
Tvorba hmotnostne spektrometrických spektrálnych knižníc nádorových bunečných línií
Building Mass Spectrometry Spectral Libraries of Human Cancer Cell Lines
Background:
Cancer research often focuses on protein quantification in model cancer cell lines and cancer tissues. SWATH (sequential windowed acquisition of all theoretical fragment ion spectra), the state of the art method, enables the quantification of all proteins included in spectral library. Spectral library contains fragmentation patterns of each detectable protein in a sample. Thorough spectral library preparation will improve quantitation of low abundant proteins which usually play an important role in cancer.
Aim:
Our research is focused on the optimization of spectral library preparation aimed at maximizing the number of identified proteins in MCF-7 breast cancer cell line. First, we optimized the sample preparation prior entering the mass spectrometer. We examined the effects of lysis buffer composition, peptide dissolution protocol and the material of sample vial on the number of proteins identified in spectral library. Next, we optimized mass spectrometry (MS) method for spectral library data acquisition.
Conclusion:
Our thorough optimized protocol for spectral library building enabled the identification of 1,653 proteins (FDR < 1%) in 1 µg of MCF-7 lysate. This work contributed to the enhancement of protein coverage in SWATH digital biobanks which enable quantification of arbitrary protein from physically unavailable samples. In future, high quality spectral libraries could play a key role in preparing of patient proteome digital fingerprints.
Key words:
biomarker – mass spectrometry – proteomics – digital biobanking – SWATH – protein quantification
This work was supported by the project MEYS – NPS I – LO1413.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE recommendation for biomedical papers.
Submitted:
7. 5. 2016
Accepted:
9. 6. 2016
Autoři:
J. Faktor; P. Bouchal
Působiště autorů:
Regionální centrum aplikované molekulární onkologie, Masarykův onkologický ústav, Brno
Vyšlo v časopise:
Klin Onkol 2016; 29(Supplementum 4): 54-58
Kategorie:
Původní práce
doi:
https://doi.org/10.14735/amko20164S54
Souhrn
Východiská:
Kvantifikácia proteínov v modelových nádorových bunečných líniách a v nádorových tkanivách je často predmetom onkologického výskumu. Metóda SWATH (sequential windowed acquisition of all theoretical fragment ion spectra), novinka v kvantitatívnej onkoproteomike, získava čoraz väčšiu popularitu, pretože umožňuje kvantifikáciu všetkých proteínov zaznamenaných v spektrálnej knižnici. Spektrálna knižnica obsahuje fragmentačné informácie o každom detekovateľnom proteíne vo vzorke. Dôkladná príprava spektrálnej knižnice výrazne vylepší kvantifikáciu obzvlášť nízkoabundantných proteínov, ktoré často zohrávajú výnamnú úlohu v nádorových ochoreniach.
Cieľ:
V tomto článku sa venujeme optimalizácii prípravy spektrálnej knižnice s cieľom maximalizovať počet kvantifikovateľných proteínov v modelovej bunečnej línii MCF-7 odvodenej od karcinómu prsníka. Experiment je zameraný na optimalizáciu postupu prípravy vzorky pred vstupom do hmotnostného spektrometru, kde bol skúmaný vplyv zloženia lyzačného pufru, vplyv postupu rozpúšťania peptidov a vplyv materiálu mikroskúmaviek na počet proteínov v spektrálnej knižnici. V neposlednom rade boli optimalizované aj parametre merania na hmotnostnom spektrometri pre získavanie dát do spektrálnej knižnice.
Záver:
Dôslednou optimalizáciou postupu sa podarilo pripraviť spektrálnu knižnicu obsahujúcu fragmentačné informácie o 1 653 proteínoch (FDR < 1%) z 1 µg lyzátu MCF-7. Hlavným prínosom optimalizácie postupu prípravy spektrálnej knižnice je rozšírenie pokrytia proteómu vo SWATH digitálnych biobankách umožňujúcich kvantifikáciu ľubovoľného proteínu vo fyzicky už nedostupných vzorkách. Kvalitné spektrálne knižnice by mohli v budúcnosti zohrávať kľúčovú úlohu pri tvorbe digitálnych fingerprintov proteómu pacientov.
Kľúčové slová:
nádorové biomarkery – hmotnostná spektrometria – proteomika – digitálna biobanka – SWATH – kvantifikácia proteínov
Úvod
Rakovina patrí v dnešnej dobe k veľmi častým ochoreniam a v roku 2012 bola celosvetovo príčinou 8,2 milióna úmrtí [1]. Rakovinové ochorenia sú spôsobované genómovými zmenami a aj zmenami v biochemických dráhach, ktoré vedú k nekontrolovanému deleniu buniek a v pokročilejších štádiách onemocnenia k metastázovaniu. Včasná diagnostika a cielená liečba zameraná na charakteristické proteíny môže znížiť úmrtnosť spôsobenú dôsledkom rakovinových ochorení [2]. Preto sa onkologický výskum zameriava na hľadanie nových rakovinových biomarkerov a terapeutických cieľov a onkoproteomika, zaoberajúca sa výskumom proteínov zmenených v súvislosti s rakovinovými ochoreniami, sa stala jeho nezastupiteľným odvetvím [3]. Pre onkoproteomiku sú kľúčové metódy využívajúce techniky hmotnostnej spektrometrie (mass spectrometry – MS), ktoré popisujú zmeny v koncentračnom zastúpení proteínov v závislosti na vonkajších alebo vnútorných podnetoch. Novinkou v oblasti cielenej onkoproteomiky je metóda SWATH (sequential windowed acquisition of all theoretical fragment ion spectra), ktorá bola vyvinutá za účelom spoľahlivej kvantifikácie všetkých detekovateľných proteínov v jednej analýze [4]. Výhodou pre onkologický výskum je možnosť tvorby digitálnych spektrálnych biobánk biologického materiálu, ktoré môžu obsahovať súbory tkanív, ale aj bunečných línií. Digitálny spektrálny biobanking v princípe umožňuje kvantifikáciu vybraného proteínu aj niekoľko rokov po zmeraní dát a výmenu dát medzi laboratóriami, čo významne urýchľuje výskum [5]. Pre extrakciu SWATH kvantitatívnych dát je nevyhnutné použiť spektrálnu knižnicu obsahujúcu informácie o proteínoch/peptidoch zastúpených vo vzorke. Podmienkou je, aby v spektrálnej knižnici mal každý proteín reprezentujúci peptid priradený retenčný čas a fragmentačné spektrum produktových iónov s ich intenzitami [6]. Kvantifikácia proteínov, ktoré nie sú v spektrálnej knižnici, je nemožná napriek tomu, že ich spektrá môžu byť prítomné v SWATH dátach. Dôkladná optimalizácia prípravy spektrálnej knižnice má teda zásadný význam pre množstvo kvantifikovateľných proteínov vo SWATH biobanke. Cieľom nášho výskumu bolo nájsť vhodné podmienky pre prípravu spektrálnej knižnice modelovej bunečnej línie MCF-7 odvodenej od karcinómu prsníka. Prvotnou úlohou bolo nájsť vhodné podmienky bunečnej lýzy, kde sa prihliadalo na kompatibilitu lyzačného pufru s MS. V ďalšom kroku boli optimalizované podmienky na rozpúšťanie peptidov pred MS analýzou a bola porovnaná adsorpcia peptidov na rôzne povrchy stien mikroskúmaviek. Najväčší vplyv na počet proteínov obsiahnutých v spektrálnej knižnici má správne nastavenie parametrov MS metódy. Preto sme sa zamerali na optimalizáciu minimálnej intenzity prekurzorového peptidu vybraného na fragmentáciu a doby vylúčenia prekurzorového peptidu. Ukázalo sa, že dôkladnou optimalizáciou postupu prípravy vzorky a MS metódy na hmotnostnom spektrometri TripleTOF 5600+ sa dá podstatne zvýšiť počet proteínov obsiahnutých v spektrálnej knižnici, čo nám rozširuje možnosti kvantifikácie proteínov zastúpených v nízkych koncentráciách. Tieto proteíny sú obzvlášť zaujímavé pre onkologický výskum.
Materiál a metódy
Príprava lyzátu bunečnej línie MCF-7 a proteolytické štiepenie proteínov
Bunky získané z MCF-7 bunečnej línie boli 30 min lyzované v lyzačnom pufre zloženom z 8 M močoviny, 0,1 M Tris/HCl, pH 8,5 a v pufre zloženom z 0,05% dodecylsulfátu sodného (SDS) a 0,5 M trietylammonium bikarbonátu. Po skončení lýzy bol lyzát centrifugovaný 30 min/14 000 g/5 °C. Metódou RC-DC (Bio-Rad, USA) bola zmeraná koncentrácia proteínu v supernatante a 100 µg proteínového lyzátu bolo odobraných na proteolytické štiepenie. Proteolytické štiepenie a purifikácia peptidov z MCF-7 boli prevedené podľa Pernikářová et al [7].
„Acetonitrilová extrakcia“ peptidov pred separáciou pomocou kvapalinovej chromatografie (LC) spojenej s MS detekciou (LC-MS)
K 15 µg peptidovej vzorky z MCF-7 lyzátu bolo pridaných 15 µl pufru zloženého z 51% acetonitrilu (AcN), 0,05% trifluoroctovej kyseliny (TFA) v H2O. Vzorka bola dôkladne premiešaná tak, aby sa rozpustili všetky peptidy na stenách skúmavky. Následne bola vzorka sonikovaná počas 5 min a opäť dôkladne premiešaná. Vzorka bola centrifugovaná 10 min/10 000 g/25 °C. Po prepipetovaní do novej skúmavky bol odparený acetonitril v Speed-Vacu tak, aby sme získali zmes peptidov v nanášacom pufre zloženom z 2% AcN, a 0,05% TFA v H2O.
Štandardné rozpúšťanie peptidovej vzorky pred LC-MS analýzou
K 15 µg peptidovej vzorky z MCF-7 lyzátu bolo pridaných 15 µl pufru zloženého z 2% AcN, 0,05% TFA v H2O. Vzorka bola dôkladne premiešaná tak, aby sa rozpustili všetky peptidy na stenách skúmavky. Následne bola vzorka sonikovaná počas 5 min a opäť dôkladne premiešaná. Vzorka bola centrifugovaná 10 min/10 000 g/25 °C.
Mikroskúmavky použité na porovnanie adsorbcie peptidov na povrch
Zriedený roztok peptidovej vzorky MCF-7 bol napipetovaný do štyroch mikroskúmaviek s rôznym povrchom tak, aby bolo možné nastrieknuť 0,05 µg peptidov na kolonu. Použité boli polypropylénové mikroskúmavky so silanizovaným povrchom kat. č. 910-00096 (Eksigent, USA), dve mikroskúmavky s poly - propylénovým povrchom „Red“ kat. č. 548-3120 (WVR, USA), „Orange“ kat. č. 548-0120 (WVR, USA) a mikroskúmavka so skleneným inzertom kat. č. 02-FIRVG (Thermo, USA). Mikroskúmavky so vzorkou boli umiestnené v autosamplere temperovanom na teplotu 8 °C.
Parametre LC-MS analýzy pre tvorbu spektrálnej knižnice
Vzorky boli separované na chromatografe Eksigent Ekspert nanoLC 400 system (SCIEX, USA), ktorý bol priamo spojený s hmotnostným spektrometrom TripleTOF 5600+ (SCIEX, Kanada).
Peptidy boli zachytené a odsolené na trap kolone µ-precolumn, 30 µm i.d., 5 mm dĺžka, C18 PepMap 100, 5 µm veľkosť častíc, 100 Å veľkosť pórov (Thermo Scientific, USA). Následne boli peptidy eluované na analytickú kolonu s náplňou 3C18-CL-120, 75 µm i.d., 150 mm dĺžka, 5 µm veľkosť častíc, 120 Å veľkosť pórov (Eksigent, USA). Mobilná fáza A bola zložená z 0,1% (v/v) kyseliny mravčej (FA) v H2O a mobilná fáza B z 0,1% (v/v) FA v AcN.
Nastavenie nano-kvapalinového chromatografu pre separáciu peptidov spektrálnej knižnice
Elúcia z analytickej kolony začínala na 2 % mobilnej fázy B počas 1 min. Percentuálny podiel mobilnej fázy lineárne stúpal na 12 % B počas 5 min, ďalších 114 min podiel B lineárne vzrástol na 35 % a v nasledujúcich 2 min vzrástol až na 80 % B, na hodnote 80 % B zostal počas 18 min a potom podiel mobilnej fázy B lineárne klesal na 2 % počas 2 min. Podiel mobilnej fázy B zostal na hodnote 2 % počas ďalších 38 min. Prietok mobilnej fázy bol 300 nl/min.
Nastavenie MS metódy pre identifikáciu proteínov (MS/MS) v spektrálnej knižnici
Hmotnostný spektrometer TripleTOF 5600+ pracoval v dáta dependentnom móde. V ionizačnom zdroji (nano-elektrospreji) bol dusík použitý ako zmlžovací plyn a teplota aj tok sušiaceho plynu boli nastavené na hodnoty 150 °C a 12 °C. Ionizačné napätie bolo nastavené na hodnotu 2700 V. MS spektrum z analyzátoru doby prieletu (TOF-MS) bolo zmerané v každom cykle nasledované fragmentáciou 20 najintenzívnejších prekurzorových iónov a zmeraním spektier ich produktových iónov. Minimálna intenzita prekurzorového iónu bola optimalizovaná. Pri tejto optimalizácii boli použité hodnoty 50 impulzov za sekundu (cps), 100 cps a 250 cps. Predmetom optimalizácie bola aj hodnota času vylúčenia zmeraných prekurzorových iónov, použité boli hodnoty 8 s, 12 s, 20 s, 30 s a 40 s. Akumulačný čas prekurzorového iónu bol 100 ms.
Analýza dát – tvorba spektrálnej knižnice
Dáta získané MS/MS metódou boli prehľadané pomocou programu ProteinPilot 4.5 (SCIEX, Kanada) proti ľudskej referenčnej databáze (Uniprot 2013_09) [8]. Parametre prehľadávania boli nastavené nasledovne – trypsín (proteolytický enzým), karbamidometyl (fixná modifikácia), FDR analýza bola prevedená proti obrátenej databáze (Uniprot 2013_09) [8].
Výsledky a diskusia
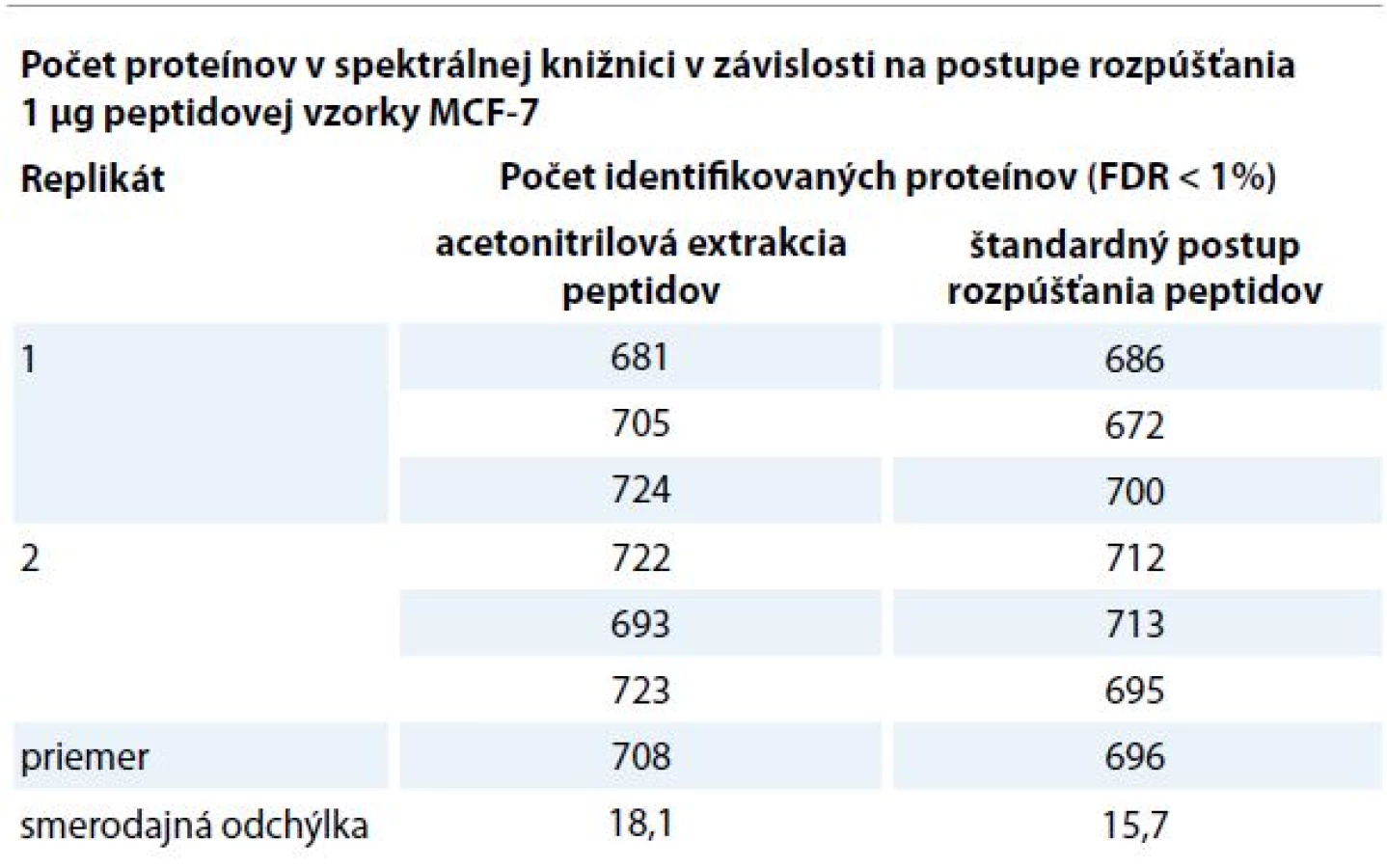
Optimalizáciou podmienok prípravy spektrálnej knižnice bunečnej línie MCF-7 (obr. 1) môžeme významne vylepšiť pokrytie kvantifikovaného proteómu a zvýšiť tak kvalitu spektrálnej knižnice (tab. 1–3) a aj kvantitatívnych dát extrahovaných zo SWATH datasetov. Prvým krokom pri príprave spektrálnej knižnice je extrakcia proteínov z bunečnej línie pomocou lyzačného pufru. Pri extrakcii proteínov je nutné použiť lyzačné pufry kompatibilné s MS, prípadne pred MS analýzou odstrániť interferujúce látky použité pri lýze [9]. Metóda štiepenia proteínov na filtri (filter aided sample preparation – FASP) [10] umožňuje použitie detergentu SDS, prípadne močoviny v lyzačnom pufre. Po pridaní lyzátu filter zabezpečí výmenu do MS kompatibilného pufru. Ukázalo sa, že lyzát pripravený v lyzačnom pufre zloženom z 8 M močoviny 0,5 M Tris/HCl, pH 8,5 poskytuje vyšší signál celkového iónového prúdu a aj viac identifikovaných proteínov v porovnaní s lyzátom pripraveným v 0,05% SDS. Vyšší počet identifikácií a celkový MS signál z lyzátu pripraveného v močovinovom lyzačnom pufre môže byť výsledkom vyššej lyzačnej a solubilizačnej efektivity. Nižší signál u MCF-7 v SDS lyzačnom pufre zrejme spôsobuje SDS zachytené na stenách filtra, ktoré po vymytí do vzorky môže negatívne interferovať počas LC-MS analýzy [11]. Po lýze, FASP proteolytickom štiepení, odsolení a zakoncentrovaní biologickej vzorky sa získava koncentrát obsahujúci zmes peptidov, ktorý sa pred analýzou rozpúšťa v nanášacom pufre (2% AcN + 0,05% TFA v H2O). Nanášací pufer musí rozpúšťať celé spektrum peptidov, efektivita závisí od postupu rozpúšťania peptidov a od zloženia nanášacieho pufru [12]. Porovnaný bol klasický postup rozpúšťania peptidovej vzorky z MCF-7, kde sa k lyzátu pridáva nanášací pufer s následným miešaním a postup acetonitrilovej extrakcie peptidov. Acetonitrilová extrakcia peptidov mierne zvýšila počet identifikácií v spektrálnej knižnici na 716 (FDR < 1%) oproti klasickému postupu (tab. 1). Postup acetonitrilovej extrakcie peptidov umožňuje efektívnejšie uvoľnenie hydrofóbnych peptidov zo stien skúmavky dôsledkom použitia vyššieho podielu organickej fázy (50% AcN) v porovnaní s klasickou metódou rozpúšťania v 2% AcN. Roztok peptidov v nanášacom pufre je následne premiestnený do LC-MS systému v mikroskúmavkách. Nežiaducim efektom je adsorbcia peptidov na steny mikroskúmavky, kde sa na nepolárne plastické povrchy počas státia v autosampleri adsorbujú najmä hydrofóbne peptidy [13]. Na trhu sú k dispozícii mikroskúmavky vyrobené z rôznych materiálov, ktoré by mali minimalizovať adsorbciu peptidov. V súčasnosti sa často používajú sklenené mikroskúmavky, mikroskúmavky so silanizovaným povrchom, prípadne s povrchom potiahnutým polypropylénom alebo polyetylénom [14]. Porovnaním mikroskúmaviek sme dospeli k záveru, že použitý materiál nemá zásadný vplyv na signál a počet identifikovaných proteínov v spektrálnej knižnici (tab. 2). Ukázalo sa, že optimalizácia parametrov metódy na ovládanie LC-MS prístroja má naopak podstatný vplyv na rozsah a kvalitu spektrálnej knižnice. V rámci optimalizácie MS/MS metódy sme hľadali najlepšie nastavenie minimálnej intenzity peptidového (prekurzorového) iónu použitého na fragmentáciu a dobu jeho vylúčenia (tab. 3). Optimalizáciou minimálnej intenzity prekurzorového píku sme zamedzili fragmentáciu prekurzorov na úrovni šumu a zároveň sme zamedzili vylúčenie prekurzorov pochádzajúcich z proteínov zastúpených v nízkych koncentráciách, ktoré sú často predmetom onkologického výskumu. Optimálna doba vylúčenia prekurzorového iónu ďalej rozšírila počet identifikovaných proteínov. Vylúčenie fragmentovaných prekurzorových iónov zabraňuje opakovanej fragmentácii prekurzorového iónu s rovnakým m/z, a umožňuje tak fragmentáciu menej zastúpených prekurzorov. Po optimalizácii prípravy peptidovej vzorky z bunečnej línie MCF-7 a parametrov MS metódy bola vytvorená spektrálna knižnica obsahujúca 1 653 (FDR < 1%) proteínov z 1 µg lyzátu MCF-7 bunečnej línie, čo predstavuje 8,1 % zo všetkých 20 197 recenzovaných proteínov obsiahnutých v ľudskej databáze proteínov Swiss-Prot [15].



Záver
S využitím rôznych lyzačných podmienok, výberom vhodných mikroskúmaviek ako aj optimalizáciou MS metódy sme dosiahli vyšší počet identifikovaných proteínov a vytvorili sme kompletnejšiu spektrálnu knižnicu pre nádorovú bunečnú líniu MCF-7. Nový postup tvorby spektrálnej knižnice bude ďalej aplikovaný pri SWATH kvantifikácii proteínov v biologických vzorkách, kde významne ovplyvní kvalitu vyextrahovaných kvantitatívnych dát a pokrytie celkového proteómu. Rozšírenie spektrálnej knižnice umožní kompletnejší náhľad na reguláciu proteínov zastúpených v nízkych koncentráciách, ktoré sú obyčajne najzaujímavejšie pre onkologický výskum, a tým zjednoduší hľadanie nových biomarkerov a terapeutických cieľov. Kompletnejšie spektrálne knižnice prispejú k rozšíreniu a prehĺbeniu poznatkov v onkologickom výskume.
Práce byla podpořena projektem MŠMT – NPU I – LO1413.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Mgr. Jakub Faktor
Regionální centrum aplikované molekulární onkologie
Masarykův onkologický ústav
Žlutý kopec 7
656 53 Brno
e-mail: jakub.faktor@mou.cz
Obdrženo: 7. 5. 2016
Přijato: 9. 6. 2016
Zdroje
1. Torre LA, Bray F, Siegel RL et al. Global cancer statistics, 2012. CA Cancer J Clin 2015; 65 (2): 87–108. doi: 10.3322/caac.21262.
2. Weigelt B, Peterse JL, van’t Veer LJ. Breast cancer metastasis: markers and models. Nat Rev Cancer 2005; 5 (8): 591–602.
3. Cho WC, Cheng CH. Oncoproteomics: current trends and future perspectives. Expert Rev Proteomics 2007; 4 (3): 401–410.
4. Gillet LC, Navarro P, Tate S et al. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics 2012; 11 (6): O111.016717. doi: 10.1074/mcp.O111.016717.
5. Faktor J, Michalova E, Bouchal P. pSRM, SWATH and HRM – targeted proteomics approaches on TripleTOF 5600+ mass spectrometer and their applications in oncology research. Klin Onkol 2014; 27 (Suppl 1): S110–S115. doi: 10.14735/amko20141S110.
6. Schubert OT, Gillet LC, Collins BC et al. Building high-quality assay libraries for targeted analysis of SWATH MS data. Nat Protoc 2015; 10 (3): 426–441. doi: 10.1038/nprot.2015.015.
7. Pernikářová V, Sedláček V, Potěšil D et al. Proteomic responses to a methyl viologen-induced oxidative stress in the wild type and FerB mutant strains of Paracoccus denitrificans. J Proteomics 2015; 125 : 68–75. doi: 10.1016/j.jprot.2015.05.002.
8. ftp.uniprot.org [homepage on the Internet]. European Bioinformatics Institute (EBI), United Kingdom, c2002–2015 [updated 2015 March 14; cited June 9]. Available from: ftp://ftp.uniprot.org/pub/databases/uniprot/previous_releases/release-2013_09/.
9. Gundry RL, White MY, Murray CI et al. Preparation of proteins and peptides for mass spectrometry analysis in a bottom-up proteomics workflow. Curr Protoc Mol Biol 2009; Chapter 10: Unit 10.25. doi: 10.1002/0471142727.mb1025s88.
10. Wiśniewski JR, Zougman A, Nagaraj N et al. Universal sample preparation method for proteome analysis. Nat Methods 2009; 6 (5): 359–362. doi: 10.1038/nmeth. 1322.
11. Pellerin D, Gagnon H, Dubé J et al. Amicon-adapted enhanced FASP: an in-solution digestion-based alternative sample preparation method to FASP. F1000Research 2015; 4 : 140. doi: 10.12688/f1000research.6529.1.
12. Peterson A, Hohmann L, Huang L et al. Analysis of RP-HPLC loading conditions for maximizing peptide identifications in shotgun proteomics. J Proteome Res 2009; 8 (8): 4161–4168. doi: 10.1021/pr9001417.
13. Duncan MR, Lee JM, Warchol MP. Influence of surfactants upon protein/peptide adsorption to glass and polypropylene. Int J Pharm 1995; 120 (2): 179–188.
14. Stejskal K, Potěšil D, Zdráhal Z. Suppression of peptide sample losses in autosampler vials. J Proteome Res 2013; 12 (6): 3057–3062. doi: 10.1021/pr400183v.
15. UniProt.org [homepage on the Internet]. European Bioinformatics Institute (EBI), United Kingdom, c2002–2015 [updated 2015 March 14; cited 2016 Aug 2]. Available from: http://www.uniprot.org/uniprot/query=*&fil=reviewed%3Ayes+AND+organism%3A%22Homo+sapiens+%28Human%29+%5B9606%5D %22.
Štítky
Dětská onkologie Chirurgie všeobecná OnkologieČlánek vyšel v časopise
Klinická onkologie

2016 Číslo Supplementum 4
- Diosmin a hesperidin: Co ukazuje farmakokinetika?
- Zkušenosti s axitinibem v léčbě metastatického renálního karcinomu
- Evaluace skóre m7-FLIPI u pacientů s folikulárním lymfomem odhalila slibný prediktivní biomarker k určení vhodné chemoterapie
- Neuropatie výrazně zpomaluje proces hojení u povrchových ran
- Zeolit-jodový komplex pomáhá v péči o infikované rány
Nejčtenější v tomto čísle
- Úloha PD-1/PD-L1 signalizace v protinádorové imunitní odpovědi
- Nemalobuněčný karcinom plic – od imunobiologie k imunoterapii
- Nádorové buňky jako dynamický systém – molekulární a fenotypové změny v průběhu vzniku, progrese a šíření nádoru
- Molekulární podstata kancerogeneze epiteliálních ovariálních karcinomů
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy