
A Study of CNVs As Trait-Associated Polymorphisms and As Expression Quantitative Trait Loci
We conducted a comprehensive study of copy number variants (CNVs) well-tagged by SNPs (r2≥0.8) by analyzing their effect on gene expression and their association with disease susceptibility and other complex human traits. We tested whether these CNVs were more likely to be functional than frequency-matched SNPs as trait-associated loci or as expression quantitative trait loci (eQTLs) influencing phenotype by altering gene regulation. Our study found that CNV–tagging SNPs are significantly enriched for cis eQTLs; furthermore, we observed that trait associations from the NHGRI catalog show an overrepresentation of SNPs tagging CNVs relative to frequency-matched SNPs. We found that these SNPs tagging CNVs are more likely to affect multiple expression traits than frequency-matched variants. Given these findings on the functional relevance of CNVs, we created an online resource of expression-associated CNVs (eCNVs) using the most comprehensive population-based map of CNVs to inform future studies of complex traits. Although previous studies of common CNVs that can be typed on existing platforms and/or interrogated by SNPs in genome-wide association studies concluded that such CNVs appear unlikely to have a major role in the genetic basis of several complex diseases examined, our findings indicate that it would be premature to dismiss the possibility that even common CNVs may contribute to complex phenotypes and at least some common diseases.
Published in the journal:
. PLoS Genet 7(2): e32767. doi:10.1371/journal.pgen.1001292
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001292
Summary
We conducted a comprehensive study of copy number variants (CNVs) well-tagged by SNPs (r2≥0.8) by analyzing their effect on gene expression and their association with disease susceptibility and other complex human traits. We tested whether these CNVs were more likely to be functional than frequency-matched SNPs as trait-associated loci or as expression quantitative trait loci (eQTLs) influencing phenotype by altering gene regulation. Our study found that CNV–tagging SNPs are significantly enriched for cis eQTLs; furthermore, we observed that trait associations from the NHGRI catalog show an overrepresentation of SNPs tagging CNVs relative to frequency-matched SNPs. We found that these SNPs tagging CNVs are more likely to affect multiple expression traits than frequency-matched variants. Given these findings on the functional relevance of CNVs, we created an online resource of expression-associated CNVs (eCNVs) using the most comprehensive population-based map of CNVs to inform future studies of complex traits. Although previous studies of common CNVs that can be typed on existing platforms and/or interrogated by SNPs in genome-wide association studies concluded that such CNVs appear unlikely to have a major role in the genetic basis of several complex diseases examined, our findings indicate that it would be premature to dismiss the possibility that even common CNVs may contribute to complex phenotypes and at least some common diseases.
Introduction
Although genome wide association studies (GWAS) have considerably advanced our knowledge of the genetics of complex traits, they fail to account for the bulk of the overall heritability [1]. Structural variation, including copy number variants (CNVs), may account for some of the missing heritability, but a comprehensive study of the contribution of these variants to complex traits genetics is generally lacking. One of the main challenges of integrating CNVs into GWAS is the reliability of their assay, but improvements in genotyping platforms and the development of algorithms for the analysis of CNVs increasingly facilitate the investigation of the role of CNVs in disease susceptibility [2]. Given the effect of adjacent CNVs on SNP intensity data, early SNP arrays were designed with the consequence that assays from many CNV loci were excluded, but a newer generation of arrays have been implemented that combine SNP assays and copy number probes optimized for copy-number measurement for target regions of known CNVs [3]. New algorithms are also facilitating higher resolution maps of common CNVs (those that segregate at an allele frequency >5%).
Partly due to these technological and analytic developments, the search for links between CNVs and disease susceptibility has been actively pursued in recent years. Structural variation has of course long been known to influence such Mendelian disorders as Williams-Beuren syndrome or Charcot-Marie Tooth neuropathy Type 1A, but more recently there has been widespread interest in the study of CNVs as mediators of more common complex diseases. The contribution of structural variation to certain neurodevelopmental disorders such as schizophrenia and autism spectrum disorder has been investigated by recent studies of de novo copy number variation [4]. A common 20 kb deletion upstream of IRGM in perfect linkage disequilibrium (r2 = 1.0) with the SNP most strongly associated with Crohn's disease in the region has recently been shown to alter IRGM regulation, which in turn is known to affect autophagy, consistent with the deletion polymorphism being the causal allele [5]. Similarly, a 45 kb deletion polymorphism in NEGR1 has been shown to be the likely causal variant for the reproducible association with body mass index in humans, demonstrating a neuronal influence on body weight regulation [6]. These discoveries and similar developments suggest that the interpretation of known disease associations continues to be fraught with challenges, as CNVs may be in linkage disequilibrium (LD) with associated variants, and that the identification of causal variants requires an approach that considers both SNPs and CNVs at associated loci.
The recent release [7] of a reference map of human CNVs in the HapMap populations promises to shed light on the role of these variants in disease pathogenesis. On the other hand, a recent comprehensive study [8] involving 16,000 cases of eight common diseases and 3000 shared controls strongly suggests that CNVs are likely to make a relatively minor contribution to disease susceptibility. In the present study, we approached the investigation of this important topic by exploring evidence of enrichment of CNVs among trait-associated loci in disease susceptibility and in disease classes, as represented by the NHGRI catalog of reproducible associations from genome-wide association scans [9], for certain classes of CNVs, and by conducting a comprehensive functional characterization of CNVs as expression quantitative trait loci (eQTLs). The former seeks to evaluate the genetic information contained in CNVs in regard to disease risk and other complex traits; the latter assesses the functional impact of CNVs on the transcriptome, through which they may contribute to or cause disease phenotypes and other complex traits [10].
Results
Tagged CNVs (tCNVs) are enriched for eQTLs and cis-acting eQTLs
Table 1 summarizes the CNVs included in our study. Because of the ease with which SNPs can be utilized in enrichment studies appropriately conditioned on minor allele frequency, we utilized tCNVs (CNVs tagged by SNPs) for many of these studies, but did not otherwise consider them to be a special class of CNVs.

At an expression p value threshold of 10−4, tCNVs from the recently released study (N = 3,432) of CNVs in 16,000 cases and 3,000 controls [8] were evaluated for their effect on gene expression. We first considered tCNVs that are tagged at r2≥0.80. The tag SNPs for these CNVs enable SNP-based analyses and simulation studies. We identified 1,714 such SNPs tagging CNVs (corresponding to 1,761 tCNVs), of which 532 are eQTLs at this threshold; collectively, these SNPs tagging CNVs were found to target 2,215 distinct transcripts. Restricting our focus to eQTLs that regulate transcript levels in cis (defined as within 4 MB of target transcript), we observed a highly significant cis eQTL enrichment (p<0.001) from a simulation procedure (N = 1000) used to empirically generate the null distribution using randomly generated sets of variants of the same set size and matching minor allele frequency distribution as the SNPs tagging CNVs (see Materials and Methods). These SNPs tagging CNVs are significantly more likely to be associated with altered expression of genes in their vicinity than frequency-matched SNPs (see Figure 1). Specifically, 24 such SNPs tagging CNVs are acting in cis, whereas the expected count is 10.7 (SD = 3.2). On the other hand, the observed count of trans-acting eQTLs is 508 while expected count is 488.9 (SD = 17.6).

We identified 621 SNPs tagging CNVs corresponding to 626 tCNVs that are perfectly tagged (r2 = 1.0). Of these 621, 185 predict the expression of at least one transcript, whereas, based on simulations, the expected count is 166.5 (SD = 9.9). This observation implies a statistically significant (p = 0.024) enrichment for eQTLs among this class of CNVs. Restricting our focus to eQTLs that regulate transcript levels locally, we observed 10 such cis-acting tCNVs. This observed count is, as before, a significant enrichment (p<0.001) for cis-acting regulators compared with expected count (see Materials and Methods) of 3.5 (SD = 1.9) (see Figure S1).
To determine whether our observations are dependent on the p value threshold used to define an eQTL, we explored evidence of eQTL enrichment among the tCNVs (r2≥0.80) at p<10−6. Simulations yielded an expected eQTL count of 19.7 (SD = 4.5) whereas observed count is 28, yielding a statistically significant enrichment (p = 0.036). Expected cis-acting eQTL count is 1.9 (SD = 1.4) while observed count is 8. These findings demonstrate that the observed enrichment holds robustly at lower expression thresholds. There are 2.5 times as many trans eQTLs as cis eQTLs among the tCNVs at this threshold.
Using Gene Ontology (GO) enrichment [11], we found the target genes of these expression associated CNVs (eCNVs) to show a highly significant overrepresentation for certain biological processes including immune response (p = 2.3e-4, adjusted p-value 0.034) and regulation of apoptosis (p = 8e-6, adjusted p-value 0.015). Relative to other genic CNVs, the observation on the enrichment of the target genes of eCNVs for response to stimuli continues to hold (p = 9.9e-3, adjusted p-value 0.05).
tCNVs are enriched for eQTL hotspots
Notably, we identified 22 tCNVs (r2 = 1) that predict the transcript level of 10 or more genes. These CNVs are regulating the expression of distal transcripts. By performing simulations on frequency-matched SNPs as the tCNVs, we observed an enrichment (expected count = 14.7, SD = 3.8) for CNVs controlling the expression of 10 or more, often distal, transcripts (See Figure S1). We identified 2 CNVs CNVR4596.1 (chrom10 : 4698559–4700493) and CNVR1045.1 (chrom2 : 165722938–165725072) that predict the expression of at least 100 transcripts. CNVR4596.1 does not contain any genes; on the other hand, CNVR1045.1 overlaps with SCN3A. Additional CNVs CNVR3307.1 (chrom7 : 17780194–17781249), CNVR3500.1 (chrom7 : 97233946–97240537), and CNVR7062.1 (chrom17 : 27130696–27131638) predict the expression of at least 50 transcripts. The CNV region CNVR3307.1 contains several SNPs (e.g., rs2723520, rs1404418, and rs1404419) in strong LD, which individually predict the expression of more than 80 transcripts.
We identified a CNV (CNVR2845.27 at chrom6 : 32710664–32743652) in perfect LD with the replicated type 1 diabetes SNP rs9272346 predicting the expression of at least 20 transcripts, mostly in the HLA region; a CNV (CNVR2845.40 at chrom6 : 32735154–32737954) is tagged (r2 = 0.95) by the same type 1 diabetes SNP. A tCNV CNVR2845.46 is also in strong LD (r2 = 0.93) with a replicated multiple sclerosis SNP rs3135388 and predicts the expression of 20 transcripts (of which the largest expression association p value is ∼10−33).
CNVs harbor a high proportion of regulatory SNPs
Since the deletion/duplication of multiple regulatory SNPs may result in aberrant transcription or disease, we evaluated the presence of SNP eQTLs within CNVs. We identified 33 CNVs harboring 50 or more SNP eQTLs that predict the expression of a transcript (Table S1 lists these CNVs based on the number of SNP eQTLs within); these CNVs show an average SNP eQTL density of 6 per 10 kb.
We observed 1,306 CNVs (out of the 3,432 CNVs included in the WTCCC study) containing at least one SNP eQTL. Notably, CNVs that harbor a greater number of SNP eQTLs tend to be less well-tagged; this observation holds robustly after accounting for CNV minor allele frequency, which shows little correlation with the number of SNP eQTLs within the CNV (corr = 0.005). In particular, of the top 100 WTCCC CNVs ranked according to the number of SNP eQTLs within, only 31 are well-tagged (r2≥0.80) and 56 are tagged at r2<0.30.
SCAN: SNP and CNV annotation as eQTLs
Given the observed functional relevance of tCNVs as regulatory polymorphisms, we comprehensively and systematically characterized the recent genome-wide survey of CNVs in HapMap lymphoblastoid cell lines (LCLs) for their role in expression. We expanded our SCAN database [12], which to date serves results only on expression associated SNPs, to include eQTL mappings of HapMap CNVs to transcriptional expression (assayed by the Affymetrix exon array) in the full set of HapMap CEU (Caucasians from UT, USA) and YRI (Yoruba people from Ibadan, Nigeria) populations (See Materials and Methods). This online catalog of expression associated CNVs (eCNVs), publicly available at http://www.scandb.org, should serve as an important resource in helping to inform our understanding of complex traits.
Of the CNVs in HapMap [7] showing association with a transcript (at p value threshold of 10−4) in the CEU and YRI samples, 31.1% and 33.1% respectively are predicting the expression of 5 or more transcripts. This is a much higher proportion than is the case for the well-tagged CNVs in the WTCCC CNV study (12.8%).
Reproducible trait associations are enriched for tCNVs
Among trait-associated SNPs as represented in the NHGRI catalog, we found a significant overrepresentation (p = 0.01) for tCNVs (r2≥0.80). Figure 2 shows the distribution of the number of tag SNPs generated from 1000 random sets of SNPs matching the minor allele frequency of the trait-associated SNPs. Table 2 shows the observed overlap between the trait-associated SNPs and the tCNVs (r2≥0.80). Enrichment analysis of disease classes shows that the observed tCNV enrichment for reproducible trait associations holds for autoimmune disorders and in quantitative metabolic traits.

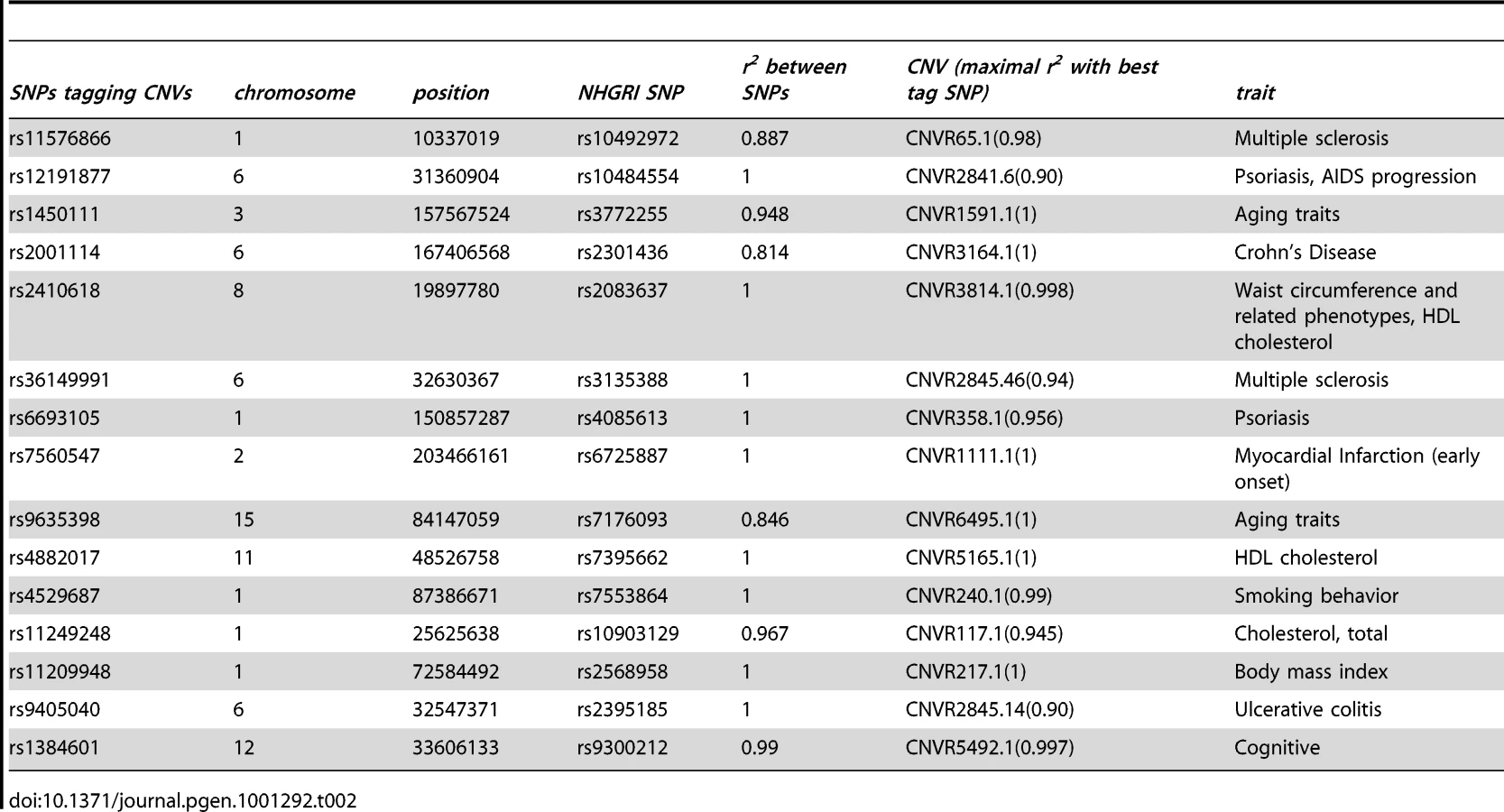
Three HLA region CNVs, namely CNVR2841.20, CNVR2845.14, and CNVR2845.46, were recently shown to be associated with Crohn's disease, rheumatoid arthritis, and type 1diabetes, respectively [8]. Our expression study confirms that the last two of these (tagged at r2>0.90) are eQTLs that regulate the expression of multiple genes in this region; in contrast, the first CNV shows no evidence for being an eQTL. A previously identified Crohn's disease associated locus CNVR2647.1 [5] on chromosome 5, 22 kb upstream of the IRGM gene, is a trans eQTL for SHISA4 (p = 3e-05). Two replicated SNPs associated with HDL cholesterol are tagging (r2≥0.99) nearby CNVs, CNVR3814.1 (chrom8 : 19898924–19899800) and CNVR5165.1 (chrom11 : 48557432–48560877). Our study shows that both CNVs are eQTLs, with CNVR5165.1 targeting, distally, the olfactory receptor gene OR6J1. The CNV CNVR3814.1 is also well-tagged (r2 = 0.913) by a SNP reproducibly associated with triglycerides and is an eQTL targeting ARF4, which has been shown, in mice, to be involved in metabolic disorders [13].
Discussion
Some studies have observed a differential taggability for common CNVs; these loci, it is argued, are less likely to be in strong LD (r2>0.8) with flanking markers than are frequency-matched SNPs. Several explanations have been proposed to account for this observed differential taggability, such as lower SNP density in regions near CNVs and the higher mutation rates of certain CNVs (producing greater allelic diversity nearby) than those of SNPs [2], [14], [15]. This observed difference in the magnitude of LD between CNVs and SNPs relative to LD among SNPs impacts, through its effect on allelic association, our ability to assess the phenotypic influence of CNVs. On the other side of this controversy, the recent genome-wide study of an extensive catalog of CNVs in 16,000 cases of eight common diseases has argued that CNVs are generally well-tagged by SNPs. Indeed, among 2 - and 3 - class CNVs that passed QC and had MAF>10%, the study found that nearly 80% were tagged by SNPs at r2>0.80. Consequently, replication of association results for CNVs can be conducted in an independent sample set by the use of tag SNPs. In this study, we set out to conduct a study of CNVs by analyzing their effect on gene expression and their association with disease susceptibility and other traits. The CNVs that are well-tagged by SNPs, which we call tCNVs, facilitate SNP-based simulation studies to evaluate enrichment. We proceeded to test whether these CNVs were disproportionately more likely to be functional than frequency-matched SNPs, as trait-associated loci or, under the assumption that few trait-associated polymorphisms are likely to alter the composition of gene products, as eQTLs influencing phenotype by altering gene regulation. Our study found that CNV-tagging SNPs are enriched for cis eQTLs, and, furthermore, that reproducible trait associations show an overrepresentation of tCNVs relative to frequency-matched SNPs. While the tagged CNVs are particularly easy to investigate in enrichment studies, we found that the proportion of eQTLs (at p value threshold of 10−4) in the non-WTCCC CNVs (39%) is higher than in the well-tagged WTCCC CNVs (30%). Given these strong findings on the functional relevance of CNVs, we created a comprehensive online resource of expression associated CNVs in the HapMap populations to supplement our earlier studies on SNP eQTLs.
CNVs can affect phenotype in several ways [16]. Genes fully covered by CNVs may contribute to disease through a duplication or deletion event. Copy number variant breakpoints may disrupt the expression of genes that overlap CNVs. On the other hand, we have identified two CNVs at considerable distance (in trans) from their targets controlling transcript abundance as potential master regulators. Another CNV contains multiple regulatory elements which are each predicting the expression of at least 80 transcripts; the deletion of such important regulatory elements is likely to profoundly alter gene transcription. Importantly, we observed that 1,306 CNVs (out of the 3,432 CNVs included in the WTCCC study) harbor at least one SNP eQTL (defined at p value threshold of 10−4). Given our earlier observation that tCNVs are enriched for expression-associated CNVs (eCNVs), it is interesting to ask whether those CNVs not well-tagged by SNPs have interesting properties with respect to gene regulation. Of the CNVs that are tagged at r2<0.30, 44% harbor SNP eQTLs.
Previous studies [7], [15], [16] have reported that genes that undergo dosage differences due to the presence (proximity) of CNVs show an enrichment for genes involved in immune response and response to external biotic stimuli. We identified an olfactory receptor gene OR6J1 and a gene DAD1 (defender against cell death 1), both on chromosome 14, that are trans-regulated genes for the tCNV CNVR5165.1 on chromosome 11. We found an overrepresentation for target genes involved in the calibrated molecular response to stimulus (whether chemical stimulus or potential internal or invasive threat). Our novel observation in this regard is that previously reported enrichment for genes relevant for molecular-environmental interactions generalizes to the target genes for tCNVs as eCNVs.
The recent WTCCC CNV study concluded that common CNVs that can be typed on existing platforms are unlikely to have a major role in the genetic basis of complex diseases [8]. The same study reported that there was no enrichment of association signals among CNVs involving exonic deletions. Our findings recommend caution in assessments of the contribution of CNVs to the genetics of complex traits. Even under the assumption that most common CNVs are well tagged by SNPs and therefore interrogated by existing SNP GWAS, reproducible trait associations are enriched for these CNVs compared to random expectation. The prominence of such CNVs among reproducible trait associations with autoimmune disorders and metabolic traits suggests that these variants may indeed contribute to certain disease classes; alternatively, they may act in conjunction with other variants such as SNPs to confer susceptibility. Importantly, these CNVs are disproportionately more likely to predict transcript levels than frequency-matched SNPs, and they are more likely to affect many different gene expression traits as master regulatory polymorphisms. An important issue to address is whether the enrichment of tCNVs as eQTLs and as disease-associated SNPs are correlated. Note that the probability that a random SNP is found in the NHGRI catalog increases from 0.062% in the set of HapMap SNPs to about 0.5% (more than 5-fold) in the set of well-tagged CNVs that are not eQTLs and to 2% in the tCNVs that are eQTLs.
It should be noted that the WTCCC CNVs included in our study reflect certain limitations. Indeed, as the WTCCC study itself explicitly noted [8], a large proportion (nearly 60%) of the candidate list of putative CNVs could not be reliably assigned copy number classes from the combination of experimental assay and analytical approaches; it is estimated that only about half of these are not polymorphic [8]. Particularly, nearly 6,500 such putative polymorphisms were excluded from subsequent analyses, as they were called with a single copy number class. It is of course possible that our conclusions may not generalize to these CNVs. Furthermore, eQTL mapping in microarray-based studies in LCLs is likely to yield only a subset of the eQTLs that will be identified using more refined methods in a variety of human tissues. The present study also has little conclusive to say about low frequency variants. Despite these current limitations, the annotation system we implemented in SCAN should prove useful to other investigators and seeks to be as comprehensive as possible by providing functional information for the most extensive map of CNVs to date from the recent population-based genome-wide survey [7]. Since the MAF spectrum of the NHGRI catalog of trait-associated SNPs from published GWAS is quite different from that of the SNPs on the genotyping platforms used to conduct these GWAS, we performed our enrichment analyses while conditioning on the MAF distribution. There are other potential representational biases in the NHGRI catalog of reported variants that may have affected our studies. The enrichment in disease classes relative to other classes of traits, for example, may be the result of increased power (e.g., due to greater sample sizes) for these categories.
Since gene expression is intermediate to other complex phenotypes, a global view of the influence of CNVs on the transcriptome may lead to a better understanding of their role in disease susceptibility. While we identified, as perhaps expected, CNVs located in the HLA region associated with multiple expression phenotypes and with various autoimmune disorders, we also observed, strikingly, several non-HLA CNVs that regulate multiple transcripts, including 2 (one on chromosome 10 and the other on chromosome 2) associated with more than 100 expression traits. Using the most extensive population-based CNV map available [7], we observed a greater proportion of master regulatory CNVs than observed among the well-tagged CNVs. Collectively, all these findings reinforce the importance of considering all types of variation to elucidate the genetic architecture of complex traits.
Materials and Methods
Gene expression dataset using Affymetrix Exon Array
The mRNA expression was assayed in HapMap lymphoblastoid cell lines (LCLs) in 87 CEU and 89 YRI using the Affymetrix GeneChip Human Exon 1.0 ST Array [18]. This dataset is available in the public repository Gene Expression Omnibus (Accession No: GSE9703). Measurements on the exon array are done both at the exon-level and at the gene-level. The exon array includes 1.4 million probesets and profiles nearly 13,000 transcript clusters. The transcript clusters include a set of probesets containing all known exons and 5′ and 3′ UTR regions in the genome so that probes interrogate entire gene regions and not just 3′ UTRs (in contrast to other arrays).
SCAN: A database of eQTL SNPs and CNVs
The results of our eQTL studies, previously on single base polymorphisms [10], [17], [18], in HapMap LCLs have been made publicly available in an online database SCAN [12]. To evaluate the influence of CNVs on the transcriptome, we performed linear regression on over 13,000 transcript clusters with reliable expression – namely, the log2 transformed normalized expression intensity is greater than 6 for at least 80% of the samples – and the CNVs assayed in HapMap LCLs. We downloaded genotype information for these CNVs from the recently released reference catalog of HapMap CNVs [7]. All eQTL analyses were done in both CEU and YRI samples. We extended the SCAN database to include a catalog of expression-associated CNVs in the HapMap populations.
NHGRI database of trait-associated SNPs
The National Human Genome Research Institute (NHGRI) curates the results of published GWAS and makes them publicly available in an online catalog of SNP-trait associations. This catalog provides a useful resource for the investigation of genomic characteristics of trait-associated SNPs and of the role of common variants in common disease etiology [9]. The version we utilized for our present study was retrieved on June 29, 2009. SNP-trait associations included in the catalog are those with p values<1.0×10−5, and generally only one SNP within a gene or a high LD region is included unless there is evidence of an independent association. To evaluate whether our observation on the significant enrichment among tagged CNVs for trait-associated SNPs is driven by certain traits or disease classes, we considered separately various disease types, including neuropsychiatric disorders, cancers, and autoimmune disorders.
Simulation studies
We conducted simulations to evaluate enrichment for eQTLs among the tCNVs included in our study. To empirically generate the null distribution of no enrichment, we randomly generated sets of SNPs of matching minor allele frequency as the original list of SNPs tagging the tCNVs, as previously described [10]. To enable us to perform simulations conditional on MAF, we constructed non-overlapping MAF bins, each of width 0.05, using the MAFs of the SNPs in the HapMap CEU samples. The null sets were drawn from the combined platform SNPs (Affymetrix 6.0 and Illumina 1M) as well as from the entire set of HapMap CEU SNPs. The observed count is then compared to the empirically generated distribution to get an empirical p value for the enrichment.
A catalog of Wellcome Trust CNVs
A survey of 3,432 WTCCC CNVs [8] shows that their minor allele frequency distribution differs markedly from the distribution of reproducible trait-associated SNPs as represented in the NGHRI catalog (See Figure S2). Of these, nearly 80% with minor allele frequency at least 10% are well tagged (r2≥0.8). Roughly 89% of the tagged CNVs (r2≥0.8) are less than 10 kb in length, and 26% are less than 1 kb long (See ). Table S2 shows the median length (in log10) of the WTCCC CNVs.
The 3,432 CNVs included in this study passed the WTCCC quality control filters out of the 4,326 that were called with multiple classes [8].
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. ManolioTA
CollinsFS
CoxNJ
GoldsteinDB
HindorffLA
2009 Finding the missing heritability of complex diseases. Nature 461 747 753
2. McCarrollSA
KuruvillaFG
KornJM
CawleyS
NemeshJ
2008 Integrated detection and population-genetic analysis of SNPs and copy-number variation. Nature Genetics 40 1166 1174
3. McCarrollSA
2008 Extending genome-wide association studies to copy-number variation. Human Molecular Genetics. Hum Mol Gen 17 R2 R135 42
4. WalshT
McClellanJM
McCarthySE
AddingtonAM
PierceSB
2008 Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 320 5875 539 43
5. McCarrollSA
HuettA
KuballaP
ChilewskiSD
LandryA
2008 Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn's disease. Nature Genetics 40 9 1107 12
6. WillerCJ
SpeliotesEK
LoosRJF
LiS
LindgrenCM
2009 Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nature Genetics 41 1 25 34
7. ConradDF
PintoD
RedonR
FeukL
GokcumenO
2010 Origins and functional impact of copy number variation in the human genome. Nature 464 7289 704 12
8. WTCCC 2010 Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature 464 7289 713 20
9. HindorffLA
SethupathyP
JunkinsHA
RamosEM
MehtaJP
2009 Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA 106 23 9362 7
10. NicolaeDL
GamazonE
ZhangW
DuanS
DolanME
2010 Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet 6 e1000888 doi:10.1371/journal.pgen.1000888
11. HuangDW
ShermanBT
LempickiRA
2009 Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nature Protoc 4 1 44 57
12. GamazonER
ZhangW
KonkashbaevA
DuanS
KistnerEO
2010 SCAN: SNP and copy number annotation. Bioinformatics 26 2 259 62 doi:10.1093/bioinformatics/btp644
13. LiS
ChenX
ZhangH
LiangX
XiangY
2009 Differential expression of microRNAs in mouse liver under aberrant energy metabolic status. J Lipid Res 50 9 1756 65
14. RedonR
IshikawaS
FitchKR
FeukL
PerryGH
2006 Global variation in copy number in the human genome. Nature 411 199 204
15. McCarrollSA
BradnerJE
TurpeinenH
VolinL
MartinPJ
2009 Donor-recipient mismatch for common gene deletion polymorphisms in graft-versus-host disease. Nature Genetics 41 1341 1344
16. FeukL
CarsonAR
SchererSW
2006 Structural variation in the human genome. Nat Rev Genet 7 85 97
17. DuanS
HuangRS
ZhangW
BleibelWK
RoeCA
2008 Genetic architecture of transcript-level variation in humans. Am J Hum Genet 82 5 1101 13
18. ZhangW
DuanS
BleibelWK
WiselSA
HuangRS
2009 Identification of common genetic variants that account for transcript isoform variation between human populations. Hum Genet 125 1 81 93
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 2
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Meta-Analysis of Genome-Wide Association Studies in Celiac Disease and Rheumatoid Arthritis Identifies Fourteen Non-HLA Shared Loci
- MiRNA Control of Vegetative Phase Change in Trees
- Risk Alleles for Systemic Lupus Erythematosus in a Large Case-Control Collection and Associations with Clinical Subphenotypes
- The Cardiac Transcription Network Modulated by Gata4, Mef2a, Nkx2.5, Srf, Histone Modifications, and MicroRNAs
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy