
Identification and Functional Analysis of the Vision-Specific BBS3 (ARL6) Long Isoform
Bardet-Biedl Syndrome (BBS) is a heterogeneous syndromic form of retinal degeneration. We have identified a novel transcript of a known BBS gene, BBS3 (ARL6), which includes an additional exon. This transcript, BBS3L, is evolutionally conserved and is expressed predominantly in the eye, suggesting a specialized role in vision. Using antisense oligonucleotide knockdown in zebrafish, we previously demonstrated that bbs3 knockdown results in the cardinal features of BBS in zebrafish, including defects to the ciliated Kupffer's Vesicle and delayed retrograde melanosome transport. Unlike bbs3, knockdown of bbs3L does not result in Kupffer's Vesicle or melanosome transport defects, rather its knockdown leads to impaired visual function and mislocalization of the photopigment green cone opsin. Moreover, BBS3L RNA, but not BBS3 RNA, is sufficient to rescue both the vision defect as well as green opsin localization in the zebrafish retina. In order to demonstrate a role for Bbs3L function in the mammalian eye, we generated a Bbs3L-null mouse that presents with disruption of the normal photoreceptor architecture. Bbs3L-null mice lack key features of previously published Bbs-null mice, including obesity. These data demonstrate that the BBS3L transcript is required for proper retinal function and organization.
Published in the journal:
. PLoS Genet 6(3): e32767. doi:10.1371/journal.pgen.1000884
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1000884
Summary
Bardet-Biedl Syndrome (BBS) is a heterogeneous syndromic form of retinal degeneration. We have identified a novel transcript of a known BBS gene, BBS3 (ARL6), which includes an additional exon. This transcript, BBS3L, is evolutionally conserved and is expressed predominantly in the eye, suggesting a specialized role in vision. Using antisense oligonucleotide knockdown in zebrafish, we previously demonstrated that bbs3 knockdown results in the cardinal features of BBS in zebrafish, including defects to the ciliated Kupffer's Vesicle and delayed retrograde melanosome transport. Unlike bbs3, knockdown of bbs3L does not result in Kupffer's Vesicle or melanosome transport defects, rather its knockdown leads to impaired visual function and mislocalization of the photopigment green cone opsin. Moreover, BBS3L RNA, but not BBS3 RNA, is sufficient to rescue both the vision defect as well as green opsin localization in the zebrafish retina. In order to demonstrate a role for Bbs3L function in the mammalian eye, we generated a Bbs3L-null mouse that presents with disruption of the normal photoreceptor architecture. Bbs3L-null mice lack key features of previously published Bbs-null mice, including obesity. These data demonstrate that the BBS3L transcript is required for proper retinal function and organization.
Introduction
Visual impairment and blindness have far reaching implications for society. Hundreds of individually rare, but collectively common Mendelian disorders can cause blindness. One of these disorders is a heterogeneous syndromic form of retinal degeneration, Bardet-Biedl Syndrome (BBS, OMIM 209900). This pleiotropic disorder is characterized by retinal degeneration, obesity, polydactyly, renal abnormalities, hypogenitalism and cognitive impairment [1]–[4]. Additionally, BBS is associated with an increased incidence of hypertension, diabetes mellitus and heart defects [1],[2],[5]. Although there is variability in the ocular phenotype between individuals, BBS patients typically present with early and progressive photoreceptor degeneration, leading to both central and peripheral vision loss by the third decade of life [1], [6]–[13].
To date, 14 genes (BBS1-14) have been implicated in BBS [14]–[28]. Analysis of mouse models of BBS (Bbs1M390R/M390R, Bbs2−/−, Bbs4−/− and Bbs6−/−) reveals that these mice have major components of the human phenotype including retinal degeneration, obesity, renal cysts and neurological deficits [29]–[32]. Multiple lines of evidence suggest that BBS phenotypes involve cilia dysfunction in a range of tissues, including the retina. The vertebrate retina contains photoreceptors, highly polarized cells with a modified cilium (connecting cilium) that joins the photosensitive outer segment (OS) to the protein synthesizing inner segment (IS). The connecting cilium transports cellular components from the IS to the OS that are necessary for the structure and function of the OS [33],[34]. Intraflagellar transport (IFT) proteins are important in this intraphotoreceptor transport process as they play a key role in both assembly and maintenance of photoreceptor cells [35]–[37]. Loss of IFT genes in vertebrates leads to abnormal OS development, retinal degeneration and mislocalization of photopigments [36],[38],[39].
Retina phenotypes observed in Bbs-null mice are similar to those seen with loss of IFT genes, indicating that BBS proteins play a role in transporting proteins through the connecting cilium into the OS of the photoreceptor. For instance, characterization of the retinal phenotype in the mouse model has shown that photoreceptor death is preceded by mislocalization of rhodopsin [29]–[32],[40]. Recent work with two independently generated Bbs4-null mice indicates that Bbs4 proteins play an important role in establishing both correct structure as well as proper transport of phototransduction proteins [40],[41]. In the zebrafish model system, individual knockdown of bbs genes results in defects in the ciliated Kupffer's Vesicle (KV) and delayed retrograde transport within the melanosome [26],[42],[43]. Moreover, work in Caenorhabditis elegans has shown that bbs1, bbs3, bbs5, bbs7 and bbs8 localize to the basal body of ciliated cells and are involved in IFT [23],[44]. Taken together, these data strongly support a role for BBS proteins in intracellular transport and cilia; thus further substantiating a critical role for BBS genes in the specialized connecting cilium of the photoreceptor for cell maintenance and function.
BBS3 is a member of the Ras superfamily of small GTP-binding proteins, which is subdivided into ADP-ribosylation factor (ARF) and ARF-like (ARL) subgroups [45]. The precise function of ARL proteins is unknown, but it has been proposed that they play a role in membrane and/or vesicular trafficking [45]. Work in C. elegans indicates that ARL6 is specifically expressed in ciliated cells and undergoes IFT along the ciliary axoneme [17]. Here we report the identification of a second transcript of BBS3, BBS3L, and determine that the BBS3L protein product plays an important role in eye structure and function. BBS3L is evolutionally conserved and is unique among BBS gene products as it is expressed predominantly in the eye, suggesting a specialized role in vision. We have established both mouse and zebrafish models to study the function of BBS3L, and determined that BBS3L is specifically required for retinal organization and function.
Results
Identification of a second BBS3 transcript in human, mouse, and zebrafish
Expressed sequence tag (EST) data for human BBS3 was compared to the known coding region of the gene. Although most of the ESTs were virtually identical to the BBS3 reference sequence, a few were found to contain 13 extra base pairs. Interestingly, all ESTs that contained this alternative sequence originated from retina or whole eye libraries, suggesting that this second longer transcript, BBS3L, has an expression pattern that is limited to the eye.
BBS3L results from differential splicing that leads to the inclusion of a 13 base pair exon and a shift in the open reading frame generating different C-terminal regions (Figure 1A). The striking conservation of the C-terminal region of the long isoform in human, mouse and zebrafish strongly suggests that bbs3L has functional relevance (Figure 1B). To determine if the bbs3 and bbs3L transcripts have similar tissue-specific expression in other species, RT-PCR of zebrafish and mouse tissues was performed. Zebrafish bbs3 is expressed in all adult tissues examined, while bbs3L expression is limited to the eye (Figure 1C). Similar tissue expression patterns for Bbs3 and Bbs3L were seen in the mouse with the addition of low levels of Bbs3L mRNA expression in the brain (Figure S1). A developmental profile in zebrafish embryos reveals that while bbs3 is expressed throughout development, bbs3L is not expressed until 48 hours post fertilization (hpf). This is a time when retinal neuroepithelial cells are exiting the cell cycle and differentiating into photoreceptor cells, the light sensing cells of the retina [46] (Figure 1D).

Knockdown of bbs3 results in characteristic BBS phenotypes
To determine the functional role of bbs3L in development and to distinguish the individual roles of the two bbs3 protein products, we utilized antisense oligonucleotide mediated gene knockdown (morpholinos, MO) in zebrafish. Two independent MOs were utilized: one targeting the splice junction specific to the long transcript (bbs3 long MO) and the other a previously described MO targeting both transcripts (bbs3 aug MO) through blocking of the translational start site [43] (Figure 2A). RT-PCR was used to determine the knockdown efficiency of the bbs3 long MO on staged embryos and demonstrated knockdown of the long transcript through at least 5 days post fertilization (dpf) (Figure 2B).

Knockdown of bbs function in zebrafish generates two prototypical defects: reduction of the size of the Kupffer's vesicle (KV) as well as retrograde transport defects [26],[42],[43]. As previously demonstrated, alterations in the formation of the ciliated KV was the earliest observable phenotype resulting from knockdown of both bbs3 transcripts by the bbs3 aug MO [43]. At the 8–10 somite stage (12–14 hpf) in wild-type and control injected embryos the KV has formed in the posterior tailbud. The KV diameter is approximately 50 µm and is larger than the width of the notochord (Figure 2C and 2D). Injection of the bbs3 aug MO resulted in a reduction of KV size to a width less than that of the notochord (Figure 2E). Knockdown of both bbs3 transcripts by the aug MO results in a statistically significant increase in embryos with KV defects (Fisher's exact test, p<0.001) (Figure 2F). Of note, injection of the bbs3 long MO does not lead to KV defects (Figure 2F).
The second prototypical phenotype observed in bbs MO-injected embryos (morphants) is delayed trafficking of melanosomes. Zebrafish are able to adapt to their surroundings through intracellular trafficking of melanosomes within melanophores in response to light and hormonal stimuli [47]–[50]. To test the rate of this movement, 5-day old zebrafish were dark adapted, to maximally disperse the melanosomes (Figure 2G and 2H) and then treated with epinephrine to chemically stimulate the retrograde transport of melanosomes [42],[43],[51] (Figure 2I). Wild-type and control injected embryos show rapid movement of melanosomes to a perinuclear location averaging 1.4 minutes, whereas bbs3 aug MO injected embryos demonstrated a statistically significant delay averaging 2.1 minutes (ANOVA with Tukey, p<0.01) (Figure 2J). In contrast, the rate of melanosome movement in bbs3 long knockdown embryos is statistically the same as control embryos, averaging 1.5 minutes (Figure 2J).
To test for MO-specificity as well as differential function of the two bbs3 transcripts, human RNAs of both BBS3 and BBS3L were used. Embryos co-injected with a combination of MO and RNA were evaluated for suppression of MO induced KV and melanosome transport defects. Co-injection of BBS3 RNA with the aug MO did not rescue the KV defect but was sufficient to suppress the melanosome transport delay; however, co-injection of the aug MO with BBS3L RNA was not able to suppress either MO-induced defect (Table S1). Myc-tagged BBS3 and BBS3L RNA injection and Western blot analysis confirmed expression of the protein out to 5 dpf (data not shown). Taken together, our data demonstrates that bbs3L plays a role independent from KV formation and melanosome transport and that human BBS3 can partially compensate for the loss of zebrafish bbs3.
bbs3 long knockdown causes a vision defect in zebrafish
Since BBS patients develop retinitis pigmentosa and the bbs3L transcript is differentially expressed in the eye, we sought to functionally test the role of bbs3 in vision. The zebrafish retina develops rapidly; at 60 hpf the retina is fully laminated and by 3 dpf zebrafish larvae are visually responsive [52]–[54]. Zebrafish elicit a characteristic escape response when exposed to rapid changes in light intensity and this startle response can be used as an assay for vision function [53]. In this assay, the behavior of a 5-day old larvae was monitored in response to short blocks of a bright, stable light source [53] (t = 0, Figure 3A). The typical response, a distinct C-bend and a change in swimming direction, is scored over a series of 5 trials, timed 30 seconds apart (t = 139ms, Figure 3A). Uninjected embryos respond on average 3.09 times (Figure 3B, Table 1 and Video S1). Cone-rod homeobox (crx) gene knockdown was used as a control for vision impairment as loss of this gene is known to affect photoreceptor formation in zebrafish [55],[56]. crx knockdown embryos respond an average of 1.28 times (ANOVA with Tukey, p<0.01). Knockdown using either the bbs3 aug or bbs3 long MO resulted in a statistically significant (ANOVA with Tukey, p<0.01) reduction in the number of responses (1.81 and 1.77 times respectively) compared to controls, indicating vision impairment (Figure 3B, Table 1 and Video S2). These data support a key role for bbs3L in vision function.


To functionally test the specific role of both bbs3 and bbs3L in vision, rescue experiments were performed. To investigate whether bbs3 could compensate for loss of bbs3L, wild-type human BBS3 RNA was co-injected with the bbs3 long MO. Although BBS3 RNA was sufficient to suppress the melanosome transport delays associated with bbs3 aug morphant embryos (Table S1), BBS3 RNA was insufficient to rescue the vision impairment induced by loss of only bbs3L (Figure 3B and Table 1). Conversely, co-injection of BBS3L RNA with the bbs3 aug MO was sufficient to rescue the vision defect (ANOVA with Tukey, p<0.01) (Figure 3B and Table 1). Based on these rescue experiments, bbs3L is necessary and sufficient for vision function.
hBBS3L is sufficient to rescue green opsin mislocalization in bbs3 morphant zebrafish
Previous work has demonstrated that Bbs1 M390R knockin, Bbs2, Bbs4 and Bbs6 mutant mice initially form photoreceptors; however, the photoreceptors subsequently show a mislocalization of rhodopsin, a photopigment protein, to the cell bodies of the outer nuclear layer (ONL) and undergo progressive photoreceptor degeneration [29]–[32],[40]. By gross histology, wild-type, bbs3 aug and bbs3 long morphant zebrafish embryo retinas displayed a fully laminated retina at 5 dpf (data not shown). Ganglion cell outgrowth and optic nerve formation was evaluated using the ath5:GFP [Tg(atoh7:GFP)] transgenic line, a marker of ganglion cell and axon outgrowth [57]. We found that gross retinal ganglion axon trajectories were not perturbed in bbs3 aug or long morphants (data not shown).
While the overall architecture of the retina appeared morphologically normal at 5 dpf, we investigated photopigment localization in bbs3 morphants. Photopigments are known to localize to the outer segment of the zebrafish photoreceptor; therefore, we assessed opsin localization using an antibody specific to green cone opsin [58]. In the wild-type retina, green opsin is found in the outer-segment of the green cone photoreceptor (Figure 4A). In bbs3 aug and bbs3 long morphants green opsin expression was not restricted to the outer segments of the photoreceptors; rather, green opsin was also detected in the cell bodies of the outer nuclear layer throughout the entire retina (Figure 4B and 4E).

To determine whether there is a functional difference between BBS3 and BBS3L in its ability to rescue the green opsin localization in the photoreceptors of MO-injected embryos rescue experiments were performed. The first question we addressed was if BBS3L RNA was sufficient to rescue green opsin localization in morphant embryos. Expression of wild-type human BBS3L RNA led to improved green opsin localization in both bbs3 aug and bbs3L morphant embryos (Figure 4D and 4G). The percentage of cells mislocalizing green opsin was quantified and indeed BBS3L RNA was able to statistically rescue the green opsin defect in bbs3 aug morphants (Fisher's exact test, p<0.01) (Figure 4H and Table 1). We next investigated whether BBS3 could compensate for loss of bbs3L in the zebrafish retina. Co-injection of wild-type human BBS3 RNA failed to rescue green opsin localization in bbs3 aug and bbs3L morphant embryos (Figure 4C, 4F, and 4H and Table 1). These data are consistent with the vision startle response rescue data and supports the hypothesis that BBS3L has an eye specific role. Moreover, these data support a specific role for bbs3L in the retina and for localization of proteins within the photoreceptor cell.
Bbs3 is expressed in ganglion and photoreceptor cells in mouse and human retinas
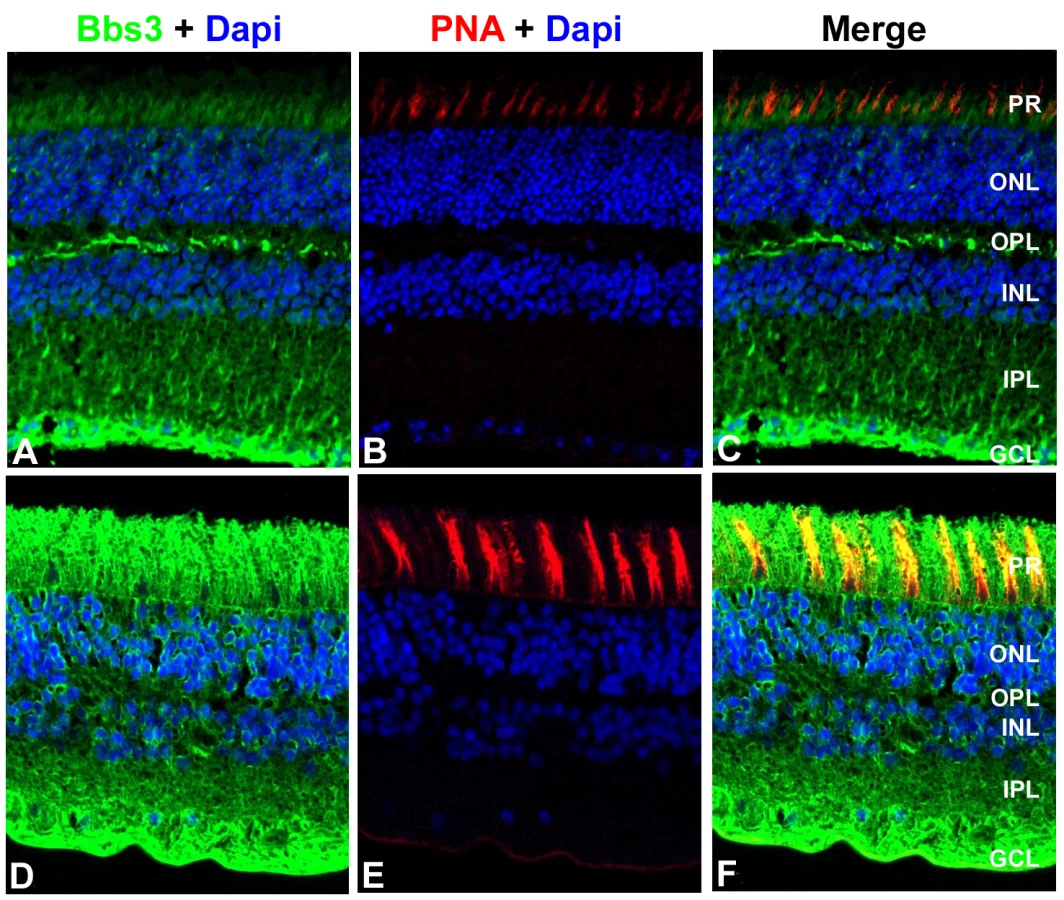
A polyclonal antibody against a central region of the mouse Bbs3 peptide, which is conserved across human and mouse, was generated to recognize both isoforms of Bbs3. Cellular localization of Bbs3 was assessed in donor human and mouse retinal tissue. Immunohistochemistry was performed on transverse cryosections from adult human and adult mouse eyes using the Bbs3 antibody. Staining revealed expression of Bbs3 (green) in the ganglion cell layer and the nerve fiber layer as well as the photoreceptor cells of both mouse (Figure 5A) and human retinal tissue (Figure 5D). Additionally, peanut agglutinin (PNA, red) was used as a marker for cone outer segments in both mouse (Figure 5B) and human retinal sections (Figure 5E). The merge represents the co-localization of Bbs3 (green) and PNA (red) in the photoreceptor cells of both mouse (Figure 5C) and human (Figure 5F). The specificity of the BBS3 antibody for immunohistochemistry was confirmed through peptide blocking of the antibody on wild-type mouse retina (Figure S2).
Bbs3L−/− mice display structural abnormalities
To characterize the effects of loss of Bbs3L on mammalian photoreceptors a targeted knockin of the long form of Bbs3 was carried out by altering the splice donor and acceptor sites flanking exon 8, leading to the exclusion of exon 8 upon homologous recombination (Figure 6A). This approach leads to the preservation of Bbs3 expression in the Bbs3L-null mice. RT-PCR confirmed the generation and transmission of the Bbs3L allele in +/ − and −/− mice (Figure S3). Unlike previously generated BBS knockout mice, which are obese by 7 months of age, Bbs3L−/− mice do not become obese (data not shown) [29]–[32]. This supports the idea that Bbs3L function is restricted to the retina and is consistent with the zebrafish knockdown studies.

Gross histological examination of 8-month-old wild-type and homozygous (Bbs3L−/−) mutant mice revealed that while all cell layers were present (Figure 6B and 6E), the inner segments of the photoreceptors were disrupted in a majority of the mutant mice as compared to wild-type (Figure 6C and 6F). In wild-type mice, the inner segment layer is arranged in a parallel array; while in the Bbs3L-null mice the parallel arrangement of the IS was eccentric with individual inner segments randomly oriented. Additionally, immunohistochemistry with the Bbs3 antibody (green), which recognizes the endogenous Bbs3 protein that is still present, and rhodopsin (red) in Bbs3L−/− mice further confirms inner segment disorganization in Bbs3L−/− mutant mice compared to wild-type (Figure 6D and 6G).
Discussion
The present study identifies and characterizes the eye-enriched transcript BBS3L using both the zebrafish and mouse model systems. While typical BBS genes are ubiquitously expressed and lead to multiple phenotypes in human, mice and zebrafish, BBS3L expression is restricted to the eye and serves as a useful tool for understanding the specific pathophysiology of BBS proteins in blinding diseases. By knockdown in zebrafish, we find that bbs3L is required for visual function and localization of the photopigment green cone opsin; however, bbs3L is dispensable for the cardinal features of BBS in zebrafish, including reduced KV and delayed melanosome transport. Moreover, BBS3L RNA, but not BBS3 RNA, is sufficient to rescue both the vision defect as well as green opsin localization. These data provide strong evidence that bbs3L is specifically required for retinal organization and function.
Immunohistochemistry using an antibody that recognizes both Bbs3 and Bbs3L indicates strong expression of the protein in the ganglion cell layer, nerve fiber layer and photoreceptor cells in both human and mouse retinas. By using this antibody on Bbs3L-null mouse retinas, we can deduce that Bbs3 is expressed in both the photoreceptors and ganglion cells. This is consistent with expression data indicating that Bbs3L is enriched in the retina.
We have previously demonstrated that knockdown of bbs genes in the zebrafish leads to KV defects and melanosome transport delays [26],[42],[43]. As previously reported, knockdown of bbs3 using the aug morpholino yields both KV and melanosome transport defects; however, knockdown of only bbs3L does not affect the KV or melanosome transport. The lack of these cardinal features is not surprising given that bbs3L is not expressed at the KV stage and that in adult zebrafish the long transcript is only expressed in the eye. Since bbs3 and bbs3L are identical except for the splicing of the last exon, we cannot technically knockdown bbs3 alone without affecting bbs3L. However, based on rescue data, bbs3 knockdown alone appears to be responsible for both the KV and melanosome transport defects seen with the aug morpholino. Importantly, bbs3 and bbs3L do not seem to be functionally interchangeable. Forced expression of BBS3L RNA, at a time and place where the endogenous transcript is not present, does not rescue the cardinal features of BBS in the zebrafish that result from knockdown of both transcripts. Moreover, over-expression of BBS3 in the whole embryo cannot restore vision loss resulting from the knockdown of only bbs3L. It should be noted that melanosome transport is evaluated after the vision startle assay; therefore, we know that over-expressed BBS3 is functional at the time of the vision assay. Although bbs3 may have some effect on vision that is below the detection level of our assay, we have demonstrated that bbs3L function is both necessary for vision and sufficient to rescue vision loss in the zebrafish.
Similar to the zebrafish results, a Bbs3L-null mouse lacks the observed phenotypes of previously published Bbs-null mice, such as obesity [29]–[32]. The effect of Bbs3 in the mouse retina may be more significant as Bbs3L-null mice present with only a variable mild disruption of the normal architecture. This indicates that in the mouse retina, Bbs3 is able to partially compensate for loss of Bbs3L. Moreover, the difference in phenotype between zebrafish and mouse could potentially be due to the ratio of cones and rods found in each model system. One hypothesis is that bbs3L plays a major functional role in cones, but only a minor role in rods. At the stages examined in the zebrafish, cones are the only functional photoreceptors in the retina, whereas mice have a rod-dominated retina [59],[60]. These attributes are important to consider when looking at the role of BBS in human disease progression, as humans rely on their fovea, a specialized cone-dominant structure in the center of the macula, for visual acuity. Continued characterization of the Bbs3L-null mouse may shed more light on this difference between the mouse and zebrafish system, as well as elucidate a more definitive role for BBS3L in the retina.
Taken together, these date demonstrate that the BBS3L transcript is specifically required for retinal organization and function. While we have identified a second transcript of BBS3, a gene known to cause BBS, we would not expect patients with mutations affecting only BBS3L to present with BBS. Based on our findings in both a zebrafish and mouse model of BBS3L, patients with mutations in BBS3L alone would present with a non-syndromic retinal disease, characterized by photoreceptor dysfunction and death. Indeed, recent homozygosity mapping of a consanguineous Saudi family has identified a missense mutation in BBS3 that leads to non-syndromic RP [61]. Functional characterization of this mutation in the zebrafish may provide additional clues to the role of BBS3 in the eye. Thus this eye specific transcript, BBS3L, will serve as a useful tool for understanding the pathophysiology of other blinding diseases. In addition, our data indicate that expression of BBS3L, rather than BBS3, would be needed for gene therapy aimed at treatment of blindness in BBS3 patients.
Materials and Methods
Ethics statement
All animal work in this study was approved by the by the University Animal Care and Use Committee at the University of Iowa.
EST
Expressed sequence tag (EST) data for human and mouse BBS3 was downloaded from NCBI and compared to the known coding region as represented by the NCBI reference sequence (NM_177976.1 and NM_032146.3 for human and NM_019665.3 for mouse).
Danio rerio
RT–PCR
RNA was extracted from a pool of 10–20 embryos at the following stages: 8–12 somites, 24, 36, 42, 48, 60, 72, 96 hpf and 5 dpf. Additionally, RNA was extracted from the following adult tissues: fat, brain, heart, whole eye and retina. cDNA was synthesized using oligo dT primers and bbs3 primer pair 1 recognizing both bbs3 transcripts were used to evaluate expression. β-actin expression served as a control.
Primers:
bbs3 primer pair 1-F: 5′-AAGGACAAACCATGGCATATC-3′
bbs3 primer pair 1-R: 5′-TTACGTTTTCATCGCTCTGAT-3′
β-actin-F: 5′-TCAGCCATGGATGATGAAAT-3′
β-actin-R: 5′-GGTCAGGATCTTCATGAGGT-3′
Morpholino injections and knockdown efficiency
Antisense morpholinos (MO) were designed and purchased from Gene Tools.
bbs3_aug [43]: AGCTTGTCAAAAAGCCCCATTTGCT
bbs3_long: ATTTCAGCTTCAGTACTTACAGTGC
control MO bbs3_6mis: AaCTTGTgAAAtAGCgCCATaTGaT
crx: GGCTGCTTTATGTAGGACATCATTC
MOs (12 ng) were air-pressure-injected into one - to four-cell staged embryos. Transcript knockdown efficiency was assessed by RT-PCR as described above using bbs3 long splice-blocking morphants at the following stages: 8–12 somites, 72 hpf and 5 dpf. Primers recognizing both bbs3 (bbs3 primer pair 1) transcripts were used to assess knockdown efficiency.
Human BBS3 cloning and RNA synthesis
Wild-type human BBS3 and BBS3L constructs were generated by TA cloning into the Gateway vector system (Invitrogen), and subsequently subcloned into Gateway expression vectors with a C-terminal mCherry or myc tag (generous gift from Chien and Lawson Lab).
Primers:
-hBBS3-F: ATGGGATTGCTAGACAGACTTTC
-hBBS3-R: TGTCTTCACAGTCTGGATCTG
-hBBS3L-R: TCTTTTCATGTCTTCACAGTC
For rescue experiments, MO-resistant C-terminally tagged mCherry RNA was synthesized using the mMessage mMachine transcription kit (Ambion). hBBS3 or hBBS3L RNA (8 pg) was co-injected with the appropriate MO into one - to four-cell staged embryos.
Analysis of Kupffer's Vesicle
Embryos with KVs smaller than the width of the notochord (less than approximately 50 µm in diameter) were considered reduced, while embryos in which KVs could not be morphologically identified were scored as absent. Live embryos were photographed on a stereoscope with a Zeiss Axiocam camera.
Melanosome transport assay
The melanosome transport assay was performed as previously described [26],[42],[43]. Dark-adapted 5-day-post fertilization larvae were treated with epinephrine (50 mg/ml, Sigma, E4375) added to egg water [62] for a final concentration of 500 µg/ml. Melanosome retraction time was monitored under the microscope. Live embryos were photographed on a stereoscope with a Zeiss Axiocam camera.
Vision startle response assay
A visually evoked startle response behavioral assay was modified from a previously described assay [53]. Prior to experimentation, 5-day-old zebrafish larvae were light adapted for 1 hour. A visual stimulus was applied by performing rapid changes (approximately 1 second) in white light intensity through abruptly opening and closing the shutter located between the light source and the animal. An abrupt movement of the zebrafish within one second of visual stimuli application was scored as a positive response. After performing five visual stimuli trials spaced at 30 seconds apart, the mechanical stimulus response was evaluated by probing embryos with the tip of a blunt needle. Embryos that failed to respond to the mechanical stimulation, although rare, were not included in the analysis.
Immunohistochemistry
Five day post-fertilized larvae were fixed overnight at 4°C with 4% paraformaldehyde (PFA) prepared in BT buffer (4% sucrose, 0.1M CaCl2 in 0.1M PO4, pH 7.3). Embryos were rinsed with phosphate-buffer saline (PBS) and infiltrated at 4°C with 15% sucrose, 30% sucrose and overnight in 100% optimal cutting temperature compound (OCT, Sakura). Embryos were cryosectioned at −21°C. Sections were collected at 12 µm and were allowed to dry for 1 hour at 25°C. The tissues were incubated with blocking solution (5% normal donkey serum, 0.1% tween-20, 1% DMSO in PBS) for 2 hours and then incubated overnight at 4°C with mouse-anti-green cone opsin diluted in blocking solution (1∶500, generous gift from the Hyde lab). Following washes with PBDT (PBS, 1% DMSO, 0.1% tween-20) sections were incubated for 1.5 hours at 25°C with goat-anti-mouse Alexa 488 (1∶400, Molecular Probes) diluted in blocking solution. Nuclei were counterstained with To-Pro3 (1∶1000, Molecular Probes) diluted in PBS. Sections were mounted in Vectashield mounting medium (Vector Laboratories) and analyzed using a Leica SP2 laser confocal microscope system with 63× magnification and 3x zoom. Images are representative of maximum projections of multiple focal planes (z-series).
Green opsin cell counts
The ratio of mislocalized green opsin cells to total green opsin positive cells was determined from a 12 µm thick central retina image taken of a single eye. Mislocalization of green opsin was defined as the presence of green opsin present in the outer nuclear layer (ONL) of the retina. The number of independent fish retinas counted per group were as follows: wt n = 9, bbs3 aug MO n = 10, bbs3 long MO n = 15, bbs3 aug MO+ hBBS3L RNA n = 5, bbs3 long MO+hBBS3 RNA n = 5. Scorers were masked to the genotype of the embryos.
Mus musculus
Generation of Bbs3L mutant mice
A targeting plasmid was constructed by amplifying the 5′ and 3′ regions of Bbs3L using genomic DNA isolated from the 129/SvJ mouse strain. The consensus splice sites were ablated to alter the slice donor (5′ - CTGCTGTCACAAAAAACAGTACACTAAGTATCTG-3′) and splice acceptor (5′ - CAGATACTTAGTGTACTGTTTTTTGTGACAGCAG-3′) sites flanking exon 8, the exon responsible for the long transcript. Following mutagenesis, these regions were cloned into the targeting vector pOSDUPDEL (a gift from O. Smithies, University of North Carolina, Chapel Hill, NC, USA). The targeting construct was linearized with NotI and electroporated into R1 embryonic stem (ES) cells (129 X 1/SvJ3 129S1/Sv). Double selection of ES cells was carried out for the presence of the neomycin gene (Neo) and the absence of the thymidine kinase gene (TK). To identify Bbs3L-targeted ES cells, G418-resistnt clones were screened for by PCR. One ES cell line was used to produce chimeras that were bred with C57BL/6J mice to generate Bbs3L heterozygous (Bbs3L+/−) mice on a mixed background. These mixed background offspring were evaluated for germline transmission by PCR, and the resulting Bbs3L heterozygotes (Bbs3L+/−) crossed to pure 129/SvEv mice for seven generations to enrich for the pure 129/SvEv background. Heterozygous mice were intercrossed and the progeny genotyped by PCR using primers to identify the presence of the targeted allele. Presence of the wild-type and mutant allele was determined by using a three primer pool: forward primer specific to the wild-type allele (5′-TTGGAGATTTGTCTCCCTCTG-3′), forward primer specific to the mutant allele (5′-GCTACCCGTGATATTGCTGAA-3′) and a reverse primer that recognizes both alleles (5′-AAAAGGGCATAAAAGCACCTC-3′).
Histological analysis of Bbs3L−/− mice
Enucleated eyes were fixed in 4% PFA in PBS (pH 7.4). Following 2–4 hours of fixation, the anterior chamber and lens of the eye was removed and the eyecup allowed to fix further overnight at 4°C in 4% PFA. The eyecups were rinsed with PBS and cryoprotected through a series of 5%:20% sucrose incubations (2∶1, 1∶1, 1∶2) before an overnight incubation at 4°C in 20% sucrose. Eyecups were infiltrated with 2 parts 20% sucrose in 1 part OCT (Sakura) for thirty minutes at room temperature. Sections were collected at 7 µm and were allowed to dry for at least 1 hour at 25°C. Gross morphology of the retinas was evaluated with hematoxylin/eosin staining.
Immunohistochemistry of Bbs3L−/− mice
Immunohistochemistry was performed on cryosections from Bbs3L+/+ and Bbs3L−/− mouse retinas. The sections were blocked with bovine serum albumin (BSA, 1 mg/ml) in PBS for 15 min and then incubated for 1 hour at room temperature with either rabbit anti-mouse Bbs3 (1∶100), biotinylated peanut agglutinin (1∶100, PNA, Vector Laboratories) or monoclonal mouse rhodopsin (1∶1000, RET-P1, NeoMarker) in PBS. Following washes with PBS, sections were incubated for 30 minutes at room temperature with a species specific secondary antibody: goat-anti-rabbit Alexa 488 (1∶200, Molecular Probes), Texas Red Avidin D (1∶200, Vector Laboratories) or goat-anti-mouse Alexa 546 (1∶200, Molecular Probes). Nuclei were counter stained with either 4′, 6-diamidino-2-phentlindole (DAPI, Molecular Probes) or To-Pro-3 (1∶1000, Molecular Probes). Sections were mounted in Aqua Mount (Lerner Laboratories) and analyzed using either an Olympus BX-41 microscope with a SPOT RT digital camera (Diagnostic Instruments) or a Bio-Rad 1024 confocal microscope system. Images from the confocal are representative of multiple focal planes (z-series).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. GreenJS
ParfreyPS
HarnettJD
FaridNR
CramerBC
1989 The cardinal manifestations of Bardet-Biedl syndrome, a form of Laurence-Moon-Biedl syndrome. N Engl J Med 321 1002 1009
2. HarnettJD
GreenJS
CramerBC
JohnsonG
ChafeL
1988 The spectrum of renal disease in Laurence-Moon-Biedl syndrome. N Engl J Med 319 615 618
3. BardetG
1995 On congenital obesity syndrome with polydactyly and retinitis pigmentosa (a contribution to the study of clinical forms of hypophyseal obesity). 1920. Obes Res 3 387 399
4. BiedlA
1995 A pair of siblings with adiposo-genital dystrophy. 1922. Obes Res 3 404
5. ElbedourK
ZuckerN
ZalzsteinE
BarkiY
CarmiR
1994 Cardiac abnormalities in the Bardet-Biedl syndrome: echocardiographic studies of 22 patients. Am J Med Genet 52 164 169
6. LeysMJ
SchreinerLA
HansenRM
MayerDL
FultonAB
1988 Visual acuities and dark-adapted thresholds of children with Bardet-Biedl syndrome. Am J Ophthalmol 106 561 569
7. RiiseR
1987 Visual function in Laurence-Moon-Bardet-Biedl syndrome. A survey of 26 cases. Acta Ophthalmol Suppl 182 128 131
8. JacobsonSG
BorruatFX
ApathyPP
1990 Patterns of rod and cone dysfunction in Bardet-Biedl syndrome. Am J Ophthalmol 109 676 688
9. BealesPL
WarnerAM
HitmanGA
ThakkerR
FlinterFA
1997 Bardet-Biedl syndrome: a molecular and phenotypic study of 18 families. J Med Genet 34 92 98
10. CarmiR
ElbedourK
StoneEM
SheffieldVC
1995 Phenotypic differences among patients with Bardet-Biedl syndrome linked to three different chromosome loci. Am J Med Genet 59 199 203
11. RiiseR
AndreassonS
BorgastromMK
WrightAF
TommerupN
1997 Intrafamilial variation of the phenotype in Bardet-Biedl syndrome. Br J Ophthalmol 81 378 385
12. FultonAB
HansenRM
GlynnRJ
1993 Natural course of visual functions in the Bardet-Biedl syndrome. Arch Ophthalmol 111 1500 1506
13. HeonE
WestallC
CarmiR
ElbedourK
PantonC
2005 Ocular phenotypes of three genetic variants of Bardet-Biedl syndrome. Am J Med Genet A 132A 283 287
14. MykytynK
NishimuraDY
SearbyCC
ShastriM
YenHJ
2002 Identification of the gene (BBS1) most commonly involved in Bardet-Biedl syndrome, a complex human obesity syndrome. Nat Genet 31 435 438
15. NishimuraDY
SearbyCC
CarmiR
ElbedourK
Van MaldergemL
2001 Positional cloning of a novel gene on chromosome 16q causing Bardet-Biedl syndrome (BBS2). Hum Mol Genet 10 865 874
16. ChiangAP
NishimuraD
SearbyC
ElbedourK
CarmiR
2004 Comparative genomic analysis identifies an ADP-ribosylation factor-like gene as the cause of Bardet-Biedl syndrome (BBS3). Am J Hum Genet 75 475 484
17. FanY
EsmailMA
AnsleySJ
BlacqueOE
BoroevichK
2004 Mutations in a member of the Ras superfamily of small GTP-binding proteins causes Bardet-Biedl syndrome. Nat Genet 36 989 993
18. MykytynK
BraunT
CarmiR
HaiderNB
SearbyCC
2001 Identification of the gene that, when mutated, causes the human obesity syndrome BBS4. Nat Genet 28 188 191
19. LiJB
GerdesJM
HaycraftCJ
FanY
TeslovichTM
2004 Comparative genomics identifies a flagellar and basal body proteome that includes the BBS5 human disease gene. Cell 117 541 552
20. KatsanisN
BealesPL
WoodsMO
LewisRA
GreenJS
2000 Mutations in MKKS cause obesity, retinal dystrophy and renal malformations associated with Bardet-Biedl syndrome. Nat Genet 26 67 70
21. SlavotinekAM
StoneEM
MykytynK
HeckenlivelyJR
GreenJS
2000 Mutations in MKKS cause Bardet-Biedl syndrome. Nat Genet 26 15 16
22. BadanoJL
AnsleySJ
LeitchCC
LewisRA
LupskiJR
2003 Identification of a novel Bardet-Biedl syndrome protein, BBS7, that shares structural features with BBS1 and BBS2. Am J Hum Genet 72 650 658
23. AnsleySJ
BadanoJL
BlacqueOE
HillJ
HoskinsBE
2003 Basal body dysfunction is a likely cause of pleiotropic Bardet-Biedl syndrome. Nature 425 628 633
24. NishimuraDY
SwiderskiRE
SearbyCC
BergEM
FergusonAL
2005 Comparative genomics and gene expression analysis identifies BBS9, a new Bardet-Biedl syndrome gene. Am J Hum Genet 77 1021 1033
25. StoetzelC
LaurierV
DavisEE
MullerJ
RixS
2006 BBS10 encodes a vertebrate-specific chaperonin-like protein and is a major BBS locus. Nat Genet 38 521 524
26. ChiangAP
BeckJS
YenHJ
TayehMK
ScheetzTE
2006 Homozygosity mapping with SNP arrays identifies TRIM32, an E3 ubiquitin ligase, as a Bardet-Biedl syndrome gene (BBS11). Proc Natl Acad Sci U S A 103 6287 6292
27. StoetzelC
MullerJ
LaurierV
DavisEE
ZaghloulNA
2007 Identification of a novel BBS gene (BBS12) highlights the major role of a vertebrate-specific branch of chaperonin-related proteins in Bardet-Biedl syndrome. Am J Hum Genet 80 1 11
28. LeitchCC
ZaghloulNA
DavisEE
StoetzelC
Diaz-FontA
2008 Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat Genet 40 443 448
29. MykytynK
MullinsRF
AndrewsM
ChiangAP
SwiderskiRE
2004 Bardet-Biedl syndrome type 4 (BBS4)-null mice implicate Bbs4 in flagella formation but not global cilia assembly. Proc Natl Acad Sci U S A 101 8664 8669
30. NishimuraDY
FathM
MullinsRF
SearbyC
AndrewsM
2004 Bbs2-null mice have neurosensory deficits, a defect in social dominance, and retinopathy associated with mislocalization of rhodopsin. Proc Natl Acad Sci U S A 101 16588 16593
31. FathMA
MullinsRF
SearbyC
NishimuraDY
WeiJ
2005 Mkks-null mice have a phenotype resembling Bardet-Biedl syndrome. Hum Mol Genet 14 1109 1118
32. DavisRE
SwiderskiRE
RahmouniK
NishimuraDY
MullinsRF
2007 A knockin mouse model of the Bardet-Biedl syndrome 1 M390R mutation has cilia defects, ventriculomegaly, retinopathy, and obesity. Proc Natl Acad Sci U S A 104 19422 19427
33. YoungRW
1967 The renewal of photoreceptor cell outer segments. J Cell Biol 33 61 72
34. BesharseJC
HorstCJ
1990 The photoreceptor connecting cilium. A model for for the transition zone.
BloodgoodRA
Ciliary and Flagellar Membranes New York Plenum Publishing Corp 389 417
35. KrockBL
PerkinsBD
2008 The intraflagellar transport protein IFT57 is required for cilia maintenance and regulates IFT-particle-kinesin-II dissociation in vertebrate photoreceptors. J Cell Sci 121 1907 1915
36. PazourGJ
BakerSA
DeaneJA
ColeDG
DickertBL
2002 The intraflagellar transport protein, IFT88, is essential for vertebrate photoreceptor assembly and maintenance. J Cell Biol 157 103 113
37. Luby-PhelpsK
FogertyJ
BakerSA
PazourGJ
BesharseJC
2008 Spatial distribution of intraflagellar transport proteins in vertebrate photoreceptors. Vision Res 48 413 423
38. TsujikawaM
MalickiJ
2004 Intraflagellar transport genes are essential for differentiation and survival of vertebrate sensory neurons. Neuron 42 703 716
39. SukumaranS
PerkinsBD
2009 Early defects in photoreceptor outer segment morphogenesis in zebrafish ift57, ift88 and ift172 Intraflagellar Transport mutants. Vision Res 49 479 489
40. Abd-El-BarrMM
SykoudisK
AndrabiS
EichersER
PennesiME
2007 Impaired photoreceptor protein transport and synaptic transmission in a mouse model of Bardet-Biedl syndrome. Vision Res 47 3394 3407
41. SwiderskiRE
NishimuraDY
MullinsRF
OlveraMA
RossJL
2007 Gene expression analysis of photoreceptor cell loss in bbs4-knockout mice reveals an early stress gene response and photoreceptor cell damage. Invest Ophthalmol Vis Sci 48 3329 3340
42. YenHJ
TayehMK
MullinsRF
StoneEM
SheffieldVC
2006 Bardet-Biedl syndrome genes are important in retrograde intracellular trafficking and Kupffer's vesicle cilia function. Hum Mol Genet 15 667 677
43. TayehMK
YenHJ
BeckJS
SearbyCC
WestfallTA
2008 Genetic interaction between Bardet-Biedl syndrome genes and implications for limb patterning. Hum Mol Genet
44. BlacqueOE
ReardonMJ
LiC
McCarthyJ
MahjoubMR
2004 Loss of C. elegans BBS-7 and BBS-8 protein function results in cilia defects and compromised intraflagellar transport. Genes Dev 18 1630 1642
45. PasqualatoS
RenaultL
CherfilsJ
2002 Arf, Arl, Arp and Sar proteins: a family of GTP-binding proteins with a structural device for 'front-back' communication. EMBO Rep 3 1035 1041
46. HuM
EasterSS
1999 Retinal neurogenesis: the formation of the initial central patch of postmitotic cells. Dev Biol 207 309 321
47. SkoldHN
AspengrenS
WallinM
2002 The cytoskeleton in fish melanophore melanosome positioning. Microsc Res Tech 58 464 469
48. BarralDC
SeabraMC
2004 The melanosome as a model to study organelle motility in mammals. Pigment Cell Res 17 111 118
49. MarksMS
SeabraMC
2001 The melanosome: membrane dynamics in black and white. Nat Rev Mol Cell Biol 2 738 748
50. BlottEJ
GriffithsGM
2002 Secretory lysosomes. Nat Rev Mol Cell Biol 3 122 131
51. NascimentoAA
RolandJT
GelfandVI
2003 Pigment cells: a model for the study of organelle transport. Annu Rev Cell Dev Biol 19 469 491
52. SchmittEA
DowlingJE
1999 Early retinal development in the zebrafish, Danio rerio: light and electron microscopic analyses. J Comp Neurol 404 515 536
53. EasterSSJr
NicolaGN
1996 The development of vision in the zebrafish (Danio rerio). Dev Biol 180 646 663
54. BranchekT
1984 The development of photoreceptors in the zebrafish, brachydanio rerio. II. Function. J Comp Neurol 224 116 122
55. LiuY
ShenY
RestJS
RaymondPA
ZackDJ
2001 Isolation and characterization of a zebrafish homologue of the cone rod homeobox gene. Invest Ophthalmol Vis Sci 42 481 487
56. ShenYC
RaymondPA
2004 Zebrafish cone-rod (crx) homeobox gene promotes retinogenesis. Dev Biol 269 237 251
57. MasaiI
LeleZ
YamaguchiM
KomoriA
NakataA
2003 N-cadherin mediates retinal lamination, maintenance of forebrain compartments and patterning of retinal neurites. Development 130 2479 2494
58. VihtelicTS
DoroCJ
HydeDR
1999 Cloning and characterization of six zebrafish photoreceptor opsin cDNAs and immunolocalization of their corresponding proteins. Vis Neurosci 16 571 585
59. BilottaJ
SaszikS
SutherlandSE
2001 Rod contributions to the electroretinogram of the dark-adapted developing zebrafish. Dev Dyn 222 564 570
60. YoungRW
1985 Cell differentiation in the retina of the mouse. Anat Rec 212 199 205
61. Abu SafiehL
AldahmeshM
ShamseldinH
HashemM
ShaheenR
2009 Clinical and Molecular Characterization of Bardet-Biedl Syndrome in Consanguineous Populations: The Power of Homozygosity Mapping. J Med Genet
62. WesterfieldM
1993 The Zebrafish book. A guide for the laboratory use of zebrafish (Brachydanio rerio). Eugene, OR University of Oregon Press
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 3
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Deciphering Normal Blood Gene Expression Variation—The NOWAC Postgenome Study
- Transgenic Rat Model of Neurodegeneration Caused by Mutation in the Gene
- Papillorenal Syndrome-Causing Missense Mutations in / Result in Hypomorphic Alleles in Mouse and Human
- Fatal Cardiac Arrhythmia and Long-QT Syndrome in a New Form of Congenital Generalized Lipodystrophy with Muscle Rippling (CGL4) Due to Mutations
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy