
Nádorové buňky jako dynamický systém – molekulární a fenotypové změny v průběhu vzniku, progrese a šíření nádoru
Cancer Cells as Dynamic System – Molecular and Phenotypic Changes During Tumor Formation, Progression and Dissemination
Dynamic, punctual and perfectly coordinated cellular response to internal and external stimuli is a crucial prerequisite for adaptation of mammalian cells to all changes that occur during cellular development under physiological conditions. Hijacking this ability is characteristic for tumor cells that are capable to adapt to unfavorable conditions which contribute to the formation and development of cancer during the process of tumor formation and progression. By changing key mechanisms, malignant cells can avoid cell death and thus allow development and spread of the tumor. The changes at the genetic level are manifested by various phenotypic characteristics, through which tumor cells are able to escape defense mechanisms, to acquire resistance to treatment, to invade and to create secondary tumors. In recent years, one of the most studied properties include changes in energy metabolism, when tumor cells specifically control reprogramming of the main metabolic pathways for their own benefit and to satisfy their increased needs not only for energy, but also for building materials required for increased proliferation. To adapt to extracellular conditions, it is necessary that cells undergo morphological changes, where modifications in the cell shape through reorganization of cytoskeletal filaments allow tumor cells to increase their invasiveness and other aggressive features. Clarifying these changes together with understanding of the switch in the genetic program within cancer cells, which allows them to overcome different stages of differentiation from cancer stem cells to fully differentiated cells, would be an important prerequisite for identification of the cancer cell “weaknesses” and may lead to improved cancer treatment. The ability of tumor cells to alter the rules of their own organism thus represents an important challenge for oncological research.
Key words:
cellular reprogramming – cancer cell plasticity – cancer metabolism – tumor heterogeneity – cytoskeleton remodeling – metastasis – oncogenesis
This work was supported by the project MEYS – NPS I – LO1413.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE recommendation for biomedical papers.
Submitted:
3. 7. 2016
Accepted:
11. 8. 2016
Autoři:
L. Sommerová; E. Ondroušková; R. Hrstka
Působiště autorů:
Regionální centrum aplikované molekulární onkologie, Masarykův onkologický ústav, Brno
Vyšlo v časopise:
Klin Onkol 2016; 29(Supplementum 4): 6-11
Kategorie:
Přehled
doi:
https://doi.org/10.14735/amko20164S6
Souhrn
Dobře načasovaná, přesná a dokonale koordinovaná reakce buňky na vnitřní i vnější podněty představuje základní předpoklad pro úspěšnou adaptaci savčích buněk na všechny změny, které se dějí za fyziologických podmínek v průběhu buněčného vývoje. Zneužít těchto schopností umí právě nádorové buňky, které jsou tak schopné se přizpůsobit stresovým podmínkám, které představují důležitý předpoklad pro vznik a vývoj nádoru. Změnou klíčových mechanizmů se nádorovým buňkám podaří vyhnout buněčné smrti, a tak je umožněn vývoj a šíření nádoru. Změny na genetické úrovni se manifestují celou řadou fenotypových projevů, díky kterým jsou nádorové buňky schopny uniknout obranným mechanizmům organizmu, získat rezistenci k léčbě, invadovat, zakládat dormantní ložiska či vytvářet sekundární nádory. Mezi jednu ze základních vlastností studovaných v posledních letech patří změna energetického metabolizmu, kdy si nádorová buňka cíleně reguluje přeprogramování hlavních metabolických drah k vlastnímu prospěchu a uspokojení zvýšených nároků nejen na energii, ale také na stavební materiály, které jsou nutné pro zajištění zvýšené buněčné proliferace. Přizpůsobení se okolním podmínkám je nutné i na morfologické úrovni, kdy změna tvaru buněk pomocí reorganizace cytoskeletálních vláken umožní nádorovým buňkám zvýšit jejich agresivitu a invazivní vlastnosti. Pochopení těchto změn spolu s porozuměním přepínání genetického programu v nádorových buňkách umožňujícího jejich přechod mezi různými diferenciačními stadii od nádorových kmenových buněk až po plně diferencované buňky by tak mohl vést k odhalení „slabých míst“ nádorových buněk a přispět tak k efektivitě léčby nádorových onemocnění. Schopnost nádorových buněk měnit pravidla vlastního organizmu tak představuje obrovskou výzvu pro onkologický výzkum.
Klíčová slova:
přeprogramování buněk – plasticita nádorových buněk – nádorový metabolizmus – heterogenita nádoru – remodelace cytoskeletu – metastázy – onkogeneze
Úvod
Transformace normálních buněk na buňky nádorové sestává z mnoha kroků společně vedoucích ke změně buněčné struktury a funkce. V roce 2000 bylo popsáno šest základních vlastností nádorových buněk: 1. nezávislost na tvorbě růstových faktorů, 2. neomezený replikační potenciál, 3. zvýšená proliferace, 4. rezistence k buněčné smrti, 5. schopnost indukovat angiogenezi, 6. zvýšená invazivita a tvorba metastáz [1]. Nedávno k nim přibylo několik dalších, jako je změna energetického metabolizmu, únik před imunitním systémem, nádorem vyvolaný zánět či genomová nestabilita. Projevem těchto vlastností tak nádorové buňky získávají svůj agresivní až devastující fenotyp. Další významnou vlastností nádorových buněk je schopnost rychle se přizpůsobit změnám okolního prostředí [2]. Příkladem je proces epiteliálně-mezenchymální tranzice (EMT), v rámci kterého dochází ke změně epiteliálního typu buněk na mezenchymální, čímž se nejen zvyšuje migrační potenciál, ale i buněčná plasticita nádorových buněk [3]. V nádorových buňkách též dochází k metabolickým změnám směrem k energeticky méně výhodné anaerobní glykolýze [4]. Popsané vlastnosti jsou spojené se změnami v expresi onkogenů a nádorových supresorových genů.
Změny v buněčném metabolizmu
Nádorové buňky jsou charakterizované schopností nekontrolovaného buněčného dělení, k čemuž je zapotřebí dostatečné množství živin a energie. Vybalancování zvýšených buněčných požadavků je dosaženo transformací buněčného metabolizmu. První popis metabolických změn v průběhu tvorby nádoru pochází z 30. let 20. století, kdy Otto Warburg pozoroval, že na rozdíl od normálních je v nádorových buňkách posílen glukózový metabolizmus [5]. Deregulace metabolizmu glukózy dle některých vědců představuje hlavní předpoklad vzniku nádoru a význam nádorového metabolizmu dokládá i skutečnost, že metabolické buněčné přeprogramování bylo zařazeno mezi 10 základních vlastností nádorových buněk [6]. Warburgův efekt představuje změnu v metabolizmu buněk charakterizovanou zvýšeným příjmem glukózy, která je proměněna na pyruvát a místo následného vstupu do Krebsova cyklu je konvertována na laktát. Tento metabolický proces je běžně uplatňován při hypoxických podmínkách, kdy má buňka nedostatek kyslíku, nicméně nádorové buňky preferují zpracování glukózy na laktát i za přítomnosti kyslíku. Aerobní glykolýza, jak se tento jev nazývá, probíhá v cytozolu bez účasti mitochondrií. Jedná se o rychlý zdroj energie v podobě ATP. Proces je však méně účinný, a nádorové buňky se tedy stávají silně závislé na zvýšeném příjmu glukózy [7]. Této vlastnosti se využívá k diagnostickým účelům, kdy se značený analog glukózy, 2-deoxy-2-18fluoro-D-glukóza, aplikuje pacientovi při vyšetření pozitronovou emisní tomografií (18F-FDG-PET) k hledání rychle proliferujících primárních nádorů a metastáz [8].
Pro nádorovou buňku je nutné podstoupit metabolické přeprogramování, aby si udržela selektivní výhodu spočívající v buněčném růstu a proliferaci. Musí však najít způsob, jakým získávat nejen energii, ale také stavební materiál pro biosyntézu nukleových kyselin, proteinů, lipidů a kofaktorů, které umožní udržet buněčnou homeostázu v podmínkách zvýšeného buněčného stresu (obr. 1) [9].
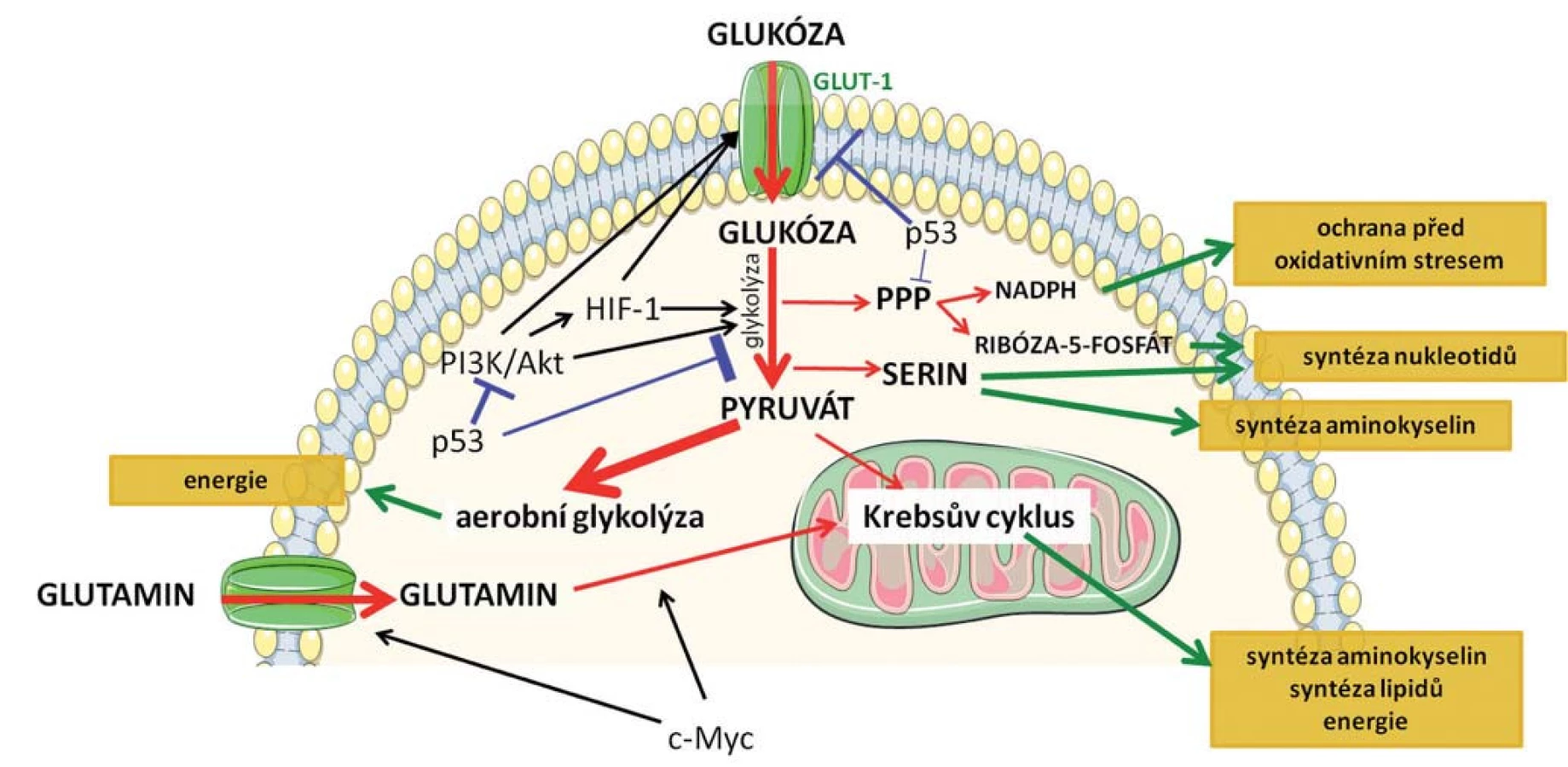
Po přijetí buňkami může glukóza vstoupit do procesu glykolýzy vedoucího k produkci ATP nebo může být přesměrována do tzv. pentózofosfátové dráhy (PPP). Během tohoto procesu je produkován ribóza-5-fosfát, který představuje substrát pro syntézu nukleotidů, nukleových kyselin a NADPH (redukovaný nikotinamidadenindinukleotid fosfát) [10]. Vlivem zrychleného metabolizmu a zvýšenou proliferací v porovnání s normálními buňkami dochází v nádorových buňkách často k navození oxidativního stresu vyvolaného přítomností radikálů. Pokud nejsou radikály včas odstraněny, dochází k poškození mitochondriální i jaderné DNA, k oxidaci proteinů a lipidů a tím k narušení jejich funkčnosti. NADPH slouží buňce ke zmírnění a k ochraně před oxidativním stresem a zvýšená syntéza tohoto kofaktoru pomocí PPP je tedy pro nádorovou buňku zásadní [11].
Meziprodukty glykolýzy se dále uplatňují při metabolizmu serinu, který je nezbytný pro syntézu nukleotidů, lipidů a jiných aminokyselin. Zvýšená exprese enzymu PHGDH (fosfoglycerát dehydrogenáza), který katalyzuje přeměnu 3-fosfoglycerátu získaného glykolýzou na serinový prekurzor, byla prokázána u karcinomu mléčné žlázy [12] a melanomu [12,13]. Kromě glukózy jsou nádorové buňky závislé na glutaminu, jenž se podílí na zrychlené proliferaci. Tato aminokyselina tedy musí být silně zastoupena v kultivačním médiu při studiu nádorových buněk in vitro. Glutamin představuje alternativní zdroj pro Krebsův cyklus, kdy po vstupu do buňky je postupně přeměněn na glutamát a následně na acetyl-CoA, který se uplatňuje v biosyntéze lipidů a je též zdrojem energie. Navíc se jedná o donor dusíku pro biosyntézu nukleotidů a bílkovin [14]. Metabolizmus glutaminu je spojen se zvýšenou expresí transkripčního faktoru c-Myc. c-Myc reguluje membránové transportéry pro glutamin a zároveň indukuje expresi enzymu glutaminázy 1 nutného pro lepší příjem a zpracování glutaminu a výraznější tvorbu prekurzorů nezbytných k syntéze biomolekul [15,16].
Mezi klíčové signální dráhy, které se podílejí na regulaci metabolických změn, patří fosfoinositid-3-kináza (PI3K) /Akt signalizace [17]. Aktivací PI3K/Akt dochází ke zvýšení glukózového příjmu díky zvýšené expresi povrchových receptorů GLUT1 a aktivaci hlavních enzymů účastnících se degradace glukózy (hexokináza a fosfofruktokináza 1) [15]. Aktivace PI3K/Akt signalizace v průběhu nádorového procesu tak představuje zásadní stimul vedoucí ke změněnému energetickému metabolizmu nádorové buňky.
Rozšiřující se poznatky o nádorovém metabolizmu vedou k odhalení nových funkcí klíčových a dobře prostudovaných onkoproteinů (c-Myc, proteiny uplatňující se při PI3K/Akt signalizaci) a nádorových supresorů. Nejvýznamnějším inhibitorem karcinogeneze je protein p53, často označovaný jako „strážce genomu“ [18]. Poměrně nedávno byla zjištěna jeho nová úloha, a to v regulaci metabolizmu. Na úrovni transkripce p53 reprimuje expresi glukózových transportérů a snižuje tak příjem glukózy [19]. Současně p53 snižuje i intracelulární koncentraci fruktóza-2,6-bisfosfátu, čímž dochází nejen k redukci glykolýzy, ale i k inhibici PI3K/Akt signalizace a potlačení metabolizmu glukózy [20]. Protein p53 negativně ovlivňuje i PPP, čímž redukuje produkci ribóza-5-fosfátu a NADPH, klíčových faktorů podporujících růst a dělení nádorových buněk [21].
Přeprogramování metabolizmu je regulováno také hypoxií, kdy dochází ke stabilizaci jedné z podjednotek HIF-1 (hypoxií inducibilní faktor 1) komplexu. HIF-1 zvyšuje expresi GLUT-1 (glukózových transportérů 1) na buněčném povrchu, enzymu hexokinázy a laktátdehydrogenázy, čímž stimuluje přeměnu glukózy na laktát [22]. Inhibicí konverze pyruvátu na acetyl-CoA pomocí pyruvát - dehydrogenáza kinázy 1 [23] HIF-1 blokuje Krebsův cyklus v metabolizmu glukózy [23].
Buněčný metabolický převrat může být způsoben celou řadou faktorů, vč. aktivace onkogenů, represe nádorových supresorových genů, dysfunkce mitochondriální DNA (mtDNA) a deregulace řady signálních drah [24]. Navzdory zvýšené snaze porozumět pozměněnému metabolizmu v nádorové buňce však stále zůstává otázkou, zdali je příčinou, či pouhým následkem nádorového procesu. Je však vědecky doloženo, že metabolické přeprogramování je nutné pro úspěšný vývoj nádorové buňky. Zesílení glykolýzy, pentózové fosfátové dráhy, metabolizmu glutaminu a dalších metabolických pochodů poskytne buňce nejen energii, ale také důležité stavební materiály pro biosyntézu aminokyselin, nukleotidů a lipidů, a tím je transformované buňce umožněno rychleji proliferovat a navozovat svůj nádorový charakter. Metabolické změny v nádorových buňkách představují nejen atraktivní cíl pro vývoj nových diagnostických přístupů, ale poskytují i nové možnosti v souvislosti s cílenou terapií.
Remodelace cytoskeletu v průběhu nádorové progrese
Metastázy představují hlavní příčinu úmrtí na nádorová onemocnění [25]. Důležitý krok při metastazování představuje proces epiteliálně-mezenchymální tranzice, kdy spolu se změnou adhezivních a migračních vlastností nádorové buňky podstupují morfologickou proměnu. Epiteliální buňky primárního nádoru, které mají apiko-bazální polaritu, se při EMT morfologicky mění, přičemž získávají mezenchymální fenotyp typický pro fibroblasty. Přechod buněk z jednoho místa na druhé a tvorba metastáz tak vyžaduje komplexní a dramatickou reverzibilní změnu buněčného cytoskeletu. Během progrese a šíření nádoru musí nádorové buňky dynamicky reagovat na extracelulární i intracelulární stimuly a v nádoru tak dochází ke změnám v expresi a organizaci proteinů, které jsou významnou součástí buněčného cytoskeletu. Konkrétně se jedná o 1. mikrofilamenta (aktin), 2. intermediární filamenta a 3. mikrotubuly. Alterace cytoskeletu zvyšují buněčnou pohyblivost, kontraktilnost a spolu s aktivací různých signálních kaskád přispívají ke zvýšené agresivitě nádorových buněk.
I když jednotlivé složky cytoskeletu účinkují synergicky, aktin bývá charakterizován jako „motor“ (driving force) buněčné migrace [26]. Aktinová mikrofilamenta regulují morfologii buněk, jejich kompartmentarizaci a udržují buněčnou polaritu. Zároveň se podílejí na regulaci buněčné signalizace. Aktinová filamenta přestavují přepínač mechanických stimulů přijatých buňkou pomocí integrinů (mechanoreceptorů) z vnějšího prostředí a převádí je na biochemické signály aktivací např. Erk a Akt signalizace [27]. Pro epiteliální buňky je charakteristická kortikální aktinová struktura, zatímco pro mezenchymální buňky je typická organizace aktinových filament do stresových vláken. Aktin ovlivňuje mezibuněčné pevné spoje, kde se adherentní protein E-cadherin váže na cytoskelet. Rozrušením adherentních spojů dochází k translokaci β-kateninu, který následně spustí WNT signalizaci a remodelaci cytoskeletu, jenž potencuje metastatický proces [28]. Změna v cytoskeletu tak umožní buňkám uniknout z původního místa a vede ke vzniku metastáz. Aktin se v buňkách vyskytuje ve dvou izoformách – β a γ, přičemž jejich funkce a distribuce se liší [29]. β-aktin zajišťuje buněčné spoje a kontrakce, zatímco γ-aktin je zodpovědný za tvorbu lamelárních, kortikálních a lamelipodních struktur v migrujících buňkách. V neoplasticky transformovaných buňkách jsou hladiny β-aktinu snížené. V podstatě platí pravidlo, že čím je transformační potenciál silnější, tím intenzivněji vznikají disperzní struktury, které se rozprostírají po celé buňce, zatímco v netransformovaných tvoří shluky v oblasti mezibuněčných spojů [29]. Co do organizace existují v buňce dva typy aktinu: 1. monomerní globulární G-aktin a 2. polymerní filamentární F-aktin. Proces polymerizace a rozpadu aktinových vláken závisí na koncentraci aktinu v buňce. Pro změnu morfologie a zvýšenou migraci a invazi nádorových buněk je klíčovou právě dynamická změna aktinových vláken, kdy se G-protein mění na stresová vlákna F-aktinu a dochází k formaci tzv. vedoucího konce (leading edge) [30].
K vytvoření komplexnější architektury se aktinová filamenta vážou s aktin asociovanými proteiny, např. actin-nucleating factors (ANFs) a/nebo nucleation promoting factors (NPFs), které regulují polymerizaci aktinu a umožňují vytvářet komplexní struktury, jako jsou filopodia, lamelipodia a invadopodia. Tyto tři struktury představují výběžky cytoplazmatické membrány a jsou nezbytné k zajištění adhezivity, migrace a invazivity nádorových buněk [31–33].
Intermediární filamenta (IF) udržují funkčnost plazmatické membrány a správný buněčný tvar. Rozeznáváme šest různých typů. Hlavním typem filament epiteliálních buněk jsou keratinová filamenta, tzv. IF typu I. V endoteliálních, mezenchymálních a hematopoetických buňkách převládají IF typu III, tzv. vimentinová filamenta, která zároveň slouží jako diagnostický marker pro epiteliální buňky, které prošly EMT. V průběhu EMT totiž dochází ke snížení exprese keratinu, který je nahrazen vimentinem, a tato změna je spojená s indukcí migračních schopností buněk [34]. Vazba keratinu na desmozomy (adhezivní mezibuněčná spojení) umožňuje silnou mezibuněčnou adhezi a udržuje tak epiteliální strukturu, která představuje bariéru pro indukci EMT. I když samotná ztráta keratinu není dostatečná pro vyvolání změn spojených s EMT, snížená hladina keratinu 8 a 18 představuje významný diagnostický marker spojený se zvýšenou migrací a invazivitou endometriálních a hepatocelulárních karcinomů [35,36]. Při změně exprese proteinů intermediárních filament směrem ke zvýšené expresi vimentinu buňky získávají mezenchymální buněčný tvar a zvýšený migrační potenciál [37].
Třetí složkou cytoskeletu jsou mikrotubuly (α a β tubulin), které se podílejí na udržení polarity buňky. Buněčná polarita je klíčovou vlastností, která určuje tvar buňky, asymetrickou lokalizaci proteinů a organel, reguluje dělení buněk a vede k udržení integrity tkání. Ztráta polarity je považovaná za jeden ze znaků maligní transformace buněk, neboť polární uspořádaní buňky slouží jako bariéra proti vzniku metastáz a potlačuje nekontrolovanou proliferaci buněk. Epiteliální buňky jsou charakterizované svojí apiko-bazální polaritou, která je daná mikrotubuly a při proměně epiteliálních buněk v buňky mezenchymální schopné migrovat pak dochází k prostorové reorganizaci. Ve všech epiteliálních buňkách vycházejí mikrotubuly tvořené z polymerů α a β tubulinu z centrozomů umístěných těsně pod apikální stranou buněčné membrány a směrují k bazální straně buněk, zatímco v mezenchymálních buňkách jsou mikrotubuly organizované od buněčného jádra radiálně směrem k povrchu ve směru pohybu buňky [38,39]. Mnohé nádorové supresory indukují stabilizaci mikrotubulů a regulaci epiteliálního fenotypu. Jedním z následků inaktivace nádorového supresoru v průběhu vývoje a progrese nádoru je tedy i narušení epiteliální polarizace, čímž je umožněn vznik agresivnějších nádorů. Mezi pět takových nejlépe popsaných nádorových supresorů patří APC (adenomatous polyposis coli), RASSF1A (Ras associated domain family 1A), VHL (von Hippel-Lindau), E-cadherin a merlin. Jejich inaktivace má za následek destabilizaci mikrotubulů vedoucí ke změnám v cytoskeletu, která vede ke zvýšení migračních vlastností buňky a rovněž k intracelulárním translokacím onkogenních molekul [39].
Buněčná migrace je obvykle spouštěna různými chemoatraktanty, které se vážou na membránové receptory a indukují signalizaci, která se projevuje cytoskeletálními změnami. Mezi nejvíce prostudované signální proteiny, které spouštějí cytoskeletální remodelaci v nádorových buňkách, patří proteiny WASP (Wiskott-Aldrich syndrome protein) /Arp2/3, LIM-kinázy/kofilin a kontraktin. Zvýšená aktivace těchto signalizací byla popsaná u řady nádorů – např. u karcinomu prsu, kolorektálního a hepatocelulárního karcinomu a úzce souvisí s fenotypem nádorových buněk [32,40].
Buněčné přeprogramování – nádorová plasticita
Buněčná plasticita představuje schopnost některých buněk změnit svůj fenotyp. První experimentální důkaz přeměny buněčného fenotypu byl získán v roce 1971, kdy Pierce et al pomocí světelné a elektronové mikroskopie v kombinaci s radioaktivním značením prokázali změnu nediferencovaných buněk na buňky diferencované [41]. Navození změny fenotypu umožní nádorovým buňkám rychle reagovat na různé podněty, které by mohly ovlivnit buněčné chování. Mezi vnitřní stimuly, které mohou vést k indukci změny fenotypu, patří změna genové exprese vyvolaná radioterapií nebo chemoterapií. Příkladem terapií vyvolané plasticity u nádorových buněk jsou leukemické buňky, které se vlivem ozáření proměnily na buňky s podobnými charakteristikami, jako mají leukemické kmenové buňky, jež jsou odolné vůči léčbě [42], nebo buňky karcinomu prsu, které se fenotypově mění z diferencovaných na buňky mezenchymální s vlastnostmi nádorových kmenových buněk v rámci odpovědi na chemoterapii [43,44]. Na změně fenotypu a přechodu mezi různými typy buněk se však podílejí i vnější podněty. Nejvýznamnější stimuly z vnějšího prostředí představují vzájemná interakce mezi buňkou a extracelulární matrix [45] a přítomnost rozdílných typů buněk v nádorovém stromatu.
Nejběžnějším příkladem buněčné plasticity je embryogeneze, při níž epiteliální buňky podstoupí transformaci na buňky mezenchymální. Tyto změny v adhezivitě a buněčném chování umožní buňkám vycestovat a vytvořit ostatní orgány [46]. Po dosažení místa určení buňky přejdou zpět k původnímu fenotypu, který jim umožní usadit se a vytvořit nové orgány. Analogický proces jako buňky embryonální adoptovaly i nádorové buňky. Nazývá se epiteliálně-mezenchymální tranzice, resp. mezenchymálně-epiteliální tranzice a je považován za klíčový při vzniku nádorových metastáz [47].
Buněčná plasticita představuje nástroj, kterým buňky mění vlastní identitu a získávají vlastnosti, které odpovídají jiným buněčným typům. Nádorovým buňkám pomáhá získat výhodu v metastazování, agresivnějším průběhu onemocnění a v úniku při cílené léčbě. Proces tumorigeneze, kdy se normální buňky promění na nádorové, je stejně tak možný pouze v případě, pokud buňky iniciující vznik nádorů mají plastické schopnosti, a tedy schopnost přeprogramovat svou vlastní identitu. Jako příklad úplné transdiferenciace nádorových buněk na jiný typ buněk slouží vytvoření vaskulárních mimiker. Bylo prokázáno, že vlivem hypoxie došlo u glioblastomů ke změně nádorových buněk na buňky endoteliální, které byly rezistentní k anti-VEGF terapii, a přispívaly tak ke zvýšení agresivity onemocnění [48].
Závěr
Buněčná a molekulární plasticita představuje klíčovou vlastnost buněk i při rozvoji mnohobuněčného organizmu. Nádorové buňky si tuto vlastnost zachovávají a stávají se tak nejen agresivnější, ale posilují i svou odolnost vůči léčbě. Tradičně se předpokládá, že dceřiné buňky kmenových buněk projdou progresivními a jednosměrnými změnami v genové expresi a tím dochází k jejich buněčné diferenciaci. Avšak tento koncept byl zavržen v důsledku nedávného objevu, který prokázal, že stačí pouze změna v expresi čtyř klíčových transkripčních faktorů (mezi které patří Oct-3/4, Sox2, KLF4 a c-Myc) [49,50], která spustí kaskádu změn vedoucích k rychlejšímu přeprogramování tisíce genů, což má za následek změnu buněčného fenotypu, kdy se buňky diferencované transformují na buňky podobné kmenovým buňkám, nazvaným indukované pluripotentní kmenové buňky [51].
Dediferenciace a následná rediferenciace nádorových buněk potencuje růst nádoru a zvyšuje jeho agresivitu vzhledem k již existujícím genetickým modifikacím. Bylo získáno několik důkazů o pozitivním vlivu nádorové plasticity na nádor, např. ztráta citlivosti leukemických buněk k imatinibu [52], nebo když vlivem hypoxie došlo k vytvoření dediferencovaných buněk, které následně vykazovaly vyšší tumorigenicitu [53]. Nicméně buněčné přeprogramování nemusí nutně vést jen ke zvýšení nádorového potenciálu. Některé studie naopak dokazují, že správnou kombinací aktivace a represe genů může být přeprogramovatelnost využita i k terapeutickým účelům [54]. Porozumění přeprogramovávacímu procesu nádorových buněk tudíž představuje značně perspektivní úkol na poli nádorové biologie a mohlo by vytvořit základ pro nové terapeutické strategie v boji s nádorovými onemocněními.
Práce byla podpořena projektem MŠMT – NPU I – LO1413.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Mgr. Lucia Sommerová
Regionální centrum aplikované molekulární onkologie
Masarykův onkologický ústav
Žlutý kopec 7
656 53 Brno
e-mail: lucia.sommerova@mou.cz
Obdrženo: 3. 7. 2016
Přijato: 11. 8. 2016
Zdroje
1. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100 (1): 57–70.
2. Klein CA. Selection and adaptation during metastatic cancer progression. Nature 2013; 501 (7467): 365–372. doi: 10.1038/nature12628.
3. Friedl P, Alexander S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell 2011; 147 (5): 992–1009. doi: 10.1016/j.cell.2011.11.016.
4. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer 2011; 11 (2): 85–95. doi: 10.1038/nrc2981.
5. Warburg O. On the origin of cancer cells. Science 1956; 123 (3191): 309–314.
6. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144 (5): 646–674. doi: 10.1016/j.cell. 2011.02.013.
7. Granja S, Pinheiro C, Reis RM et al. Glucose addiction in cancer therapy: advances and drawbacks. Curr Drug Metab 2015; 16 (3): 221–242.
8. Kelloff GJ, Hoffman JM, Johnson B et al. Progress and promise of FDG-PET imaging for cancer patient management and oncologic drug development. Clin Cancer Res 2005; 11 (8): 2785–2808.
9. Schulze A, Harris AL. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature 2012; 491 (7424): 364–373. doi: 10.1038/nature11706.
10. Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol 2011; 27 : 441–464. doi: 10.1146/annurev-cellbio-092910-154237.
11. Patra KC, Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci 2014; 39 (8): 347–354. doi: 10.1016/j.tibs.2014.06.005.
12. Possemato R, Marks KM, Shaul YD et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 2011; 476 (7360): 346–350. doi: 10.1038/nature10350.
13. Mullarky E, Mattaini KR, Vander Heiden MG et al. PHGDH amplification and altered glucose metabolism in human melanoma. Pigment Cell Melanoma Res 2011; 24 (6): 1112–1115. doi: 10.1111/j.1755-148X.2011.00919.x.
14. DeBerardinis RJ, Lum JJ, Hatzivassiliou G et al. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 2008; 7 (1): 11–20. doi: 10.1016/j.cmet.2007.10.002.
15. DeBerardinis RJ, Mancuso A, Daikhin E et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A 2007; 104 (49): 19345–19350.
16. Dang CV. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb Perspect Med 2013; 3 (8): pii: a014217. doi: 10.1101/cshperspect.a014217.
17. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009; 324 (5930): 1029–1033. doi: 10.1126/science.1160809.
18. Lane DP. Cancer. p53, guardian of the genome. Nature 1992; 358 (6381): 15–16.
19. Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res 2004; 64 (7): 2627–2633.
20. Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer 2009; 9 (10): 691–700. doi: 10.1038/nrc2715.
21. Jiang P, Du W, Wang X et al. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol 2011; 13 (3): 310–316. doi: 10.1038/ncb2172.
22. Denko NC. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer 2008; 8 (9): 705–713. doi: 10.1038/nrc2468.
23. Kim JW, Tchernyshyov I, Semenza GL et al. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 2006; 3 (3): 177–185.
24. Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer‘s Achilles‘ heel. Cancer Cell 2008; 13 (6): 472–482. doi: 10.1016/j.ccr.2008.05.005.
25. Schroeder A, Heller DA, Winslow MM et al. Treating metastatic cancer with nanotechnology. Nat Rev Cancer 2012; 12 (1): 39–50. doi: 10.1038/nrc3180.
26. Chan E, Saito A, Honda T et al. The acetylenic tricyclic bis (cyano enone), TBE-31 inhibits non-small cell lung cancer cell migration through direct binding with actin. Cancer Prev Res (Phila) 2014; 7 (7): 727–737. doi: 10.1158/1940-6207.CAPR-13-0403.
27. Müller P, Langenbach A, Kaminski A et al. Modulating the actin cytoskeleton affects mechanically induced signal transduction and differentiation in mesenchymal stem cells. PLoS One 2013; 8 (7): e71283. doi: 10.1371/journal.pone.0071283.
28. Schlessinger K, Hall A, Tolwinski N. Wnt signaling pathways meet Rho GTPases. Genes Dev 2009; 23 (3): 265–277. doi: 10.1101/gad.1760809.
29. Shagieva GS, Domnina LV, Chipysheva TA et al. Actin isoforms and reorganization of adhesion junctions in epithelial-to-mesenchymal transition of cervical carcinoma cells. Biochemistry (Mosc) 2012; 77 (11): 1266–1276. doi: 10.1134/S0006297912110053.
30. Nürnberg A, Kitzing T, Grosse R. Nucleating actin for invasion. Nat Rev Cancer 2011; 11 (3): 177–187. doi: 10.1038/nrc3003.
31. Mattila PK, Lappalainen P. Filopodia: molecular architecture and cellular functions. Nat Rev Mol Cell Biol 2008; 9 (6): 446–454. doi: 10.1038/nrm2406.
32. Yamaguchi H, Condeelis J. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim Biophys Acta 2007; 1773 (5): 642–652.
33. Leong HS, Robertson AE, Stoletov K et al. Invadopodia are required for cancer cell extravasation and are a therapeutic target for metastasis. Cell Rep 2014; 8 (5): 1558–1570. doi: 10.1016/j.celrep.2014.07.050.
34. Helfand BT, Chang L, Goldman RD. The dynamic and motile properties of intermediate filaments. Annu Rev Cell Dev Biol 2003; 19 : 445–467.
35. König K, Meder L, Kröger C et al. Loss of the keratin cytoskeleton is not sufficient to induce epithelial mesenchymal transition in a novel KRAS driven sporadic lung cancer mouse model. PLoS One 2013; 8 (3): e57996. doi: 10.1371/journal.pone.0057996.
36. Fortier AM, Asselin E, Cadrin M. Keratin 8 and 18 loss in epithelial cancer cells increases collective cell migration and cisplatin sensitivity through claudin1 up-regulation. J Biol Chem 2013; 288 (16): 11555–11571. doi: 10.1074/jbc.M112.428920.
37. Lang SH, Hyde C, Reid IN et al. Enhanced expression of vimentin in motile prostate cell lines and in poorly differentiated and metastatic prostate carcinoma. Prostate 2002; 52 (4): 253–263.
38. Müsch A. Microtubule organization and function in epithelial cells. Traffic 2004; 5 (1): 1–9.
39. Hernandez P, Tirnauer JS. Tumor suppressor interactions with microtubules: keeping cell polarity and cell division on track. Dis Model Mech 2010; 3 (5–6): 304–315. doi: 10.1242/dmm.004507.
40. Sahai E. Mechanisms of cancer cell invasion. Curr Opin Genet Dev 2005; 15 (1): 87–96.
41. Pierce GB, Wallace C. Differentiation of malignant to benign cells. Cancer Res 1971; 31 (2): 127–134.
42. Lee GY, Shim JS, Cho B et al. Stochastic acquisition of a stem cell-like state and drug tolerance in leukemia cells stressed by radiation. Int J Hematol 2011; 93 (1): 27–35. doi: 10.1007/s12185-010-0734-2.
43. Almendro V, Kim HJ, Cheng YK et al. Genetic and phenotypic diversity in breast tumor metastases. Cancer Res 2014; 74 (5): 1338–1348. doi: 10.1158/0008-5472.CAN-13-2357-T.
44. Almendro V, Cheng YK, Randles A et al. Inference of tumor evolution during chemotherapy by computational modeling and in situ analysis of genetic and phenotypic cellular diversity. Cell Rep 2014; 6 (3): 514–527. doi: 10.1016/j.celrep.2013.12.041.
45. Lipkin G. Plasticity of the cancer cell: implications for epigenetic control of melanoma and other malignancies. J Invest Dermatol 2008; 128 (9): 2152–2155. doi: 10.1038/jid.2008.69.
46. Acloque H, Adams MS, Fishwick K et al. Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Invest 2009; 119 (6): 1438–1449. doi: 10.1172/JCI38019.
47. Thiery JP, Acloque H, Huang RY et al. Epithelial-mesenchymal transitions in development and disease. Cell 2009; 139 (5): 871–890. doi: 10.1016/j.cell.2009.11.007.
48. Soda Y, Marumoto T, Friedmann-Morvinski D et al. Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc Natl Acad Sci U S A 2011; 108 (11): 4274–4280. doi: 10.1073/pnas.1016030108.
49. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006; 126 (4): 663–676.
50. Yamanaka S. Induction of pluripotent stem cells from mouse fibroblasts by four transcription factors. Cell Prolif 2008; 41 (Suppl 1): 51–56. doi: 10.1111/j.1365 - 2184.2008.00493.x.
51. Yu J, Vodyanik MA, Smuga-Otto K et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007; 318 (5858): 1917–1920.
52. Carette JE, Pruszak J, Varadarajan M et al. Generation of iPSCs from cultured human malignant cells. Blood 2010; 115 (20): 4039–4042. doi: 10.1182/blood-2009-07-231 845.
53. Mathieu J, Zhang Z, Zhou W et al. HIF induces human embryonic stem cell markers in cancer cells. Cancer Res 2011; 71 (13): 4640–4652. doi: 10.1158/0008-5472.CAN-10-3320.
54. Miyoshi N, Ishii H, Nagai K et al. Defined factors induce reprogramming of gastrointestinal cancer cells. Proc Natl Acad Sci U S A 2010; 107 (1): 40–45. doi: 10.1073/pnas.0912407107.
Štítky
Dětská onkologie Chirurgie všeobecná OnkologieČlánek vyšel v časopise
Klinická onkologie

2016 Číslo Supplementum 4
- Profesionální expozice azbestu a riziko vzniku karcinomu plic
- Plazmatický fibrinogen jako možný prognostický marker u nemetastatického renálního karcinomu
- Aktuální možnosti léčby mnohočetného myelomu
- Proces hojení ran krok za krokem a co ho může zkomplikovat
- Účinnost ceftazidimu/avibaktamu v léčbě infekcí způsobených enterobakteriemi produkujícími karbapenemázy
Nejčtenější v tomto čísle
- Úloha PD-1/PD-L1 signalizace v protinádorové imunitní odpovědi
- Nemalobuněčný karcinom plic – od imunobiologie k imunoterapii
- Nádorové buňky jako dynamický systém – molekulární a fenotypové změny v průběhu vzniku, progrese a šíření nádoru
- Molekulární podstata kancerogeneze epiteliálních ovariálních karcinomů
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy