
Transgenic Rat Model of Neurodegeneration Caused by Mutation in the Gene
TDP-43 proteinopathies have been observed in a wide range of neurodegenerative diseases. Mutations in the gene encoding TDP-43 (i.e., TDP) have been identified in amyotrophic lateral sclerosis (ALS) and in frontotemporal lobe degeneration associated with motor neuron disease. To study the consequences of TDP mutation in an intact system, we created transgenic rats expressing normal human TDP or a mutant form of human TDP with a M337V substitution. Overexpression of mutant, but not normal, TDP caused widespread neurodegeneration that predominantly affected the motor system. TDP mutation reproduced ALS phenotypes in transgenic rats, as seen by progressive degeneration of motor neurons and denervation atrophy of skeletal muscles. This robust rat model also recapitulated features of TDP-43 proteinopathies including the formation of TDP-43 inclusions, cytoplasmic localization of phosphorylated TDP-43, and fragmentation of TDP-43 protein. TDP transgenic rats will be useful for deciphering the mechanisms underlying TDP-43–related neurodegenerative diseases.
Published in the journal:
. PLoS Genet 6(3): e32767. doi:10.1371/journal.pgen.1000887
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1000887
Summary
TDP-43 proteinopathies have been observed in a wide range of neurodegenerative diseases. Mutations in the gene encoding TDP-43 (i.e., TDP) have been identified in amyotrophic lateral sclerosis (ALS) and in frontotemporal lobe degeneration associated with motor neuron disease. To study the consequences of TDP mutation in an intact system, we created transgenic rats expressing normal human TDP or a mutant form of human TDP with a M337V substitution. Overexpression of mutant, but not normal, TDP caused widespread neurodegeneration that predominantly affected the motor system. TDP mutation reproduced ALS phenotypes in transgenic rats, as seen by progressive degeneration of motor neurons and denervation atrophy of skeletal muscles. This robust rat model also recapitulated features of TDP-43 proteinopathies including the formation of TDP-43 inclusions, cytoplasmic localization of phosphorylated TDP-43, and fragmentation of TDP-43 protein. TDP transgenic rats will be useful for deciphering the mechanisms underlying TDP-43–related neurodegenerative diseases.
Introduction
TAR DNA-binding protein (TDP-43) is a highly conserved ribonucleoprotein that is encoded by the TDP gene and can bind to RNA, DNA, and proteins [1]-[3]. In mammals, the primary transcript of the TDP gene can be alternatively spliced to generate 11 mRNA molecules. The major splice variant is full-length and encodes TDP-43 [4]. While the functions of this complex molecule remain largely unknown, ubiquitinated and phosphorylated TDP-43 accumulates in the nucleus and cytoplasm of affected cells in sporadic amyotrophic lateral sclerosis (ALS) and frontotemporal lobe degeneration (FTLD) [5],[6]. TDP-43 resides predominately in the nucleus and its translocation to the cytoplasm appears to be an early event in the pathological process underlying sporadic ALS [7]. At the end-stages of sporadic ALS and FTLD, C-terminal fragments of TDP-43 are remarkably increased in the brain [5],[6], but the full-length protein remains the major species in spinal cord [8], suggesting that regional differences exist in the metabolism and pathological mechanisms of TDP-43. Although TDP-43 proteinopathies have been identified in a wide range of neurodegenerative diseases including sporadic ALS, FTLD, Alzheimer's disease, and dementia with Lewy bodies [5]–[9], TDP-43 inclusions have not been detected in familial ALS caused by mutation of the SOD1 and FUS genes [10]–[13]. These findings imply that TDP-43 proteinopathy is common to neurodegenerative diseases and that divergent pathological processes may underlie sporadic and familial cases of ALS.
Mutations in the TDP gene segregate with ALS and FTLD associated with motor neuron disease (FTLD-MND) in geographically unrelated families [14]–[18], suggesting that TDP mutation is pathogenic in a subset of neurodegenerative diseases. Transient expression of the mutant, but not the normal, human TDP gene leads to apoptotic death of spinal motor neurons in chicken embryos [15]. In Drosophila melanogaster, depletion of the TDP homolog results in deficient locomotor activity and defects at neuromuscular junctions (NMJs) [19]. Suppression of TDP gene expression induces cell death in cultured neuroblastoma cells [20]. Previous studies indicate that mutation of the TDP gene is neurotoxic and that normal TDP-43 is important to cellular function; however, how mutations in the TDP gene cause neurodegeneration remains unknown.
To study the consequences of TDP mutation in an intact system, we expressed a mutant form of the human TDP gene in rats, which were chosen over mice because they are the preferred animals for pharmacological studies. Overexpression of a mutant, but not the normal, human TDP gene caused widespread neurodegeneration, which predominantly affected the motor system. Transgenic rats that constitutively or conditionally expressed a mutant form of human TDP with a valine-to-methionine substitution at position 337 (M337V) developed similar phenotypes at early ages, the phenotypes that were characterized by motor neuron degeneration accompanied by astrocyte and microglial activation in the spinal cord.
Results
Constitutive expression of a mutant, but not the normal, human TDP gene causes early death in transgenic founder rats
TDP-43 is widely expressed in mammalian tissues [21]. To mimic the expression profile of the endogenous TDP gene, we extracted the minimal human TDP gene (mini TDP gene) from a BAC clone and discarded the excess flanking sequences. The mini human TDP transgene contains essential elements for regulating transgene expression but does not carry unwanted genes into transgenic rats (Figure 1A). Among all known mutations in the TDP gene, the M337V substitution is found in geographically unrelated families and thus is an excellent representative of TDP gene mutations [15],[17]. We introduced the M337V mutation into the mini TDP transgene using a recombineering technique [22]. Using pronuclear injection, we generated three transgenic founders (two males: founders 1 and 2; one female: founder 3) that robustly expressed the miniTDP43M337V transgene (Figure 1A–1C and 1F). The mutant TDP transgenic founders were indistinguishable from their nontransgenic littermates at birth; however, they soon lost mobility and died at postnatal ages. Founder 3 died at the age of 10 days. Founder 2 showed weakness in the limbs at the age of 13 days and became paralyzed by the age of 18 days. Founder 1 showed weakness in a forelimb at the age of 21 days and became paralyzed by the age of 29 days. We examined founder 1 using immunohistochemistry and observed a reduction in motor neurons in the ventral horn of the lumbar spinal cord (Figure 1F). Since none of the mutant TDP (miniTDP43M337V) transgenic rats survived to sexual maturity, mutant TDP transgenic lines could not be established. In parallel, we generated two transgenic founder rats that carried the normal human TDP transgene (miniTDP43wt), which had an identical DNA composition as miniTDP43M337V except that it lacked the M337V mutation (Figure 1A–1E). The miniTDP43wt transgenic rats expressed human TDP-43 protein at levels comparable to those detected in the miniTDP43M337V transgenic founder rats but did not develop paralysis by the age of 200 days. These findings suggest that the disease phenotypes observed in the miniTDP43M337V transgenic founder rats result from toxicity of the TDP gene mutation.

Temporal expression of a mutant human TDP gene in postnatal rats causes progressive paralysis
Since constitutive expression of a mutant human TDP gene caused a severe phenotype in transgenic founders, we used a tetracycline (Tet) regulatory system to express the mutant TDP transgene in a controlled manner. In this way, we could establish transgenic rat lines expressing the human TDP transgene with a pathogenic mutation. The Tet-off system is commonly used in transgenic studies and is comprised of only two elements — a Tet-controlled transactivator (tTA) and a tTA-activated promoter (TRE) [23]. Using pronuclear injection, we established two transgenic lines (line number corresponds to transgene copy) that carry 7 or 16 copies of the TRE-TDP-43M337V transgene under the control of the TRE promoter (Figure 2A). The transcriptional activator, tTA, is inactive in the presence of the Tet derivative, Doxycycline (Dox), allowing for inactivation of a TRE promoter-controlled gene through Dox administration in the bigenic rats that carry the TRE-TDP-43M337V and the tTA transgenes (Figure 2A). In the absence of Dox, tTA constantly activates the TRE-TDP-43M337V transgene, producing an expression pattern that is indistinguishable from constitutive transgene expression [24].

Constitutive expression of the miniTDP-43M337V transgene caused postnatal death in the transgenic founder rats (Figure 1), suggesting that the mutant TDP gene is highly toxic. To test whether the severe phenotype observed in the constitutive transgenic rats could be reproduced in conditional transgenic rats, we produced the TRE-TDP-43M337V and tTA double transgenic rats by crossing the TRE-TDP-43M337V transgenic lines with a tTA transgenic line that expresses the tTA transgene at levels sufficient to vigorously activate tTA reporter genes [24]. To obtain a constitutive pattern of transgene expression, we allowed the TRE-TDP-43M337V transgene to be expressed from early embryogenesis by withholding Dox treatment. Consistent with findings in constitutive transgenic rats (Figure 1), expression of the TRE-TDP-43M337V transgene from early embryonic stages caused severe phenotypes in the conditional transgenic rats of line 16 (Figure 2B). Transgenic rats of line 16 became paralyzed and died by postnatal day 20 (P20). The similarity in phenotypes between the constitutive and conditional transgenic rats indicates that the observed defects did not result from an insertional mutation.
Expression of the TDP-43M337V transgene from early embryogenesis caused early death in transgenic rats, making functional analysis of this model a challenge. To facilitate analysis of motor function, we added Dox to the drinking water (50 µg/ml) of breeding rats to suppress transgene expression during embryonic development. We then withdrew Dox at 4 days before delivery to allow for recovery of transgene expression in postnatal rats. As a result, the transgene was not expressed in newborn pups but was fully expressed in postnatal rats by P10 (Figure S1). The TRE-TDP-43M337V transgenic rats of line 16 showed a rapid progression of disease phenotypes, exhibiting limb weakness by P20 and paralysis before P35 (Figure 2E). In contrast, the TRE-TDP-43M337V transgenic rats of line 7 showed a later onset and a slower progression of similar phenotypes (Figure 2C–2E). Disease progression in line 7 could be divided into four distinct stages [25]: the nonsymptomatic stage, disease onset, the paralysis stage, and the disease end stage. Disease onset was defined as an unrecoverable reduction in running time on a rotating Rotarod. The paralysis stage was defined as visible dragging of a limb. The disease end stage was defined as paralysis in two or more limbs. Postnatal rats aged 21 days were subjected to a Rotarod test to determine disease onset (Figure 2D). Since transgenic rats of line 16 developed early paralysis and had a rapid disease progression, determining the time of disease onset for this high-copy line was technically difficult. Transgenic rats of line 16 showed limb weakness by an age of 20 days and became paralyzed in the legs by an age of 35 days, with no sexual dimorphism existing in the rate of disease progression (Figure 2E). In contrast, transgenic rats of line 7 displayed sexual dimorphism in the time of disease onset and in the rate of disease progression (Figure 2D and 2E). Sexual dimorphism in phenotypic onset has also been observed in an ALS animal model expressing mutant human SOD1 genes [26]–[29]. The disease phenotypes observed in the mutant TDP (TRE-TDP-43M337V) transgenic rats were not observed in normal TDP transgenic rats (miniTDP-43WT) by an age of 200 days, though these rats expressed the human TDP transgene at comparable levels as TRE-TDP-43M337V rats (Figure 2B–2E). An examination of TRE-TDP-43M337V transgenic offspring revealed that, consistent with findings in miniTDP-43M337V transgenic founders (Figure 1), the disease phenotypes in these animals were related to mutation of the TDP gene.
Axon terminals are the primary targets of degeneration caused by mutation of the TDP gene
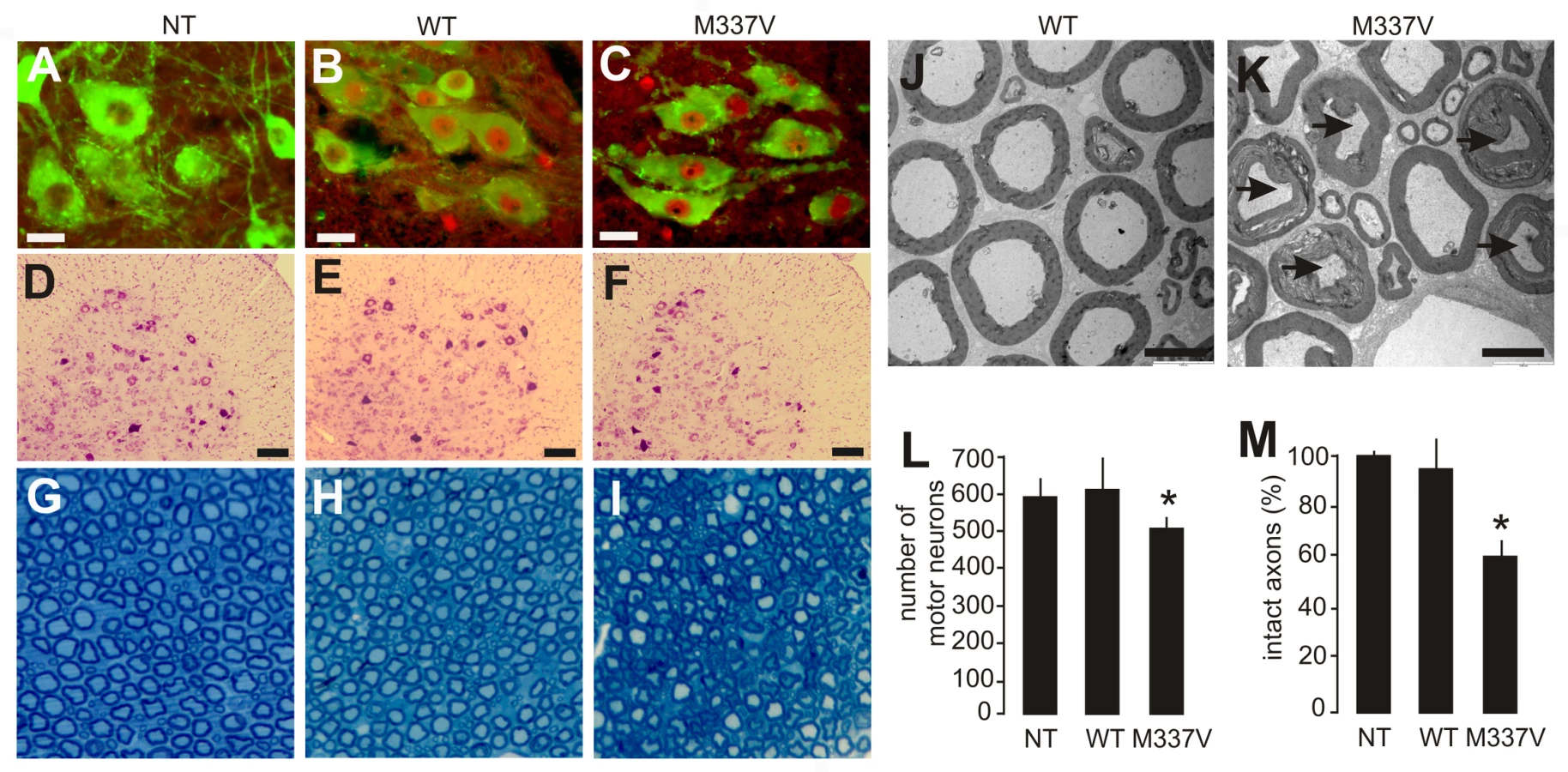
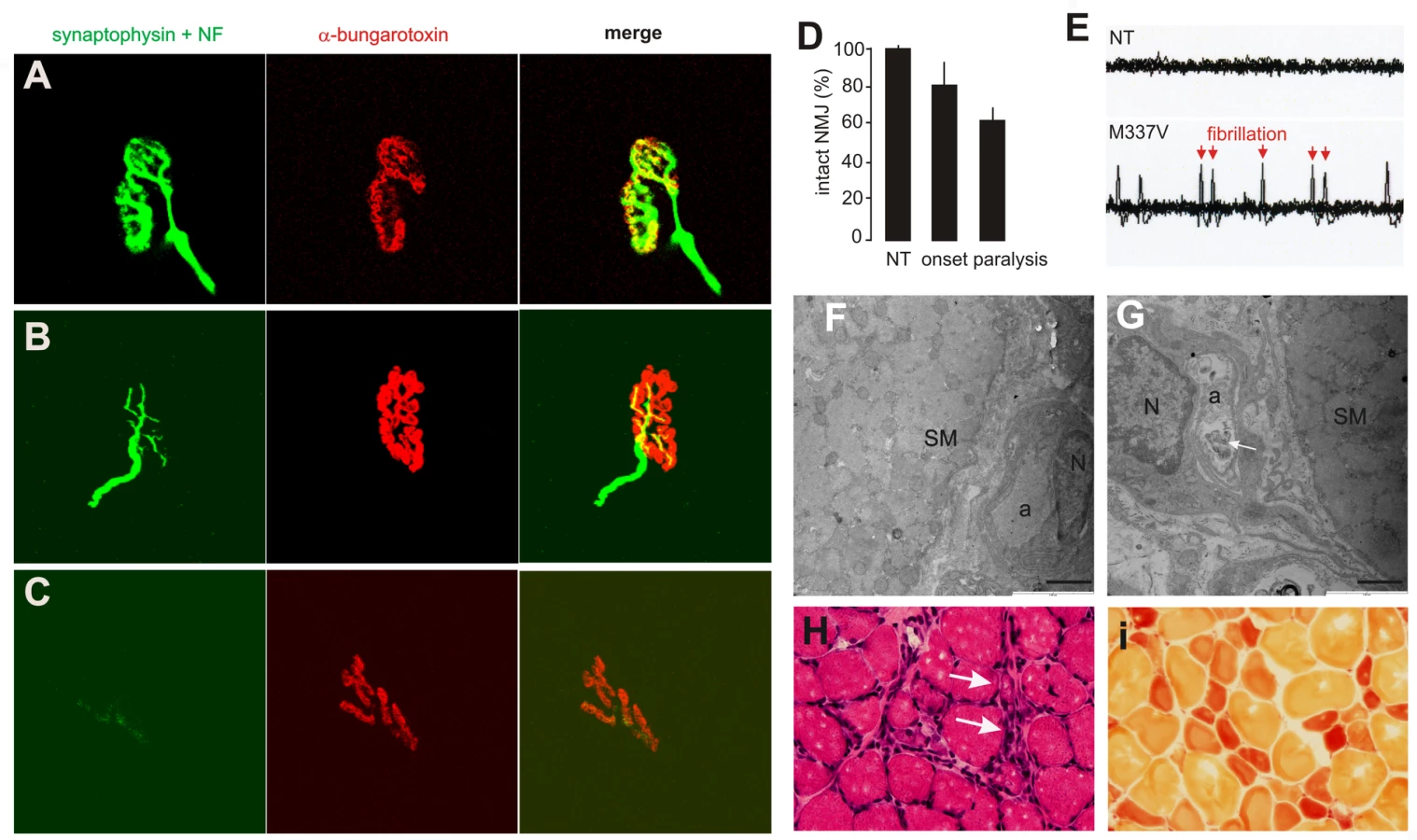
Anatomical analysis revealed that motor neurons in the spinal cord robustly expressed the human TDP transgene (Figure 3A–3C). The number of spinal motor neurons was significantly reduced in mutant TDP transgenic rats but not in normal TDP transgenic rats (Figure 3D–3F and 3I), although the mutant and normal TDP transgenic rats expressed human TDP-43 at comparable levels (Figure 2B). Large-caliber neurons were preferentially affected in mutant rats at the end stages of disease (Figure 3D–3F and 3L). During the paralysis stage, degenerating axons were clearly visible in the ventral (Figure 3G–3I and 3M) and dorsal roots (Figure S2), with motor axons of the corticospinal track also being affected (Figure S3). Confocal microscopy revealed that denervation of synaptic endplates in skeletal muscle occurred at disease onset (Figure 4B and 4D) and worsened at the end stage of disease (Figure 4C and 4D). Electron microscopy confirmed that, in the mutant transgenic rats, degeneration of motor neuron axons occurred at disease onset (Figure 4F and 4G); however, no loss of motor neurons was detected in the mutant TDP transgenic rats at this time. These findings suggest that axon terminals are the primary targets of degeneration associated with pathogenic mutation of TDP. In the mutant TDP transgenic rats, denervation of skeletal muscle fibers was confirmed by electromyography, which detected frequent fibrillation potentials—a characteristic of muscle denervation and regeneration (Figure 4E). As results of denervation, groups of skeletal muscles were atrophied (Figure 4H and 4I). These pathological changes were correlated with progressive paralysis in the mutant transgenic rats (Figure 2C–2E).
Neurodegeneration is accompanied by glial reactivity
Silver staining revealed that, in mutant TDP transgenic rats, spinal motor neurons degenerated during end-stage disease (Figure 5A and 5B). A previous study showed that transient expression of the mutant, but not the normal human TDP gene, causes apoptotic death in the spinal cord of chicken embryos [15]. Consistent with the finding from this transient transfection study [15], motor neurons in the spinal cord underwent apoptosis in paralyzed transgenic rats (Figure 5C and 5D). Studies of mutant SOD1 mice suggest that glial cells play an important role in ALS pathogenesis [28], [30]–[33]. Therefore, we examined glial reactions in our paralyzed rats. We found that astrocytes and microglia were increased around the motor neurons in the spinal cord (Figure 6A–6D). The finding suggests that a glial reaction occurs in response to motor neuron degeneration.


Neurodegeneration is not restricted to motor neurons at end stages of disease
TDP-43 inclusions are found in the brain and spinal cord of patients with sporadic ALS, FTLD, Alzheimer's disease, or dementia with Lewy bodies [5]–[9], suggesting that TDP-43 proteinopathies are common to neurodegenerative diseases. Pathogenic mutations in the TDP gene have been identified not only in ALS, but also in FTLD-MND [14]–[18]. Degeneration associated with mutations in the TDP gene may not be restricted to motor neurons. Indeed, silver staining revealed that neurodegeneration occurred in the cortex, hippocampus, and cerebellum of mutant transgenic rats with end stages of disease (Figure 7A–7F) but not in those with earlier stages of disease (Figure 2D and data not shown). Nevertheless, degenerating neurons were not detected in the substantia nigra of paralyzed rats (data not shown), despite the fact that transient overexpression of the normal human TDP gene in rats has been shown to induce a loss of dopaminergic neurons in this brain region [34]. Neuropathological findings were correlated with phenotypic expression in mutant TDP transgenic rats (Figure 2, Figure 3, Figure 4). Toxicity of the pathogenic TDP gene mutation was not restricted to motor neurons, though these neurons were affected by the mutation to a greater degree than all the other neuron types examined.

Phosphorylated TDP accumulates in affected cells in TDP transgenic rats
Phosphorylated TDP-43 inclusions are a signature pathological feature of sporadic ALS and FTLD [5], [6], [35]–[37]. To detect phosphorylated TDP-43 inclusions in our transgenic rats, we tested a polyclonal antibody specific to phosphorylated TDP-43 on brain sections of FTLD patients and TDP transgenic rats. This phospho-TDP-43 antibody detected cytoplasmic accumulation of phosphorylated TDP-43 in FTLD patients, but not in control subjects (Figure 8A). Similarly, phosphorylated TDP-43 was diffusely distributed in affected neurons in transgenic rats expressing the mutant or normal human TDP transgene (Figure 8D). We generated a polyclonal antibody recognizing both phosphorylated and non-phosphorylated human TDP-43 (Figure 1C–1F) and detected a robust expression of the human TDP transgene in transgenic rats (Figure 8B and 8C). TDP-43 was diffusely distributed in the nucleus and cytoplasm of cells within transgenic rats (Figure 8). However, TDP-43 inclusions were detected rarely, being present only in the cortex (Figure 8B) and not in the spinal cord (Figure 8C) of transgenic animals.

Immunohistochemistry revealed that typical ubiquitin-positive inclusions were not present in the spinal cords of normal or mutant TDP transgenic rats, though the intensity of ubiquitin immunostaining was greater in these animals than in nontransgenic rats (Figure S4). Since TDP-43 inclusions were rare in transgenic rats, even at end-stage disease, we further examined TDP-43 ubiquitination using immunoprecipitation combined with immunoblotting analysis. Ubiquitinated TDP-43 was detected in the mutant TDP transgenic rats (Figure S4). Immunoblotting revealed that a small amount of TDP-43 fragments (less than 43 kDa) was present in TDP transgenic rats (Figure 2B and Figure S5). TDP-43 fragments were detected in urea tissue extracts from rats at the paralysis stage, but not in extracts from those at disease onset (Figure S5). The finding suggests that the solubility of the small TDP-43 fragment is reduced as the disease progresses.
Discussion
Expression of the human TDP gene containing a M337V substitution reproduced the phenotypes of ALS in rats. That is, these animals exhibited progressive degeneration of motor neurons and denervation atrophy of skeletal muscles. In this transgenic rat model, neurodegeneration was not restricted to motor neurons and could be seen in other types of neurons including cortical neurons, hippocampal neurons, and cerebellar neurons. However, TDP mutation affected motor neurons earlier and more severely than other neurons in the central nervous system (CNS). This robust rat model also recapitulated features of TDP-43 proteinopathies, including the formation of TDP-43 inclusions, cytoplasmic localization of phosphorylated TDP-43, and fragmentation of TDP-43.
While our transgenic rat model developed the phenotypes of ALS, it displayed degeneration of CNS neurons other than motor neurons at the end stages of the disease. Our findings in mutant TDP transgenic rats do not necessarily contradict observations in ALS patients. ALS is traditionally thought to affect only motor neurons, but recent studies showed that neurons other than motor neurons also degenerate in ALS [38]. This point is strikingly illustrated by the observation in some ALS patients who live with the disease much longer than the average disease duration [38]–[40]. Moreover, some ALS and FTLD cases share symptoms and pathological characteristics [41]. Although mutations of the TDP gene are primarily associated with ALS [14]–[17], a recent study found that a novel mutation in the TDP gene is associated with FTLD-MND [18], suggesting that the toxicity (if any) of the TDP gene mutation is not restricted to motor neurons [18]. Further studies are warranted to ascertain whether a correlation exists between the pathological changes induced by TDP mutation and TDP-43 proteinopathies observed in sporadic ALS and FTLD. The fact that TDP-43 proteinopathy is observed in a wide range of neurodegenerative diseases suggests that mutations in the TDP gene are generally neurotoxic [5], [6], [9], [42]–[45]. Neurodegenerative diseases may share common pathological mechanisms, with a certain subgroup of neurons being predominantly affected under each disease condition. Our mutant TDP transgenic rat is a robust model of neurodegeneration caused by mutation of the TDP gene.
Many features of TDP-43 proteinopathies were reproduced in our TDP transgenic rats. Redistribution, phosphorylation, and aggregation of TDP-43 are all hallmarks of sporadic FTLD and ALS [5],[44],[45]. A recent clinical study showed that TDP-43 redistribution appears to be an early event in TDP-43 proteinopathy [7], suggesting that TDP-43 redistribution underlies the pathogenesis of neurodegeneration. Our results showed that phosphorylated TDP-43 was diffusely distributed in the cytoplasm and nucleus of affected cells in paralyzed mutant TDP transgenic rats as well as in non-paralyzed, normal TDP transgenic rats. The presence of phosphorylated TDP-43 in normal TDP transgenic rats does not exclude the possibility that TDP-43 phosphorylation contributes to pathogenesis induced by TDP mutation. Specifically, TDP mutation may impart toxicity by enhancing the normal functions of the gene. For example, mutation of the LRRK2 gene causes Parkinson's disease by enhancing (at least partially) the kinase activity of LRRK2 [46],[47]. Gene mutations can be classified into three types based on their effect on protein function: gain of function, loss of function, and dominant negative effect. Pathogenic mutation of the TDP gene may cause disease through any one of these three effects on protein function. Resolving the nature of the TDP gene mutation will require a more sophisticated model such as a knockin mouse. TDP-43 inclusions and fragmentation were rarely observed and were present only at end-stage disease, suggesting that these pathologies may be consequence of, rather than a cause of, neurodegeneration in TDP transgenic rats. C-terminal truncated products of TDP-43 are thought to result from caspase cleavage of full-length TDP-43 [48]. Accordingly, C-terminal fragmentation of TDP-43 is likely a consequence, instead of a cause, of neurodegeneration because caspase activation is a terminal feature of cell death. In addition, we cannot exclude the possibility that overexpression of the TDP transgene interferes with rat development, since the mutant TDP transgenic rats died at postnatal ages.
Typical ALS has a late onset and rapidly progresses [12], [13], [17], [29], [49]–[51]. In contrast, mutant TDP transgenic rats developed paralysis at early ages, with the paralysis being similar to that seen in ALS. Early onset of disease phenotypes in our rat model likely results from toxicity of the TDP gene mutation, as evidenced by the following three findings. First, paralysis and lethality were observed in the mutant miniTDP43M337V transgenic founders, but not in the normal miniTDP43WT transgenic founders. Second, paralysis and neurodegeneration were observed in the inducible mutant TDP transgenic rats, but not in offspring of the constitutive normal TDP transgenic rats, despite the fact that both lines exhibited comparable expression of the human TDP transgenes. Third, similar phenotypes were observed in the constitutive mutant TDP transgenic founders and in the inducible mutant TDP transgenic offspring. One transgenic founder rat carried only six copies of the mini mutant TDP transgene and developed paralysis in postnatal age. The copy number of the mutant TDP transgene that is required for phenotypic expression in transgenic rats is much lower than the copy threshold of mutant SOD1 transgenes [52],[53]. To activate the inducible mutant TDP transgene, we used a low-copy tTA transgenic line that carries only two copies of the tTA transgene [24]. Therefore, expression levels of the TDP transgene in the inducible transgenic rats were comparable to those in the constitutive normal TDP transgenic rats. Transgenic rats expressing the mutant TDP gene displayed a wider range of neurodegeneration than transgenic rodents expressing mutant SOD1 genes [29], [52]–[54], with neurodegeneration predominantly affecting the motor system. Such unrestricted toxicity of the TDP gene mutation may lead to an early onset of the disease. In some aspects, phenotypes observed in our transgenic rats are similar to those detected in transgenic mice that express the human TDP gene with a A315T substitution [55]. In these rodent models, both upper and lower motor neurons are affected and TDP-43 inclusions are rare. However, our rat model developed paralysis at postnatal ages and experienced a rapid disease progression, while the mutant TDP transgenic mice develop disease phenotypes during middle age and have varying disease durations [55]. Different mutations in the TDP gene and different animal species may contribute to phenotypic variation between the rat and mouse models.
Our findings in TDP transgenic rats indicate that mutation of the TDP gene is highly toxic in rodents, though the nature of the pathogenic mutation in the TDP gene remains to be resolved. Since deletion of the TDP gene in Drosophila causes defects at NMJs [19], the possibility that the TDP gene mutation produces a dominant-negative effect cannot be excluded. Although the nature of TDP gene mutation will need to be determined using a more sophisticated model, our TDP transgenic rats will be useful for mechanistic study of TDP-43-related neurodegenerative diseases.
Materials and Methods
Ethics statement
Animal use followed NIH guidelines. The animal use protocol was approved by the Institutional Animal Care and Use Committees (IACUC) at Thomas Jefferson University. The Committee for Oversight of Research Involving the Dead at the University of Pittsburgh School of Medicine approved the use of human tissue from the University of Pittsburgh ALS Tissue Bank. Age-matched tissue sections from two FTLD and two non-neurological disease controls were used for the study.
Transgene constructs
The 22-kb mini human TDP gene was extracted from a BAC clone (RP11-829B14), and a M337V substitution was introduced into the mini TDP gene by homologous recombination in Escherichia coli [22]. The normal and mutant mini TDP transgenes were linearized by restriction digestion, purified from agarose gels, and then used to produce transgenic rats through microinjection. To generate Tet-regulatable TDP transgenic rats, we PCR-amplified the human TDP-43 ORF from a human brain cDNA pool (Invitrogen) and generated a mutant carrying the M337V substitution using site-directed mutagenesis (Stratagene). The mutated human TDP-43 cDNA gene was inserted downstream of a tTA-dependent promoter that was constructed by fusing seven tetracycline-responsive elements (TRE) with a minimal cytomegalovirus promoter (TRE-miniCMV). To enhance gene splicing and expression, we inserted the first intron of the human ubiquitin C gene between the TRE-miniCMV promoter and the TDP-43 ORF [24].
Transgenic animal production
Linearized miniTDP43 and TRE-TDP43 transgenes were injected into the pronuclei of fertilized eggs of Sprague-Dawley rats. The injected eggs were then transplanted into pseudopregnant females for embryonic development [56]. Transgenic founders carrying miniTDP-43 transgenes were analyzed for disease phenotypes. Transgenic founders carrying TRE-TDP-43 transgenes were crossed with CAG-tTA transgenic rats to produce double transgenic offspring, which were analyzed for transgene expression and disease phenotypes. The TDP transgenic rats were identified by PCR amplification of the human TDP gene using the following primer pair: 5′-TGCGGGAGTTCTTCTCTCAG (forward) and 5′-AGCCACCTGGATTACCACCA (reverse). The copy number of the transgene was determined by quantitative PCR using two primer pairs. The first primer pair was designed to amplify a DNA fragment of the same composition from both the human and the rat TDP gene: 5′-TGAGCCCATTGAAATACCATC-3′ and 5′-TACACTGAGACACTGGATTC. The second primer pair was designed to amplify the rat prolactin gene as an internal control: 5′-CCTCTATGAACGAAACCCAC-3′ and 5′-CTTCCGGCTAATCCA CAATG-3′.
Antibody production
A rabbit polyclonal antibody was produced by Genemed Company. Rabbits were immunized with the synthetic peptide, (N-terminal)-EDELREFFSQYGDVM. Antiserum was then affinity-purified using a peptide-conjugated column (Pierce).
Immunohistochemistry
Under deep anesthesia, animals were transcardially perfused with 1X PBS (pH 7.4) and then with 4% paraformaldehyde (PFA) dissolved in 1X PBS buffer. The brain, spinal cord, and gastrocnemius muscle of perfused animals were collected and further fixed in the same fixative overnight. Some tissue blocks were embedded in paraffin and sectioned into 10 µm-thick slices. Paraffin-embedded sections were treated with 10 mM sodium citrate buffer (pH 6.0) to retrieve antigens for immunostaining. Paraffin-embedded coronal sections of the brain and transverse sections of the spinal cord were deparaffinized and immunostained with human TDP-43-specific antibody (1∶1,000; made in house) or a phospho-TDP-43-specific antibody (1∶1,000; COSMO Bio Co., TIP-PTD-P02). Immunostaining was visualized using an ABC kit in combination with diaminobenzidine (Vector). The immunostained sections were lightly counterstained with hematoxylin to display nuclei. After antigen retrieval, paraffin-embedded sections of the lumbar spinal cord were immunostained for human TDP-43 (1∶300) and ChAT (goat antiserum; Millipore). Immunofluorescence staining for human TDP-43 (red) and ChAT (green) was visualized using a Nikon fluorescence microscope, and images were acquired using a Nikon digital camera. Paraffin-embedded sections of the gastrocnemius muscle were stained with hematoxylin and eosin (H&E) to visualize tissue structures.
For NMJ detection, gastrocnemius muscles were fixed in 4% PFA for 2 h and sectioned on a cryostat into 100 µm-thick sections. Serial sections of the muscles were incubated with α-bungarotoxin (Invitrogen) for 30 min, washed in PBS three times, incubated overnight with mouse monoclonal antibodies to neurofilament (Sigma) and synaptophysin (Millipore), and then incubated for 1 h with a secondary antibody (FITC goat anti-mouse IgG1; Jackson Immunology).
For detection of apoptotic cells and glial cells, 4% PFA-fixed lumbar spinal cords were cut into three sets of 10 µm-thick serial sections on a cryostat. Every first section was incubated with TUNEL staining reagent (Millipore) and goat anti-ChAT antibody. Every second section was incubated with the ChAT antibody and mouse anti-GFAP. Every third section was incubated with the ChAT antibody and mouse anti-CD68 antibody. Sections were then incubated with appropriately labeled secondary antibodies. The antibodies were purchased from Millipore. Images were captured using a Zeiss LSM510 META confocal system. The NMJ was reconstructed using z-stack projections produced from serial scanning every 1 µm.
Esterase histochemistry
Fresh gastrocnemius muscle was snap-frozen in liquid nitrogen and cut into 12 µm-thick sections on a cryostat. Nonspecific esterase activity was detected using the α-napthyl acetate protocol. Denervated muscle fibers were stained a red-brown color, with normal fibers displaying a yellow-to-brown color.
Silver staining
Degenerating neurons were visualized using the Bielschowski silver-staining method as well as the FD NeuroSilver kit (FD Neurotechnologies, Baltimore, MD). For the Bielschowski silver method, paraffin-embedded spinal cord was transversely cut into 10 µm-thick sections. For staining using the FD NeuroSilver kit, 40 µm-thick coronal sections were obtained by slicing through the forebrain and cerebellum using a cryostat and then stained per the manufacturer's instructions.
Toluidine blue staining and electron microscopy
Rats were anesthetized and perfused with a mixture of 4% PFA and 2% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4). The L3 and L4 ventral and dorsal roots were removed and post-fixed in the same fixative at 4°C overnight. The roots were then further fixed in 1% osmium tetroxide in 0.1 M phosphate buffer (pH 7.4) for 1 h. The well-fixed tissues were dehydrated in graded ethanol and embedded in Epon 812 (Electron Microscopic Sciences, Fort Washington, PA). Thin sections (80 nm) were then stained with uranyl acetate and lead citrate for observation under a transmission electron microscope (Hitachi H7500-I). For toluidine staining, roots were transversely cut into 1 µm-thick sections. Axons in the nerve roots were examined in the semi-thin sections under a light microscope (Olympus AX70).
Cresyl violet staining and stereological cell counting
A 1-mm central segment of the L3 spinal cord was cut into 30-µm thick sections using a cryostat. Every third section was stained with cresyl violet and mounted in sequential order (rostral-caudal). Neurons with a diameter larger than 25 µm were counted in the ventral horns on both sides. The number of targeted neurons was estimated using a fractionator-based unbiased stereology software program (Stereologer), which was run on a PC computer that was attached to a Nikon 80i microscope with a motorized XYZ stage (Prior). At low magnification (40x), the targeting area was outlined, and a random sampling grid was created. At 1000× magnification, an optical dissector probe was randomly generated by the program in the designated area. The presence of clearly definable neurons was noted according to defined inclusion and exclusion limits of the dissector. This process was repeated on all selected sections. The total number of defined neurons was calculated by the software based on values obtained from random counts.
Electromyography
Animals were anesthetized during electromyography (EMG) examination. The fibrillation potential of the gastrocnemius muscle was recorded with an EMG instrument (CMS6600; COTEC Inc.) using a 27-gauge monopolar needle electrode and a 29-gauge reference needle electrode. During recording, a sub-dermal ground electrode was placed in the forelimb. Spontaneous electrical activity of selected skeletal muscle was recorded for 2 min.
Statistical analysis
The number of defined neurons in the ventral horn was compared between groups of transgenic rats. The difference in the number of neurons was analyzed using an unpaired t test. The null hypothesis was rejected at the 0.05 level.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. BurattiE
BrindisiA
GiombiM
TisminetzkyS
AyalaMY
2005 TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J Biol Chem 280 37572 37584
2. AbhyankarMM
UrekarC
ReddiPP
2007 A novel CpG-free vertebrate insulator silences the testis-specific SP-10 gene in somatic tissues: role for TDP-43 in insulator function. J Biol Chem 282 36143 36154
3. BoseKJ
WangFI
HungL
TarnYW
ShenKC
2008 TDP-43 overexpression enhances exon 7 inclusion during the survival of motor neuron pre-mRNA splicing. J Biol Chem 283 28852 28859
4. WangYH
WangFI
BoseJ
ShenKC
2004 Structural diversity and functional implications of the eukaryotic TDP gene family. Genomics 83 130 139
5. NeumannM
SampathuMD
KwongKL
TruaxCA
MicsenyiCM
2006 Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314 130 133
6. AraiT
HasegawaM
AkiyamaH
IkedaK
NonakaT
2006 TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351 602 611
7. GiordanaTM
PiccininiM
GrifoniS
MarcoDG
VercellinoM
2009 TDP-43 Redistribution Is an Early Event in Sporadic Amyotrophic Lateral Sclerosis. Brain Pathol 20 351 360
8. NeumannM
KwongKL
LeeBE
KremmerE
FlatleyA
2009 Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol 117 137 149
9. AraiT
MackenzieRI
HasegawaM
NonokaT
NiizatoK
2009 Phosphorylated TDP-43 in Alzheimer's disease and dementia with Lewy bodies. Acta Neuropathol 117 125 136
10. MackenzieIR
BigioEH
IncePG
GeserF
NeumannM
2007 Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol 61 427 434
11. TanCF
EguchiH
TagawaA
OnoderaO
IwasakiT
2007 TDP-43 immunoreactivity in neuronal inclusions in familial amyotrophic lateral sclerosis with or without SOD1 gene mutation. Acta Neuropathol 113 535 542
12. KwiatkowskiJTJr
BoscoAD
LeclercLA
TamrazianE
VanderburgRC
2009 Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323 1205 1208
13. VanceC
RogeljB
HortobagyiT
VosDJK
NishimuraLA
2009 Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323 1208 1211
14. KabashiE
ValdmanisNP
DionP
SpiegelmanD
McConkeyJB
2008 TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 40 572 574
15. SreedharanJ
BlairPI
TripathiBV
HuX
VanceC
2008 TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319 1668 1672
16. DeerlinMV
LeverenzBJ
BekrisML
BirdDT
YuanW
2008 TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol 7 409 416
17. RutherfordJN
ZhangJY
BakerM
GassMJ
FinchAN
2008 Novel Mutations in TARDBP (TDP-43) in Patients with Familial Amyotrophic Lateral Sclerosis. PLoS Genet 4 e1000193 doi:10.1371/journal.pgen.1000193
18. BenajibaL
BerLI
CamuzatA
LacosteM
Thomas-AnterionC
2009 TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Annals of Neurology 65 470 474
19. FeiguinF
GodenaKV
RomanoG
D'AmbrogioA
KlimaR
2009 Depletion of TDP-43 affects Drosophila motoneurons terminal synapsis and locomotive behavior. FEBS Lett 583 1586 1592
20. IguchiY
KatsunoM
NiwaJ
YamadaS
SoneJ
2009 TDP-43 depletion induces neuronal cell damage through dysregulation of Rho family GTPases. J Biol Chem 284 22059 22066
21. OuHS
WuF
HarrichD
Garcia-MartinezFL
GaynorBR
1995 Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol 69 3584 3596
22. WarmingS
CostantinoN
CourtLD
JenkinsAN
CopelandGN
2005 Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res 33 e36
23. YamamotoA
HenR
DauerTW
2001 The ons and offs of inducible transgenic technology: a review. Neurobiol Dis 8 923 932
24. ZhouH
HuangC
YangM
LandelPC
XiaYP
2009 Developing tTA Transgenic Rats for Inducible and Reversible Gene Expression. Int J Biol Sci 2 171 181
25. XiaGX
ZhouH
HuangY
XuZ
2006 Allele-specific RNAi selectively silences mutant SOD1 and achieves significant therapeutic benefit in vivo. Neurobiol Dis 23 578 586
26. BruijnIL
BecherWM
LeeKM
AndersonLK
JenkinsAN
1997 ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 18 327 338
27. BruijnIL
HouseweartKM
KatoS
AndersonLK
AndersonDS
1998 Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 281 1851 1854
28. ClementMA
NguyenDM
RobertsAE
GarciaLM
BoilleeS
2003 Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice, Science 302 113 117
29. GurneyEM
PuH
ChiuYA
CantoDCM
PolchowYC
1994 Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264 1772 1775
30. BoilleeS
YamanakaK
LobsigerSC
CopelandGN
JenkinsAN
2006 Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312 1389 1392
31. D GiorgioPF
CarrascoAM
SiaoCM
ManiatisT
EgganK
2007 Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat Neurosci 10 608 614
32. NagaiM
ReBD
NagataT
ChalazonitisA
JessellMT
2007 Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci 10 615 622
33. YamanakaK
ChunJS
BoilleeS
Fujimori-TonouN
YamashitaH
2008 Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci 11 251 253
34. TatomBJ
WangBD
DaytonDR
SkalliO
HuttonLM
2009 Mimicking Aspects of Frontotemporal Lobar Degeneration and Lou Gehrig's Disease in Rats via TDP-43 Overexpression. Mol Ther 17 607 613
35. CairnsJN
NeumannM
BigioHE
HolmEI
TroostD
2007 TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol 171 227 240
36. SeelaarH
SchelhaasJH
AzmaniA
KustersB
RossoS
2007 TDP-43 pathology in familial frontotemporal dementia and motor neuron disease without Progranulin mutations. Brain 130 1375 1385
37. BrandmeirNJ
GeserF
KwongLK
ZimmermanE
QianJ
2008 Severe subcortical TDP-43 pathology in sporadic frontotemporal lobar degeneration with motor neuron disease. Acta Neuropathol 115 123 131
38. NishihiraY
TanFC
HoshiY
IwanagaK
YamadaM
2009 Sporadic amyotrophic lateral sclerosis of long duration is associated with relatively mild TDP-43 pathology. Acta Neuropathol 117 45 53
39. MachidaY
TsuchiyaK
AnnoM
HagaC
ItoT
1999 Sporadic amyotrophic lateral sclerosis with multiple system degeneration: a report of an autopsy case without respirator administration. Acta Neuropathol 98 512 515
40. TsuchiyaK
SanoM
ShiotsuH
AkiyamaH
WatabikiS
2004 Sporadic amyotrophic lateral sclerosis of long duration mimicking spinal progressive muscular atrophy exists: additional autopsy case with a clinical course of 19 years. Neuropathology 24 228 235
41. LilloP
HodgesRJ
2009 Frontotemporal dementia and motor neurone disease: overlapping clinic-pathological disorders. J Clin Neurosci 16 1131 1135
42. RohnTT
2008 Caspase-cleaved TAR DNA-binding protein-43 is a major pathological finding in Alzheimer's disease. Brain Res 1228 189 198
43. Nakashima-YasudaH
UryuK
RobinsonJ
XieXS
HurtigH
2007 Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol 114 221 229
44. KwongKL
NeumannM
SampathuMD
LeeMV
TrojanowskiQJ
2007 TDP-43 proteinopathy: the neuropathology underlying major forms of sporadic and familial frontotemporal lobar degeneration and motor neuron disease. Acta Neuropathol 114 63 70
45. KwongKL
UryuK
TrojanowskiQJ
LeeMV
2008 TDP-43 proteinopathies: neurodegenerative protein misfolding diseases without amyloidosis. Neurosignals 16 41 51
46. MacLeodD
DowmanJ
HammondR
LeeteT
InoueK
2006 The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron 52 587 593
47. SmithWW
PeiZ
JiangH
DawsonLV
DawsonMT
2006 Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci 9 1231 1233
48. ZhangJY
XuFY
DickeyAC
BurattiE
BaralleF
2007 Progranulin mediates caspase-dependent cleavage of TAR DNA binding protein-43. J Neurosci 27 10530 10534
49. KabashiE
DaoudH
RiviereBJ
ValdmanisNP
BourgouinP
2009 No TARDBP mutations in a French Canadian population of patients with Parkinson disease. Arch Neurol 66 281 282
50. DengXH
HentatiA
TainerAJ
IqbalZ
CayabyabA
1993 Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science 261 1047 1051
51. RosenRD
SiddiqueT
PattersonD
FiglewiczAD
SappP
1993 Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362 59 62
52. HowlandSD
LiuJ
SheY
GoadB
MaragakisJN
2002 Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc Natl Acad Sci U S A 99 1604 1609
53. NagaiM
AokiM
MiyoshiI
KatoM
PasinelliP
2001 Rats expressing human cytosolic copper-zinc superoxide dismutase transgenes with amyotrophic lateral sclerosis: associated mutations develop motor neuron disease. J Neurosci 21 9246 9254
54. WongCP
PardoAC
BorcheltRD
LeeKM
CopelandGN
1995 An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 14 1105 1116
55. WegorzewskaI
BellS
CairnsNJ
MillerTM
BalohRH
2009 TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci USA 106 18809 18814
56. FilipiakEW
SaundersLT
2006 Advances in transgenic rat production. Transgenic Res 15 673 686
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 3
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Deciphering Normal Blood Gene Expression Variation—The NOWAC Postgenome Study
- Transgenic Rat Model of Neurodegeneration Caused by Mutation in the Gene
- Papillorenal Syndrome-Causing Missense Mutations in / Result in Hypomorphic Alleles in Mouse and Human
- Fatal Cardiac Arrhythmia and Long-QT Syndrome in a New Form of Congenital Generalized Lipodystrophy with Muscle Rippling (CGL4) Due to Mutations
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy