
Genome-Wide Transcript Profiling of Endosperm without Paternal Contribution Identifies Parent-of-Origin–Dependent Regulation of
Seed development in angiosperms is dependent on the interplay among different transcriptional programs operating in the embryo, the endosperm, and the maternally-derived seed coat. In angiosperms, the embryo and the endosperm are products of double fertilization during which the two pollen sperm cells fuse with the egg cell and the central cell of the female gametophyte. In Arabidopsis, analyses of mutants in the cell-cycle regulator CYCLIN DEPENDENT KINASE A;1 (CKDA;1) have revealed the importance of a paternal genome for the effective development of the endosperm and ultimately the seed. Here we have exploited cdka;1 fertilization as a novel tool for the identification of seed regulators and factors involved in parent-of-origin–specific regulation during seed development. We have generated genome-wide transcription profiles of cdka;1 fertilized seeds and identified approximately 600 genes that are downregulated in the absence of a paternal genome. Among those, AGAMOUS-LIKE (AGL) genes encoding Type-I MADS-box transcription factors were significantly overrepresented. Here, AGL36 was chosen for an in-depth study and shown to be imprinted. We demonstrate that AGL36 parent-of-origin–dependent expression is controlled by the activity of METHYLTRANSFERASE1 (MET1) maintenance DNA methyltransferase and DEMETER (DME) DNA glycosylase. Interestingly, our data also show that the active maternal allele of AGL36 is regulated throughout endosperm development by components of the FIS Polycomb Repressive Complex 2 (PRC2), revealing a new type of dual epigenetic regulation in seeds.
Published in the journal:
. PLoS Genet 7(2): e32767. doi:10.1371/journal.pgen.1001303
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001303
Summary
Seed development in angiosperms is dependent on the interplay among different transcriptional programs operating in the embryo, the endosperm, and the maternally-derived seed coat. In angiosperms, the embryo and the endosperm are products of double fertilization during which the two pollen sperm cells fuse with the egg cell and the central cell of the female gametophyte. In Arabidopsis, analyses of mutants in the cell-cycle regulator CYCLIN DEPENDENT KINASE A;1 (CKDA;1) have revealed the importance of a paternal genome for the effective development of the endosperm and ultimately the seed. Here we have exploited cdka;1 fertilization as a novel tool for the identification of seed regulators and factors involved in parent-of-origin–specific regulation during seed development. We have generated genome-wide transcription profiles of cdka;1 fertilized seeds and identified approximately 600 genes that are downregulated in the absence of a paternal genome. Among those, AGAMOUS-LIKE (AGL) genes encoding Type-I MADS-box transcription factors were significantly overrepresented. Here, AGL36 was chosen for an in-depth study and shown to be imprinted. We demonstrate that AGL36 parent-of-origin–dependent expression is controlled by the activity of METHYLTRANSFERASE1 (MET1) maintenance DNA methyltransferase and DEMETER (DME) DNA glycosylase. Interestingly, our data also show that the active maternal allele of AGL36 is regulated throughout endosperm development by components of the FIS Polycomb Repressive Complex 2 (PRC2), revealing a new type of dual epigenetic regulation in seeds.
Introduction
Seed development is a tightly regulated process that is controlled, both before and after fertilization and requires tight coordination of parental gene expression [1]. A paradigm for the importance of balanced parental contribution is the observation that certain genes in the developing offspring of flowering plants are exclusively or preferentially expressed from only one of the two parental genomes, a phenomenon called genomic imprinting that has also been observed in mammals [2], [3]. The relevance of parent-of-origin effects was first found in interploidy crosses [4]. Typically, an increase in the paternal genome results in larger seeds, while the opposite is observed if the maternal gene dosage is higher than normal [5]. This is in agreement with the parental conflict theory, which implies that fathers direct maximal amount of maternal resources to their own offspring and thereby promote growth. Mothers on the other hand would seek to distribute the resources equally among all their offspring, and balance their resource between themselves and their offspring. Thus, maternal factors are thought to dampen growth [6].
In mammals, imprinted genes are often involved in growth control [7]–[10]. In Arabidopsis, the endosperm is the major tissue regulating the flow of nutrients to the embryo, and is therefore a likely site for parent-of-origin dependent gene expression.
Imprinting results from differences in epigenetic marks, involving DNA methylation and post-translational modifications of histones on the parental alleles [11], [12]. Trimethylation of lysine 27 on histone H3 (H3K27me3) leading to repression of gene expression, has been found to be a particularly important imprinting mechanism in plants. In Arabidopsis seeds, H3K27me3 mark is set by the FIS Polycomb Repressive Complex 2 (PRC2), which consists of at least four components; the histone methyltransferase MEDEA (MEA), FERTILIZATION INDEPENDENT SEED 2 (FIS2), FERTILIZATION INDEPENDENT ENDOSPERM (FIE), and MULTICOPY SUPPRESSOR OF IRA 1 (MSI1). The corresponding genes were identified in screens for autonomous endosperm development, indicating that the FIS complex acts as a repressor of endosperm development prior to fertilization [13]–[17].
An equally important regulatory mechanism in imprinting is DNA methylation resulting from the activity of several different methyltransferase enzymes, where each has specificity for cytosine (C) in certain sequence contexts. So far, imprinting has been shown to be under the influence of MET1, the major Arabidopsis maintenance DNA methyltransferase involved in CG-methylation [11], [18]–[20]. DNA demethylation can be achieved either by a passive process i.e. the repression of MET1 expression [21], [22], or by an active mechanism involving DNA glycosylase enzymes such as DME [23]. Several lines of evidence show that DME, which is expressed in the central cell of the female gametophyte, is necessary for maternal-specific gene expression in the endosperm [11], [18], [19], [24].
So far, only about a dozen genes in Arabidopsis have been identified to have parental-specific gene expression, and they illustrate different modes of imprinting [3]. MEA, ARABIDOPSIS FORMIN HOMOLOGUE 5 (AtFH5) and PHERES 1 (PHE1) are imprinted by the action of FIS PRC2, where only the latter is paternally expressed [13], [25]–[31]. FIS2, FLOWERING WAGENINGEN (FWA) and MATERNALLY EXPRESSED PAB C-TERMINAL (MPC) are all maternally expressed and regulated by the dual action of MET1 and DME [11], [19], [24], [32]–[34]. Recently, five novel imprinted genes, HOMEODOMAIN GLABROUS 3 (HDG3), HDG8, HDG9, At5g62110 and ATMYB3R2 were identified by differential DNA methylation in embryo and endosperm [35].
In comparison to Arabidopsis, more than 100 genes have been shown to have a uniparental or preferential parental expression pattern in mammals [36]–[39]. This suggests that additional genes in Arabidopsis are imprinted. Furthermore, the low number of known imprinted genes in plants precludes the identification of general principles in this kind of gene expression control and thus, the identification of further imprinted genes is pivotal. Moreover, the targets of imprinted genes, as well as genomic pathways and regulatory modules influenced by imprinted genes are largely unknown.
Here, we have designed a microarray strategy for the identification of seed regulators by exploiting the cdka;1 mutation. Using this approach, we have identified a cluster of previously uncharacterized AGAMOUS-LIKE (AGL) Type-I MADS-box transcription factors that are downregulated in endosperm with no paternal contribution. Here, we report that AGL36 is imprinted by the dual action of MET1 and DME. In addition, AGL36 is regulated throughout endosperm development in its maternal expression cycle by the Polycomb FIS-complex, thereby identifying a novel mode of regulation for imprinted genes.
Results
cdka;1 is a tool to identify key seed regulators
Here we have used cdka;1 as a tool to identify factors sensitive to the vital parental gene balance in the endosperm. In heterozygous cdka;1 mutants, the second pollen mitosis is either missing or is severely delayed. However, mutant pollen can successfully fertilize the egg cell while leaving the central cell unfertilized [40], [41]. A detailed analysis by Aw and colleagues has revealed that a second sperm cell is delivered to the central cell, but that karyogamy does not take place [42]. Although not properly fertilized, the majority of the central cells in cdka;1 fertilized ovules (70–90%) are triggered to initiate endosperm proliferation [40], [42], [43]. Thus, fertilization by cdka;1 sperm cells creates a unique situation where endosperm initially develops without any paternal contribution (in the following also referred to as cdka;1P). The endosperm, however remains under-developed, and ultimately the seed aborts, further demonstrating the importance of the paternal contribution to the endosperm for proper seed development. Since activation of maternal alleles by loss of maternal FIS PRC2 could rescue seed lethality [43], we hypothesized that the disturbance of parental gene balance in the endosperm is the main cause leading to developmental arrest of cdka;1P at 3–4 days after pollination (DAP).
To identify factors and mechanisms sensitive to such an imbalance in gene dosage in the endosperm and with that likely key regulators of seed development, we performed microarray transcript profiling of cdka;1 fertilized seeds at 3 DAP (Figure S1A). Due to the heterozygous nature of the cdka;1 mutant line used, a transcript that is absent in cdka;1p seeds will lead to a reduction of maximal 50% in the genome profiling experiment. For example, genes that are only expressed from the paternal genome would show such reduced expression levels (Figure S1B). Likewise, maternally expressed genes that require activation by a paternally expressed gene(s) would be downregulated (Figure S1C), whereas genes that are acted upon by paternally expressed repressors were expected to be upregulated in the microarray screen (Figure S1D).
When we compared the transcriptional profiles of Ler x cdka;1 versus Ler x Col seeds 3 DAP, we detected 17223 nuclear genes that were expressed in all biological replicates of both mutant (cdka;1 set) and wild-type (WT set) seed profiles. Our result is in good agreement with a set of genes identified by Goldberg & Harada laboratories (GH) in globular stage seeds of Arabidopsis Ws-0 plants as 68% of our genes were also identified by GH, and our gene set included >90% of the GH globular seed gene set (Figure 1A; http://seedgenenetwork.net, [44]).

To further validate the quality of our dataset, we examined the expression pattern of genes known to be preferentially expressed from the paternal allele. To date, only three genes have been identified that show a predominant paternal expression pattern; PHE1, HDG3 and At5g62110, where all three genes were found to be downregulated in our arrays (Figure S1E), supporting our working hypothesis that paternally expressed genes can be detected amongst downregulated genes. In addition, out of seven imprinted maternally expressed genes present in our microarray sets, four were also detected as downregulated (Figure S1E). This could reflect required activation by paternal factors (Figure S1C), or be a result of more complex deregulation in response to change in gene dosage. To exclude array artifacts we tested all down-regulated genes by means of real-time PCR and could confirm their deregulation (Figure 1B).
Due to the background noise in the microarray experiment, modest but reproducible downregulation of arithmetic ratios (ar) ranging from 0.5 to 1.0 will produce False Discovery Rates (FDR, see materials and methods) with insignificant q values. Since the absence of paternally expressed genes was the simplest hypothesis to account for downregulation, we defined a functional limit for screening purposes that allowed us to detect two out of three known paternally expressed genes in the array. Both PHE1 and HDG3 are detected at q values of 0.35 and a downregulation cutoff of 0.8 (ar). Consequently these values were chosen and used to filter the microarray data.
Using these criteria, a set of 602 genes was extracted (q≤0.35 and ar ≤0.8), subsequently called Down 0.8. For upregulation, we worked with two gene sets. For the first set, Up 1.2, we used parameters equivalent to the downregulated set (q≤0.35 and ar ≥1.2), which resulted in a set of 1030 genes. For the second set, Up 1.5, resulting in 323 genes, we chose ar ≥1.5, a threshold for deregulation commonly used in genome-wide expression studies (Table S3).
To test whether the deregulated genes could preferentially be attributed to a certain seed structure, we compared our data to gene sets expressed in different seed regions and compartments of globular stage seeds using data generated by Goldberg & Harada (GH) laboratories available at http://seedgenenetwork.net [44]. The overlap between the upregulated gene sets and the GH embryo, seed coat and endosperm was significantly lower than expected for independent sets of genes, indicating that among the upregulated genes we preferentially find those that are below the detection limit of the GH analyses. However looking at the downregulated genes, the picture was different. While we found slightly less overlap than expected by chance for the GH embryo set, the overlap was clearly larger than expected by chance for GH seed-coat (1.2<2.7e−07) and even more significant for the GH endosperm (rf = 1.3, p<2.0e−13, Figure S2A, S2B).
Lack of the paternal genome results in the downregulation of a group of MADS-box Type-I Mγ transcription factors
In order to functionally classify the deregulated gene sets according to their molecular functions we used the GO Slim classification system (Figure 1C). Only for the GO Slim term “Transcription factor activity” we find a higher percentage and significant over-representation of both up - and down-regulated groups when compared to all genes on the array/all genes expressed. Since key regulators of seed development are likely to be transcription factors (TF), we analyzed this class in detail.
When comparing the fraction of deregulated genes among the different TF families, the Mγ MADS-box transcription factors clearly stood out with more than 60% of the seed expressed members being downregulated in Ler x cdka;1 arrays (Figure S3A, S3B). We therefore focused on this MADS Type-I class for further analysis. Searches in publically available expression databases (www.genevestigator.com, Figure S4) revealed that all identified genes were exclusively expressed in the seed and predominantly in the endosperm. From the identified Type-I Mγ MADS-box genes, we selected AGL36 for further in depth analysis (Figure S4). AGL36 was the previously undescribed Mγ candidate that interacted with the highest number of described AGLs in a Y2H screen performed by de Folter et al [45]. Both AGL36 and PHE1 have been shown to interact with AGL62, which plays a major role in endosperm development [45], [46]. Within the Mγ class, AGL36 clusters together with AGL34 and AGL90 [47], which are both also detected as downregulated in our microarray experiment (Figure S4). AGL36 shares 85.7% and 84% nucleotide identity with AGL34 and AGL90, respectively (Figure S8). On the amino acid level this results in of 80.2% similarity of AGL36 with AGL34 and 83.9% similarity with AGL90.
AGL36 is only expressed from its maternal allele
Real-time PCR measurement of AGL36 relative expression level three days after pollination (3 DAP) in Ler ovules fertilized with either Col or cdka;1 pollen confirmed that AGL36 expression was reduced in cdka;1 fertilized seeds, (27% when normalized towards ACT11, and 36% when normalized towards GAPA) compared to wild-type seeds (Figure 2A).

To determine whether AGL36 has parental-specific expression, we took advantage of an AGL36 Single Nucleotide Polymorphism (SNP) existing between the Col and Ler ecotypes. This SNP allows the PCR product of Col cDNA to be digested by AlwNI, leaving the Ler cDNA PCR product intact (Figure 2B). We performed reciprocal crosses between Col and Ler ecotypes, and analyzed the digested RT-PCR fragments on an Agilent Bioanalyzer Lab-on-a-Chip, allowing accurate measurement of fragment sizes and their concentrations. When Colmaternal is crossed with Lerpaternal, we only detected the Col bands (165 bp+234 bp) after AlwNI digestion, indicating only maternal expression (Figure 2C). Similarly, in the reciprocal cross when Lermaternal is fertilized with Colpaternal pollen, the cDNA PCR digest resulted only in an undigested band (399 bp) originating from Ler, indicative of maternal expression (Figure 2C). This testified that AGL36 was only expressed from the maternal genome after fertilization and thus identified as a novel imprinted gene.
AGL36 is imprinted throughout early seed development
AGL36 expression level in wild-type seeds (Ler x Col) at different stages of seed development was monitored over a period of 12 days after pollination. Initially, a low expression level was detected (1 DAP), followed by a rapid increase and subsequent peak in AGL36 expression at 4 DAP, when the embryo is at the late globular stage of development, before declining (Figure 3A). At the embryo heart stage, corresponding to 6 DAP, AGL36 expression had decreased to similar levels as 1 DAP. To address whether AGL36 imprinting is maintained throughout its expression cycle, we performed a SNP analysis of the RT-PCR product obtained from Ler x Col crosses harvested during 1 to 12 DAP (Figure 3B). We found that AGL36 expression is originating from the maternal genome (Ler) throughout the experiment. By plotting the molarities of the maternal band obtained by Agilent Bioanalyzer, an expression profile closely identical to the pattern obtained in the real-time PCR analysis was found (Figure 3C).

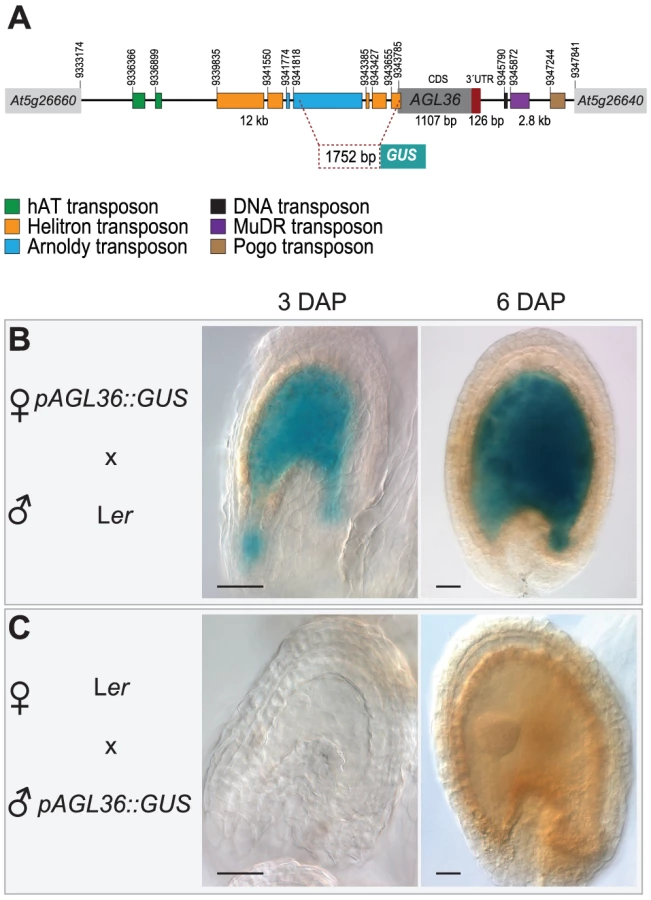
To rule out that the observed maternal expression is due to expression of AGL36 in the ovule integument, which is a maternal tissue, we generated a reporter construct consisting of 1752 bp of the AGL36 promoter region fused to a GUS reporter (pAGL36::GUS) (Figure 4A). Single-copy lines carrying this construct were used in reciprocal crosses with wild-type Ler and Col plants to examine GUS expression at 3 and 6 DAP. When inherited maternally, pAGL36::GUS expression in the seed was indeed found to be restricted only to the fertilization product (Figure 4B, Figure S7D). In the reciprocal cross, when pAGL36::GUS was inherited from the paternal genome, no GUS expression was detected, (Figure 4C, Figure S7E). Consistent with the SNP analysis, this demonstrated that AGL36 was imprinted and only maternally active throughout its expression cycle. Furthermore, the 1.7 Kb promoter fragment used in this analysis appears to be sufficient to confer parent-of-origin specific expression of the reporter.
AGL36 is not required for seed survival
To further investigate the biological function of AGL36, we screened the Koncz T-DNA collection for insertions [48]. We identified a mutant line, agl36-1, harboring a single T-DNA insertion 16 bp upstream of the AGL36 ATG start codon (Figure S5A). The agl36-1 line showed Mendelian segregation of the T-DNA insertion, as 75% of the plants were resistant to Hygromycin (N = 1025, χ2 = 0,83, Table S1).
To test the transmission through the male and female gametes directly, reciprocal crosses of both hemizygous and homozygous agl36-1 mutant plants with wild-type plants were performed (Table S1). In a reciprocal cross, a hemizygous mutant will segregate 50% of the T-DNA resistance marker if the disrupted gene is not vital for gametophyte transmission or function. Thus, gametophyte requirement can be scored directly as reduced frequency of resistant plants [49]. In reciprocal crosses with agl36-1, no transmission distortion through female or male gametophytes could be observed (N = 661, χ2 = 0,13 and N = 1015, χ2 = 0,00 respectively, Table S1).
The position of the T-DNA insertion in agl36-1 predicts AGL36 expression failure, and indeed real-time PCR analyses of 3 DAP seeds of homozygous agl36-1−/− plants compared to Col wild-type indicate a 1000-fold AGL36 downregulation in the mutant seeds (Figure S5B). In line with an imprinted and maternal-only expression of AGL36, close to 50% reduction of the transcript level was observed in 3 DAP hemizygous agl36-1+/− seeds (Figure S5B). We thereby concluded that agl36-1 represents a loss-of-function allele of AGL36.
Although depletion of AGL36 did not interfere with the fitness of the mutant allele in our experimental system, we have shown that AGL36 is specifically expressed from the maternal allele in the fertilization product, in a time frame between 2 and 6 DAP. To investigate whether this was reflected morphologically or developmentally in the developing seed, we compared embryo and endosperm development in wild-type and homozygous agl36-1−/− seeds within the AGL36 expression time frame.
After fertilization of the egg and the central cell, the endosperm in Arabidopsis undergoes three syncytial rounds of nuclear divisions before the first asymmetric division of the zygote that creates the apical embryo proper and the basal suspensor that connects the embryo proper and the maternal tissue (Figure S5C). At the 2 DAP stage, no obvious difference could be observed between wild-type and agl36-1−/− seeds, both typically harboring a 1–2 cell embryo proper and a 16–32 nucleated endosperm (Figure S5C, left section). The embryo continues to divide through radial, longitudinal and transverse divisions to produce the so-called globular stage at 4 DAP (Figure S5C, middle section). The endosperm also undergoes 3–4 syncytial nuclear divisions and remains uncellularized as cell proliferation at the upper half of the embryo forms the cotyledon primordia at the so-called heart stage at 6 DAP (Figure S5C, right section). Although the main AGL36 expression peak occurs during this time frame, no obvious deviation between wild-type and agl36-1−/− could be observed at these stages. Similarly, using an endosperm specific pFIS2::GUS reporter [33], a wild-type endosperm division pattern was observed in agl36-1+/− seeds (Figure S5D).
MET1 is required for AGL36 imprinting
The majority of imprinted, maternally expressed genes identified in Arabidopsis so far have been shown to be paternally silenced by mechanisms involving symmetric CG methylation, maintained by MET1 [11], [18], [19]. Although not directly linked to imprinting, methylation can also be directed by CHROMOMETHYLASE 3 (CMT3) that has specificity for CNG, and members of the DOMAINS REARRANGED METHYLTRANSFERASE (DRM) family; DRM1 and DRM2, that are mainly responsible for asymmetric CHH methylation [50]. In order to address the involvement of DNA methylation in the regulation of paternal AGL36 expression, we performed SNP analyses of 3 DAP ovules from reciprocal crosses with mutants that have been shown to be involved in DNA methylation. In the SNP RT-PCR analysis of mutant pollen crossed to wild-type, paternal AGL36 expression is expected if the tested mutants are involved in AGL36 imprinting.
CMT3 DNA methylation has been reported to be guided to specific sites by KRYPTONITE (KYP) H3K9 methylation [51]. When mutant cmt3-7 and kyp-2 pollen were crossed to Col wild-type plants, no difference in AGL36 expression was observed (Figure 5A). In the reciprocal cross with cmt3-7 also no difference could be detected compared to wild-type expression (Figure S6).

DRM1 and DRM2 are mainly responsible for asymmetric DNA CHH methylation [50] and rely on small interfering RNAs, processed by ARGONAUTE4 (AGO4), for target template guidance [52]. In our assays, fertilization by pollen lacking DRM1;DRM2 and pollen lacking AGO4 had no effect on the AGL36 expression pattern (Figure 5A). Likewise, AGL36 expression in the reciprocal cross was identical to wild-type (Figure S6).
DECREASE IN DNA METHYLATION1 (DDM1) is involved in maintenance of DNA methylation [53]. In our SNP RT-PCR analyses where mutant ddm1-2 pollen was used to fertilize wild-type ovules, paternal AGL36 expression was not activated (Figure 5A). In summary, CMT3, KYP, DRM1;DRM2, AGO4 and DDM1 appear not to be involved in the establishment nor maintenance of AGL36 imprinting (Figure 5A, Figure S6).
However, paternal AGL36 expression was detected when plants hemizygous for the met1-4 mutation were used as pollen donor in crosses with wild-type Ler (Figure 5B). In the reciprocal cross, using met1+/− as the maternal partner, no AGL36 expression from the paternal genome could be observed (Figure 5B). Furthermore, we performed crosses using pollen from homozygous met1-4 parents. When first generation homozygous met1 plants were used as pollen donor on wild-type plants, prominent AGL36 expression from the paternal Col genome could be observed (Figure 5B). This strongly suggests that the repression of the paternal copy of AGL36 is lifted due to the met1-4 mutation, and that MET1 is required for maintaining paternal inactivation of AGL36. In the reciprocal crosses, only expression from the maternal genome could be detected, both in the heterozygous and the homozygous met1-4 situation, further substantiating the requirement of MET1 in the male germline in order to maintain AGL36 imprinting (Figure 5B). Maternal AGL36 expression levels using homozygous met1-4 as the maternal cross partner appeared to be equal to maternal levels in the reciprocal crosses (Figure 5B). This opens for the interpretation that DNA methylation is not required for the regulation of maternal AGL36 expression.
Silencing of vegetative AGL36 expression involves MET1
In public expression databases, AGL36 is reported to be expressed in the seed and more precisely in the endosperm [54] (Figure S4). In order to monitor AGL36 expression in vegetative tissues and its dependence on DNA methylation, we performed a real-time PCR experiment on vegetative tissues from reciprocal Ler x Col crosses and homozygous met1-4 tissues. In biological replicates of progenies from both reciprocal crosses, weak AGL36 expression ranging from 1–6% of the seed expression level could be detected in seedlings, leaves and flowers (Figure 6A). This showed that AGL36 was expressed throughout the plant life cycle, although at very low levels. In the same experiment, we monitored expression in met1-4 tissues. AGL36 expression levels were 50–90-fold higher in met1-4 leaves compared to seed expression levels (Figure 6A). In a direct comparison, expression levels were elevated 2000-fold in homozygous met1-4 leaves compared to wild-type Col x Ler leaves (Figure 6B). In flowers, the upregulation was more than 20-fold in met1-4 compared to wild-type Col x Ler flowers (Figure 6C). In conclusion, these data showed that silencing of AGL36 in vegetative tissues involves MET1, suggesting that the absence of maintenance DNA methylation elevates vegetative AGL36 expression beyond the maternal expression levels found in seeds.

AGL36 is biparentally expressed in vegetative tissues
In order to investigate the parental expression pattern of AGL36 in vegetative tissues, we performed SNP analyses of flowers from F1 hybrids of Ler and Col reciprocal crosses. In both reciprocal crosses, AGL36 appeared to be expressed equally from the parental Ler and Col genomes, indicating biparental expression in flowers (Figure 6D). This indicates that parental-specific expression, i.e. imprinting of AGL36, as expected, only takes place in the seed and that a low basal biparental expression is present throughout the plant life cycle. Interestingly, biallelic expression in flowers suggests that further silencing of AGL36 takes place in the male germline before uniparental expression in the seed (Figure 6D).
AGL36 is controlled by DEMETER
According to our data, the action of MET1 suppresses AGL36 expression throughout the vegetative phase and this suppression is maintained in the fertilization product through the male germline. AGL36 imprinting thus requires specific activation of the maternal allele. DNA demethylation by DME has previously been shown to mediate maternal-specific gene expression in the endosperm [11], [18], [19], [24], and we therefore investigated AGL36 expression in dme-6 mutant plants. Since dme cannot be maintained in a homozygous state, we harvested siliques of dme-6+/− heterozygous plants pollinated with Col pollen at 3 and 6 DAP. We monitored the relative expression by means of real-time PCR using FWA and FIS2 as controls. At 3 DAP, both controls were downregulated by 69±0.09% and 53±0.30% respectively (Figure 6E), in line with a lack of functional DME in 50% of the seeds in heterozygous dme-6+/− plants. AGL36 was downregulated in a similar manner as FIS2 (41±0.20%), suggesting that DME is indeed involved in early activation of the maternal AGL36 allele.
Expression of maternal AGL36 is regulated by the PRC2 FIS-complex
We also tested the expression of FWA and FIS2 in 6 DAP samples and found that their downregulation were sustained as predicted (Figure 6E). However, to our surprise AGL36 expression in dme-6+/− seeds was elevated more than 50-fold (Figure 6E). This result was unexpected, and implicated a more intricate regulation of AGL36.
DME is required for the activation of MEA, the core histone H3K27 methyltransferase (HMTase) of the PRC2 FIS-complex [46], [55], [56]. To determine whether PRC2 FIS is involved in the regulation of AGL36, we analyzed the relative expression of AGL36 over time (1 to 12 DAP) in mea mutant seeds compared to wild-type (Figure 7A). While AGL36 expression in wild-type seeds was at its maximum at 4 DAP, we observed that AGL36 expression in mea seeds surpassed the maximum levels of wild-type at 4 DAP, and reached its highest levels at around 6 DAP. At this point, the AGL36 relative expression in mea mutant seeds was approximately 40-fold higher than wild-type expression at the same stage, and 7-fold higher than the maximum AGL36 level found in wild-type seeds at 4 DAP (Figure 7A). Our data thus indicate that the FIS-complex is indeed a repressor of AGL36 expression, and could also explain the elevated AGL36 expression level in 3 DAP dme-6+/− seeds (Figure 6E). In line with these findings, we found highly elevated AGL36 relative expression levels in mutant seeds from three different mutant alleles of mea (Figure 7C). Similar results were also obtained with mutants of other components of the FIS PRC2 complex (FIS2, FIE and MSI1, data not shown).

To investigate whether FIS activity was exerted on the maternal and/or paternal allele of AGL36, we performed SNP analyses on the RT-PCR product of AGL36 obtained from mea mutant plants (in Ler background) pollinated with Col wild-type pollen. We found that AGL36 is expressed only from its maternal allele in the mea background throughout the duration of our experiment (Figure 7B). In comparison to the expression pattern in wild-type (Figure 3B), strong ectopic maternal expression was also observed at 9 and 12 DAP stages. No paternal expression could be observed in these stages. By plotting the molarities of the maternal band detected by the Agilent Bioanalyzer, an expression profile for the maternal allele could be generated (Figure 7B, lower panel). This demonstrated that in the absence of MEA, AGL36 expression continues to increase after 4 DAP, and although the intensity decreases from 6 DAP, high level of AGL36 is maintained at 12 DAP. Hence, the FIS-complex represses the maternal allele of AGL36 after the 3 DAP stage.
To further substantiate that maternal AGL36 expression is regulated by the maternal action of MEA, we crossed mea mutant plants with pollen expressing the pAGL36::GUS reporter line. Here, no obvious activation of the paternal transgene could be observed at 3 DAP (Figure S7A). Surprisingly, at 6 DAP, corresponding to embryo heart stage, weak expression of the paternal copy in the embryo could be found (Figure S7A). In addition, we performed reciprocal crosses with the pAGL36::GUS reporter line in mutant mea background. When the transgene was contributed from the female side in mea background, a GUS signal was found in 3 DAP stages that increased drastically up to 6 DAP (Figure S7B). In the reciprocal cross however, no expression could be observed (Figure S7C).
The E(z) class of H3K27 histone methyltransferases (HMTases) in Arabidopsis consists of MEA, SWINGER (SWN) and CURLY LEAF (CLF) that participate in different PRC2 complexes. To test whether AGL36 repression is a specific function of FISMEA PRC2, we analyzed AGL36 expression in homozygous swn-4 and clf-2 seeds. For mutants of both HMTases values similar to the wild-type situation were found, and in conclusion AGL36 appear to be specifically regulated by FISMEA PRC2 (Figure 7C).
In summary, maternal AGL36 expression appears to be repressed specifically by the maternal action of FIS PRC2.
PRC2 acts on a subset of MET1/DME–regulated genes
For all genes known to be imprinted by PRC2, the FIS-complex is involved in the repression of the silenced allele [25]-[27], [30], [56]. Our data suggest that silencing of the paternal AGL36 allele requires MET1 whereas the maternal allele is activated by DME. Modulation of female AGL36 expression by PRC2 thus represents a novel mechanism in this type of gene expression system, and adds an additional level of parent-of-origin specific gene expression to the scheme. In order to investigate if this regulation applies to other genes imprinted by the dual action of MET1/DME [11], [18], [19], we analyzed the relative expression levels of FWA, FIS2, AGL36 and MPC in a mea mutant. At 3 DAP expression levels were unchanged or slightly downregulated (0.40–0.99) for all genes tested (Figure 7D). However, while the expression of FWA and FIS2 remained stable at 6 DAP, AGL36 and MPC levels were elevated up to 80-fold (Figure 7D). Thus, genes imprinted by means of MET1/DME can be divided in two classes based on their dependence of FIS PRC2 for additional regulation of the expressed allele. Whereas one class appears not to be regulated by FIS PRC2, the other class depends on the action of the FIS-complex for developmental regulation of its expression.
Discussion
We have performed genome-wide microarray transcript profiling of seeds with only maternal endosperm as a screening method to identify novel regulators of seed development. Previous experiments have shown that a paternal genomic contribution is essential in wild-type Arabidopsis plants for successful seed development. Thus, our working hypothesis was that in the absence of the paternal genome in the endosperm, key regulators of seed development are not present or not effectively transcribed.
Using selection criteria that allowed for the identification of known paternally expressed genes, we extracted a set of downregulated genes that significantly overlapped with a set of endosperm expressed genes identified by Goldberg & Harada laboratories. The GO-Slim term Transcription factor activity was overrepresented in both down - and up-regulated gene sets, and a closer analysis revealed a striking overrepresentation of the Type-I Mγ MADS-box class among the downregulated transcription factors. With the selection criteria used, each detected gene could be a false positive at a probability of 0.35 at the highest, and thus a thorough examination of candidate genes, as performed in this report for AGL36, will be required.
MADS-box transcription factors play important roles in developmental control and signal transduction pathways in most if not all eukaryotes [57]. They are divided into two groups: the very well studied Type-II group (46 genes) including the MIKC class with important regulators such as AGAMOUS, and the Type-I group (61 genes), on which there is very limited information related to function [54], [58], [59]. Emerging data suggest that Type-I MADS-box genes differ from Type-II genes by being involved in female gametophyte and seed development [46], [60]–[62]. In addition they were found to be only weakly expressed, and most members of this group contain no introns [63].
A comprehensive interaction study with members of the Arabidopsis MADS-box protein family by de Folter and colleagues indicated a complex network of interactions between these proteins (Figure S4). It revealed for instance that PHE1 interacts with AGL62, which in turn interacts with both AGL36 and AGL80. AGL62 itself is regulated by the FIS-complex, and functions as a suppressor of endosperm cellularization [46], [59]. PHE1 and AGL36 on the other hand both interact with AGL28. In addition, mutant analysis has shown that AGL80 function is required for the expression of DME in the central cell, and is therefore an upstream regulator of FIS PRC2 [60]. Moreover, AGL61 is required for central cell development, and there is evidence that a heterodimerization between AGL61 and AGL80 is necessary for AGL61 translocation to the nucleus [59], [62]. PHE1 expression is upregulated in A. thaliana (At) x A. arenosa (Aa) incompatible hybrids due to loss of maternal PHE1 silencing, and introgression of phe1 could improve seed viability in semi-compatible 4xAt x 2xAa crosses [64]. In A. thaliana, expression of a PHE1 antisense construct (MEApromoter::asPHE1) could partially restore the seed abortion phenotype in mea mutants [29]. Peculiarly, PHE1 loss-of-function has no phenotypic effect in A. thaliana [56]. However, given the high sequence similarity within the Mγ class of Type-I MADS-box factors, it is possible that MEApromoter::asPHE1 silenced not only PHE1 but also many other Mγ class genes. Taken together, it seems likely that additional Type-I MADS-box factors are upregulated in mea mutants and a collective downregulation by antisense PHE1 would thus restore some of the defects in mea.
In the cluster of Type-I AGL proteins identified in our screen we also found a large overlap with genes recently shown to be upregulated in incompatibly balanced At x Aa crosses compared to semi-compatible At x Aa maternal excess crosses (AGL35, AGL36, PHE1, PHE2, AGL62, AGL90) [65]. In accordance, mutations of both AGL62 and AGL90 partially restore seed lethality in incompatibly balanced At x Aa crosses, accompanied with selective transmission of the mutant alleles. This array of genes was also found to be upregulated in a PRC2 fis2 mutant [65]. In addition, AGL36, AGL62, AGL90 and PHE1 were commonly upregulated in transcriptional profiles of At paternal excess crosses using tetraploid or unreduced jason (jas) pollen [66].
Together with these recent findings, the network of interactions with AGL62 (AGL36, PHE1, PHE2, AGL90) and PHE1 (AGL40, AGL62) and interactors of these proteins (AGL40, AGL45 and AGL90) strongly suggest that the here identified cluster of Type-I AGL proteins plays key roles in parent-of-origin dependent regulation of seed development. An in-depth study of different members of this group will therefore be of great value in understanding this process, and aid the identification of novel imprinted genes.
AGL36 imprinting requires MET1
Here, we report that AGL36 is a novel imprinted gene that is only expressed from its maternal allele in the endosperm. Silencing of the paternal allele requires the action of MET1, as paternal expression is restored in met1 mutants.
In public high-density DNA methylation maps prepared from wild-type seedlings (http://signal.salk.edu), both the AGL36 transcribed region and the 5′and 3′regulatory regions are decorated by CG methylation. In line with this, AGL36 was expressed at very low levels in vegetative tissues. Transcript levels however, were highly elevated in the absence of MET1, in accordance with the virtual absence of CG methylation in met1 mutants (http://signal.salk.edu) [67].
AGL36 is expressed from both parental alleles at low levels in vegetative tissues, which show that AGL36 imprinting occurs specifically in the endosperm. Other imprinted genes in Arabidopsis have been shown to have biallelic expression in the embryo and other vegetative tissues [11], [34], [68]. However, for most imprinted genes this issue is not clarified [3]. Since paternal AGL36 expression is absent in the seed, it suggests that further silencing of AGL36 takes place by entry into the male germline. Moreover, silencing in the female germline must be lifted to allow AGL36 expression in the seed. Alternatively, maintenance methylation and further silencing do not take place on the AGL36 gene in the female gametophyte. The majority of previously described imprinted genes are regulated by a dual switch of methylation and demethylation involving MET1 and DME [11], [18]–[20], [35]. Here we have shown that AGL36 expression is reduced in a dme mutant, indicating that DME has an activating function towards AGL36. In accordance with this, mutants of CMT3, KYP, AGO4, DDM1 and DRM1/2 had no effect on paternal AGL36 expression suggesting that maintenance and repression by MET1 and activation by DME is sufficient for AGL36 imprinting.
In our SNP analyses, a weak paternal signal was observed only at the 2DAP stage. This was interpreted as an artifact since the signal was absent both before and after this stage. If this is a real paternal signal, it suggests an alternative hypothesis where silencing is achieved in the endosperm post fertilization. Further analyses are however required to support this.
In two recent studies, the genome-wide methylation profile of the seed was dissected by comparing cytosine methylation in wild-type embryos to wild-type and dme endosperm. This showed that endosperm development, and hence the activity of endosperm-specific genes, is marked by an extensive demethylation of the maternal genome, especially at specific transposon sequences [35], [69]. According to the Zilberman Lab Genome Browser (http://dzlab.pmb.berkeley.edu/browser/), such demethylation indeed takes place in the 5′and 3′regulatory regions of AGL36. Methylation patterns are regained in the dme mutant, supporting our data that AGL36 is maternally activated through the action of DME.
In an elegant approach by Gehring and colleagues, novel imprinted genes have recently been identified by the prediction of Differentially Methylated Regions (DMRs) between embryo and endosperm. In support of our findings, significant DMRs were also mapped to 5′and 3′region regions of AGL36 [35].
Imprinting could be demonstrated in transgenic pAGL36::GUS seeds, thus indicating that the 1752 bp promoter fragment used is sufficient for parent-of-origin specific expression. The genomic environment of imprinted genes is highly correlated with transposable elements (TE), and imprinting has been postulated to be an evolutionary byproduct of silencing of invading transposons [23], [69], [70]. For instance, methylation of a SINE-related tandem repeat structure in the 5′-region correlates with FWA expression [32], [71], and DMRs in MEA, PHE1, HDG3 and HDG9 map to TE [35]. In line with this, a variety of remnants of TE reside in both the 5′and 3′ regulatory regions of AGL36 (Figure 4A). The 1752 bp pAGL36::GUS promoter fragment harbors remnants of Helitrons and parts of an Arnoldy TE. An 800 bp DMR maps immediately (78 bp) upstream of the AGL36 transcriptional start site overlapping the Helitron TEs ([35], Mary Gehring, personal communication). Clearly, the 1752 bp 5′region is sufficient for basal AGL36 imprinting, and similar to the abovementioned examples, AGL36 DMRs map to TE. Further investigations will be needed to elucidate the role and the mechanisms of additional 5′and 3′DMRs as well as the involvement of small RNAs in AGL36 imprinting [72].
The PRC2 FIS-complex regulates maternal expression of AGL36
Distinct from the expression pattern of AGL36 that subsides at the time of cellularization in wild-type seeds, AGL36 maternal expression in mea mutant seeds was highly elevated and sustained throughout seed development. Recently, Walia et al. also reported AGL36 upregulation obtained in five days old seeds from selfed fis2+/− plants [65]. Our results show that FIS-complex mediated repression acts exclusively on the expression of the maternal allele of AGL36. The paternal allele was efficiently silenced throughout endosperm development.
Surprisingly, weak paternal pAGL36::GUS expression could be observed in 6 DAP early heart stage embryos when the mother was homozygous for mea. MEA has been shown to have biallelic expression in the embryo [28], and thus the observed paternal expression in hemizygous mea embryos is not caused by the lack of functional MEA. This could hint to dosage-dependent regulation of paternal AGL36 expression by MEA, directly or indirectly, but in lack of further experiments this remains speculation.
Different PRC2 complexes can regulate common genes [30]. However, in mutants of CLF and SWN, the paralogues of MEA, no significant effect on AGL36 expression levels was found, indicating that AGL36 regulation is specific to PRC2FIS. H3K27 trimethylation mediates PRC2s repressive function, and in a whole-genome assay for H3K27 methylation more than 4400 target genes were detected [73] (Daniel Bouyer, personal communication). AGL36 was however not part of this set of genes. Since this material was obtained from seedlings and may not reflect the situation in the seed, it is not known whether AGL36 is a direct target of H3K27 trimethylation.
AGL36 identifies a dual regulation mechanism by DME and the FIS PRC2-complex
Repression of the maternal AGL36 allele identifies a novel means of dual epigenetic regulation of imprinted genes. In this scenario, the expressed maternal AGL36 allele is antagonistically activated by DME and repressed by PRC2FIS. To our knowledge, this is the first report of an imprinted gene where the expressed allele is concurrently regulated by a repressive epigenetic mark.
We asked whether this type of regulation was specific for AGL36 by investigating the fis mutant for expression of three other imprinted genes that are activated by DME. We found that these genes fall into two distinct groups; FWA and FIS2 which were largely unaffected by the lack of FIS, and MPC along with AGL36 which showed strong upregulation. This suggests that additional PRC2 regulation of DME-activated alleles defines a common mechanism that applies to a subset of imprinted genes.
In Arabidopsis, three imprinted genes, MEA, PHE1 and AtFH5 are known to have their silenced allele repressed by PRC2FIS, and two of these genes, MEA and PHE1 are additionally regulated by DNA methylation [55]. In these cases however, the repressed allele is silenced by PRC2 whereas the active allele is regulated by DNA methylation [74]. Here, we show that AGL36 defines a novel type of regulation where the same allele is activated by DME and repressed by PRC2FIS in a sequential fashion. This suggests that maternal AGL36 expression after DME activation needs to be dampened and developmentally regulated by PRC2FIS, in accordance with the strong AGL36 expression observed in hypomethylated met1−/− plants. Interestingly, DME is required to activate both PRC2MEA and AGL36, and is thus a key player in developmental tuning of parent-of-origin specific AGL36 expression.
The role of AGL36 in seed development
AGL36 was identified in our transcript profiling as a downregulated gene when the paternal contribution to the endosperm was absent. A simple hypothesis to account for this regulation would be that AGL36 is under the control of one or more paternally expressed factor(s) that activate the maternal allele of AGL36. The identity of such factor(s) remain unknown, and was not approached in this work, but a simple prediction from this hypothesis is that AGL36 would be upregulated in paternal excess interploidy crosses. In a recent report, AGL36 is indeed upregulated in such crosses, as well as in crosses with unreduced diploid jas pollen [66]. Such parental cross-talk is however likely to involve complex genetic and epigenetic regulatory mechanisms, and the mechanism that cause the observed transcriptional response of AGL36 and other previously described imprinted genes in cdka;1p seeds remains to be clarified.
In our study, we have shown that AGL36 is only maternally expressed. Our current model suggests that the paternal allele is silenced by the action of MET1 and the maternal allele activated by DME (Figure 8). In addition, we have also shown that PRC2FIS regulates the expression of the maternal AGL36 allele at the transition between proliferation and cellularization (Figure 8).

Although AGL36 is identified as a novel target of the imprinting machinery in Arabidopsis, we have limited knowledge about its function during plant and seed development. Since expression of AGL36 and its interacting partners coincide with the transition of endosperm from proliferation to differentiation, we speculate that it plays an important role in this process. This is in agreement with recent findings [65], showing that suppression of an AGL cluster including AGL36 is critical for successful transition of endosperm from syncytial to cellularized stage.
In this work we have identified a novel imprinted gene that is controlled by a novel type of dual epigenetic regulation in the seed. This underscores the importance of further investigations to identify imprinted genes in order to unravel the complex network of epigenetic regulation of parent-of-origin effects in seed development.
Materials and Methods
Plant strains and growth conditions
All plant lines used in these experiments were obtained from the Nottingham Arabidopsis Stock Centre (NASC) unless otherwise stated. The mutant lines cdka;1-1 (SALK_106809; [40], [41]), ddm1-2 (a kind gift from E. Richards; [53]), dme-6 (GK-252E03-014577; Figure S9), mea-8 (SAIL_55_C04; [75]), mea-9 (SAIL_724_E07; Figure S9), met1-4; (SAIL_809_E03; [76] and swn-4 (SALK_109121; [77]) were in the Col accession. The mutant lines ago4-1 (N6364; [78]), clf-2 (N290; [79]), cmt3-7 (N6365; [80]), fis1 (a kind gift from A. Chaudhury; [14]) and kyp-2 (N6367; [51] were in the Ler accession. The drm1;drm2 (N6366; [81]) line was in the Ws-2 accession. Mutants used in this study were genotyped using gene-specific and T-DNA specific primers as described in Table S2. The ddm1-2 mutant line was genotyped by an allele-specific PCR test using dCAPS primers DDM1f and ddm1-2Rsa, as described by [68], allowing digestion of the PCR fragment of the ddme1-2 allele with RsaI restriction endonuclease, generating a ∼130 bp band.
We obtained the agl36-1 allele from the Koncz collection [48]. Allele-specific PCR, using the primers HOOK1 (left border T-DNA primer) and AGL36-AS2-KONCZ (genomic AGL36 primer), was carried out to verify the T-DNA insertion, followed by sequencing analysis of the PCR product using the HOOK1 primer. The left border of the insertion was verified to be 16 bp upstream of the ATG start codon of AGL36. In addition, there is an 11 bp long DNA filler located between the genomic sequence and the T-DNA sequence.
Arabidopsis seeds were surface-sterilized using EtOH, bleach and Tween20 prior to plating out on MS-2 plates [82] supplemented with 2% Sucrose, containing the correct selection when necessary. Seeds on the MS-2 plates were stratified at 4°C O.N before they were incubated for 14 days at 18°C to germinate. The seedlings were then put on soil and grown in long day conditions (16 hr light) at 18°C.
Seed isolation, RNA extraction, and cDNA synthesis
To increase tissue specificity, siliques were cut open and seeds were isolated directly in tubes containing pre-chilled ceramic beads (Roche MagNA Lyser Green Beads). Isolated tissues were stored at −80°C. Homogenization was performed by the addition of Lysis buffer containing β-ME (Sigma Spectrum Plant Total RNA Kit) directly to the samples, followed by 3×15 second intervals of homogenization using a MagNA Lyser Instrument (Roche). To prevent RNA degradation, samples were chilled on ice two minutes between each homogenization interval. After the last homogenization step, the samples were centrifuged at 4°C for one minute prior to the transfer of the lysate to a new 1.5 ml tube. RNA extraction was performed according to the Sigma Plant Total RNA Kit protocol, except that all centrifugation steps were done at 4°C and not at room temperature as indicated in the protocol. RNA was eluted in 50 µl volume. cDNA was synthesized by first preparing the RNA for real-time PCR by treatment with DNase I (Sigma) followed by Reverse Transcription using Oligo(dT) and SuperScript III Reverse Transcriptase (Invitrogen) according to the manufacturer's protocols. The synthesized cDNA was purified utilizing QIAquick PCR Purification kit (Qiagen) and eluted in 30 µl volume prior to measurement of cDNA concentration using a NanoDrop 1000 Spectrophotometer.
Microarray analysis
Plants were grown and seeds isolated as described above. Total RNA was isolated as described above. For microarray analysis, three biological replicas were generated, each consisting of approximately 35 hand-pollinated siliques from ten different plants. The microarray experiment was conducted by the NARC Microarray Service in Trondheim. Microarray slides were printed by the Norwegian Microarray Consortium (Trondheim, Norway). A custom made Arabidopsis chip with 32567 unique 70-mer oligo probes was used in the experiments. Total RNA (15 mg) and Super-Script III reverse transcriptase (Invitrogen) were used in a reverse transcription reaction. A 3DNA Array 350 kit with Cy3 - and Cy5-labelled dendrimers (Genisphere Inc.) was used for labeling. Hybridizations were performed in a Slide Booster Hybridization Station (Advalytix), and the slides were washed according to the manufacturers' descriptions (Genisphere and Advalytix). The slides were scanned at 10 mm resolution on a G2505B Agilent DNA microarray scanner (Agilent Technologies). The resulting image files were processed using GenePix 5.1 software (Axon Instruments). Spots identified as not found or manually flagged out as bad were filtered out. Spots with more than 50% saturated pixels were also excluded. The data sets were log-transformed and normalized using the print-tip Loess approach [83]. Within-array replicated measurements for the same gene were merged by taking the average between the replicates. The data were then scaled so that all array data sets had the same median absolute deviation. The differentially expressed genes were identified using the Limma software package [84]. The resulting set of p-values were used to compute the q-values as described [85].
The microarray data generated in this publication have been deposited in NCBI's Gene Expression Omnibus and are accessible through GEO Series accession number GSE24809 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE24809).
Bioinformatics analyses
We defined the following sub-sets for our microarray data (see Table S3): All expressed = all genes having a present call (17223 genes); Down 0.8 = in Ler x cdka;1 downregulated genes with q≤0.35 and arithmetic ratio (ar) ≤0.8 (602 genes); Up 1.5 = in Ler x cdka;1 upregulated genes with q≤0.35 and ar ≥1.5 (323 genes); Up 1.2 = in Ler x cdka;1 upregulated genes with q≤0.35 and ar ≥1.2 (1030 genes). The q-value is the false discovery rate (FDR) of the p-value, and was adjusted with Storey's q-procedure [85]. The threshold for analysis was set to q≤0.35 since this value detected paternally expressed genes at an arithmetic ratio (ar) ≤0.8. A functional classification was done at http://www.arabidopsis.org/tools/bulk/go/ using the GO-Slim Molecular Function classification system. For the detailed transcription factor analysis we used the Transcription Factor (TF) classification from the Arabidopsis transcription factor database (AtTFDB) hosted on the Arabidopsis Gene Regulatory Information Server (AGRIS, http://arabidopsis.med.ohio-state.edu/AtTFDB). The MADS TFs were sub-grouped as in de Folter et al [45]. We compared our microarray data with seed expression data generated by the Goldberg & Harada laboratories, available at http://seedgenenetwork.net/analyze?project=Arabidopsis.
For data comparison a reference set of genes was used that contained all genes covered by the Operon chip used in our study (Arabidopsis thaliana 34K NARC serie 8; GEO Platform GPL11051GPL) and the Affimetrix chip used by Goldberg & Harada (Ath1, GEO Platform GPL198). For the Ath1 chip we used the annotation provided by Goldberg & Harada available at http://seedgenenetwork.net/media/Arab_Final_Annotations_09-07-07_completed.txt. For the operon chip we used the current TAIR 9.0 annotation. From these annotations all AGIs for nuclear genes were extracted and the overlap was calculated. This reference set contained 22130 genes.
We used the reference set overlap of the following Goldberg/Harada datasets for comparison: GH seed = call all present and experiment in Arabidopsis ATH1 Array/Arabidopsis/Globular Stage/Seed; GH seed coat = call all present and experiment in Arabidopsis ATH1 Array/Arabidopsis/Globular Stage/Chalazal Seed Coat or Arabidopsis ATH1 Array/Arabidopsis/Globular Stage/General Seed Coat; GH endosperm = call all present and experiment in Arabidopsis ATH1 Array/Arabidopsis/Globular Stage/Chalazal Endosperm or Arabidopsis ATH1 Array/Arabidopsis/Globular Stage/Micropylar Endosperm or Arabidopsis ATH1 Array/Arabidopsis/Globular Stage/Peripheral Endosperm; GH embryo = call all present and experiment in Arabidopsis ATH1 Array/Arabidopsis/Globular Stage/Embryo Proper.
Venn diagrams were generated using the VENN diagram generator designed by Tim Hulsen at http://www.cmbi.ru.nl/cdd/biovenn/ [86]. The test for statistical significance of the overlap between two groups of genes was calculated by using software provided by Jim Lund accessible at http://elegans.uky.edu/MA/progs/overlap_stats.html.
Plasmid construction
To generate the pAGL36::GUS construct we utilized the Gateway cloning technology (Gateway; Invitrogen). The promoter region (÷1740–12) spanning the ATG start codon was amplified using the attB sequence containing primers attB1-pAGL36-AS7 and attB2-pAGL36-S4 (Table S2), and cloned into the pMDC162 GUS-vector [87]. The resulting construct, after checking the DNA sequence, was introduced to Col ecotype by Agrobacterium tumefaciens mediated transformation using the floral-dip method [88].
β-Glucuronidase expression analysis and histology
Histochemical assays were performed after a modified protocol from Grini et al. (2002) by incubating the tissues in staining buffer (2 mM X-Gluc; 50 mM NaPO4, pH 7.2; 2 mM K4Fe(CN)6 x 3H2O; 2 mM K3Fe(CN)6; 0.1% Triton) overnight at 37°C before the reaction was terminated using 50% EtOH. The tissues were cleared and mounted on slides according to Grini et al. (2002), and inspected using an Axioplan 2 Carl Zeiss Microscope. Images were acquired with an AxioCam HRc Carl Zeiss camera and processed with AxioVs40 V 4.5.0.0 software.
Real-time quantitative PCR
Real-time PCR was performed using a Light-cycler LC480 instrument (Roche) according to the manufacturer's protocol. To ensure high PCR efficiency and to avoid undesired primer dimers, all oligonucleotide pairs were initially tested by melting point analysis using SYBR Premix Ex Taq (TaKaRa). To obtain higher level of gene specificity, probe-based real-time PCR with confirmed primers were performed using Universal Probe Library (UPL) hydrolysis probes (Roche) in combination with Premix Ex Taq (TaKaRa).
For AGL36 real-time PCR, we used primers AGL36-160-LP and AGL36-160-RP, which gave a 60 bp amplicon (Table S2). Comparison of the sequences of the coding region and the 3′UTR of AGL36 with AGL34 and AGL90, revealed more than 85% and 84% sequence similarity respectively between these genes (Figure S8). To ensure that the abovementioned primers are only amplifying AGL36, we cloned the obtained amplicon from four independent reactions into the pCR2.1 vector (Invitrogen), and subsequently sequenced two clones of each construct with M13-Forward and M13-Reverse primers. Sequence results revealed exclusive and specific AGL36 amplification.
ACTIN11 (ACT11), a housekeeping gene that is strongly expressed in the developing ovules [89], was shown in a preliminary analysis not to be affected by our experimental conditions (data not shown), and was therefore selected as a reference gene. GLYCERALDEHYDE-3-P DEHYDROGENASE A-SUBUNIT (GAPA) was used as an additional reference gene. The oligo sequences, their amplicons and appropriate UPL probes are shown in Table S2.
Real-time PCR of all samples and reference controls were performed in two independent biological replicates and repeated at least two times (technical replicas) unless otherwise stated. The PCR efficiency was determined independently for all replicates (biological and technical) by series of dilutions (100 ng, 50 ng, 20 ng, 5 ng template/rxn) for each experiment. This allowed us to obtain the efficiency for each single reaction. Calculations of relative expression ratios were performed according to a model described by Pfaffl [90] with minor exceptions. Since we had efficiency for all reactions (four values for each calculation corresponding to Etarget-sample, Etarget-standard, Ereference-sample and Ereference-standard), we calculated the average Etarget and Ereference values from the standards and the samples, ending up with two E-values that we could use in the formula described by Pfaffl.
Single Nucleotide Polymorphism analysis
RNA was isolated and cDNA synthesized and purified as described above. Polymorphisms between various ecotypes were identified using TAIR Genome Browser (www.arabidopsis.org) and/or the Arabidopsis SNP Sequence Viewer tool provided by the Salk Institute Genomic Analysis Laboratory (http://natural.salk.edu/cgi-bin/snp.cgi). A selected region spanning the SNP of interest was amplified by PCR using TaKaRa Ex Taq DNA polymerase applying 100 ng template per reaction, and the following PCR parameters in a 50 µl reaction: 94°C-3 min, 35×(94°C-1 min, 58°C-30 sec, 72°C-1 min/kb), 72°C-5 min, 4°C-∞. Parental-specific expression based on SNP was determined by setting up an appropriate restriction digest. For AGL36 SNP analysis, 20 µl of the SNP PCR reaction was digested with 15 U of AlwNI at 37°C for a duration of 2.5 hrs, followed by a 20 min inactivation at 65°C. For the FWA control SNP, due to the absence of a restriction site in the SNP region in both Col and Ler ecotypes, dCAPS primers were used, generating a NheI restriction site in the Col ecotype. The obtained amplicons for both ecotypes were digested with NheI [11].
In cases where the detected SNP did not result in digestion in neither ecotype, a primer sequence was designed to introduce a base exchange adjacent to the SNP, leading to restriction digestion of one of the ecotypes. The obtained amplicon for both ecotypes were then treated with the appropriate restriction enzyme. In all experiments either genomic DNA or cDNA from wild-type plants from both ecotypes used in the study was used as controls for the presence or absence of digestion. The digested samples were analyzed using DNA-1000-LabOnChip and 2100 Bioanalyzer (Agilent Technologies).
To rule out that the primers used for AGL36 SNP PCR (AGL36-SP7-SNP and AGL36-ASP6-SNP) (Table S2) would amplify the highly similar AGL90, we oriented the AGL36-SP7-SNP primer such that it was located in a region that was annotated as intron in AGL90 but not in AGL36 (Figure S8). First, the presence of the intron in AGL90 was confirmed by amplifying the intron-flanking region (AGL90-SP1-subcloning and AGL90-ASP2-subcloning primers (Table S2)), and comparing the size differences obtained between the genomic PCR and cDNA PCR. Due to high sequence similarity, we suspected to amplify both AGL36 and AGL90 in these PCR reactions. To distinguish between these two amplicons, we took advantage of the presence of two unique restriction sites (MslI and BspBI) in the amplified region of AGL36 that are absent in AGL90.
Sequence comparison between the abovementioned AGL36-SNP primers and AGL34 showed that there was approximately 70% and 91% sequence similarity between the primers and the AGL34 gene. However, if these primers were functional in amplifying AGL34, they would result in a smaller amplicon than AGL36 amplicon (373 bp versus 399 bp respectively). This difference could easily be detected using a DNA-1000-LabOnChip. Our SNP data only showed the expected 399 bp band, verifying that AGL34 was not amplified using the above primers. The paternally imprinted FWA gene was used as a positive control by utilizing primers FWA-RTf and FWA-dNheI (Table S2) for PCR amplification followed by NheI restriction digest.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. NowackMK
UngruA
BjerkanKN
GriniPE
SchnittgerA
2010 Reproductive cross-talk: seed development in flowering plants. Biochem Soc Trans 38 604 612
2. FeilR
BergerF
2007 Convergent evolution of genomic imprinting in plants and mammals. Trends Genet 23 192 199
3. JullienPE
BergerF
2009 Gamete-specific epigenetic mechanisms shape genomic imprinting. Curr Opin Plant Biol 12 637 642
4. LinBY
1984 Ploidy Barrier to Endosperm Development in Maize. Genetics 107 103 115
5. ScottRJ
SpielmanM
BaileyJ
DickinsonHG
1998 Parent-of-origin effects on seed development in Arabidopsis thaliana. Development 125 3329 3341
6. HaigD
WestobyM
1991 Genomic Imprinting in Endosperm: Its Effect on Seed Development in Crosses between Species, and between Different Ploidies of the Same Species, and Its Implications for the Evolution of Apomixis. Philosophical Transactions: Biological Sciences 333 1 13
7. DeChiaraTM
EfstratiadisA
RobertsonEJ
1990 A growth-deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II gene disrupted by targeting. Nature 345 78 80
8. FilsonAJ
LouviA
EfstratiadisA
RobertsonEJ
1993 Rescue of the T-associated maternal effect in mice carrying null mutations in Igf-2 and Igf2r, two reciprocally imprinted genes. Development 118 731 736
9. LauMM
StewartCE
LiuZ
BhattH
RotweinP
1994 Loss of the imprinted IGF2/cation-independent mannose 6-phosphate receptor results in fetal overgrowth and perinatal lethality. Genes Dev 8 2953 2963
10. LeightonPA
IngramRS
EggenschwilerJ
EfstratiadisA
TilghmanSM
1995 Disruption of imprinting caused by deletion of the H19 gene region in mice. Nature 375 34 39
11. KinoshitaT
MiuraA
ChoiY
KinoshitaY
CaoX
2004 One-way control of FWA imprinting in Arabidopsis endosperm by DNA methylation. Science 303 521 523
12. LawrenceRJ
EarleyK
PontesO
SilvaM
ChenZJ
2004 A concerted DNA methylation/histone methylation switch regulates rRNA gene dosage control and nucleolar dominance. Mol Cell 13 599 609
13. GrossniklausU
Vielle-CalzadaJP
HoeppnerMA
GaglianoWB
1998 Maternal control of embryogenesis by MEDEA, a polycomb group gene in Arabidopsis. Science 280 446 450
14. ChaudhuryAM
MingL
MillerC
CraigS
DennisES
1997 Fertilization-independent seed development in Arabidopsis thaliana. Proc Natl Acad Sci U S A 94 4223 4228
15. OhadN
MargossianL
HsuYC
WilliamsC
RepettiP
1996 A mutation that allows endosperm development without fertilization. Proc Natl Acad Sci U S A 93 5319 5324
16. KohlerC
HennigL
BouveretR
GheyselinckJ
GrossniklausU
2003 Arabidopsis MSI1 is a component of the MEA/FIE Polycomb group complex and required for seed development. Embo J 22 4804 4814
17. GuittonAE
PageDR
ChambrierP
LionnetC
FaureJE
2004 Identification of new members of Fertilisation Independent Seed Polycomb Group pathway involved in the control of seed development in Arabidopsis thaliana. Development 131 2971 2981
18. TiwariS
SchulzR
IkedaY
DythamL
BravoJ
2008 MATERNALLY EXPRESSED PAB C-TERMINAL, a novel imprinted gene in Arabidopsis, encodes the conserved C-terminal domain of polyadenylate binding proteins. Plant Cell 20 2387 2398
19. JullienPE
KinoshitaT
OhadN
BergerF
2006 Maintenance of DNA methylation during the Arabidopsis life cycle is essential for parental imprinting. Plant Cell 18 1360 1372
20. AdamsS
VinkenoogR
SpielmanM
DickinsonHG
ScottRJ
2000 Parent-of-origin effects on seed development in Arabidopsis thaliana require DNA methylation. Development 127 2493 2502
21. JohnstonAJ
MatveevaE
KirioukhovaO
GrossniklausU
GruissemW
2008 A Dynamic Reciprocal RBR-PRC2 Regulatory Circuit Controls Arabidopsis Gametophyte Development. Curr Biol 18 1680 1686
22. JullienP
MosqunaA
IngouffM
SakataT
OhadN
2008 Retinoblastoma and its binding partner MSI1 control imprinting in Arabidopsis. PLoS Biol 6 e194 doi:10.1371/journal.pbio.0060194
23. GehringM
ReikW
HenikoffS
2009 DNA demethylation by DNA repair. Trends Genet 25 82 90
24. ChoiY
GehringM
JohnsonL
HannonM
HaradaJJ
2002 DEMETER, a DNA glycosylase domain protein, is required for endosperm gene imprinting and seed viability in Arabidopsis. Cell 110 33 42
25. BarouxC
GagliardiniV
PageDR
GrossniklausU
2006 Dynamic regulatory interactions of Polycomb group genes: MEDEA autoregulation is required for imprinted gene expression in Arabidopsis. Genes Dev 20 1081 1086
26. Fitz GeraldJN
HuiPS
BergerF
2009 Polycomb group-dependent imprinting of the actin regulator AtFH5 regulates morphogenesis in Arabidopsis thaliana. Development 136 3399 3404
27. JullienPE
KatzA
OlivaM
OhadN
BergerF
2006 Polycomb group complexes self-regulate imprinting of the Polycomb group gene MEDEA in Arabidopsis. Curr Biol 16 486 492
28. KinoshitaT
YadegariR
HaradaJJ
GoldbergRB
FischerRL
1999 Imprinting of the MEDEA polycomb gene in the Arabidopsis endosperm. Plant Cell 11 1945 1952
29. KohlerC
HennigL
SpillaneC
PienS
GruissemW
2003 The Polycomb-group protein MEDEA regulates seed development by controlling expression of the MADS-box gene PHERES1. Genes Dev 17 1540 1553
30. MakarevichG
LeroyO
AkinciU
SchubertD
ClarenzO
2006 Different Polycomb group complexes regulate common target genes in Arabidopsis. EMBO Rep 7 947 952
31. ScottRJ
SpielmanM
2006 Genomic imprinting in plants and mammals: how life history constrains convergence. Cytogenet Genome Res 113 53 67
32. KinoshitaY
SazeH
KinoshitaT
MiuraA
SoppeWJ
2007 Control of FWA gene silencing in Arabidopsis thaliana by SINE-related direct repeats. Plant J 49 38 45
33. LuoM
BilodeauP
DennisES
PeacockWJ
ChaudhuryA
2000 Expression and parent-of-origin effects for FIS2, MEA, and FIE in the endosperm and embryo of developing Arabidopsis seeds. Proc Natl Acad Sci U S A 97 10637 10642
34. TiwariS
SpielmanM
DayRC
ScottRJ
2006 Proliferative phase endosperm promoters from Arabidopsis thaliana. Plant Biotechnol J 4 393 407
35. GehringM
BubbKL
HenikoffS
2009 Extensive demethylation of repetitive elements during seed development underlies gene imprinting. Science 324 1447 1451
36. IslesAR
HollandAJ
2005 Imprinted genes and mother-offspring interactions. Early Hum Dev 81 73 77
37. MorisonIM
RamsayJP
SpencerHG
2005 A census of mammalian imprinting. Trends Genet 21 457 465
38. ReikW
LewisA
2005 Co-evolution of X-chromosome inactivation and imprinting in mammals. Nat Rev Genet 6 403 410
39. WoodAJ
OakeyRJ
2006 Genomic imprinting in mammals: emerging themes and established theories. PLoS Genet 2 e147 doi:10.1371/journal.pgen.0020147
40. NowackMK
GriniPE
JakobyMJ
LafosM
KonczC
2006 A positive signal from the fertilization of the egg cell sets off endosperm proliferation in angiosperm embryogenesis. Nat Genet 38 63 67
41. IwakawaH
ShinmyoA
SekineM
2006 Arabidopsis CDKA;1, a cdc2 homologue, controls proliferation of generative cells in male gametogenesis. Plant J 45 819 831
42. AwSJ
HamamuraY
ChenZ
SchnittgerA
BergerF
2010 Sperm entry is sufficient to trigger division of the central cell but the paternal genome is required for endosperm development in Arabidopsis. Development 137 2683 2690
43. NowackMK
ShirzadiR
DissmeyerN
DolfA
EndlE
2007 Bypassing genomic imprinting allows seed development. Nature 447 312 315
44. LeBH
ChengC
BuiAQ
WagmaisterJA
HenryKF
2010 Global analysis of gene activity during Arabidopsis seed development and identification of seed-specific transcription factors. Proc Natl Acad Sci U S A 107 8063 8070
45. de FolterS
ImminkRG
KiefferM
ParenicovaL
HenzSR
2005 Comprehensive interaction map of the Arabidopsis MADS Box transcription factors. Plant Cell 17 1424 1433
46. KangIH
SteffenJG
PortereikoMF
LloydA
DrewsGN
2008 The AGL62 MADS domain protein regulates cellularization during endosperm development in Arabidopsis. Plant Cell 20 635 647
47. ParenicovaL
de FolterS
KiefferM
HornerDS
FavalliC
2003 Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis: new openings to the MADS world. Plant Cell 15 1538 1551
48. RiosG
LossowA
HertelB
BreuerF
SchaeferS
2002 Rapid identification of Arabidopsis insertion mutants by non-radioactive detection of T-DNA tagged genes. Plant J 32 243 253
49. GriniPE
SchnittgerA
SchwarzH
ZimmermannI
SchwabB
1999 Isolation of ethyl methanesulfonate-induced gametophytic mutants in Arabidopsis thaliana by a segregation distortion assay using the multimarker chromosome 1. Genetics 151 849 863
50. CaoX
JacobsenSE
2002 Locus-specific control of asymmetric and CpNpG methylation by the DRM and CMT3 methyltransferase genes. Proc Natl Acad Sci U S A 99 Suppl 4 16491 16498
51. JacksonJP
LindrothAM
CaoX
JacobsenSE
2002 Control of CpNpG DNA methylation by the KRYPTONITE histone H3 methyltransferase. Nature 416 556 560
52. ChanSW
ZilbermanD
XieZ
JohansenLK
CarringtonJC
2004 RNA silencing genes control de novo DNA methylation. Science 303 1336
53. JeddelohJA
StokesTL
RichardsEJ
1999 Maintenance of genomic methylation requires a SWI2/SNF2-like protein. Nat Genet 22 94 97
54. DayRC
HerridgeRP
AmbroseBA
MacknightRC
2008 Transcriptome analysis of proliferating Arabidopsis endosperm reveals biological implications for the control of syncytial division, cytokinin signaling, and gene expression regulation. Plant Physiol 148 1964 1984
55. JullienPE
BergerF
2010 Parental genome dosage imbalance deregulates imprinting in Arabidopsis. PLoS Genet 6 e1000885 doi:10.1371/journal.pgen.1000885
56. KohlerC
PageDR
GagliardiniV
GrossniklausU
2005 The Arabidopsis thaliana MEDEA Polycomb group protein controls expression of PHERES1 by parental imprinting. Nat Genet 37 28 30
57. BeckerA
TheissenG
2003 The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Mol Phylogenet Evol 29 464 489
58. KofujiR
SumikawaN
YamasakiM
KondoK
UedaK
2003 Evolution and divergence of the MADS-box gene family based on genome-wide expression analyses. Mol Biol Evol 20 1963 1977
59. BemerM
Wolters-ArtsM
GrossniklausU
AngenentGC
2008 The MADS domain protein DIANA acts together with AGAMOUS-LIKE80 to specify the central cell in Arabidopsis ovules. Plant Cell 20 2088 2101
60. PortereikoMF
LloydA
SteffenJG
PunwaniJA
OtsugaD
2006 AGL80 is required for central cell and endosperm development in Arabidopsis. Plant Cell 18 1862 1872
61. ColomboM
MasieroS
VanzulliS
LardelliP
KaterMM
2008 AGL23, a type I MADS-box gene that controls female gametophyte and embryo development in Arabidopsis. Plant J 54 1037 1048
62. SteffenJG
KangIH
PortereikoMF
LloydA
DrewsGN
2008 AGL61 interacts with AGL80 and is required for central cell development in Arabidopsis. Plant Physiol 148 259 268
63. De BodtS
RaesJ
FlorquinK
RombautsS
RouzeP
2003 Genomewide structural annotation and evolutionary analysis of the type I MADS-box genes in plants. J Mol Evol 56 573 586
64. JosefssonC
DilkesB
ComaiL
2006 Parent-dependent loss of gene silencing during interspecies hybridization. Curr Biol 16 1322 1328
65. WaliaH
JosefssonC
DilkesB
KirkbrideR
HaradaJ
2009 Dosage-dependent deregulation of an AGAMOUS-LIKE gene cluster contributes to interspecific incompatibility. Curr Biol 19 1128 1132
66. ErilovaA
BrownfieldL
ExnerV
RosaM
TwellD
2009 Imprinting of the polycomb group gene MEDEA serves as a ploidy sensor in Arabidopsis. PLoS Genet 5 e1000663 doi:10.1371/journal.pgen.1000663
67. ZhangX
YazakiJ
SundaresanA
CokusS
ChanSW
2006 Genome-wide High-Resolution Mapping and Functional Analysis of DNA Methylation in Arabidopsis. Cell 126 1189 1201
68. YadegariR
KinoshitaT
LotanO
CohenG
KatzA
2000 Mutations in the FIE and MEA genes that encode interacting polycomb proteins cause parent-of-origin effects on seed development by distinct mechanisms. Plant Cell 12 2367 2382
69. HsiehTF
IbarraCA
SilvaP
ZemachA
Eshed-WilliamsL
2009 Genome-wide demethylation of Arabidopsis endosperm. Science 1451 1454
70. BarlowDP
1993 Methylation and imprinting: from host defense to gene regulation? Science 260 309 310
71. ChanSW
ZhangX
BernatavichuteYV
JacobsenSE
2006 Two-step recruitment of RNA-directed DNA methylation to tandem repeats. PLoS Biol 4 e363 doi:10.1371/journal.pbio.0040363
72. MosherRA
MelnykCW
KellyKA
DunnRM
StudholmeDJ
2009 Uniparental expression of PolIV-dependent siRNAs in developing endosperm of Arabidopsis. Nature 460 283 286
73. ZhangX
ClarenzO
CokusS
BernatavichuteYV
PellegriniM
2007 Whole-genome analysis of histone H3 lysine 27 trimethylation in Arabidopsis. PLoS Biol 5 e129 doi:10.1371/journal.pbio.0050129
74. MakarevichG
VillarC
ErilovaA
KohlerC
2008 Mechanism of PHERES1 imprinting in Arabidopsis. Journal of Cell Science 121 906 912
75. NgoQA
MooreJM
BaskarR
GrossniklausU
SundaresanV
2007 Arabidopsis GLAUCE promotes fertilization-independent endosperm development and expression of paternally inherited alleles. Development 134 4107 4117
76. SazeH
Mittelsten ScheidO
PaszkowskiJ
2003 Maintenance of CpG methylation is essential for epigenetic inheritance during plant gametogenesis. Nat Genet 34 65 69
77. WangD
TysonMD
JacksonSS
YadegariR
2006 Partially redundant functions of two SET-domain polycomb-group proteins in controlling initiation of seed development in Arabidopsis. Proc Natl Acad Sci U S A 103 13244 13249
78. ZilbermanD
CaoX
JacobsenSE
2003 ARGONAUTE4 control of locus-specific siRNA accumulation and DNA and histone methylation. Science 299 716 719
79. GoodrichJ
PuangsomleeP
MartinM
LongD
MeyerowitzEM
1997 A Polycomb-group gene regulates homeotic gene expression in Arabidopsis. Nature 386 44 51
80. LindrothAM
CaoX
JacksonJP
ZilbermanD
McCallumCM
2001 Requirement of CHROMOMETHYLASE3 for maintenance of CpXpG methylation. Science 292 2077 2080
81. CaoX
JacobsenSE
2002 Role of the Arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr Biol 12 1138 1144
82. MurashigeT
SkoogF
1962 A rewised medium for rapid growth and bioassays with tobacco tissue cultures. Physiologia Plantarum 15 473 497
83. YangYH
DudoitS
LuuP
LinDM
PengV
2002 Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res 30 e15
84. SmythGK
2004 Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3 1 25
85. StoreyJD
2002 A direct approach to false discovery rates. Journal of the Royal Statistical Society: Series B (Statistical Methodology) 64 479 498
86. HulsenT
de VliegJ
AlkemaW
2008 BioVenn - a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics 9 488
87. CurtisMD
GrossniklausU
2003 A gateway cloning vector set for high-throughput functional analysis of genes in planta. Plant Physiol 133 462 469
88. BentA
2006 Arabidopsis thaliana floral dip transformation method. Methods Mol Biol 343 87 103
89. HuangS
AnYQ
McDowellJM
McKinneyEC
MeagherRB
1997 The Arabidopsis thaliana ACT11 actin gene is strongly expressed in tissues of the emerging inflorescence, pollen, and developing ovules. Plant Mol Biol 33 125 139
90. PfafflMW
2001 A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29 e45
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 2
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Meta-Analysis of Genome-Wide Association Studies in Celiac Disease and Rheumatoid Arthritis Identifies Fourteen Non-HLA Shared Loci
- MiRNA Control of Vegetative Phase Change in Trees
- Risk Alleles for Systemic Lupus Erythematosus in a Large Case-Control Collection and Associations with Clinical Subphenotypes
- The Cardiac Transcription Network Modulated by Gata4, Mef2a, Nkx2.5, Srf, Histone Modifications, and MicroRNAs
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy