
Nemoci žlučových cest z pohledu patologa: Přínos jaterní biopsie v rutinní diagnostické praxi
Cholangiopathies from a Pathologist‘s Perspective: The Role of Liver Biopsy in Routine Diagnostic Practice
The biliary tree comprises a three-dimensional network of intrahepatic and extrahepatic ducts lined by biliary epithelium (cholangiocytes). The bile ducts and cholangiocytes may be affected by a broad spectrum of disorders collectively referred to as cholangiopathies. These conditions are classified based on anatomical aspects (predominantly affecting small or large bile ducts), aetiopathogenesis (immune-mediated, toxic and drug-induced, ischaemic, infectious, genetically determined, or neoplastic), and the predominant morphological pattern of injury (inflammatory or non-inflammatory). While abnormalities in medium-sized and large bile ducts are typically detected using radiological methods, the diagnosis of small duct cholangiopathies continues to rely primarily on histopathological evaluation of liver tissue.
This review summarises the key morphological features of the most clinically significant cholangiopathies, focusing on histopathological changes observed in liver biopsy.
Keywords:
Bile ducts – cholangitis – cholangiocytes – cholangiopathies – liver biopsy
Autoři:
Ivana Jusková 1,2; Andrea Vajsová 1,3; Ondřej Fabián 1,4; Eva Sticová 1,5
Působiště autorů:
Pracoviště klinické a transplantační patologie, Institut klinické a experimentální medicíny, Praha
1; Ústav histologie 3. lékařské fakulty Univerzity Karlovy, Praha
2; Ústav patologie 1. lékařské fakulty Univerzity Karlovy a Všeobecné fakultní nemocnice, Praha
3; Ústav patologie a molekulární medicíny 3. lékařské fakulty Univerzity Karlovy a Fakultní Thomayerovy nemocnice, Praha
4; Ústav patologie 3. lékařské fakulty Univerzity Karlovy a Fakultní nemocnice Královské Vinohrady, Praha
5
Vyšlo v časopise:
Čes.-slov. Patol., 61, 2025, No. 3, p. 118-134
Kategorie:
Přehledový článek
Souhrn
Žlučové cesty jsou tvořeny trojrozměrným systémem intrahepatálních a extrahepatálních žlučovodů vystlaných biliárními epiteliemi (cholangiocyty). Žlučovody a cholangiocyty mohou být postiženy celou řadou onemocnění souhrnně označovaných jako cholangiopatie. Tyto poruchy lze klasifikovat na základě anatomických aspektů (cholangiopatie převážně malých a převážně velkých žlučovodů), dále dle etiopatogeneze (imunitně podmíněné, toxiny a léky indukované, ischemické, infekční, geneticky podmíněné a neoplastické) či dle převládajících morfologických charakteristik poškození (zánětlivé a nezánětlivé). Zatímco změny středně velkých a velkých žlučovodů jsou obvykle identifikovány za pomocí radiodiagnostických metod, v diagnostice postižení malých intrahepatálních žlučovodů hraje stále významnou roli histopatologické vyšetření jaterní tkáně.
Tento přehledový článek shrnuje klíčové morfologické rysy klinicky nejvýznamnějších cholangiopatií s důrazem na histopatologické změny identifikovatelné v jaterní biopsii.
Adresa pro korespondenci:
MUDr. Ivana Jusková
Pracoviště klinické a transplantační patologie Institut klinické a experimentální medicíny Vídeňská 1958/9, 140 21 Praha 4
Tel.: 2360552030, fax: 236053020
E-mail: ivana.juskova@ikem.cz
Klíčová slova:
cholangitida – žlučové cesty – cholangiocyty – cholangiopatie – jaterní biopsie
Žlučový strom je trojrozměrný systém vývojově, anatomicky i funkčně odlišných luminizovaných struktur vystlaných biliárními epiteliemi zvanými cholangiocyty. Anatomicky lze žlučovody rozdělit na intrahepatální a extrahepatální. Intrahepatální žlučovody zahrnují systém žlučových kapilár (canaliculi biliferi) a Heringových kanálků, dále interlobulární žlučovody o průměru 15–100 µm, septální žlučovody s průměrem 100–300 µm a segmentální a lobární vývody, jejichž průměr dosahuje až 800 µm. Extrahepatální žlučovody tvoří pravý a levý jaterní vývod (ductus hepaticus dexter et sinister), které se spojují do společného jaterního vývodu (ductus hepaticus communis). Na společný ductus hepaticus pak navazuje ductus cysticus a ductus choledochus, který ústí do duodena prostřednictvím Vaterovy papily (schéma 1) (1).
Žlučové cesty a jejich výstelka plní celou řadu důležitých fyziologických funkcí. Kromě modifikace složení žluči jsou tyto struktury součástí lokálních obranných mechanismů a imunitně zprostředkovaných reakcí a podílejí se rovněž na procesech buněčné signalizace či regenerace (1–3).
Žlučovody a cholangiocyty mohou být postiženy celou řadou patologických procesů, které souhrnně označujeme jako cholangiopatie (4).
Společným projevem většiny cholangiopatií je cholestáza, která je definována jako porucha tvorby a sekrece žluči nebo selhání schopnosti organismu dodat do duodena dostatečné množství žluči odpovídajícího složení (5). Typickými klinickými projevy cholestázy bývají žloutenka (ikterus) a svědění kůže (pruritus). Na úrovni biochemické se cholestáza projevuje zvýšením sérové aktivity enzymů alkalické fosfatázy (ALP), γ-glutamyltransferázy (GGT), dále hypercholesterolémií, zvýšením hladiny solí žlučových kyselin a případně vzestupem bilirubinu v séru. Tento stav označujeme jako biochemická cholestáza. Jako morfologickou cholestázu označujeme soubor strukturálních změn v postižené jaterní tkáni, a to jak na úrovni makroskopické, tak na úrovni světelně-mikroskopické a případně subcelulární, elektron-mikroskopické (viz níže) (5). Mezi morfologickým nálezem a klinickými či biochemickými projevy cholestázy může existovat diskrepance. Platí to zejména pro časná stadia biliárních onemocnění či pro stavy spojené pouze s částečnou obstrukcí žlučových cest.
V těchto případech mohou pacienti vykazovat biochemické známky cholestázy, nicméně na morfologické úrovni může být nález poměrně diskrétní. Někdy naopak morfologická změna předchází rozvoji biochemických změn, případně histopatologické známky cholestázy přetrvávají delší dobu po odeznění cholestatické ataky (6).
Diagnostika poruch extrahepatálních a větších intrahepatálních žlučovodů je primárně doménou radiodiagnostických metod, jako jsou magnetická rezonance (MRCP) či endoskopická cholangiopankreatografie (ERCP). Histopatologické vyšetření jaterní tkáně získané perkutánní nebo transjugulární biopsií obvykle poskytuje jen nepřímé informace o existenci poruchy žlučové drenáže. Naopak u cholangiopatií postihujících menší intrahepatální žlučovody zůstává jaterní biopsie jedním z významných diagnostických nástrojů.
KLASIFIKACE CHOLANGIOPATIÍ
Nemoci žlučového stromu lze klasifikovat z několika hledisek, a to zejména dle anatomických aspektů, etiologie, patogeneze či dle dominujících morfologických znaků (4,6).
Z anatomického hlediska je nejjednodušší dělení chorobných procesů na ty, které postihují především extrahepatální a větší intrahepatální (hilové, lobární) žlučovody, a na cholangiopatie převážně malých intrahepatálních (interlobulárních, septálních) žlučovodů. Obě kategorie se však často prolínají a u celé řady jednotek nacházíme patologické změny na žlučovodech malých i velkých kalibrů.
Na základě etiopatogeneze lze cholangiopatie dělit na imunitně zprostředkované, vyvolané chemickými látkami (včetně léků a léčivých přípravků), ischemické, infekční, geneticky podmíněné a v širším slova smyslu i neoplastické. Tab. 1 poskytuje základní, byť zdaleka ne vyčerpávající přehled nejčastějších patologických procesů postihujících velké a malé žlučovody (4,6,7).
Z morfologického hlediska rozdělujeme nemoci žlučovodů do dvou velkých kategorií, a to na zánětlivé procesy (cholangitidy) a nezánětlivé cholangiopatie. Cholangitidy pak lze dále dělit podle převažujícího složení zánětlivého infiltrátu a případné přidružené fibrózy na cholangitidy neutrofilní, lymfocytární, pleomorfní, granulomatózní či fibrotizující (6,8).
OBECNÉ MORFOLOGICKÉ ASPEKTY CHOLANGIOPATIÍ VELKÝCH ŽLUČOVODŮ
Jak již bylo uvedeno v předchozím textu, diagnostika cholangiopatií postihujících žlučovody velkých kalibrů je doménou především zobrazovacích vyšetření (7). Histologické vyšetření jaterní tkáně získané perkutánní či transjugulární jaterní biopsií zpravidla nezastihne vlastní patologický proces a obvykle prokáže jen morfologické následky narušené drenáže žluče, případně upozorní na možné komplikace a přidružené chorobné procesy (9).
Iniciální morfologické změny mohou být u chorobných procesů postihujících žlučovody větších kalibrů poměrně nenápadné. Obvykle se jedná o známky cholestázy, která bývá akcentována v zóně 3 jaterního acinu, kolem centrální vény (obr. 1A). Akutní cholestáza se manifestuje ve formě kapének nazelenalé barvy v cytoplazmě hepatocytů (intracelulární bilirubinostáza), případně i v kanalikulech (hepatokanalikulární bilirubinostáza). Hepatocyty bývají zvětšené, se světlou cytoplazmou. Při delším trvání nacházíme i tzv. cholestatické či biliární rozety, struktury tvořené 3 a více hepatocyty koncentricky uspořádanými kolem dilatovaného žlučového kanálku (obr. 1B). V průběhu cholestázy rovněž dochází k extravazaci žluče do sinusoidálního prostoru, kde je fagocytována makrofágy (pigmentofágy). Tyto makrofágy bývají pozitivní v PAS reakci a často přetrvávají v jaterní tkáni delší dobu i po úplném odeznění cholestatické epizody. Lytická nekróza skupiny cholestatických hepatocytů spolu s lokálním nahromaděním žluče a pěnitých makrofágů pak vede ke vzniku biliárního infarktu (obr. 2). Velké žlučové infarkty jsou obecně považovány za diagnostický znak extrahepatální obstrukce, ačkoliv menší ložiska biliární nekrózy se mohou vyskytnout i u některých forem těžké intrahepatální cholestázy (9).

Tab. 1. Přehled nejčastějších patologických procesů postihujících velké a malé žlučovody.
|
Kategorie |
Nemoci postihující velké žlučovody |
Nemoci postihující malé žlučovody |
|
Imunitně zprostředkované |
Primární sklerozující cholangitida, IgG4-asociovaná cholangitida, sarkoidóza |
Primární biliární cholangitida, překryvné syndromy s autoimunní hepatitidou, primární sklerozující cholangitida malých žlučovodů, rejekční cholangiopatie, reakce štěpu proti hostiteli |
|
Infekční |
Bakteriální cholangitida, parazitární infekce, virové infekce (HIV, Covid 19) |
Virová hepatitida s postižením malých žlučovodů |
|
Genetické |
Cystická fibróza, biliární atrézie |
Alagilleův syndrom, fibropolycystická choroba, deficit ABCB4/ MDR3 |
|
Léky a toxiny |
Léky a toxiny indukovaná cholangitida velkých žlučovodů |
Duktopenie indukovaná léky, cholangitis malých žlučovodů |
|
Ischemické |
Ischemická cholangiopatie, sekundární sklerozující cholangiopatie kriticky nemocných |
Ischemické poškození malých žlučovodů (např. při trombofilních stavech) |
|
Neoplastické a paraneoplastické |
Cholangiokarcinom, intraduktální papilární neoplazie, metastázy, hematologická onemocnění |
Cholangiokarcinom malých žlučovodů, paraneoplastická duktopenie |

Při delším trvání poruchy žlučové drenáže se často objevují i morfologické známky stázy cholátů, při které dochází k poškození jaterní tkáně především akumulací bezbarvých solí žlučových kyselin a rovněž k rozvoji fibrózy. Na mikroskopické úrovni je stáza cholátů vyjádřena nejvíce v periportálních hepatocytech (zóna 1 jaterního acinu), které se mění ve velké buňky se světlou cytoplazmou (tzv. péřovitá degenerace, anglicky feathery degeneration), v níž můžeme prokázat depozita mědi, metaloproteinu (měď vázajícího proteinu) a rovněž inkluze Malloryho-Denkova hyalinu (obr. 3). K vizualizaci metaloproteinu lze použít barvení orceinem či Schmorlovou reakcí, depozita mědi lze barvit rhodaninem (9). Schmorlova reakce však není specifická pouze pro metaloproteiny – pozitivní barvení vykazuje také lipofuscin, který se v játrech ukládá především v zóně 3 jako perinukleární granulární depozita. Na rozdíl od metaloproteinu se lipofuscin vyznačuje silnou autofluorescencí a je patrný již v barvení hematoxylinem-eozinem jako žlutohnědý pigment (10-12).


Malloryho-Denkovy hyalinní inkluze odpovídají abnormálně poskládaným degradovaným proteinům buněčného cytoskeletu, zejména intermediárním filamentům, a jsou obvykle identifikovatelné již v běžném barvení hematoxylinem a eozinem, kdy se jeví jako růžová hrudkovitá depozita na pozadí světlé cytoplazmy jaterních buněk (obr. 3). V případě nejistoty pak můžeme k jejich potvrzení použít některé z imunohistochemických metod (primární protilátka proti ubiquitinu či některým cytokeratinům, např. CK8/18) (9).
Při protrahovaných cholestatických procesech se rovněž setkáváme s tzv. cholangiocelulární metaplazií hepatocytů, při které hepatocyty částečně mění svůj fenotyp a exprimují cytokeratiny typické pro biliární epitelie, například cytokeratin 7 (obr. 4) (8,9).


Současně se změnami v lobulech dochází u poruch žlučové drenáže způsobené patologií v oblasti velkých a středně velkých žlučovodů i ke změnám v portálních polích. Ta jsou v akutní fázi zvětšená, edematózní a často prostoupená řídkou smíšeně zánětlivou celulizací se zastoupením neutrofilních leukocytů. Poměrně záhy rovněž dochází na rozhraní portálního pole a přilehlého periportálního parenchymu ke změnám, které souhrnně označujeme jako biliární interface aktivita. Tento pojem zahrnuje periportální duktulární proliferaci provázenou řídkou neutrofilní celulizací (tzv. neutrofilní cholangiolitida), ke které se přidávají morfologické známky stázy cholátů (obr. 5). Duktulární proliferace a doprovodná zánětlivá odpověď narušují limitující ploténku a hrají významnou roli při vzniku a progresi biliární fibrózy (4,6,9). Při déletrvající poruše žlučové drenáže se portální pole postupně cípatě rozšiřují a dochází k tvorbě inkompletních a později i kompletních porto-portálních vazivových sept. V extrémním případě pak může dojít až k rozvoji tzv. biliární cirhózy, kdy vazivová septa obklopují regeneráty jaterního parenchymu nepravidelného tvaru, což na makroskopické i mikroskopické úrovni vytváří dojem skládačky puzzle (obr. 6) (13,14).
U chronických biliárních procesů je fibrogeneze vyjádřena v jaterním parenchymu poměrně nerovnoměrně (9,14). Na tento fakt je třeba brát zřetel při interpretaci změn v limitovaném vzorku získaném jaterní biopsií a histopatologický nález je vhodné vždy korelovat s výsledky neinvazivních zobrazovacích metod, jako je elastografie či Fibroscan.

Tab. 2. Přehled nejčastějších příčin syndromu mizejících žlučovodů v závislosti na věku.
|
Věk |
Kategorie |
Příklad onemocnění |
|
Dětský věk |
Atrézie intra či extrahepatálních žlučovodů |
Alagilleův syndrom, biliární atrézie |
|
Familiární intrahepatální cholestáza s cholangiopatií |
Deficit ABCB4/MDR3, neonatální sklerozující cholangitida |
|
|
Fibropolycystická onemocnění (ciliopatie) |
Autozomálně dominantní a autozomálně recesivní polycystické onemocnění ledvin a jater, kongenitální jaterní fibróza |
|
|
Primárně necholestatická multisystémová onemocnění |
Deficit α1-antitrypsinu |
|
|
Dospělý věk |
Imunitně zprostředkovaná onemocnění |
Primární biliární cholangitis, primární sklerozující cholangitis, chronická rejekce jaterního štěpu, reakce štěpu proti hostiteli |
|
Cévní onemocnění |
Ischemická cholangiopatie, portální biliopatie |
|
|
Infekční onemocnění |
Virová infekce (CMV), kryptosporidióza |
|
|
Poškození léky nebo toxiny |
Antibiotika, nesteroidní antirevmatika, antikonvulzíva, antidepresiva, anabolické steroidy, antineoplastická terapie, biologika |
|
|
Nádorová onemocnění a paraneoplastické procesy |
Hodgkinův lymfom (paraneoplazie), histiocytóza z Langerhansových buněk, systémová mastocytóza |
|
|
Jiné/idiopatické příčiny |
Idiopatická duktopenie dospělých, sarkoidóza |
CHOLANGIOPATIE MALÝCH ŽLUČOVODŮ
Podobně jako v případě onemocnění žlučovodů větších kalibrů, i u cholangiopatií postihujících malé žlučovody bývají v počátečních stadiích změny často vyjádřeny pouze ložiskově. Tato skutečnost klade velký důraz na reprezentativnost vyšetřovaného vzorku. V ideálním případě by jaterní biopsie měla mít délku alespoň 15 mm a měla by obsahovat minimálně 10 portálních polí (15).
Kromě obecných známek cholestázy popsaných v předchozím textu, lze u cholangiopatií malých žlučovodů identifikovat patognomické strukturální změny přímo na žlučovodech zastižitelných jaterní biopsií. Může se jednat o zánět malého žlučovodu, degenerativní, případně senescentní změny biliárních epitelií a v extrémním případě může jít až o úplnou destrukci a zánik vývodu (8,9). Jako duktopenie neboli syndrom mizejících žlučovodů je označován patologický stav charakterizovaný zánikem žlučovodu v minimálně 50 % portálních polí. Tento závažný syndrom, se kterým se setkáváme u dětí i dospělých pacientů, může být důsledkem celé řady různých etiologických faktorů (tab. 2) (9,16,17).
MORFOLOGICKÉ CHARAKTERISTIKY KLINICKY NEJVÝZNAMNĚJŠÍCH CHOLANGIOPATIÍ
V následujícím textu bude podrobněji popsáno několik klinicko-patologických jednotek, s nimiž se patolog v rutinní praxi setkává nejčastěji. Vzácnější cholangiopatie budou zmíněny pouze okrajově či v rámci diferenciální diagnózy.
IMUNITNĚ PODMÍNĚNÉ CHOLANGIOPATIE
Primární sklerozující cholangitida
Primární sklerozující cholangitida (PSC) je chronické cholestatické onemocnění charakterizované zánětem, progresivní fibrózou a postupným zánikem extrahepatálních i intrahepatálních žlučovodů, spojené s výrazným rizikem rozvoje biliární cirhózy, selhání jater a cholangiokarcinomu (18,19).
Onemocnění je častější u mužů, s poměrem výskytu mezi pohlavími přibližně 2 : 1. První příznaky se obvykle objevují mezi 30. a 40. rokem života. Více než polovina pacientů s PSC má také přidružené onemocnění, především idiopatický střevní zánět (PSC-IBD) (20-22).
PSC může zpočátku probíhat asymptomaticky a bývá diagnostikována při náhodném záchytu zvýšených sérových hodnot cholestatických jaterních enzymů ALP a GGT. U symptomatických pacientů dominuje únava, svědění kůže a epizody akutní cholangitidy s bolestmi v pravém podžebří, horečkou a žloutenkou (18).
Pro diagnostiku onemocnění, zejména klasických forem s postižením velkých žlučovodů, mají zásadní význam zobrazovací vyšetření. Typickým nálezem na MRCP či ERCP je obraz „růžence“ s nepravidelným střídáním stenotických a dilatovaných úseků na žlučovodech větších kalibrů (23-25). Přibližně v 5-10 % případů je však onemocnění limitováno na žlučovody, které jsou příliš malé na to, aby mohly být zachytitelné cholangiograficky. U této formy označované jako PSC malých žlučovodů (small-duct PSC, sdPSC) se diagnostika z velké části opírá o nález v jaterní punkci (23,24).
Kromě zobrazovacích metod se v diagnostice onemocnění mohou uplatnit i sérologická vyšetření. Detekovatelné autoprotilátky se nacházejí až u 97 % pacientů s PSC. Nejčastěji se jedná o protilátky proti hladkému svalstvu (ASMA) a antinukleární protilátky (ANA). Perinukleární antineutrofilní cytoplazmatické protilátky (pANCA) a anti-P-40 protilátky bývají zastiženy přibližně u 30–80 % pacientů s PSC-IBD (19,25).
Histopatologický obraz PSC
U klasických forem PSC jsou patognomické morfologické změny přítomny zejména na žlučovodech velkých a středních kalibrů. Typickým nálezem je tzv. fibro-obliterativní léze, koncentrická („cibulovitá“) periduktální fibróza, zpočátku řídká a provázená různě intenzivní zánětlivou celulizací. S postupem času fibróza progreduje, vazivo se stává denzním a dochází k segmentálnímu zúžení, a nakonec až k úplné obliteraci lumen žlučovodu. V místě zaniklého vývodu zůstává vazivová jizva (9,24,25).
Při histologickém vyšetření periferního jaterního parenchymu získaného jaterní punkcí se u pacientů s PSC obvykle zastihnou nespecifické změny odpovídající poruše žlučové drenáže s cholestázou (viz výše). Vzácněji jsou přítomny fibro-obliterativní léze (obr. 7) a jizvení s postupným zánikem malých žlučovodů, který může vyústit až do obrazu duktopenie. Tento stav je provázen těžkou cholestázou a zpravidla i pokročilou fibrózou s tvorbou porto-portálních vazivových sept a v konečné fázi s rozvojem biliární cirhózy (8,9,24).
V rámci morfologické diferenciální diagnózy PSC připadají v úvahu zejména primární biliární cholangitida se zánětlivě granulomatózní destrukcí malých žlučovodů, dále IgG4-asociovaná cholangiopatie, pro niž je kromě jizevnatého zániku žlučovodů typická i přítomnost IgG4-pozitivních plazmatických buněk, známky storiformní fibrózy a obliterativní flebitidy. Srovnání základních klinických, laboratorních a morfologických znaků těchto onemocnění uvádí tab. 3 (9,26-28).

Poměrně obtížné může být odlišení PSC od sekundárních forem sklerozující cholangitidy vzniklé na ischemickém podkladě, případně od některých léky indukovaných cholangiopatií. V těchto případech nám ke správné interpretaci změn mohou pomoci zobrazovací vyšetření jaterní vaskulatury a rovněž klinické informace či farmakologická anamnéza (26,27).
Do širší morfologické diferenciální diagnózy je rovněž nutné zahrnout Wilsonovu chorobu, která může vést k pokročilé fibróze či cirhotické remodelaci jaterního parenchymu, doprovázené variabilně vyjádřenými metabolickými a zánětlivými změnami a rovněž známkami stázy cholátů s hojnými intracelulárními depozity metaloproteinu a mědi (29).
Primární biliární cholangitida
Primární biliární cholangitida (PBC), dříve známá jako primární biliární cirhóza, je prototypem imunitně podmíněné cholangiopatie, která selektivně postihuje malé intrahepatální žlučovody. PBC se častěji vyskytuje u žen ve středním věku a bývá asociována s dalšími autoimunitními chorobami, jako je Sjögrenův syndrom, vitiligo či autoimunitní tyreoiditida (30-33). PBC se klinicky projevuje především pruritem, který může předcházet diagnostickým laboratorním změnám, a rovněž i hyperpigmentací kůže či přítomností xantomů. V laboratorním nálezu obvykle nacházíme zvýšenou aktivitu sérové ALP a GGT, dále hypercholesterolémii, hypergamaglobulinémii, zejména zvýšení IgM, a typická je rovněž pozitivita antimitochondriálních protilátek (AMA) typu M2 (32,33).
Histopatologický obraz PBC
Časné morfologické projevy PBC bývají v jaterní tkáni vyjádřeny často pouze ložiskově a je třeba po nich cíleně pátrat. V iniciálních stadiích onemocnění nacházíme portálně vázaný lymfoplazmocelulární zánětlivý infiltrát, někdy denzní a uspořádaný do periduktálních lymfatických agregátů či folikulů, často s menší či větší příměsí eozinofilních leukocytů. Zánětlivý infiltrát typicky postihuje pouze některá portální pole, jiná mohou být zcela bez zánětu či s minimální zánětlivou celulizací. Malé portální žlučovody vykazují známky lymfocytární cholangitidy, posléze dochází k rupturám bazální membrány a k rozvoji periduktální granulomatózní reakce (tzv. floridní duktální léze) (obr. 8). Zánětlivá destrukce žlučovodů může, obdobně jako v případě PSC, v konečném důsledku vést až k rozvoji duktopenie (9,34,35). Je třeba však upozornit, že duktopenie s těžkou cholestázou se u některých pacientů s PBC může rozvinou ještě před vznikem pokročilé fibrózy či cirhózy (9,35). Kromě duktopenie mohou někteří pacienti již v precirhotickém stadiu onemocnění vyvinout známky nodulární regenerativní hyperplazie s následnou portální hypertenzí (36,37).
Tab. 3. Srovnání klíčových klinických, laboratorních a radiologických rysů AIH, PBC, PSC a IgG4 sklerozující cholangitidy.
|
Kategorie |
AIH |
PBC |
PSC |
IgG4-SC |
|
Demografie |
Převaha žen (4 : 1), všechny věkové skupiny |
Převaha žen (9 : 1), obvykle >50 let |
Převaha mužů (7 : 3); typicky 40 let, často spojena s IBD |
Převaha mužů (4 : 1); dospělí, typicky >40 let |
|
Příznaky |
Asymptomatická, případně: ikterus, bolest v pravém podžebří, únava |
Běžně asymptomatická, symptomy: svědění, únava, ikterus, xantomy |
Běžně asymptomatická, symptomy: svědění, bolest břicha, ikterus |
Nebolestivý ikterus |
|
Jaterní testy |
Zvýšené ALT/AST |
Zvýšené ALP, GGT |
Zvýšené ALP, GGT |
Zvýšené ALP, GGT |
|
Dominantní imunoglobulin |
IgG |
IgM |
Mírné zvýšení IgG nebo IgM |
IgG4 |
|
Autoprotilátky |
ANA, ASMA (typ 1 AIH), LKM1 (typ 2 AIH) |
ANA, AMA |
Bez specifických asociací, často ANA/SMA pozitivita |
Bez specifických asociací, ANA může být pozitivní |
|
Cholangiografie |
Normální |
Normální |
Stenózy, obraz „růžence“ |
Striktury, nepravidelný vzhled |
|
Histologie jater |
Interface hepatitida, plazmatické buňky |
Floridní léze žlučovodů, lymfocytární cholangitida |
Koncentrická periduktální fibróza |
Periportální zánět, zvýšený počet IgG4+ buněk |
AIH – autoimunní hepatitida; PBC – primární biliární cholangitida; PSC primární sklerozující cholangitida; IgG4-SC – IgG4 asociovaná sklerozující cholangitida; IBD – idiopatický střevní zánět; ALT – alaninaminotransferáza; AST aspartátaminotransferáza; ALP – alkalická fosfatáza; GGT – gama-glutamyltransferáza; ANA – antinukleární protilátky; ASMA – protilátky proti hladkému svalu; LKM1 protilátky proti jaternímu a renálnímu mikrosomu typu 1; AMA – antimitochondriální protilátky


Změny malých žlučovodů bývají konstantně provázeny histopatologickými známkami chronické cholestázy. Již v časných stadiích lze detektovat depozita metaloproteinu a mědi v periportálních hepatocytech. Zpočátku je nález velmi diskrétní, s postupem choroby se ale cholestatické změny stávají výraznější. V pokročilých stadiích onemocnění pak často nacházíme nápadné světlé lemy kolem fibrózně rozšířených portálních polí a vazivových sept, což odpovídá známkám vystupňované stázy cholátů (obr. 9) (8,9).
Histopatologické vyšetření má význam nejen pro vlastní diagnostiku PBC, ale rovněž přináší informaci o stupni fibrotizace jaterní tkáně a o stadiu patologického procesu.
Tradiční klasifikace podle Scheuera a Ludwiga rozděluje onemocnění do čtyř stadií na základě charakteristických mikroskopických změn (tab. 4) (38,39).
Vedle těchto tradičních jednoduchých a dobře reprodukovatelných systémů byl Nakanumou a kol. navržen klasifikační systém, který je složitější, avšak přesněji odráží komplexní patogenezi onemocnění. Při hodnocení stadia choroby (stagingu) zohledňuje tento nový systém tři klíčové parametry, a to fibrózu jaterního parenchymu, zánik žlučovodů a tíži cholestázy (tab. 5) (40,41).
Tab. 4. Histopatologická stadia PBC dle Scheuera a Ludwiga.
|
Stadium |
Popis |
Typické histologické znaky |
|
1 |
Fokální destruktivní lymfocytární a/nebo granulomatózní cholangitida interlobulárních žlučovodů („floridní duktální léze“) |
Zánět nepřesahuje hranice portálního traktu |
|
2 |
Pokračující poškození žlučovodů s expanzí portálního pole |
Periportální duktulární proliferace, zánětlivá interface aktivita, periportální fibróza |
|
3 |
Přemosťující septální (porto-portální) fibróza |
Zánik malých žlučovodů, postupující fibróza |
|
4 |
Cirhóza s duktopenií a chronickou cholestázou |
Kompletní strukturální přestavba jaterní tkáně |
Tab. 5. Staging primární biliární cholangitidy podle Nakanumy.
|
Kategorie |
Skóre |
Popis |
|
Fibróza |
0 |
Žádná nebo minimální fibróza |
|
1 |
Periportální fibróza, občasná nekompletní septa |
|
|
2 |
Přemosťující septální fibróza s narušením lobulární architektoniky |
|
|
3 |
Cirhóza |
|
|
Zánik žlučovodů |
0 |
Žádný zánik žlučovodů |
|
1 |
Zánik žlučovodů v <1/3 portálních polí |
|
|
2 |
Zánik žlučovodů v 1/3-2/3 portálních polí |
|
|
3 |
Zánik žlučovodů ve >2/3 portálních polí |
|
|
Cholestáza (orcein pozitivní granulární depozita) |
0 |
Žádná depozita |
|
1 |
Periportální depozita v <1/3 portálních polí |
|
|
2 |
Periportální depozita v 1/3-2/3 portálních polí |
|
|
3 |
Periportální depozita ve >2/3 portálních polí |
Kromě stadia onemocnění se systém navržený Nakanumou zabývá i hodnocením a kvantifikací zánětlivé aktivity, tzv. gradingem (tab. 6). Zejména časnější stadia PBC bývají kromě výše popsaného ložiskového portálního zánětu s alterací malých žlučovodů provázena i různě významnou zánětlivou aktivitou na porto-lobulárním rozhraní (tzv. zánětlivou interface aktivitou, dříve označovanou rovněž jako zánětlivá piecemeal nekróza) a rovněž nekroinflamatorními změnami v lobulech. Tato tzv. zánětlivá forma PBC s hepatitickými rysy může být mylně interpretována jako překryvný (overlap) syndrom s autoimunní hepatitidou (AIH) (28,42-44). Skutečný překryvný syndrom PBC a AIH, vyžadující obvykle kromě terapie kyselinou ursodeoxycholovou i nasazení kortikoterapie, je daleko vzácnější a měl by splňovat kritéria navržená Mezinárodní skupinou pro autoimunitní hepatitidu (28,44). Tato kritéria zahrnují přítomnost alespoň dvou ze tří diagnostických znaků PBC (1–3) a alespoň dvou ze tří diagnostických znaků AIH (4–6):
- GGT ≥ 5× horní hranice normy nebo ALP ≥ 2× horní hranice normy;
- pozitivní antimitochondriální protilátky (AMA);
- floridní léze žlučovodů v histologickém obraze;
- zvýšené hodnoty alaninaminotransferázy (ALT) ≥ 5× horní hranice normy;
- sérové hladiny IgG ≥ 2× horní hranice normy nebo pozitivní protilátky proti hladkému svalstvu (SMA);
- středně těžká nebo těžká lymfocytární interface aktivita v histologickém obraze.
Kromě překryvného syndromu zahrnuje morfologická diferenciální diagnóza PBC celou řadu dalších jednotek a stavů s granulomatózní komponentou. Jedná se zejména o některá infekční onemocnění, například mykobakteriální či mykotické infekce, případně některé antropozoonózy, či léky indukovaná poškození jater. V těchto případech přispívá ke správné interpretaci nálezu v jaterní biopsii znalost výsledků mikrobiologických vyšetření a podrobná farmakologická anamnéza. Přítomnost nekaseifikujících granulomů je rovněž typická pro sarkoidózu (viz níže). Na rozdíl od PBC bývají granulomy u sarkoidózy zpravidla dobře formované a nacházíme je i mimo portální oblasti, často ve vazbě na cévní struktury. Diagnózu PBC navíc podporují sérologické nálezy (přítomnost AMA a elevace IgM), zatímco hepatobiliární sarkoidóza bývá součástí multisystémového onemocnění, provázeného zvýšenou hladinou sérového angiotenzin-konvertujícího enzymu (ACE) (8,9).
Sklerozující cholangitida asociovaná s IgG4
S IgG4 asociovaná sklerozující cholangitida je biliárním projevem poměrně vzácného systémového onemocnění, charakterizovaného zvýšenými sérovými koncentracemi IgG4 a infiltrací tkání IgG4-pozitivními plazmatickými buňkami, to vše na pozadí storiformní fibrózy a vaskulárních změn (45-49). Kromě léze žlučovodů mohou pacienti v rámci hepatobiliárního IgG4-sklerozujícího onemocnění vyvinout rovněž známky IgG4-asociované AIH či tzv. zánětlivý pseudotumor (49). Postižení žlučovodů může být provázeno projevy autoimunní pankreatitidy 1. typu, případně postižením celé řady dalších orgánů či orgánových systémů. Sklerozující cholangitida asociovaná s IgG4 typicky postihuje extrahepatální a hilové žlučovody, ale u 1/3 případů nacházíme rovněž změny na malých intrahepatálních žlučovodech (48-50).
Histopatologický obraz sklerozující cholangitidy asociované s IgG4
Pro IgG4-asociovanou cholangitidu byla na základě konsenzu doporučena následující histomorfologická kritéria (obr. 10) (50,51):
- v resekátech by mělo být přítomno více než 50 IgG4-pozitivních buněk na jedno zorné pole velkého zvětšení a v biopsiích více než 10 IgG4-pozitivních buněk na jedno zorné pole velkého zvětšení
- poměr IgG4/IgG-pozitivních plazmatických buněk by měl překročit 40 %.
IgG4-asociovaná cholangitida se stává vysoce suspektní, pokud jsou navíc splněny alespoň dvě z následujících tří podmínek:
- výrazná lymfoplazmocytární infiltrace;
- storiformní fibróza;
- obliterativní flebitida.
Tab. 6. Grading primární biliární cholangitidy podle Nakanumy.
|
Grading |
Skóre |
Popis |
|
Aktivita chronické cholangitidy (CA) |
CA 0 |
Bez významné lymfocytární infiltrace kolem žlučovodů |
|
CA 1 |
Známky chronické cholangitidy min. v jednom portálním poli |
|
|
CA 2 |
Známky chronické cholangitidy dvou a více žlučovodů |
|
|
CA 3 |
Minimálně jedno ložisko zánětlivé destrukce žlučovodu |
|
|
Aktivita hepatitidy (HA) |
HA 0 |
Bez známek interface hepatitidy, žádná nebo minimální lobulární hepatitida |
|
HA 1 |
Fokální interface hepatitida postihující 10 sousedících hepatocytů v jednom portálním poli a mírná až středně těžká lobulární hepatitida |
|
|
HA 2 |
Interface hepatitida postihující 10 hepatocytů ve dvou nebo více portálních polích a mírná až střední lobulární hepatitida |
|
|
HA 3 |
Interface hepatitida postihující alespoň 20 hepatocytů ve ≥50 % portálních polí, střední až výrazná lobulární hepatitida nebo přemosťující nekróza |

Součástí morfologického obrazu onemocnění jsou rovněž konstantně známky cholestázy se stázou cholátů (48,49).
Zvýšený počet IgG4-pozitivních plazmocytů může být přítomen i v rámci jiných zánětlivých procesů, které nesouvisejí s IgG4-asociovaným onemocněním. Typickým příkladem je plazmocelulární typ rejekce jaterního štěpu či zvýšená koncentrace IgG4-pozitivních plazmocytů v místech chronického hnisání, což je stav, se kterým se nezřídka setkáváme u pacientů s PSC. V těchto případech však obvykle nebývá splněno kritérium minimálního poměru IgG4-pozitivních buněk k celkovému počtu plazmocytů. Navíc u těchto onemocnění nebývá přítomna storiformní fibróza a zpravidla chybí i vaskulární léze (51,52).
Rejekční cholangiopatie a reakce štěpu proti hostiteli
Do spektra imunitně podmíněných cholangiopatií postihujících malé žlučovody patří také rejekční cholangiopatie a cholangiopatie v rámci reakce štěpu proti hostiteli (GVHD).
Biliární léze při akutní rejekci zprostředkované T lymfocyty se vyznačují intraepiteliální lymfocytární infiltrací, degenerativními změnami cholangiocytů a případně rupturami bazální membrány vývodů, což bývá doprovázeno známkami významné akutní cholestázy (obr. 11).
Chronická (duktopenická) rejekce je charakterizována tzv. senescentními změnami biliárních epitelií, projevujícími se eozinofilní transformací cytoplazmy a jadernou hyperchromázií s narušením buněčné polarity, a dále progresivní ztrátou interlobulárních žlučovodů vedoucí až k obrazu duktopenie (obr. 12). Výše popsané změny žlučovodů bývají u akutní i chronické rejekce provázeny i dalšími morfologickými projevy, a to zejména portální a centrolobulární rejekční celulizací a zánětem mikrovaskulatury (53-56).
U GVHD, která je závažnou komplikací allogenní transplantace kostní dřeně či kmenových buněk, se imunitně zprostředkované poškození malých žlučovodů projevuje obdobně jako u rejekce dystrofickými změnami biliární výstelky a jejich postupnou destrukcí. Tyto změny bývají zpravidla provázeny těžkou cholestázou, zánětlivá celulizace je však minimální (61,62).
Hepatobiliární projevy sarkoidózy
Sarkoidóza je systémové granulomatózní onemocnění, které může postihnout játra a žlučové cesty, a to buď izolovaně, nebo jako součást multisystémového postižení (59-61). Etiologie sarkoidózy dosud nebyla plně objasněna, předpokládá se však významná spoluúčast imunopatologických procesů (62,63).
Z morfologického hlediska je charakteristickým znakem sarkoidózy přítomnost dobře formovaných nekaseifikujících epiteloidních granulomů, které nacházíme nejen v portálních prostorech, ale často i v lobulech, obvykle ve vazbě na cévní struktury. Granulomy jsou tvořeny jednojadernými epiteloidními histiocyty s variabilní účastí obrovských mnohojaderných buněk Langhansova typu (obr. 13). Někdy vídáme splývání granulomů do větších celků, což bývá provázeno denzní fibrózou a jizvením přilehlých tkání a struktur. Tato fibrotizace jaterního parenchymu bývá u sarkoidózy vyjádřena značně nerovnoměrně a nález v jaterní biopsii je třeba vždy korelovat i s výsledky zobrazovacích vyšetření. Postižení žlučových cest s destrukcí interlobulárních žlučovodů může i u sarkoidózy dospět do stadia duktopenie (9,64,65).
Diferenciální diagnóza hepatobiliární sarkoidózy je poměrně široká a v první řadě zahrnuje infekční granulomatózní procesy, zejména mykobakteriální a mykotické infekce, případně některé antropozoonózy, dále granulomatózní autoimunitní onemocnění (PBC), léky indukované změny, granulomatózní reakce asociované s nádorovým procesem či cizím tělesem (65-67).




Nedílnou součástí histopatologického vyšetření hepatobiliárních granulomatózních procesů je, stejně jako i u jiných orgánů, vyšetření v polarizovaném světle a rovněž podrobné bakterioskopické vyšetření. K tomu slouží zejména rutinní barvení dle Ziehl-Neelsena, PAS reakce a stříbření dle Grocotta, případně lze doplnit průkaz patogenů imunohistochemickými technikami a molekulárně-genetickými metodami. Mikroskopický nález je rovněž třeba korelovat s klinickými údaji a s výsledky dalších laboratorních a zobrazovacích vyšetření, zaměřených zejména na oblast plic a mediastina (9,65).
CHOLANGIOPATIE INDUKOVANÉ LÉKY A TOXINY
Lékové poškození jater (drug induced liver injury, DILI) představuje klinicky významnou a značně heterogenní skupinu akutních i chronických poruch, které mohou věrně napodobovat primární cholangiopatie typu PBC či PSC. Mezi dobře zdokumentované následky expozice lékům a toxinům patří progresivní ztráta malých žlučovodů (duktopenie), dále cholestatická fibróza, případně až cirhóza s biliárními rysy (68-71). Některé léky selektivně poškozují velké žlučovody, avšak většina nežádoucích lékových reakcí se manifestuje pod obrazem postižení malých žlučovodů (71). Nejčastějšími léky spojenými s tímto typem poškození jsou neuroleptika, tricyklická antidepresiva, antikonvulziva, hormonální preparáty, antibiotika či nesteroidní antirevmatika (71).
V posledních letech je zvláštní pozornost věnována cholangiopatiím spojeným s inhibitory imunitních kontrolních bodů (check-point inhibitors, CPI). Tyto léky mohou způsobit cholangitidu s převahou neutrofilního nebo lymfocytárního infiltrátu, doprovázenou apoptózami a dystrofickými změnami výstelky, imitující tak některé infekční a imunitně podmíněné cholangiopatie. Dále mohou indukovat granulomatózní reakce podobné PBC nebo fibrotizující cholangitidu připomínající PSC malých žlučovodů (obr. 14). Poškození žlučovodů indukované CPI může v některých případech vést až k obrazu duktopenie (72-75). Na tuto skutečnost je třeba brát zřetel při interpretaci biliárních změn u pacientů léčených biologiky.
V některých případech je nutné zvážit i možnost genetické predispozice daného jedince k rozvoji léky indukovaných nežádoucích účinků. Klasickým příkladem mohou být hormonální preparáty, které u probandů s mutacemi v genech kódujících některé transportéry solí žlučových kyselin mohou vyvolat závažné cholestatické stavy s cholangiopatickou komponentou (76-79).
GENETICKY PODMÍNĚNÉ CHOLANGIOPATIE
Geneticky podmíněné cholangiopatie představují značně různorodou skupinu onemocnění. Postižení žlučovodů může být způsobeno poruchami embryonálního vývoje, zejména narušením procesu remodelace duktální ploténky, jak je tomu například u Caroliho choroby nebo kongenitální jaterní fibrózy, nebo dysfunkcí specifických proteinů zajišťujících transport žluči, jako u cystické fibrózy. Některé formy, například Alagillův syndrom, souvisejí s poruchou signalizačních drah klíčových pro vývoj žlučových struktur (9,80,81). Genetické vlivy hrají pravděpodobně významnou roli i u biliární atrézie (82). Různě závažná biliární léze může být dále součástí primárně necholestatických multisystémových genetických poruch, jako je cystická fibróza nebo deficit α-1 antitrypsinu (83,84).
Malformace duktální ploténky
Malformace duktální ploténky představují heterogenní soubor onemocnění vycházejících z narušené remodelace embryonální duktální ploténky, což vede k vzniku biliárních hamartomatózních lézí, segmentálních dilatací žlučovodů a cyst (obr. 15) (85,86).

Mezi klinické jednotky spojené s touto malformací patří Caroliho nemoc, která byla poprvé popsána v roce 1958 Jacquesem Carolim jako vrozená segmentální dilatace větších intrahepatálních žlučovodů. Později se ukázalo, že se toto onemocnění může kombinovat s kongenitální fibrózou jater v těchto případech se označuje jako Caroliho syndrom. Tato forma bývá provázena portální hypertenzí a mívá závažnější klinický průběh (87).
Postižení jater bývá často provázeno renálními abnormalitami, přičemž patologické změny vycházejí z genetických mutací postihujících proteiny zajišťující vývoj a funkci primárních cilií. Tato skupina onemocnění je označována jako ciliopatie (88,89). Pokroky v identifikaci příslušných genů v posledních letech výrazně přispěly k hlubšímu porozumění molekulárním mechanismům, které se na vzniku těchto poruch podílejí, a zároveň otevřely nové možnosti v oblasti diagnostiky i cílené terapie. Přehled nejčastějších jaterně-renálních ciliopatií uvádí tab. 7 (90-92).
Atrézie žlučových cest
Biliární atrézie (BA) je závažné neonatální onemocnění charakterizované progresivní destrukcí intrahepatálních a extrahepatálních žlučovodů, vedoucí k cholestáze, fibróze až cirhóze jater a k nevratnému poškození jaterních funkcí (82,93). Etiologie této poruchy není zcela objasněna, nicméně se předpokládá multifaktoriální původ zahrnující genetické predispozice, virové infekce a abnormální imunitní odpověď. Mezi možné spouštěče patří infekce viry, jako je rotavirus nebo reovirus typu 3, které mohou vyvolat imunitní reakci vedoucí k destrukci biliárního epitelu. Rovněž se diskutuje o vlivu genetických mutací a embryonální malformace, zejména v rámci syndromických forem BA (93,94).
Histopatologický obraz BA
Charakteristickým rysem onemocnění je významná redukce až úplná absence extrahepatálních žlučovodů (obr. 16). V jaterním parenchymu nacházíme v časných fázích onemocnění fibroedematózní rozšíření portálních polí s peripotální duktulární proliferací a s přítomností duktulární bilirubinostázy. Přítomny jsou i změny portální mikrovaskulatury se zmnožením arteriálních profilů a s částečnou obliterací až zánikem portální venózní větve.
Přítomen bývá smíšený zánětlivý infiltrát variabilní intenzity, který může z portálních polí přestoupit do přilehlého periportálního jaterního parenchymu. Portální změny mohou být provázeny obrovskobuněčnou (syncyciální) transformací hepatocytů a známkami lobulární cholestázy. Dalším významným znakem BA je rychlá progrese fibrózy. Cirhóza biliárního typu se u neléčených případů zpravidla rozvine do šesti měsíců a bývá provázena významnou redukcí počtu intrahepatálních žlučovodů a známkami vystupňované cholestázy s pseudoxantomatózní transformací jaterních buněk, žlučovými infarkty a s variabilní akumulací mědi a metaloproteinu (95-98).

Tab. 7. Přehled ciliopatií s hepatobiliárními projevy.
|
Porucha |
Gen |
Produkt |
|
ARPKD |
PKHD1 |
Fibrocystin/polyduktin |
|
ADPKD |
PKD1 |
Polycystin 1 |
|
PKD2 |
Polycystin 2 |
|
|
GANAB/PKD3 |
Glucosidase II alpha subunit |
|
|
PCLD |
PRKCSH |
Glucosidase II beta subunit |
|
ALG8 |
Alpha-1,3-glucosyltransferase |
|
|
SEC61B |
SEC61 translocon subunit beta |
|
|
SEC63 |
Translocon component in the ER |
|
|
LRP5 |
LDL receptor-related protein 5 |
|
|
CHF-SC |
ZFYVE19 |
Zinc finger FYVE-type containing 19 |
|
Meckelův syndrom |
TMEM67 |
Meckelin |
|
CEP290 |
Centrosomal protein 290 |
|
|
RPGRIP1L |
RPGRIP interacting protein 1-like |
ARPKD autozomálně recesivní polycystická choroba ledvin; ADPKD autozomálně dominantní polycystická choroba ledvin; PCLD – polycystická choroba jater; CHF-SC – kongenitální jaterní fibróza se sklerozující cholangitidou
Progresi onemocnění může významně zpomalit zavedení portoenterostomie dle Kasaie, při které se odstraní obliterované extrahepatální žlučovody a na oblast porta hepatis se našije část tenkého střeva, čímž se zajistí odvod žluči z jater do střevního lumen. U pacientů, u kterých tento zákrok selže nebo u nich dojde k rozvoji jaterní cirhózy, zůstává metodou volby transplantace jater (96,98).
V raných stadiích může být obtížné odlišit BA od jiných příčin neonatální cholestázy, proto je klíčová pečlivá morfologická analýza v kombinaci s klinickými a laboratorními nálezy a rovněž s výsledky zobrazovacích vyšetření (9,95,96).
Alagilleův syndrom
Alagilleův syndrom (ALGS) je autosomálně dominantní porucha s postižením celé řady orgánů a orgánových systémů. Syndrom je způsoben mutací v genech, které hrají klíčovou roli v buněčné signalizaci, nejčastěji v genu JAG1 (v 89 % případů) nebo méně často v genu NOTCH2. Vedoucími klinickými projevy jsou cholestáza obstrukčního typu, vrozené srdeční vady, oční anomálie, motýlovitý tvar obratlů a specifický obličejový fenotyp (99-101).
Histopatologické vyšetření jater zpravidla odhalí významnou redukci či úplnou absenci interlobulárních žlučovodů. Poměr interlobulárních žlučovodů k počtu portálních oblastí se u pacientů a ALGS pohybuje mezi 0,0 a 0,4, ve srovnání s 0,9–1,8 u zdravých jedinců. Redukce počtu žlučovodů bývá provázena portální zánětlivou celulizací mírného až středního stupně, obrovskobuněčnou transformací hepatocytů, známkami cholestázy a stázy cholátů a rovněž rozvojem fibrózy (obr. 17). Poměrně charakteristickým nálezem je u pacientů s ALGS chybění imunoexprese CD10 (endopeptidázy) v oblasti apikálního pólu cholangiocytů (101-103).
Cholangiopatie spojená s deficitem ABCB4
Deficit ABCB4 (MDR3) představuje klinicky významnou geneticky podmíněnou poruchu, která se projevuje poruchou sekrece fosfolipidů do žluče. Následkem je tvorba toxické žluče s detergentními účinky, což vede k narušení buněčných membrán výstelky žlučových cest a k progresivní fibróze (104,105). Mutace v ABCB4 se mohou manifestovat pod obrazem progresivní familiární intrahepatální cholestázy 3. typu (PFIC3), dále jako syndrom cholelitiázy s nízkými fosfolipidy (LPAC, rovněž Gallbladder Disease type 1), intrahepatální těhotenskou cholestázou (ITC) či jako již výše zmíněné léky indukované jaterní poškození. Do spektra poruch způsobených deficitem ABCB4 patří rovněž transientní neonatální cholestáza, fibrózní cholangiopatie malých žlučovodů a kryptogenní biliární fibróza, případně až cirhóza. Tato porucha může představovat zvýšené riziko vzniku hepatocelulárního a cholangiocelulárního karcinomu (104-107). Včasná diagnóza a terapeutická intervence, včetně použití kyseliny ursodeoxycholové, může významně zlepšit prognózu pacientů (108).
Histopatologický obraz deficitu ABCB4/MDR3
Charakteristickým mikroskopickým nálezem je u nejtěžších forem deficitu ABCB4 probíhajícího pod obrazem PFIC3 obrovskobuněčná transformace hepatocytů provázená významnou hepatokanalikulární cholestázou, dále zpravidla mírný, portálně vázaný zánětlivý infiltrát a známky pokročilejší jaterní fibrózy, případně až cirhotické přestavby. Poměrně často bývají patrny i změny na žlučovém stromu, zejména koncentrická periduktální fibróza kolem malých žlučovodů, provázená periportální duktulární proliferací. V lumen malých žlučovodů se mohou nacházet krystaly cholesterolu či drobné konkrementy (obr. 18).
U těžkých forem onemocnění projevujících se již v časném věku může k diagnóze přispět rovněž imunohistochemie prokazující sníženou či chybějící expresi proteinu MDR3 v oblasti kanalikulární membrány hepatocytů. Často však bývá exprimován nefunkční protein, což představuje významný limit pro využití imunohistochemických metod v diagnostice tohoto onemocnění, zvláště v případech s pozdní manifestací projevů (105,109).
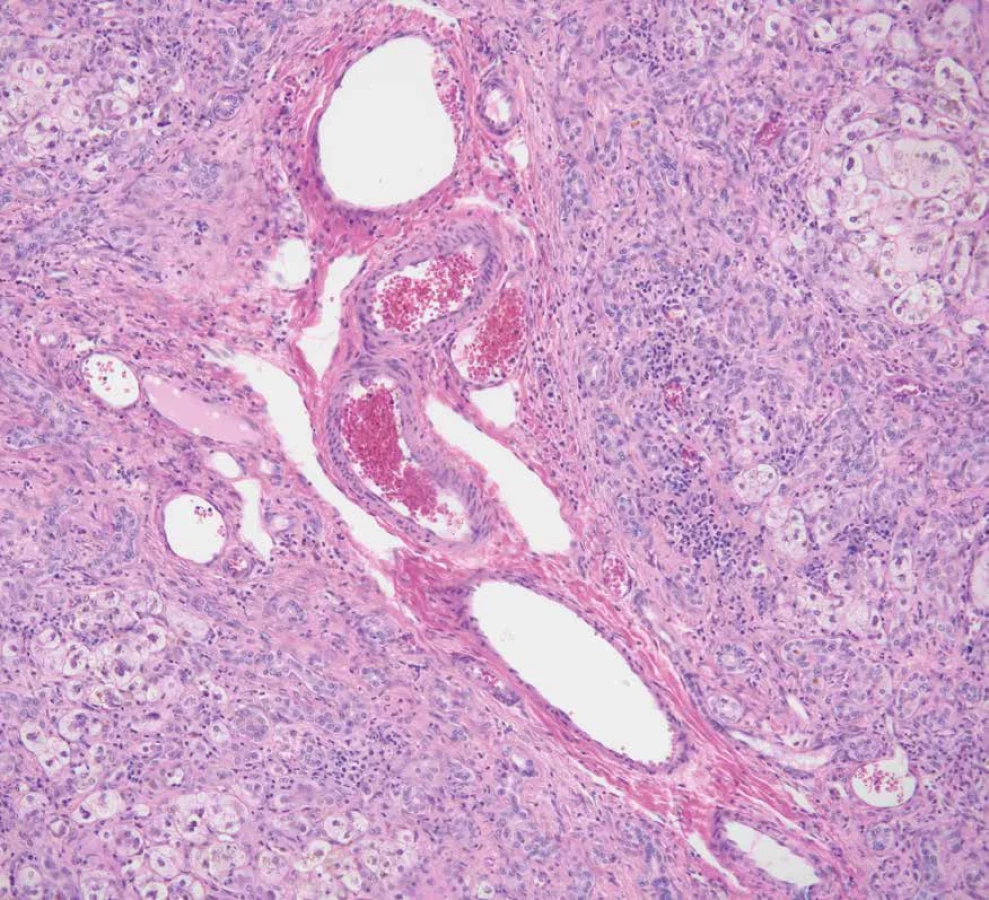

Neonatální sklerozující cholangitida
Neonatální sklerozující cholangitida (NSC) je vzácné genetické onemocnění spojené s mutacemi v genu DCDC2 kódujícím protein, který se podílí na polymerizaci mikrotubulů a zajišťuje správnou funkci primárních cilií. Klinicky se NSC manifestuje v raném novorozeneckém věku žloutenkou, hepatosplenomegalií a těžkou cholestázou, často s rychlou progresí k jaternímu selhání (110).
Histopatologické změny zahrnují koncentrickou periduktální fibrózu a destrukci malých žlučovodů. Důležitým diagnostickým nálezem je imunohistochemický průkaz absence proteinů DCDC2 a acetylovaného α-tubulinu (110,111).
INFEKČNÍ CHOLANGIOPATIE
Infekční cholangiopatie představují rozsáhlou skupinu onemocnění hepatobiliárního systému způsobených různými mikrobiálními agens. Kromě poškození žlučového stromu vlastním zánětlivým procesem hrají některé bakteriální a virové infekce klíčovou roli v progresi ischemického a imunitně zprostředkovaného poškození žlučových cest, například při GVHD nebo rejekci jaterního aloštěpu (112,113).
Akutní (ascendentní) cholangoitida
Ascendentní cholangitida je bakteriální infekce žlučových cest, která se typicky rozvíjí v důsledku poruchy žlučové drenáže, nejčastěji způsobené choledocholitiázou, benigními nebo maligními strikturami žlučovodů či přítomností biliárních stentů. Klinicky se manifestuje horečkou, bolestí v pravém podžebří a ikterem (114). Mezi nejčastější patogeny způsobující ascendentní cholangitidu patří gramnegativní a anaerobní mikroorganismy, zejména Escherichia coli, Klebsiella, Enterobacter, Pseudomonas a Citrobacter (114-116). Ve většině případů se bakterie do žlučovodů dostávají ascendentní cestou z duodena, zcela výjimečně i cestou hematogenní, z portálního řečiště. Akutní cholangitida se může rozvinout i jako iatrogenní komplikace po chirurgických či endoskopických výkonech na žlučovodech, například po ERCP (116).
Při histopatologickém vyšetření se ascendentní cholangitida vyznačuje akumulací neutrofilních leukocytů ve stěně a v lumen žlučovodů. V těžších případech může zánětlivý exsudát zcela vyplnit biliární lumen a vést k destrukci stěny vývodu s tvorbou ulcerací a nekróz. Šíření zánětu do přilehlých měkkých tkání a jaterního parenchymu vede k tvorbě abscesů, patologických dutin bez vlastní epitelové výstelky vyplněných nekrotickými hmotami a zánětlivým exsudátem s příměsí žluči.
Jaterní abscesy mohou být variabilní velikosti, solitární či mnohočetné, a v extrémních případech mohou vést k destrukci většiny jaterního parenchymu. Při déletrvajícím hnisání dochází v důsledku vystupňované fagocytózy rozpadajících se zánětlivých elementů ke zmnožení makrofágů s pěnitou cytoplazmou a k rozvoji xantogranulomatózní cholangitidy (9,114). Závažnou komplikací akutní cholangitidy je šíření zánětlivých změn na větve portální vény s rozvojem pyleflebitidy a případně i pyletrombózy (9).
Po odstranění obstrukce a úspěšné antibiotické terapii obvykle dochází k regresi zánětlivých změn. Opakovaná či dlouhotrvající expozice zánětlivým stimulům však vede ke vzniku jizevnatých striktur žlučovodů a k následnému rozvoji biliární fibrózy, případně i sekundární biliární cirhózy (115-116).
HIV a cholangiopatie
U některých pacientů s pokročilým syndromem získané imunodeficience (AIDS) bývá popisována tzv. HIV-asociovaná cholangiopatie, což je stav charakterizovaný přítomností benigních striktur žlučového stromu. Rozvoj tohoto typu cholangiopatie je spojován s oportunními infekcemi, jako jsou CMV infekce (obr. 19), infekce kryptosporidiem či Giardia lamblia (117-119).
Diagnostika onemocnění se obvykle opírá o zobrazovací metody, které odhalí stenózy extrahepatálních žlučovodů, případně papilární stenózu. K biopsii jater se přistupuje pouze v nejasných případech a histopatologický obraz zpravidla odpovídá fibrózní cholangiopatii připomínající PSC (9,117).
CMV cholangitida
CMV infekce jater se často rozvíjí v predisponovaném terénu poškozeném ischémií, infekcí či jinou noxou. Klinická a histopatologická manifestace se zpravidla liší u imunokompetentních a imunosuprimovaných pacientů, u kterých je výskyt nemoci mnohem častější (120,121).
U imunokompetentních pacientů dominuje v histopatologickém obraze portální a zejména lobulární zánětlivý infiltrát s intrasinusoidální lymfocytózou připomínající infekční mononukleózu. U imunokompromitovaných jedinců je portální zánět většinou méně vyjádřený, častěji však nacházíme virové inkluze, které představují důležitý diagnostický znak cytomegalovirové infekce. Tyto inkluze se nacházejí především v hepatocytech, biliárních epiteliích, v endoteliálních elementech a v Kupfferových buňkách a mají charakteristický vzhled. Infikované buňky jsou nápadně zvětšené, s velkým ovoidním jádrem a bazofilní intranukleární inkluzí obklopenou světlým halo, což bývá přirovnáváno k „sovímu oku“ (obr. 19). V lobulech bývají přítomny tzv. mikroabscesy tvořené drobnými kolekcemi neutrofilních leukocytů kolem zanikajících hepatocytů. Detekce CMV inkluzí může být podpořena i imunohistochemickým vyšetřením virových antigenů, které je přínosné zejména u časných nebo málo expresivních forem infekce (9,120).

Cholangiopatie spojená s COVID-19
Po těžkém průběhu onemocnění COVID-19 byl u některých pacientů popsán syndrom progredujícího poškození žlučovodů provázený výrazným zvýšením sérové aktivity ALP. Patogeneze tohoto onemocnění je komplexní a zahrnuje přímé poškození biliárního epitelu virem, ischemické poškození způsobené mikrovaskulární koagulopatií a narušení rovnováhy mezi působením toxických komponent žluče a ochrannými mechanismy žlučových cest (122,123). Při vyšetření MRCP jsou patrny změny na intrahepatálních i extrahepatálních žlučovodech s mnohočetnými benigními strikturami a se ztluštěním stěny postižených vývodů (124). Histopatologické vyšetření jaterní tkáně zpravidla odhalí edém a fibrózní rozšíření portálních polí s periportální duktulární proliferací, provázenou neutrofilní zánětlivou celulizací a těžkou parenchymální cholestázou (124,125).
ISCHEMICKÁ CHOLANGIOPATIE
Peribiliární kapilární pleteň je téměř výhradně zásobována větvemi jaterní tepny, což činí intrahepatální žlučovody – na rozdíl od jaterního parenchymu s dvojím cévním zásobením – výrazně citlivějšími na poruchy arteriální perfuze. Ischemická cholangiopatie představuje formu poškození žlučových cest vznikající v důsledku narušení arteriálního průtoku v této cévní síti, ke kterému většinou dochází při lokalizované obstrukci či přerušení přítoku krve arterií, jako například při trombóze jaterní tepny po transplantaci jater, transarteriální chemoembolizaci (TACE), radiofrekvenční ablaci nádorů nebo jako následek užívání kokainu. Vzácně se ischemická léze vyvine jako následek systémového postižení spojeného s těžkou globální hypoperfuzí, jako je například protrahovaný šokový stav (8, 126-128).
V histologickém obraze je ischemická cholangiopatie charakterizována degenerativními změnami a deskvamací biliární výstelky a v pokročilých případech nekrózou stěny žlučovodů s extravazací žluči do přilehlých měkkých tkání (obr. 20). V některých případech dochází ke vzniku patologické dutiny vyplněné žlučí, tzv. bilomu. Chronickou komplikací ischemické cholangiopatie je rozvoj jizevnatých striktur a progresivní ztráta žlučovodů malých kalibrů. Mezi časté život ohrožující komplikace patří bakteriální a plísňové infekce (9,127,128).
Sekundární sklerozující cholangitida u kriticky nemocných pacientů (SSC-CIP)
Ischemie intrahepatálních žlučovodů je považována za klíčový faktor rozvoje SSC u pacientů dlouhodobě léčených na jednotkách intenzivní péče. Kromě ischemie hrají při rozvoji SSC-CIP roli také infekce a toxické komponenty žluči (129-130). SSC-CIP se často projeví až po zotavení z primárního onemocnění, nejčastěji žloutenkou, pruritem, bolestí v pravém horním kvadrantu břicha a opakovanými biliárními infekcemi. Stav může rychle progredovat do jaterní cirhózy. Zlatým standardem diagnostiky SSC-CIP jsou zobrazovací metody, ERCP či MRCP. Mezi typické nálezy patří nepravidelné striktury a dilatace žlučovodů vyplněné odlitky inspisované žluči, s postupnou destrukcí intrahepatálních žlučových cest. Velké extrahepatální žlučovody jsou postiženy jen zřídka (129,130).
Na mikroskopické úrovni se SSC-CIP projevuje degenerací biliární výstelky interlobulárních žlučovodů, periduktálním zánětem a periportální duktulární proliferací. V jaterním parenchymu jsou přítomny známky hepatokanalikulární bilirubinostázy se žlučovými infarkty a se známkami stázy cholátů. V případě progrese onemocnění dochází k fibróznímu rozšíření portálních polí a k rychlému rozvoji septální fibrózy až cirhózy s biliárními rysy (130).

NEOPLASTICKÉ CHOLANGIOPATIE
Biliární systém může být postižen celou řadou benigních i maligních nádorových procesů (9). Podrobně se problematice nádorových lézí věnují následující příspěvky v tomto čísle časopisu, na které si dovolujeme čtenáře odkázat.
SOUHRN
Choroby žlučových cest představují heterogenní skupinu nenádorových i nádorových onemocnění s pestrým spektrem klinických příznaků, laboratorních odchylek a morfologických změn. Mnohé z těchto jednotek mají chronický a progresivní charakter a mohou vést k ireverzibilnímu poškození jaterního parenchymu s následným rozvojem jaterního selhání, které v konečném stadiu vyžaduje transplantaci jater.
Diagnostika cholangiopatií je komplexní proces založený na korelaci klinického obrazu s biochemickými ukazateli, nálezy zobrazovacích metod a v indikovaných případech také s histopatologickým vyšetřením jaterní tkáně. Biopsie jater zůstává cenným nástrojem zejména v situacích, kdy klinický či radiologický obraz není jednoznačný, při podezření na překryvné syndromy či méně časté formy cholangiopatií postihujících zejména žlučovody menších kalibrů.
Pro stanovení správné diagnózy a zvolení optimálního terapeutického přístupu je naprosto klíčová mezioborová spolupráce mezi specialisty v oblasti hepatologie, radiodiagnostiky, patologie a celé řady dalších oborů, která umožňuje integrovaný pohled na komplexní problematiku onemocnění žlučových cest.
PROHLÁŠENÍ
Autor práce prohlašuje, že v souvislosti s tématem, vznikem a publikací tohoto článku není ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou. Toto prohlášení se týká i všech spoluautorů.
Zdroje
- de Jong IEM, van den Heuvel MC, Wells RG, Porte RJ. The heterogeneity of the biliary tree. J Hepatol 2021; 75(5): 1236-1238.
- Banales JM, Huebert RC, Karlsen T, Strazzabosco M, LaRusso NF, Gores GJ. Cholangiocyte pathobiology. Nat Rev Gastroenterol Hepatol 2019; 16(5): 269-281.
- Glaser S, Francis H, Demorrow S, Lesage G, Fava G, Marzioni M, Venter J, Alpini G. Heterogeneity of the intrahepatic biliary epithelium. World J Gastroenterol 2006; 12(22): 3523-3536.
- Lazaridis KN, Strazzabosco M, Larusso NF. The cholangiopathies: disorders of biliary epithelia. Gastroenterology 2004; 127(5): 1565-1577.
- Boyer JL. Bile formation and secretion. Comprehensive Physiology. 2013;3(3):1035–1078.
- Nakanuma Y. Tutorial review for understanding of cholangiopathy. Int J Hepatol 2012; 2012 : 547840.
- Menon S, Holt A. Large-duct cholangiopathies: aetiology, diagnosis and treatment. Frontline Gastroenterol 2019; 10(3): 284-291.
- Sticova E, Fabian O. Morphological aspects of small-duct cholangiopathies: A minireview. World J Hepatol 2023; 15(4): 538-553.
- Zen Y, Hubscher SG, Nakanuma Y. Bile Duct Diseases. In: Burt AD, Ferrell LD, Hübscher SG, eds. MacSween‘s Pathology of the Liver. 7th ed. London: Elsevier; 2017 : 516-593.
- Masia R, Pratt DS, Misdraji J. A histopathologic pattern of centrilobular hepatocyte injury suggests 6-mercaptopurine-induced hepatotoxicity in patients with inflammatory bowel disease. Arch Pathol Lab Med 2012; 136(6): 618–622.
- Di Guardo G. Lipofuscin, lipofuscin-like pigments and autofluorescence. Eur J Histochem 2015; 59(1): 1–2.
- Goldfischer S, Bernstein J. Lipofuscin (aging) pigment granules of the newborn human liver. J Cell Biol 1969; 42(1): 253–261.
- Zhao J, Yue P, Mi N, et al. Biliary fibrosis is an important but neglected pathological feature in hepatobiliary disorders: from molecular mechanisms to clinical implications. Med Rev 2024; 4(4): 326-365.
- Penz-Österreicher M, Österreicher CH, Trauner M. Fibrosis in autoimmune and cholestatic liver disease. Best Pract Res Clin Gastroenterol 2011; 25(2): 245-258.
- Neuberger J, Patel J, Caldwell H, et al. Guidelines on the use of liver biopsy in clinical practice from the British Society of Gastroenterology, the Royal College of Radiologists and the Royal College of Pathology. Gut 2020;69(8): 1382-1403.
- Reau NS, Jensen DM. Vanishing bile duct syndrome. Clin Liver Dis 2008; 12(1): 203-217.
- Bessone F, Hernández N, Tanno M, Roma MG. Drug-Induced Vanishing Bile Duct Syndrome: From Pathogenesis to Diagnosis and Therapeutics. Semin Liver Dis 2021; 41(3): 331-348.
- Karlsen TH, Folseraas T, Thorburn D, Vesterhus M. Primary sclerosing cholangitis a comprehensive review. J Hepatol 2017; 67(6): 1298-1323.
- Williamson KD, Chapman RW. Primary sclerosing cholangitis: a clinical update. Br Med Bull 2015; 114(1): 53-64.
- Kim YS, Hurley EH, Park Y, Ko S. Primary sclerosing cholangitis (PSC) and inflammatory bowel disease (IBD): a condition exemplifying the crosstalk of the gut-liver axis. Exp Mol Med 2023; 55(7): 1380-1387.
- Beheshti-Maal A, Tamimi A, Iravani S, et al. PSC associated inflammatory bowel disease: a distinct entity. Expert Rev Gastroenterol Hepatol 2022; 16(2): 129-139.
- van Munster KN, Bergquist A, Ponsioen CY. Inflammatory bowel disease and primary sclerosing cholangitis: One disease or two? J Hepatol 2024; 80(1): 155-168.
- Gidwaney NG, Pawa S, Das KM. Pathogenesis and clinical spectrum of primary sclerosing cholangitis. World J Gastroenterol 2017; 23(14): 2459–2469.
- Tow CY, Chung E, Kaul B, Bhalla A, Fortune BE. Diagnostic Tests in Primary Sclerosing Cholangitis: Serology, Elastography, Imaging, and Histology. Clin Liver Dis 2024; 28(1): 157-169.
- Cazzagon N, Sarcognato S, Catanzaro E, et al. Primary Sclerosing Cholangitis: Diagnostic Criteria. Tomography 2024; 10(1): 47-65.
- Ponsioen CY. Diagnosis, Differential Diagnosis, and Epidemiology of Primary Sclerosing Cholangitis. Dig Dis 2015; 33 Suppl 2 : 134-139.
- Gardner CS, Bashir MR, Marin D, et al. Diagnostic performance of imaging criteria for distinguishing autoimmune cholangiopathy from primary sclerosing cholangitis and bile duct malignancy. Abdom Imaging 2015; 40(8): 3052-3061.
- Boberg KM, Chapman RW, Hirschfield GM, et al. Overlap syndromes: the International Autoimmune Hepatitis Group (IAIHG) position statement on a controversial issue. J Hepatol 2011; 54(2): 374-385.
- Karadag Soylu N. Histopathology of Wilson Disease. In: Liver Pathology. IntechOpen; 2021.
- Carey EJ, Ali AH, Lindor KD. Primary biliary cirrhosis. Lancet 2015; 386(10003): 1565-1575.
- Trivella J, John BV, Levy C. Primary biliary cholangitis: Epidemiology, prognosis, and treatment. Hepatol Commun 2023; 7(6): e0179.
- Shah SK, Bowlus CL. Autoimmune Markers in Primary Biliary Cholangitis. Clin Liver Dis 2024; 28(1): 93-101.
- Tanaka A, Ma X, Takahashi A, Vierling JM. Primary biliary cholangitis. Lancet 2024; 404(10457): 1053-1066.
- Tan D, Goodman ZD. Liver Biopsy in Primary Biliary Cholangitis: Indications and Interpretation. Clin Liver Dis 2018; 22(3): 579-588.
- Liu HL, Yang AY, Xiong QF, et al. Aberrant cytokeratin 7 expression by hepatocytes can predict the ductopenia grade in primary biliary cholangitis. BMC Gastroenterol 2022; 22(1): 443.
- Warnes TW, Roberts SA, Smith A, et al. Portal hypertension in primary biliary cholangitis: prevalence, natural history and histological correlates. Eur J Gastroenterol Hepatol 2021; 33(12): 1595-1602.
- Hu YF, Li SX, Liu HL, et al. Precirrhotic Primary Biliary Cholangitis with Portal Hypertension: Bile Duct Injury Correlate. Gut Liver 2024; 18(5): 867-876.
- Ludwig J, Dickson ER, McDonald GS. Staging of chronic nonsuppurative destructive cholangitis (syndrome of primary biliary cirrhosis). Virchows Arch A Pathol Anat Histopathol 1978; 379 : 103-112.
- Scheuer PJ, Lefkowitch JH. Liver biopsy interpretation. 7th ed. London: Saunders-Elsevier; 2006 : 69-84.
- Kakuda Y, Harada K, Sawada-Kitamura S, et al. Evaluation of a new histologic staging and grading system for primary biliary cirrhosis in comparison with classical systems. Hum Pathol 2013; 44(6): 1107-1117.
- Nakanuma Y, Zen Y, Harada K, et al. Application of a new histological staging and grading system for primary biliary cirrhosis to liver biopsy specimens: Interobserver agreement. Pathol Int 2010; 60(3): 167-174.
- Liang Y, Khandakar B, Hao Y, et al. Histology and clinical correlations in autoimmune hepatitis, primary biliary cholangitis, and autoimmune hepatitis-primary biliary cholangitis overlap syndrome. Ann Diagn Pathol 2023; 67 : 152178.
- Kobayashi M, Kakuda Y, Harada K, et al. Clinicopathological study of primary biliary cirrhosis with interface hepatitis compared to autoimmune hepatitis. World J Gastroenterol 2014; 20(13): 3597-3608.
- Chazouillères O. Overlap Syndromes. Dig Dis 2015; 33 Suppl 2 : 181-187.
- Sánchez-Oro R, Alonso-Muñoz EM, Martí Romero L. Review of IgG4-related disease. Gastroenterol Hepatol 2019; 42(10): 638-647.
- Vashi B, Khosroshahi A. IgG4-Related Disease with Emphasis on Its Gastrointestinal Manifestation. Gastroenterol Clin North Am 2019; 48(2): 291-305.
- Katz G, Stone JH. Clinical Perspectives on IgG4-Related Disease and Its Classification. Annu Rev Med 2022; 73 : 545-562.
- Zen Y. The Pathology of IgG4-Related Disease in the Bile Duct and Pancreas. Semin Liver Dis 2016; 36(3): 242-256.
- Lee HE, Zhang L. Immunoglobulin G4-related hepatobiliary disease. Semin Diagn Pathol 2019; 36(6): 423-433.
- Deshpande V, Zen Y, Chan JK, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol 2012; 25(9): 1181-1192.
- Kamisawa T, Nakazawa T, Tazuma S, et al. Clinical practice guidelines for IgG4-related sclerosing cholangitis. J Hepatobiliary Pancreat Sci 2019; 26(1): 9-42.
- Tanaka A. IgG4-Related Sclerosing Cholangitis and Primary Sclerosing Cholangitis. Gut Liver 2019; 13(3): 300-307.
- Demetris AJ, Bellamy C, Hübscher SG, et al. 2016 Comprehensive Update of the Banff Working Group on Liver Allograft Pathology: Introduction of Antibody-Mediated Rejection. Am J Transplant 2016; 16(10): 2816-2835.
- Dumortier J, Conti F, Scoazec JY. Posttransplant immune-mediated cholangiopathies. Curr Opin Gastroenterol 2022; 38(2):98-103.
- Rastogi A, Nigam N, Gayatri R, et al. Biliary Epithelial Senescence in Cellular Rejection Following Live Donor Liver Transplantation. J Clin Exp Hepatol 2022; 12(6): 1420-1427.
- Trussoni CE, O‘Hara SP, LaRusso NF. Cellular senescence in the cholangiopathies: a driver of immunopathology and a novel therapeutic target. Semin Immunopathol 2022; 44(4):527-544.
- Quaglia A, Duarte R, Patch D, et al. Histopathology of graft versus host disease of the liver. Histopathology 2007; 50(6): 727-738.
- Chiba T, Yokosuka O, Goto S, et al. Clinicopathological features in patients with hepatic graft-versus-host disease. Hepatogastroenterology 2005; 52(66): 1849-1853.
- Jain R, Yadav D, Puranik N, et al. Sarcoidosis: Causes, Diagnosis, Clinical Features, and Treatments. J Clin Med 2020; 9(4): 1081.
- Nicolosi S, Chernovsky M, Angoni D, et al. Gastrointestinal Manifestations of Sarcoidosis: A State-of-the-Art, Comprehensive Review of the Literature-Practical Clinical Insights and Many Unmet Needs on Diagnosis and Treatment. Pharmaceuticals (Basel) 2024; 17(9): 1106.
- Rossi G, Ziol M, Roulot D, et al. Hepatic Sarcoidosis: Current Concepts and Treatments. Semin Respir Crit Care Med 2020; 41(5): 652-658.
- Chen C, Luo N, Dai F, et al. Advance in pathogenesis of sarcoidosis: Triggers and progression. Heliyon 2024; 10(5): e27612.
- Miedema J, Cinetto F, Smed-Sörensen A, Spagnolo P. The immunopathogenesis of sarcoidosis. J Autoimmun 2024; 103247.
- Kahi CJ, Saxena R, Temkit M, et al. Hepatobiliary disease in sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis 2006; 23(2): 117-123.
- Tana C, Donatiello I, Caputo A, et al. Clinical Features, Histopathology and Differential Diagnosis of Sarcoidosis. Cells 2021; 11(1): 59.
- Kumar SN, Prasad TS, Narayan PA, Muruganandhan J. Granuloma with langhans giant cells: An overview. J Oral Maxillofac Pathol 2013; 17(3): 420-423.
- Valeyre D, Brauner M, Bernaudin JF, et al. Differential diagnosis of pulmonary sarcoidosis: a review. Front Med (Lausanne) 2023; 10 : 1150751.
- Visentin M, Lenggenhager D, Gai Z, Kullak-Ublick GA. Drug-induced bile duct injury. Biochim Biophys Acta Mol Basis Dis 2018; 1864(4): 1498-1506.
- Salas-Silva S, Simoni-Nieves A, Lopez-Ramirez J, et al. Cholangiocyte death in ductopenic cholestatic cholangiopathies: Mechanistic basis and emerging therapeutic strategies. Life Sci 2019; 218 : 324-339.
- Scoazec JY. Drug-induced bile duct injury: new agents, new mechanisms. Curr Opin Gastroenterol 2022; 38(2): 83-88.
- LiverTox. Clinical and Research Information on Drug-Induced Liver Injury [Internet]. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases; 2012. Available from: https://www.ncbi.nlm.nih. gov/books/NBK547852/
- Zen Y, Yeh MM. Hepatotoxicity of immune checkpoint inhibitors: a histology study of seven cases in comparison with autoimmune hepatitis and idiosyncratic drug-induced liver injury. Mod Pathol 2018; 31(6): 965-973.
- Zen Y, Yeh MM. Checkpoint inhibitor-induced liver injury: A novel form of liver disease emerging in the era of cancer immunotherapy. Semin Diagn Pathol 2019; 36(6): 434-440.
- Zen Y, Chen YY, Jeng YM, et al. Immune-related adverse reactions in the hepatobiliary system: second-generation check-point inhibitors highlight diverse histological changes. Histopathology 2020; 76(3): 470-480.
- Shatila M, Zhang HC, Shirwaikar Thomas A, et al. Systematic review of immune checkpoint inhibitor-related gastrointestinal, hepatobiliary, and pancreatic adverse events. J Immunother Cancer 2024; 12(11): e009742.
- Velayudham LS, Farrell GC. Drug-induced cholestasis. Expert Opin Drug Saf 2003; 2(3): 287-304.
- Kubitz R, Dröge C, Kluge S, et al. Genetic variations of bile salt transporters. Drug Discov Today Technol 2014; 12: e55-67.
- Vitale G, Mattiaccio A, Conti A, et al. Molecular and Clinical Links between Drug-Induced Cholestasis and Familial Intrahepatic Cholestasis. Int J Mol Sci 2023; 24(6): 5823.
- Luo Z, Jegga AG, Bezerra JA. Gene-disease associations identify a connectome with shared molecular pathways in human cholangiopathies. Hepatology 2018; 67(2): 676-689.
- Fabris L, Fiorotto R, Spirli C, et al. Pathobiology of inherited biliary diseases: a roadmap to understand acquired liver diseases. Nat Rev Gastroenterol Hepatol 2019; 16(8): 497-511.
- Drenth JP, Chrispijn M, Bergmann C. Congenital fibrocystic liver diseases. Best Pract Res Clin Gastroenterol 2010; 24(5): 573-584.
- Hellen DJ, Karpen SJ. Genetic Contributions to Biliary Atresia: A Developmental Cholangiopathy. Semin Liver Dis 2023; 43(3): 323-335.
- Wood W, Tinich T, Lazar L, Schooler GR, Sathe M. Cystic fibrosis hepatobiliary involvement: an update on imaging in diagnosis and monitoring. Pediatr Radiol 2024; 54(9): 1416-1427.
- Hadchouel M, Gautier M. Histopathologic study of the liver in the early cholestatic phase of alpha-1-antitrypsin deficiency. J Pediatr 1976; 89(2): 211-215.
- Desmet VJ. Ludwig symposium on biliary disorders--part I. Pathogenesis of ductal plate abnormalities. Mayo Clin Proc 1998; 73(1): 80–89.
- Desmet VJ. Congenital diseases of intrahepatic bile ducts: variations on the theme “ductal plate malformation.” Hepatology 1992; 16(4): 1069–1083.
- Kothari TH, Khara SS, Kothari DB, Schiano T. Caroli Disease. Consultant 2024; 48(5): 391.
- Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med 2011; 364(16): 1533-1543.
- Gunay-Aygun M. Liver and kidney disease in ciliopathies. Am J Med Genet C Semin Med Genet 2009; 151C(4): 296-306.
- Perugorria MJ, Bujanda L, Banales JM. More insight into the diversity of cholangiocyte ciliopathies. J Hepatol 2016; 65(6): 1083-1085.
- Kotha S, Berry P. Hepatic, pancreatic and renal manifestations of a ciliopathy. Hepatobiliary Pancreat Dis Int 2021; 20(4): 394-395.
- Roediger R, Dieterich D, Chanumolu P, et al. Polycystic Kidney/Liver Disease. Clin Liver Dis 2022; 26(2): 229-243.
- Antala S, Taylor SA. Biliary Atresia in Children: Update on Disease Mechanism, Therapies, and Patient Outcomes. Clin Liver Dis 2022; 26(3): 341-354.
- Quelhas P, Cerski C, Dos Santos JL. Update on Etiology and Pathogenesis of Biliary Atresia. Curr Pediatr Rev 2022; 19(1): 48-67.
- Davenport M, Gonde C, Redkar R, et al. Immunohistochemistry of the liver and biliary tree in extrahepatic biliary atresia. J Pediatr Surg 2001; 36(7): 1017-1025.
- Muthukanagarajan SJ, Karnan I, Srinivasan P, et al. Diagnostic and Prognostic Significance of Various Histopathological Features in Extrahepatic Biliary Atresia. J Clin Diagn Res 2016; 10(6): EC23-27.
- Vij M, Rela M. Biliary atresia: pathology, etiology and pathogenesis. Future Sci OA 2020; 6(5): FSO466.
- Higashio A, Yoshioka T, Kanamori Y, et al. Relationships Between Histopathological Findings in the Liver and Prognosis in Patients With Biliary Atresia. Clin Pathol 2022; 15 : 2632010X221132686.
- Kohut TJ, Gilbert MA, Loomes KM. Alagille Syndrome: A Focused Review on Clinical Features, Genetics, and Treatment. Semin Liver Dis 2021; 41(4): 525-537.
- Mašek J, Andersson ER. Jagged-mediated development and disease: Mechanistic insights and therapeutic implications for Alagille syndrome. Curr Opin Cell Biol 2024; 86 : 102302.
- Spinner NB, Loomes KM, Krantz ID, et al. Alagille Syndrome. In: Adam MP, Feldman J, Mirzaa GM, et al., eds. GeneReviews®. Seattle: University of Washington; 1993-2024.
- Herman HK, Abramowsky CR, Caltharp S, et al. Identification of Bile Duct Paucity in Alagille Syndrome: Using CK7 and EMA Immunohistochemistry as a Reliable Panel for Accurate Diagnosis. Pediatr Dev Pathol 2016; 19(1): 47-50.
- Gilbert MA, Loomes KM. Alagille syndrome and non-syndromic paucity of the intrahepatic bile ducts. Transl Gastroenterol Hepatol 2021; 6 : 22.
- Reichert MC, Lammert F. ABCB4 Gene Aberrations in Human Liver Disease: An Evolving Spectrum. Semin Liver Dis 2018; 38(4): 299-307.
- Sticova E, Jirsa M. ABCB4 disease: Many faces of one gene deficiency. Ann Hepatol 2020; 19(2): 126-133.
- Stättermayer AF, Halilbasic E, Wrba F, et al. Variants in ABCB4 (MDR3) across the spectrum of cholestatic liver diseases in adults. J Hepatol 2020; 73(3): 651-663.
- Hegarty R, Gurra O, Tarawally J, et al. Clinical outcomes of ABCB4 heterozygosity in infants and children with cholestatic liver disease. J Pediatr Gastroenterol Nutr 2024; 78(2): 339-349.
- Frider B, Castillo A, Gordo-Gilart R, et al. Reversal of advanced fibrosis after long-term ursodeoxycholic acid therapy in a patient with residual expression of MDR3. Ann Hepatol 2015; 14(5): 745-751.
- Morotti RA, Suchy FJ, Magid MS. Progressive familial intrahepatic cholestasis (PFIC) type 1, 2, and 3: a review of the liver pathology findings. Semin Liver Dis 2011; 31(1): 3-10.
- Girard M, Bizet AA, Lachaux A, et al. DCDC2 Mutations Cause Neonatal Sclerosing Cholangitis. Hum Mutat 2016; 37(10): 1025-1029.
- Grammatikopoulos T, Sambrotta M, Strautnieks S, et al. Mutations in DCDC2 (doublecortin domain containing protein 2) in neonatal sclerosing cholangitis. J Hepatol 2016; 65(6): 1179-1187.
- Stanca CM, Fiel MI, Kontorinis N, et al. Chronic ductopenic rejection in patients with recurrent hepatitis C virus treated with pegylated interferon alfa-2a and ribavirin. Transplantation 2007; 84(2): 180-186.
- Dhingra S, Ward SC, Thung SN. Liver pathology of hepatitis C, beyond grading and staging of the disease. World J Gastroenterol 2016; 22(4): 1357-1366.
- Li Y, Ayata G, Baker SP, Banner BF. Cholangitis: a histologic classification based on patterns of injury in liver biopsies. Pathol Res Pract 2005; 201(8–9): 565–572.
- Uno S, Hase R, Kobayashi M, Shiratori T, Nakaji S, Hirata N, et al. Short-course antimicrobial treatment for acute cholangitis with Gram-negative bacillary bacteremia. Int J Infect Dis 2017; 55 : 81–85.
- Miura F, Takada T, Kawarada Y, Nimura Y, Wada K, Hirota M, et al. Flowcharts for the diagnosis and treatment of acute cholangitis and cholecystitis: Tokyo Guidelines. J Hepatobiliary Pancreat Surg 2007; 14(1): 27–34.
- Tonolini M, Bianco R. HIV-related/AIDS cholangiopathy: pictorial review with emphasis on MRCP findings and differential diagnosis. Clin Imaging 2013; 37(2): 219-226.
- Naseer M, Dailey FE, Juboori AA, et al. Epidemiology, determinants, and management of AIDS cholangiopathy: A review. World J Gastroenterol 2018; 24(7): 767-774.
- Alsharif NM, Souleiman MM, Gunaseelan L, et al. AIDS-Associated Cryptosporidial and Cytomegalovirus Cholangiopathy. Cureus 2024; 16(7): e63963.
- Da Cunha T, Wu GY. Cytomegalovirus hepatitis in immunocompetent and immunocompromised hosts. J Clin Transl Hepatol 2021; 9(1): 106–115.
- Demetris AJ, Jaffe R, Starzl TE. A review of adult and pediatric post-transplant liver pathology. Pathol Annu 1987; 22(Pt 2): 347–386.
- Roth NC, Kim A, Vitkovski T, et al. Post-COVID-19 Cholangiopathy: A Novel Entity. Am J Gastroenterol 2021; 116(5): 1077-1082.
- Yadlapati S, Jarrett SA, Baik D, et al. COVID-19 related biliary injury: A review of recent literature. World J Gastroenterol 2023; 29(14): 2127-2133.
- Veerankutty FH, Sengupta K, Vij M, et al. Post-COVID-19 cholangiopathy: Current understanding and management options. World J Gastrointest Surg 2023; 15(5): 788-798.
- Borges VFA, Cotrim HP, Andrade ARCF, et al. COVID-19-Related Cholangiopathy: Histological Findings. Diagnostics (Basel) 2024; 14(16): 1804.
- Deltenre P, Valla DC. Ischemic cholangiopathy. Semin Liver Dis 2008; 28(3): 235-246.
- Buis CI, Hoekstra H, Verdonk RC, et al. Causes and consequences of ischemic-type biliary lesions after liver transplantation. J Hepatobiliary Pancreat Surg 2006; 13(6): 517-524.
- Goria O, Archambeaud I, Lemaitre C, et al. Ischemic cholangiopathy: An update. Clin Res Hepatol Gastroenterol 2020; 44(4): 486-490.
- Gudnason HO, Björnsson ES. Secondary sclerosing cholangitis in critically ill patients: current perspectives. Clin Exp Gastroenterol 2017; 10 : 105-111.
- Martins P, Verdelho Machado M. Secondary Sclerosing Cholangitis in Critically Ill Patients: An Underdiagnosed Entity. GE Port J Gastroenterol 2020; 27(2): 103-114.
Štítky
Patologie Soudní lékařství ToxikologieČlánek vyšel v časopise
Česko-slovenská patologie

2025 Číslo 3
Nejčtenější v tomto čísle
- Cholangiokarcinom - morfologie, imunohistochemie, genetika
- Role endoskopie v diagnostice stenóz žlučových cest
- Nemoci žlučových cest z pohledu patologa: Přínos jaterní biopsie v rutinní diagnostické praxi
- Solitární fibrózní tumor pankreatu u pacienta s nádorovou duplicitou: kazuistika
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy