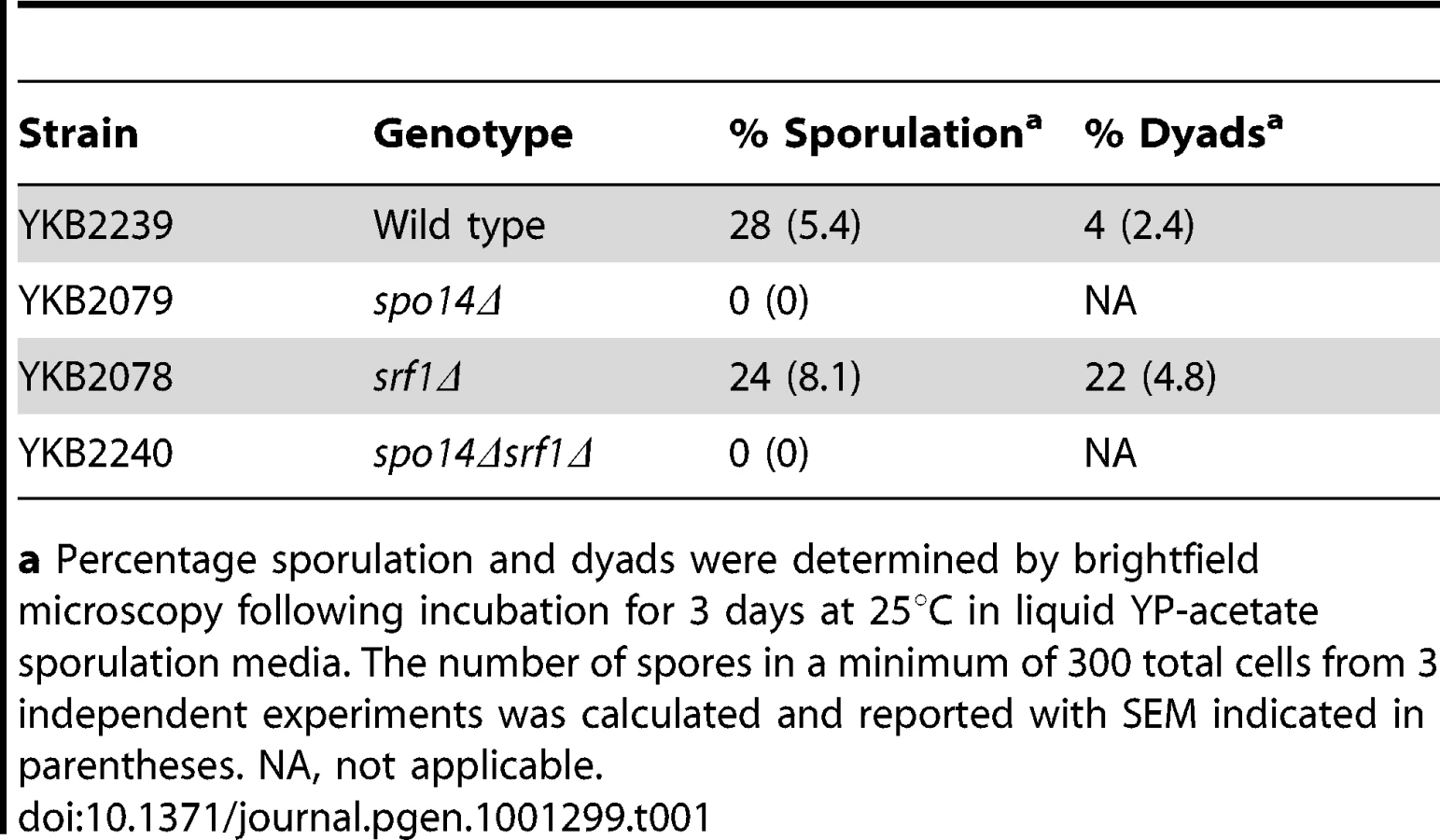
Srf1 is not essential for sporulation
Although dispensable in mitotic cells, Spo14 is strictly required for the progression of S. cerevisiae through meiosis [25]. This observation provides a simple approach to measure the effects of potential Spo14 interacting partners upon its catalytic activity during meiosis. Indeed, GCS1, which encodes an indirect regulator of Spo14 catalytic activity is essential for sporulation [26]. Therefore, we assessed whether deletion of SRF1 would result in impaired spore formation. In contrast to spo14Δ diploids that failed to sporulate, srf1Δ diploids displayed only minor impairments in sporulation which were associated with an increased frequency of dyads (Table 1). The modest effect of SRF1 on sporulation is in agreement with previous reported genome-wide studies [27], and suggests that Srf1 may have only a minor or no impact on Spo14 activity during meiosis or that Srf1 may function via PLD-independent mechanism during sporulation.
Srf1 regulates Spo14 catalytic activity in mitotic cells
Despite the limited impact on sporulation, there is a possibility that Srf1 may regulate PLD activity in mitotic cells. Therefore, we sought to examine whether Srf1 could modify Spo14 catalytic activity or localization in mitotic cells. The former possibility was directly assessed by measuring PLD activity in particulate fractions prepared from wild type and mutant strains using a previously described methodology employing a fluorescently labeled phosphatidylcholine derivative as a PLD substrate [28], [29]. Production of PA and phosphatidyl butanol (PBt), a product of transphosphatidylation, was evident in wild type particulate preparations but was completely absent in srf1Δ, spo14Δ and srf1Δspo14Δ mutant strains (Figure 5A). This result indicates that Srf1 may contribute to particulate-associated PLD catalytic activity in mitotic cells. As we have demonstrated that Spo14 physically interacts with Srf1, a predicted transmembrane protein, we sought to determine whether the deletion of SRF1 promotes the loss of Spo14 from the particulate fraction. To test this, particulate and cytosolic fractions were prepared from strains transformed with either an empty vector control or a plasmid expressing HA-tagged SPO14 [24]. The absence of PLD activity in srf1Δ strains is not a consequence of altered partitioning of Spo14 catalytic activity between particulate and cytosolic fractions in these cells as catalytic activity was absent from both fractions (Figure 5B). Furthermore, western blot analysis demonstrates that HA-Spo14 remains associated with the particulate fraction independent of Srf1 (Figure 5C). Interestingly, HA-Spo14 protein levels are consistently reduced in srf1Δ mutants (∼30% less HA-Spo14 as determined by densitometry). However, the absence of detectable PLD activity cannot be fully explained by the exclusion of Spo14 from the particulate fraction or a reduction in Spo14 protein levels (Figure 5C) thereby further implicating a biological role for Srf1 in regulating Spo14 catalytic activity during mitosis.

PLD–dependent inhibition of C16 : 0 PAF–mediated signaling and toxicity
Our findings implicating Srf1 in both buffering against the toxic effects of C16 : 0 PAF and in regulating mitotic PLD activity, underscores the importance of the PLD pathway in protecting yeast cells from C16 : 0 PAF. Therefore, we next sought to address the effects of C16 : 0 PAF on the subcellular localization and activity of Spo14. As had been previously shown [24], [30], in untreated or vehicle treated wild type (WT) cells, GFP-Spo14 displays modest peripheral and diffuse cytosolic localization (Figures 6A and S1). Deletion of SRF1 was not observed to grossly affect the subcellular localization of GFP-Spo14 in untreated or vehicle treated cells. However, addition of C16 : 0 PAF resulted in the rapid loss of GFP-Spo14 at the cell periphery with a concomitant accumulation of GFP-Spo14 at discrete foci or intracellular aggregates in 61±7% of wild type cells (Figures 6A, 6B and Figure S1). However, treatment with C16 : 0 PAF resulted in significantly fewer foci in the srf1Δ background (9±2%) suggesting Srf1 plays a role in the intracellular trafficking of Spo14 under C16 : 0 PAF treatment (Figures 6A, B S1). As a secondary method to explore the impact of C16 : 0 PAF on PLD localization we looked at the impact of C16 : 0 PAF on wild type cells expressing GFP-Q2; GFP tagged to the PA-binding domain of the transcription factor Opi1 [31]. GFP-Q2 localizes to both the periphery and nucleus in wild type cells, however in spo14Δ cells the peripheral signal is lost indicating PA and hence PLD activity is no longer concentrated at the periphery [31]. Treatment of wild type cells expressing GFP-Q2 with C16 : 0 PAF resulted in the loss of GFP-Q2 from the periphery (Figure 6C), mirroring the effect of C16 : 0 PAF upon GFP-Spo14 (Figure 6C). Together our studies support C16 : 0 PAF-mediated changes in the subcellular localization of GFP-Spo14 which are at least in part dependent upon Srf1 expression. Interestingly although C16 : 0 PAF treatment consistently resulted in the formation GFP-Spo14 aggregate structures; we never detected localization of GFP-Q2 in a similar structure. This suggests that, potentially, GFP-Spo14 aggregates that form upon PAF treatment are no longer catalytically active or alternatively that the PA produced is not accessible to GFP-Q2. Therefore we also assessed whether the C16 : 0 PAF-dependent changes in the subcellular localization of GFP-Spo14 and PA were the result of changes in Spo14 catalytic activity, partitioning within subcellular compartments or expression levels. Addition of C16 : 0 PAF was not observed to impact the catalytic activity of PLD localized to particulate fractions (Figure 6D) which is in agreement with previous in vitro findings [28]. Furthermore, C16 : 0 PAF treatment did not dissociate PLD activity (Figure 6D) or GFP-Spo14 (Figure 6E) from the particulate fraction, which suggests the C16 : 0 PAF-dependent GFP-Spo14 aggregate structures are likely still associated with a membranous compartment. Though the production of PA has previously been suggested to buffer PAF toxicity, our work further suggests that the localization of PA production is also likely of importance in regulating the deleterious effects of C16 : 0 PAF.

PLD buffers the affects of C16 : 0 PAF toxicity in Neuro-2a (N2a) cells
Our chemical genomic study clearly shows that PLD activity is essential for regulating the toxic effects of C16 : 0 PAF. To investigate whether this observation could be extended to higher eukaryotes we investigated the role of PLD activity in conferring C16 : 0 PAF resistance to the murine neuroblastoma cell line N2a, a neural cell line previously used to study PLD and Aβ effects [32], [33]. N2a cells were treated with C16 : 0 PAF or vehicle in the presence or absence of a small molecule inhibitor of PLD activity that targets both PLD1 and PLD2 with equal LC50s [34]. Treatment of N2a cells in serum-free medium with 0.1% EtOH (PAF vehicle) and 0.1% DMSO (PLD inhibitor vehicle) or with EtOH and 5 µM PLD inhibitor did not impact upon cell viability (Figure 7, inset). As expected, addition of 1 µM C16 : 0 PAF for 24 h resulted in reduced cell survival in comparison to treatment with either vehicle (Figure 7). However, treatment with C16 : 0 PAF in the presence of the PLD inhibitor resulted in a significant decrease in cell survival in comparison to treatment with C16 : 0 PAF alone or vehicle. These findings further support the role of PLD activity in regulating against C16 : 0 PAF mediated toxicity in mammalian neuroblastoma cells and indicate that a conserved mechanism for dealing with elevated levels of C16 : 0 PAF may exist within eukaryotes.

Discussion
In this study a chemical genomic screen was employed to identify the key regulators involved in buffering the toxic effects of C16 : 0 PAF and C16 : 0 lyso-PAF, lipid species previously shown to be elevated in neurons in response to oligomeric Aβ42 [1]. We identified ten deletion mutants that were sensitive to C16 : 0 PAF and five deletion mutants that were sensitive to C16 : 0 lyso-PAF as compared to wild type (Figure 2A, Table S1). The dramatically different effects on growth (Figure 1) and the minimal overlap in mutants with sensitivity to either lipid suggests that C16 : 0 PAF, C16 : 0 lyso-PAF and potentially other PAF species, impinge upon distinct cellular pathways in yeast, which parallels the distinct PAF-mediated effects that have been reported in mammalian systems [1], [35]. Such a distinction is important in light of recent evidence that aberrant metabolism, in part, underlies Aβ42 neurotoxicity with C16 : 0 PAF, but not C16 : 0 lyso-PAF or other PAF species [1], [2], [5].
Our unbiased chemical genomic approach identified the deletion mutant of SRF1 as having the most significant differential sensitivity to C16 : 0 PAF (Figure 2B). We identified a robust interaction between Srf1-TAP and Spo14 (Figure 3), whose deletion mutant is also hypersensitive to C16 : 0 PAF [Figures 2B, 4 and ref. 17]. The identification of a physical interaction between Srf1 and Spo14 is striking as only two other proteins, neither with roles in PLD function, have been reported to co-purify Spo14 in high-throughput TAP studies [20]. Furthermore, biochemical assays determined that Srf1 is required for PLD activity in mitotic cells (Figures 5 and 6). A role for Srf1 in mitotic PLD activity is also supported by genome-wide synthetic lethal genetic screens which revealed that deletion mutants of both SPO14 and SRF1 display genetic interactions with the sec14-bypass mutants CKI1 and KES1 [36]. However, in contrast to Spo14 [24], [37], [38], Srf1 is not essential for sporulation (Table 1) which suggests Srf1 is not regulating PLD activity in meiosis. Our results clearly show that Srf1-TAP can co-purify Spo14 suggesting a model where Spo14 and Srf1 form a complex in mitotic cells that is required for PLD activity and to buffer the toxicity of C16 : 0 PAF (Figure 8).

How is Srf1 regulating PLD activity? It is unlikely that the impact of Srf1 on Spo14 protein stability (Figures 5C and S1) could explain the complete absence of PLD activity in srf1Δ cells. Indeed, if Srf1 was only regulating Spo14 protein levels, then overexpression of SPO14 should have rescued the C16 : 0 PAF hypersensitivity of srf1Δ cells (Figure 4B). Additionally, it is unlikely that Srf1 is regulating PLD activity through Spo14 localization as we found that Spo14 remained associated with the particulate fraction and localized to the plasma membrane in the absence of Srf1 (Figures 5C Figure 6A, and Figure S1). How else could Srf1 be regulating Spo14? Similar to other eukaryotic PLD enzymes, Spo14 catalytic activity can be regulated by numerous mechanisms aside from changes in expression and localization. The binding of phosphoinositol phosphates, fatty acids, indirect interactions with ADP ribosylation factors (ARFs), and phosphorylation have all been demonstrated to regulate PLD activity [reviewed in 39]. Hence Srf1 may be regulating Spo14 through one of these established mechanisms. Alternatively, though we do not detect hydrolysis of phosphatidylcholine in the absence of Srf1, it is possible that some catalytic activity, possibly against other lipid targets, is still present but misregulated. Indeed this could explain why srf1Δ cells display greater sensitivity to C16 : 0 PAF than spo14Δ cells (Figure 2 and Figure 4). A different explanation for this phenomenon could be attributed to the mislocalization of Spo14 in the absence of Srf1 upon C16 : 0 PAF treatment (Figure 6). Namely, that in the absence of Srf1, the interaction of Spo14 with other cellular factors is perturbed thereby potentially serving to titrate away other cellular factors important in the response to PAF. Deletion of SPO14 in combination with deletion of SRF1 would thereby alleviate C16 : 0 PAF toxicity to that level which is observed solely in the absence of PLD activity. The exact mechanism of how Srf1 regulates Spo14 activity will require further investigation with recombinant proteins to confirm direct interaction and reconstitute the complex activity. However, our work clearly shows that Srf1 is a novel interactor and regulator of Spo14 PLD activity in mitotic cells and together Srf1 and Spo14 are necessary to buffer the toxic effects of C16 : 0 PAF (Figure 8).
Our yeast chemical genomic study and murine cell culture work indicate that the role of PLD activity in buffering the cytotoxic effects of C16 : 0 PAF is potentially conserved across species. How is PLD buffering the toxic effect of this GPC? One possibility is that PLD is rapidly inactivating C16 : 0 PAF through choline hydrolysis. Indeed, human PLD has been shown to be capable of hydrolysing lyso-PAF species [40]. However a simpler explanation is that PA (or downstream diacylglycerol (DAG)) isoforms signal inhibition of C16 : 0 PAF toxicity. Indeed, expression of the E.coli DAG kinase, which converts DAG to PA, has been shown to suppress the toxicity of lyso-PAF and PAF in wild type yeast [17]. While C16 : 0 PAF treatment is not inhibiting the PLD catalytic activity (Figure 6B and [28]), it is causing the delocalization of GFP-Spo14 and PA concentrations from the cell periphery (Figure 6 and Figure S1). Intriguingly the C16 : 0 PAF-mediated delocalization of GFP-Spo14 is dependent on Srf1 (Figure 6). This suggests that the localized generation of PA (or PAF hydrolysis) may be required to buffer the toxic effects of C16 : 0 PAF. One possibility is that delocalization of GFP-Spo14 and decreased PA levels from the periphery may induce a transcriptional response that is necessary to survive C16 : 0 PAF exposure. Indeed PA has been shown to play a direct role in the transcriptional regulation of phospholipid biosynthetic genes through the transcriptional repressor Opi1 ([31], and reviewed in [39]. Further, our chemical genomic screen supports this hypothesis as several genes with established roles in transcription were identified as being differentially sensitive to C16 : 0 PAF, including two members of the SWI/SNF chromatin remodeler complex, SNF6 and TAF14, and the transcriptional regulator UME6 (Figure 2). Although PAF has been implicated in mediating changes in gene expression, particularly those involved in responses to inflammation [41], the mechanism(s) by which these transcriptional changes occur in response to PAF are not clearly understood, nor is it known if a PAF-mediated transcriptional response is contributing to Aβ-induced neuronal toxicity.
An alternative, but not mutually exclusive hypothesis, is that PLD is buffering C16 : 0 PAF toxicity through membrane trafficking events. Our identification of an interaction between Srf1-TAP and eisosome component Pil1 [42], suggest that PLD activity may impact sites of endocytosis. However, localization of GFP-Spo14 in either untreated or PAF treated cells (Figure 6) are not reminiscent of the eisosome patches found beneath the plasma membrane [42] nor does recent genetic epistatic miniarray profiles of plasma membrane mutants implicate Spo14 in eisosome function [43]. Alternatively, despite yeast PLD's relatively minor role in vesicle budding from the Golgi and membrane trafficking [reviewed in 39], yeast PLD may become essential for lipid membrane trafficking upon C16 : 0 PAF exposure. It has recently been established that PLD1 is a negative regulator of presenilin by two independent mechanisms [32], [33]. Presenilins are a key component of the AD-associated γ-secretase complex, responsible for cleaving the amyloid precursor protein (APP) to Aβ. PLD1, but not PLD2, facilitates both APP and presenilin-1 intracellular trafficking and cell surface accumulation [33], [44], however PLD1 also interacts with presenilin inhibiting γ-secretase activity [32], thus reducing Aβ42 production. Despite this controversy, it has been suggested that inhibiting PLD1 represents a novel therapeutic approach to reducing APP and presenilin presentation at the plasma membrane and thus retard the rate of Aβ42 production [44]. In this study, our unbiased approach suggests that such inhibition may be counterproductive with respect to associated GPC metabolic defects. As intraneuronal C16 : 0 PAF levels are elevated following exposure to soluble Aβ42 oligomers [1] it may be that PLD1 can inhibit the underlying C16 : 0 PAF ER-stress pathway by reducing Aβ42 production and slowing the rate of PAF accumulation. Thus careful dissection of the impact of PLD1 on Aβ42 production and downstream GPC-mediated signaling is warranted. Here, the discovery that PLD is required to buffer the neurotoxic effect of C16 : 0 PAF suggests that therapeutic strategies modulating PLD activity may be effective in ameliorating the progression of Alzheimer's Disease pathology.
Materials and Methods
Yeast strains, genetic manipulations, plasmids, and media
The yeast strains used in this study are listed in Table 2. The MATa deletion mutant array was purchased from OpenBiosystems (catalog no. YSC1053). Deletion strains and TAP tagged SRF1 made for this study were designed using a standard PCR-mediated gene insertion technique [45], [46]. Plasmids pME962 (SPO14 LEU2 2 µ), pME940 (HA-SPO14 LEU2 CEN), pME1096 (GFP–SPO14 LEU2 2 µ) and pME1130 (GFP-SPO14K–H LEU2 2 µ) were kind gifts of J. Engebrecht [24]. Plasmid expressing GFP-Q2 was a kind gift of C. Loewen [31]. Cells were grown in standard YPD or SD medium supplemented with amino acids [47], unless otherwise described. C16 : 0 PAF and C16 : 0 lyso-PAF (L100-0025 and L101-0025, Cedarlane Canada) and resuspended in ethanol.

Chemical–genetic profiling
The MATα haploid deletion mutant array was robotically pinned in duplicate onto YPD +200 mg/L G418 plates at a density of 1536 colonies per plate using the SingerRotor HAD (Singer Instrument Company Limited) and grown for 3 days at 25°C. These plates were pinned onto YPD containing either ethanol, 120 µM C16 : 0 PAF, or 120 µM C16 : 0 lyso-PAF. Plates were incubated at 25°C and pictures taken using a Biorad Imager at 15 and 40 hours. The sensitivity of each mutant was assessed by comparing colony sizes on the treated plates to the ethanol control plates by eye and by a computer based method using AlphaEaseFC V4.0.0 (Alpha Innotech Corporation) as described [48]. The screen was performed in triplicate and any interactions identified at least 2 out of 3 times or at least once for both lipids were confirmed by quantitative growth curves. Multiple-drug resistance (MRD) genes based on published literature [49], [50] were removed from our data set.
Growth curves
Liquid growth curves were obtained for 91 strains (90 haploid knockouts plus wild type), spanning 24 plates. Each curve consisted of 45 OD readings taken at intervals of 25 minutes using Multiskan Ascent Plate Reader (Thermo Electron Corporation) and Ascent Software Version 2.6. On each plate, 6 strains were examined (wild type plus 5 others) and each strain was represented in 9 independent wells (3 replicates of the 3 treatments: ethanol, C16 : 0 lyso-PAF, and C16 : 0 PAF). In total, 1247 growth curves were analyzed (see Text S1 for more details).
Logistic growth curve analysis (LGCA)
A four-parameter logistic growth model was fit to the growth curves [51]:
where x is time and y is the OD reading, a proxy for cell density or population size. A is the starting point of growth or minimum OD reading, B is the carrying capacity or maximum OD reading, xmid is the time at 50% of total growth, and scal is approximately the time taken to go from 50 to 75% of growth.
To simultaneously account for the repeated measurement of individual wells and for the systematic effects of treatment and gene deletion on growth, we fit a mixed effects logistic growth model using the R package nlme [52], [53] (R code available upon request). Each of the four growth parameters (A, B, xmid, scal) could therefore be modeled with a combination of fixed gene deletion and/or treatment effects and random well effects (see Text S1 for details). The model was fit to the ensemble of 54 curves obtained for each plate separately. To identify chemical-genetic interactions, we focused on the xmid parameter. From the fitted model, we combined fixed effect estimates and predicted random well effects to produce a value of xmid for each growth curve, which was treated as derived data for downstream analysis.
There was evidence of non-negligible plate effects as well as interaction effects between plates and the treatments C16 : 0 lyso-PAF and C16 : 0 PAF, indicating the need for inter-plate normalization (see Text S1 for details). Due to the inclusion of wild type replicates on all plates for all 3 treatments, a normalization model could be fit to the wild type xmid values to obtain estimates of plate and plate-by-treatment interaction effects. These were then used to remove plate-related artifacts from all of the xmid values, i.e. including those for deletion mutants.
After normalization, we fit the following model for xmid:
where is the estimated/predicted and normalized value of xmid for one growth curve, corresponding to the deletion of gene g and the treatment t. The typical xmid for wild type in the ethanol reference condition is given by αwt and αg and αt are the individual effects, respectively, of deleting gene g or of treatment t. The primary parameter of interest is αg*t which captures the interaction between deletion g and treatment t. Mutants deemed hypersensitive to a single PAF species were identified based on tests of the null hypotheses that αg*L-PAF = 0 or αg*PAF = 0. Mutants deemed differentially sensitive were identified based on a test of the null hypothesis that αg*PAF−αg*L-PAF = 0. There are 90 potential gene deletions g and 3 parameters of interest (αg*L-PAF, αg*PAF, αg*PAF−αg*L-PAF), for a total of 270 tests. We did a global Bonferroni correction, i.e. multiplied p-values by 270, and thresholded at 0.04 to obtain hits (5 C16 : 0 lyso-PAF hypersensitive, 10 C16 : 0 PAF hypersensitive, 11 C16 : 0 PAF vs. C16 : 0 lyso-PAF differentially sensitive).
Srf1–TAP modified Chromatin Immunoprecipitation and mass spectrometry
Modified Chromatin Immunoprecipitation (mChIP) and mass spectrometry to identify co-purifying proteins was performed as previously described [22] from 2.1 L YPD culture of YKB2270 (OD600∼0.9) using 300 µL of Dynabeads (Invitrogen) coated with rabbit IgG (I5006, Sigma).
Dot assays
Cells were grown in YPD or minimal media at 30°C to mid-log phase and resuspended to an OD600 of 0.1 and dot assays were performed by spotting 5 µL of five-fold serial dilutions (OD600 = 0.1, 0.01, 0.001, 0.0001) onto YPD or minimal media selection plates containing the specified concentrations of ethanol, C16 : 0 lyso-PAF or C16 : 0 PAF as indicated.
Sporulation assays
Strains were grown overnight in YPD at 30°C. The following day cells were pelleted at 800 g for 5 min and washed in sterile water. An OD600 of 2.0 was resuspended in YP-acetate and incubated at 25°C for 3 days prior to microscopic examination of sporulation efficiency. The number of spores per ascus was enumerated in a minimum of 300 cells from three independent experiments and expressed as a percentage of total cells ± standard error.
Microscopy
For localization of GFP-Spo14 (pME1096) and GFP-Q2 overnight cultures of yeast cells grown at 30°C in YPD medium were re-suspended at a final OD600 of 0.2 and allowed to reach mid-log phase prior treatment and image acquisition. Similarly, wild type (YAM282-2) and srf1Δ (YKB2472) cells expressing GFP-Spo14 under the control of a copper inducible promoter were grown at 30°C and induced with 3 µM CuSO4 for 2 h to induce GFP-Spo14 expression prior to imaging or extract preparation. Cells were briefly centrifuged (800 g for 3 min), resuspended in a minimal volume of growth media, spotted onto glass slides and coverslipped prior to imaging. Images were acquired using a Leica DMI 6000 florescent microscope (Leica Microsystems GmbH, Wetzler Germany), equipped with a Sutter DG4 light source (Sutter Instruments, California, USA), Ludl emission filter wheel with Chroma band pass emission filters (Ludl Electronic Products Ltd., NY, USA) and Hamamatsu Orca AG camera (Hamamatsu Photonics, Herrsching am Ammersee, Germany). Images were acquired and analyzed using Velocity Software (Perkin Elmer).
Preparation of cell extracts
Overnight cultures of yeast strains cultured in YPD were diluted to an OD600 of 0.2 in YPD and allowed to reach mid-log growth prior to harvesting. Yeast cells were pelleted at 800 g for 5 min and washed with ice cold sterile water. Cytosolic and particulate extracts were prepared essentially as described previously [28]. Briefly, cells pellets were resuspended in 200 µL of lysis buffer (20 mM HEPES, 150 mM NaCl, 2 mM EDTA and protease inhibitor cocktail (Sigma, P-8215)) and lysed by vortexing with glass beads. Glass beads and intact cells were first removed by brief centrifugation. Particulate and cytosolic fractions were collected and separated by centrifugation at 13 000 rpm for 15 min at 4°C. Particulate fractions were resuspended in an equal volume of lysis buffer. Protein concentration was determined using Bradford reagent.
In vitro PLD assays
Cellular and particulate extracts were added in equal volume to a reaction mixture (500 mM octylglucoside, 400 mM NaCl, 60 mM HEPES (pH 7.0) and 1% v/v n-butanol) containing 200 µM BODIPY labeled glycerophosphocholine (2-decanoyl-1-(O-(11-(4,4-difluoro-5,7-dimethyl-4-bora-2a,4a-diaza-s-indacene-3-propionyl)amino)undecyl)-sn-glycero-3-phosphocholine, Invitrogen, D-3771) in the absence of exogenous PIP2, which was not observed to affect measured PLD activity (data not shown), as previously described [28]. Reactions were incubated at 30°C for 40 min prior to spotting on TLC plates (EMD chemicals, 5626-6). Products were visualized under UV light and images captured using Quantity One 4.6.1 software (Biorad) following separation in chloroform/methanol/water/acetic acid (45 : 45 : 10 : 1).
Immunoblotting
10 µg of total protein from particulate and cytosolic fractions were separated by SDS-PAGE on 5% SDS-polyacrylamide gels. Proteins were transferred to nitrocellulose at 0.8 mA/cm2 for 2 h prior to blocking overnight in 5% skim milk in TBS-T. Standard Western blotting procedures were performed using α-HA (Roche, 11667149), α-GFP (Roche, 11874460001) and peroxidase-conjugated goat α-mouse IgG (BioRad, 170-6516).
N2a cell culture and survival assay
N2a cells were maintained in DMEM/F12 media containing 10% fetal bovine serum, L-glutamine and penicillin/streptomycin. Cells were plated at 1.85×104 cells per well in 24 well plates prior to treatment. Treatment with vehicle (0.1% DMSO + 0.1% Ethanol), the small molecule PLD inhibitor N-(2-{4-[2-oxo-2,3-dihydro-1H-benzo(d)imidazol-1-yl]piperidin-1-yl}ethyl)-2-naphthamide (VUO155056, Avanti Polar Lipids, 857370) + 0.1% EtOH, C16 : 0 PAF (PAF, 1 µM) + 0.1% DMSO, or the PLD inhibitor in the presence of C16 : 0 PAF (PAF + Inh) were performed in serum free complete DMEM/F12 supplemented with 0.025% bovine serum albumin (BSA) for 24 h. Cell survival was assessed using LIVE/DEAD reagent (Invitrogen, L3224). Viable cells in each treatment condition were quantified and standardized to vehicle only treated cells. Data represent the results from the measurement of 4 fields of view from 3-4 independent wells per condition ± SEM (n = 12-16). Statistical analyses were one-way analysis of variance (ANOVA) followed by post-hoc Student-Newman-Keuls multiple comparisons test or Student's t test where only two experimental groups were analyzed.
Supporting Information
{kind=link}
Zdroje
1. RyanSD
WhiteheadSN
SwayneLA
MoffatTC
HouW
2009 Amyloid-{beta}42 signals tau hyperphosphorylation and compromises neuronal viability by disrupting alkylacylglycerophosphocholine metabolism. Proc Natl Acad Sci U S A 106 20936 20941
2. Sanchez-MejiaRO
NewmanJW
TohS
YuGQ
ZhouY
2008 Phospholipase A2 reduction ameliorates cognitive deficits in a mouse model of Alzheimer's disease. Nat Neurosci 11 1311 1318
3. KleinJ
2000 Membrane breakdown in acute and chronic neurodegeneration: focus on choline-containing phospholipids. J Neural Transm 107 1027 1063
4. SweetRA
PanchalingamK
PettegrewJW
McClureRJ
HamiltonRL
2002 Psychosis in Alzheimer disease: postmortem magnetic resonance spectroscopy evidence of excess neuronal and membrane phospholipid pathology. Neurobiol Aging 23 547 553
5. KriemB
SponneI
FifreA
Malaplate-ArmandC
Lozac'h-PillotK
2005 Cytosolic phospholipase A2 mediates neuronal apoptosis induced by soluble oligomers of the amyloid-beta peptide. FASEB J 19 85 87
6. KitaY
OhtoT
UozumiN
ShimizuT
2006 Biochemical properties and pathophysiological roles of cytosolic phospholipase A2s. Biochim Biophys Acta 1761 1317 1322
7. CalonF
LimGP
YangF
MoriharaT
TeterB
2004 Docosahexaenoic acid protects from dendritic pathology in an Alzheimer's disease mouse model. Neuron 43 633 645
8. BateC
TayebiM
WilliamsA
2010 Phospholipase A2 inhibitors protect against prion and Abeta mediated synapse degeneration. Mol Neurodegener 5 13
9. LiJ
HuJ
ShaoB
ZhouW
CuiY
2009 Protection of PMS777, a new AChE inhibitor with PAF antagonism, against amyloid-beta-induced neuronal apoptosis and neuroinflammation. Cell Mol Neurobiol 29 589 595
10. RyanSD
HarrisCS
MoF
LeeH
HouST
2007 Platelet activating factor-induced neuronal apoptosis is initiated independently of its G-protein coupled PAF receptor and is inhibited by the benzoate orsellinic acid. J Neurochem 103 88 97
11. HoonS
St OngeRP
GiaeverG
NislowC
2008 Yeast chemical genomics and drug discovery: an update. Trends Pharmacol Sci 29 499 504
12. KhuranaV
LindquistS
2010 Modelling neurodegeneration in Saccharomyces cerevisiae: why cook with baker's yeast? Nat Rev Neurosci 11 436 449
13. NielsenJ
2009 Systems biology of lipid metabolism: from yeast to human. FEBS Lett 583 3905 3913
14. NakayamaR
KumagaiH
SaitoK
1994 Evidence for production of platelet-activating factor by yeast Saccharomyces cerevisiae cells. Biochim Biophys Acta 1199 137 142
15. NakayamaR
UdagawaH
KumagaiH
1997 Changes in PAF (platelet-activating factor) production during cell cycle of yeast Saccharomyces cerevisiae. Biosci Biotechnol Biochem 61 631 635
16. NakayamaR
YunC
TamakiH
SaitoK
KumagaiH
1996 Physiological action of PAF in yeast Saccharomyces cerevisiae. Adv Exp Med Biol 416 45 50
17. ZarembergV
McMasterCR
2002 Differential partitioning of lipids metabolized by separate yeast glycerol-3-phosphate acyltransferases reveals that phospholipase D generation of phosphatidic acid mediates sensitivity to choline-containing lysolipids and drugs. J Biol Chem 277 39035 39044
18. ZarembergV
GajateC
CacharroLM
MollinedoF
McMasterCR
2005 Cytotoxicity of an anti-cancer lysophospholipid through selective modification of lipid raft composition. J Biol Chem 280 38047 38058
19. BrewerC
BoninF
BullockP
NaultMC
MorinJ
2002 Platelet activating factor-induced apoptosis is inhibited by ectopic expression of the platelet activating factor G-protein coupled receptor. J Neurochem 82 1502 1511
20. GavinAC
AloyP
GrandiP
KrauseR
BoescheM
2006 Proteome survey reveals modularity of the yeast cell machinery. Nature 440 631 636
21. KroganNJ
CagneyG
YuH
ZhongG
GuoX
2006 Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440 637 643
22. LambertJP
MitchellL
RudnerA
BaetzK
FigeysD
2009 A novel proteomics approach for the discovery of chromatin-associated protein networks. Mol Cell Proteomics 8 870 882
23. LambertJP
BaetzK
FigeysD
2010 Of proteins and DNA—proteomic role in the field of chromatin research. Mol Biosyst 6 30 37
24. RudgeSA
MorrisAJ
EngebrechtJ
1998 Relocalization of phospholipase D activity mediates membrane formation during meiosis. J Cell Biol 140 81 90
25. HonigbergSM
ConicellaC
EspositioRE
1992 Commitment to meiosis in Saccharomyces cerevisiae: involvement of the SPO14 gene. Genetics 130 703 716
26. ConnollyJE
EngebrechtJ
2006 The Arf-GTPase-activating protein Gcs1p is essential for sporulation and regulates the phospholipase D Spo14p. Eukaryot Cell 5 112 124
27. MarstonAL
ThamWH
ShahH
AmonA
2004 A genome-wide screen identifies genes required for centromeric cohesion. Science 303 1367 1370
28. EllaKM
DolanJW
MeierKE
1995 Characterization of a regulated form of phospholipase D in the yeast Saccharomyces cerevisiae. Biochem J 307 799 805
29. EllaKM
MeierGP
BradshawCD
HuffmanKM
SpiveyEC
1994 A fluorescent assay for agonist-activated phospholipase D in mammalian cell extracts. Anal Biochem 218 136 142
30. RiedelCG
MazzaM
MaierP
KornerR
KnopM
2005 Differential requirement for phospholipase D/Spo14 and its novel interactor Sma1 for regulation of exocytotic vesicle fusion in yeast meiosis. J Biol Chem 280 37846 37852
31. LoewenCJ
GasparML
JeschSA
DelonC
KtistakisNT
2004 Phospholipid metabolism regulated by a transcription factor sensing phosphatidic acid. Science 304 1644 1647
32. CaiD
NetzerWJ
ZhongM
LinY
DuG
2006 Presenilin-1 uses phospholipase D1 as a negative regulator of beta-amyloid formation. Proc Natl Acad Sci U S A 103 1941 1946
33. CaiD
ZhongM
WangR
NetzerWJ
ShieldsD
2006 Phospholipase D1 corrects impaired betaAPP trafficking and neurite outgrowth in familial Alzheimer's disease-linked presenilin-1 mutant neurons. Proc Natl Acad Sci U S A 103 1936 1940
34. ScottSA
SelvyPE
BuckJR
ChoHP
CriswellTL
2009 Design of isoform-selective phospholipase D inhibitors that modulate cancer cell invasiveness. Nat Chem Biol 5 108 117
35. RyanSD
HarrisCS
CarswellCL
BaenzigerJE
BennettSA
2008 Heterogeneity in the sn-1 carbon chain of platelet-activating factor glycerophospholipids determines pro - or anti-apoptotic signaling in primary neurons. J Lipid Res 49 2250 2258
36. FairnGD
CurwinAJ
StefanCJ
McMasterCR
2007 The oxysterol binding protein Kes1p regulates Golgi apparatus phosphatidylinositol-4-phosphate function. Proc Natl Acad Sci U S A 104 15352 15357
37. RoseK
RudgeSA
FrohmanMA
MorrisAJ
EngebrechtJ
1995 Phospholipase D signaling is essential for meiosis. Proc Natl Acad Sci U S A 92 12151 12155
38. RudgeSA
CavenaghMM
KamathR
SciorraVA
MorrisAJ
1998 ADP-Ribosylation factors do not activate yeast phospholipase Ds but are required for sporulation. Mol Biol Cell 9 2025 2036
39. MendonsaR
EngebrechtJ
2009 Phospholipase D function in Saccharomyces cerevisiae. Biochim Biophys Acta 1791 970 974
40. PaiJK
SiegelMI
EganRW
BillahMM
1988 Phospholipase D catalyzes phospholipid metabolism in chemotactic peptide-stimulated HL-60 granulocytes. J Biol Chem 263 12472 12477
41. KiharaY
IshiiS
KitaY
TodaA
ShimadaA
2005 Dual phase regulation of experimental allergic encephalomyelitis by platelet-activating factor. J Exp Med 202 853 863
42. WaltherTC
BricknerJH
AguilarPS
BernalesS
PantojaC
2006 Eisosomes mark static sites of endocytosis. Nature 439 998 1003
43. AguilarPS
FrohlichF
RehmanM
ShalesM
UlitskyI
2010 A plasma-membrane E-MAP reveals links of the eisosome with sphingolipid metabolism and endosomal trafficking. Nat Struct Mol Biol
44. OliveiraTG
Di PaoloG
2010 Phospholipase D in brain function and Alzheimer's disease. Biochim Biophys Acta 1801 799 805
45. LongtineMS
McKenzieA3rd
DemariniDJ
ShahNG
WachA
1998 Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14 953 961
46. RigautG
ShevchenkoA
RutzB
WilmM
MannM
1999 A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol 17 1030 1032
47. AbelsonJN
SimonML
GutherieC
FinkGR
2004 Guide to Yeast Genetics and Molecular Biology. California Elsevier Academic Press
48. MemarianN
JessulatM
AlirezaieJ
Mir-RashedN
XuJ
2007 Colony size measurement of the yeast gene deletion strains for functional genomics. BMC Bioinformatics 8 117
49. ParsonsAB
BrostRL
DingH
LiZ
ZhangC
2004 Integration of chemical-genetic and genetic interaction data links bioactive compounds to cellular target pathways. Nat Biotechnol 22 62 69
50. ParsonsAB
LopezA
GivoniIE
WilliamsDE
GrayCA
2006 Exploring the mode-of-action of bioactive compounds by chemical-genetic profiling in yeast. Cell 126 611 625
51. PinheiroJC
BatesDM
2000 Mixed-Effects Models in S and S-Plus; Chambers J, editor. New York Springer
52. PinheiroJC
BatesDM
DebRoyS
SarkarD
2008 nlme: Linear and Nonlinear Mixed Effects Models. R package 3.1-90 ed ed
53. Team RDC 2008 R: A Language and Environment for Statistical Computing. Vienna, Austria R Foundation for Statistical Computing
54. SikorskiRS
HieterP
1989 A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122 19 27
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 2
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Meta-Analysis of Genome-Wide Association Studies in Celiac Disease and Rheumatoid Arthritis Identifies Fourteen Non-HLA Shared Loci
- MiRNA Control of Vegetative Phase Change in Trees
- Risk Alleles for Systemic Lupus Erythematosus in a Large Case-Control Collection and Associations with Clinical Subphenotypes
- The Cardiac Transcription Network Modulated by Gata4, Mef2a, Nkx2.5, Srf, Histone Modifications, and MicroRNAs
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy