
Zmeny WHO klasifikácie lymfoidných neoplázií v kontexte revízie z roku 2016
Changes of the WHO classification of lymphoid neoplasms in the context of the 2016 revision
As a result of increasing knowledge, the validity of any tumour classification could not be unlimited. The aim of this article is to review the most important changes in the WHO classification of lymphoid neoplasms of a non-Hodgkin type that have been announced and published in relation to its revision in 2016. These changes are based on better understanding of pathogenesis and genetics of diseases, refine diagnostic criteria, reflect existence of rare forms and introduce new provisional categories of lymphoid neoplasms. WHO classification becomes more complex and the number of disease entities is increasing. However, until the the monography will be published, all changes are preliminary and incomplete, requiring work with available lymphoma literature.
Keywords:
WHO classification – lymphoid neoplasms
Autoři:
Tomáš Balhárek 1,2; Juraj Marcinek 1,2; Lukáš Plank 1,2
Působiště autorů:
Konzultačné centrum bioptickej diagnostiky ochorení krvotvorby v SR
Ústav patologickej anatómie Jesseniovej lekárskej fakulty Univerzity Komenského a Univerzitnej nemocnice v Martine
1; Konzultačné centrum bioptickej diagnostiky ochorení krvotvorby v SR
Martinské bioptické centrum, s. r. o. v Martine
2
Vyšlo v časopise:
Čes.-slov. Patol., 53, 2017, No. 3, p. 122-128
Kategorie:
Přehledový článek
Souhrn
V dôsledku pribúdajúcich poznatkov platnosť žiadnej klasifikácie nádorov nemôže byť neobmedzená. Cieľom predloženého článku je predstaviť čitateľom najdôležitejšie zmeny vo WHO klasifikácii lymfoidných neoplázií non-Hodgkinovho typu, ktoré boli avizované a publikované v súvislosti s jej revíziou, resp. aktualizáciou v roku 2016. Tieto zmeny vychádzajú z nových poznatkov o patogenéze a genetike ochorení, spresňujú diagnostické kritériá, zohľadňujú existenciu zriedkavých foriem a zavádzajú nové provizórne kategórie lymfoidných neoplázií. WHO klasifikácia sa stáva viac komplexnejšou a počet chorobných jednotiek sa zvyšuje. Do momentu knižného vydania klasifikácie však treba všetky uvedené zmeny chápať len ako predbežné a neúplné, vyžadujúce prácu s dostupnou literatúrou.
Kľúčové slová:
WHO klasifikácia – lymfoidné neoplázie
Klasifikácia lymfoidných neoplázií podľa Svetovej zdravotníckej organizácie (WHO) je postavená tak, aby umožnila definovať čo najhomogénnejšie jednotlivé kategórie nádorov, ktoré sú akceptované klinikmi a ktoré možno identifikovať na základe analýzy tkaniva fixovaného vo formalíne a zaliateho do parafínu, čo je najdostupnejší typ bioptického materiálu. Veľká časť zmien súvisí s implementáciou poznatkov o genetických abnormalitách, ktoré spresňujú diagnostiku alebo slúžia na subtypizáciu či odseparovanie niektorých lymfoidných neoplázií. Klasifikácia zohľadňuje existenciu zriedkavých foriem či umožňuje definovať nové provizórne jednotky, ktorých opodstatnenosť sa má ozrejmiť časom ich používaním v praxi. Revíziu klasifikácie ovplyvnil aj narastajúci konzervativizmus v diagnostike lymfómov, ktorý by mal zabrániť naddiagnostikovaniu lézií, ktoré sa nesprávajú ako skutočné malignity. Pre tých, ktorí sledujú vývoj v hematopatológii, by zavedené zmeny nemali byť veľkým prekvapením (1). Revízia klasifikácie je síce prezentovaná ako výsledok konsenzu medzi hematopatológmi, genetikmi a klinikmi, no už teraz je zrejmé, že nie všetky názory sú jednotné a nie všetky otázky sú zodpovedané. Tomu nasvedčujú aj pomerne neštandardné okolnosti okolo vydania predmetnej revízie, opísané v úvodných poznámkach ku klasifikácii.
ZRELÉ (PERIFÉRNE) B-BUNKOVÉ NEOPLÁZIE
Najočakávanejšími a dozaista najdôležitejšími novinkami vyplývajúcimi z poslednej revízie WHO klasifikácie sú zmeny v spektre veľkobunkových (blastických) B-lymfómov (LBCL), keďže tieto patria k najčastejším lymfoidným neopláziám, s ktorými sa v našej populácii stretávame. Nakoľko ide o extrémne heterogénnu skupinu B-bunkových neoplázií, cieľom posledných aktualizácií z rokov 2008 a 2016 bol pokus o zmysluplnú a klinicky relevantnú rizikovú subtypizáciu a stratifikáciu LBCL, formálne spadajúcich do kategórie difúznych veľkobunkových B-lymfómov (DLBCL), zostávajúcich po odseparovaní striktne definovaného Burkittovho lymfómu (BL) (2,3).
Revízia WHO klasifikácie z roku 2016 zakotvuje potrebu vykonávania imunohistochemickej fenotypovej stratifikácie DLBCL na GCB a non-GCB podtyp, ktorá sa označuje ako tzv. COO („cell of origine“) klasifikácia (3). Tá zastupuje subtypizáciu podľa pôvodu v B-bunkách zárodočných centier (GCB) alebo v aktivovaných B-bunkách (ABC), originálne postavenú na základe vyšetrovania profilu génovej expresie (GEP), ktorá nie je vhodná pre rutinnú prax (4). Na účely COO klasifikácie je s výhradami doporučený najpopulárnejší Hansovej algoritmus, založený na imunohistochemickej analýze expresie CD10, bcl-6 a MUM-1 (5), pričom treba pamätať na jeho limity a skutočnosť, že cca 10-15 % DLBCL pri jeho použití ostáva neklasifikovateľných. Subtypizácia sa má vykonávať nielen v bližšie nešpecifikovaných DLBCL, NOS, ale aj v orgánovo-špecifických a samostatných typoch DLBCL (3). COO klasifikácia pritom nie je len nezávislým prognostickým faktorom, ale súvisí s rozdielnymi molekulovými cestami patogenézy DLBCL, od čoho sa odvíja aj ich rozdielna citlivosť na liečbu (6).
Aktualizovaná klasifikácia ponecháva v platnosti všetky kategórie samostatných klinicko-patologických a orgánovo-špecifických typov DLBCL. Zo zmien v tomto spektre možno spomenúť terminologickú úpravu názvu EBV+ DLBCL starších na bližšie nešpecifikovaný EBV+ DLBCL, NOS (3), keďže tieto prípady sa môžu vyskytnúť aj u mladších pacientov (7). Spektrum EBV+ blastických B-lymfoproliferácií sa ďalej rozširuje o provizórnu jednotku tzv. EBV+ mukokutánneho vredu, čo je spravidla spontánne regredujúca kožná alebo sliznicová lézia asociovaná s iatrogénne navodenou imunosupresiou, vznikajúca ako posttransplantačné lymfoproliferatívne ochorenie alebo ako dôsledok starnutia imunitného systému (8).
Jednou z kľúčových zmien v roku 2008 bolo zavedenie provizórnej kategórie tzv. neklasifikovateľných B-lymfómov (BCLU) s črtami medzi DLBCL a BL (2,9). V rámci nich boli na základe genetických abnormalít identifikované unikátne lymfómy so súčasným výskytom prestavieb génov MYC a BCL2 a/alebo BCL6, označované aj ako lymfómy s dvomi („double-hit“, DH) či tromi zásahmi („triple-hit“, TH) (10). Tie sa spravidla vyznačujú vysokou agresivitou a extrémne zlou prognózou. Zavedenie kategórie BCLU sa časom ukázalo ako opodstatnené, lenže jej používanie v praxi nebolo konzistentné. Preto bola aktualizáciou z roku 2016 kategória BCLU eliminovaná a nahradená novou kategóriou tzv. vysokomalígnych B-lymfómov (HGBL), kde osobitnú skupinu predstavujú vysokomalígne B-lymfómy s viacerými zásahmi, označované ako HGBL s abnormalitami génov MYC a BCL2 a/alebo BCL6. Zostávajúce lymfómy s blastoidnou morfológiou alebo črtami medzi DLBCL a BL sa zaraďujú do kategórie bližšie nešpecifikovaných HGBL, NOS (3).
Uvedenou zmenou enormne vzrástol význam a potreba vyšetrovania genetických abnormalít, a to prinajmenej uvedených troch génov. To však so sebou prináša potrebu racionalizácie uvedených vyšetrení, nakoľko väčšina pracovísk nemá možnosť rutinne vykonávať genetickú analýzu uvedených génov vo všetkých prípadoch LBCL. To, ktoré prípady LBCL podrobiť FISH analýze, sa tak stalo prvou otvorenou otázkou, ktorá pravdepodobne nebude zodpovedaná ani v knižnom vydaní WHO klasifikácie. Kandidátmi na FISH analýzu stavu génov MYC, BCL2 a BCL6 sú spravidla prípady LBCL s blastoidnou alebo BL-podobnou morfológiou, GCB fenotypom, vysokou proliferačnou aktivitou (index Ki-67 >60 %) či expresiou c-myc (>40 %) a bcl-2 proteínu (>50 % buniek) (9).
Nie všetky HGBL s prestavbou MYC a BCL2 a/alebo BCL6 sú paušálne lymfómy s extrémne zlou prognózou. K črtám, ktoré by mohli zmierňovať inverzný potenciál DH/TH prestavby, by mohli patriť absencia expresie bcl-2 či c-myc proteínu, non-Ig partner prestavby MYC génu či morfológia bližšia DLBCL (9,11). Do uvedenej kategórie sa nemajú zaraďovať prípady nodulárne rastúceho folikulového lymfómu (FL) alebo lymfoblastového lymfómu (LBL) s DH/TH prestavbou (3). To zároveň otvára otázku do akej miery tolerovať občasne prítomnú expresiu TdT v DH/TH lymfómoch a kedy ich klasifikovať ako prekurzorové neoplázie.
Samotný princíp, kedy sú nové kategórie lymfómov vyčleňované na základe genetických nálezov, sa však nevyhol kritike. Odporcovia tohto prístupu argumentujú, že fenomén DH/TH genetických zmien je skôr prognostický faktor, ktorý sa vyskytuje nielen v bližšie nešpecifikovaných DLBCL, NOS, ale aj v orgánovo-špecifických DLBCL či dokonca iných B-lymfómoch. Je tiež otázne dokedy bude pokračovať takéto štiepenie kategórie DLBCL a kto bude znášať zvyšujúce sa náklady spojené s diagnostikou pribúdajúcich genetických abnormalít, ktorých konečný dopad na kvalitu života pacientov je aj tak otázny. Naopak zástancovia uvedeného princípu zdôrazňujú, že kategória HGBL aspoň čiastočne vyjasňuje heterogenitu spektra LBCL a zabraňuje nesprávnemu používaniu kategórie BCLU. Genetické abnormality menia pôvodné vlastnosti a aj vplyv miesta vzniku LBCL a zároveň dávajú možnosť, aby boli títo pacienti liečení inými, agresívnejšími liečebnými schémami.
V súvislosti so stratifikáciou a subtypizáciou DLBCL ešte treba spomenúť imunohistochemickú analýzu expresie proteínov c-myc, bcl-2 a bcl-6, na základe ktorých možno identifikovať tzv. c-myc+/bcl-2+ dvojité (DE), resp. c-myc+/bcl-2+/bcl-6+ trojité expresory (TE) (12). Fenomén DE/TE je nepriaznivým prognostickým faktorom, ktorý je nezávislý na COO klasifikácii. Treba však zdôrazniť, že DE/TE nie sú ekvivaletné DH/TH lymfómom a ani nepredstavujú samostatnú diagnostickú kategóriu DLBCL (13,14). Sú omnoho častejšie než DH/TH lymfómy, pričom v korelácii s morfológiou môžu slúžiť na skríning kandidátov vhodných na FISH analýzu.
Od DLBCL a HGBL treba odlíšiť prípady Burkittovho lymfómu (BL), ktorý je pomerne jednoznačne definovaným vysoko agresívnym B-lymfómom, geneticky charakterizovaným nekomplexnými abnormalitami karyotypu a prestavbou génu MYC vo väčšine prípadov (15). WHO klasifikácia z roku 2008 pripúšťala existenciu cca 10 % prípadov BL bez prestavby MYC génu (2). Otázka, či skutočne takéto BL existujú, nie je uzavretá. Ak existujú, potom je otázne, či ide o skutočnú negativitu alebo len neschopnosť dôkazu prestavby. V časti prípadov, ktoré morfologicky, fenotypovo a aj na základe GEP vyzerajú ako BL, ale nemajú detegovateľnú prestavbu MYC génu, boli dokázané abnormality v oblasti 11q, na čo revízia WHO klasifikácie zareagovala uvedením novej provizórnej kategórie BL-podobných („Burkitt-like“) lymfómov s abnormalitami 11q (3). Tie na rozdiel od typických BL spravidla vykazujú komplexnejšie abnormality karyotypu, nižšiu expresiu c-myc a častejšie sa vyskytujú u pediatrických pacientov (16).
Spektrum novozavedených blastických B-lymfómov nakoniec uzatvára provizózna kategória veľkobunkových B-lymfómov s prestavbou IRF4, ktoré boli odseparované od blastických FL a DLBCL (3). Ide o ďalšiu geneticky definovanú zriedkavú entitu, ktorá sa spravidla vyskytuje u detí a mladých dospelých v podobe lokalizovaného ochorenia v oblasti Waldayerovho okruhu alebo krčných lymfatických uzlín (LU). Tieto lymfómy môžu mať folikulový alebo difúzny rast, čím pripomínajú FL G3 alebo DLBCL. Majú GCB fenotyp a zvyčajne exprimujú MUM-1 a bcl-6, bez prestavby BCL2 génu. Vyžadujú liečbu, ale majú dobrú prognózu (17).
Príkladom B-bunkových neoplázií, u ktorých lepšie poznanie ich genetického pozadia prispelo k zlepšeniu a spresneniu diagnostiky sú vlasatobunková leukémia (HCL) a lymfoplazmocytový lymfóm (LPL) / Waldenströmova makroglobulinémia (WM). Tie boli totiž v minulosti považované za lymfoidné neoplázie bez špecifických či diagnosticky nápomocných genetických zmien (2). Dnes je známe, že takmer vo všetkých prípadoch klasickej HCL sa vyskytujú mutácie génu BRAF V600E (18). Tie sa tak stali dôležitým diagnostickým kritériom, ako aj efektívnym nástrojom na monitoring ochorenia po liečbe. Mutácie BRAF sa nevyskytujú u tzv. variantnej HCL (HCL-v), ktorá patrí spolu s difúznym splenickým malobunkovým B-lymfómom červenej pulpy do kategórie neklasifikovateľných splenických lymfómov a leukémií, ktoré si aj naďalej zachovávajú status provizórnej kategórie. Raritné prípady BRAF-negatívnej HCL, ktoré exprimujú gény IGVH 4-34, mávajú mutácie MAP2K1, ktoré sa tiež vyskytujú asi v polovici prípadov HCL-v (19).
V LPL/WM bola identifikovaná mutácia MYD88 L265P, ktorá sa vyskytuje vo viac ako 90 % prípadov (20). Táto mutácia síce nie je špecifická pre LPL/WM, pretože v nízkej frekvencii bola dokázaná aj u iných malobunkových i blastických B-lymfómov, ale je charakteristická a nevyskytuje sa pri plazmocytovom myelóme (21). Stala sa tak veľmi nápomocným nástrojom v rámci diferenciálnej diagnostiky plazmocytovo diferencovaných B-lymfómov, a to nielen v tkanive LU. Navyše, mutácia sa vyskytuje aj v prípadoch IgM+ monoklonovej gamapatie nejasného významu (MGUS), čím napomohla objasneniu jej úzkeho vzťahu k LPL/WM. Ďalšou genetickou abnormalitou, dokázanou v časti prípadov LPL (cca 30 %) a IgM+ MGUS (cca 20%), sú mutácie génu CXCR4 (22).
Lepšie poznanie molekulovej patogenézy ovplyvnilo aj pohľad na lymfóm z plášťových buniek (MCL). I keď vačšina MCL svojim klinickým správaním patrí medzi agresívne lymfómy, je známe, že ojedinelé prípady sa vyznačujú aspoň určitý čas neagresívnym, dokonca bezpríznakovým priebehom. Tieto sa označujú ako indolentné MCL (iMCL) (23). Podľa súčasných predstáv MCL môže vzniknúť dvomi cestami. Prvou je cesta vzniku konvenčných MCL, vznikajúcich z naivných B-buniek plášťovej zóny lymfatických folikulov bez, resp. len s minimálnymi mutáciami IGVH génov. Tieto prípady sú spravidla SOX-11+ a vyskytujú sa ako nodálne alebo extranodálne, konvenčné, blastoidné či pleomorfné MCL. Druhou cestou je cesta cez zárodočné centrá lymfatických folikulov, kde dochádza k hypermutáciám IGVH génov, pričom tieto prípady sú spravidla SOX-11-. Takto by mali vznikať prípady iMCL, ktoré sa často prezentujú ako leukemické, mimouzlinové či primárne splenické, postihujúce periférnu krv (PK), kostnú dreň (KD) a často aj slezinu (24,25). Vplyvom evolúcie sekundárnych genetických abnormalít však aj tieto indolentné formy môžu časom progredovať. Navyše ich treba odlíšiť od leukemicky prebiehajúcich klasických MCL, kde je naopak leukemizácia nepriaznivým znakom. Z literatúry je tiež známe, že pravdepodobne existujú aj nodálne SOX-11+ prípady MCL, ktoré sa môžu aspoň nejaký čas správať indolentne (26). Táto možnosť však v publikovanej verzii WHO klasifikácie komentovaná nie je, s výnimkou zmienky, že aj klasické MCL s nízkou proliferačnou frakciou sa môžu správať relatívne indolentne (3).
Kategóriou B-bunkových neoplázií, v rámci ktorej došlo k významnej úprave diagnostických kritérií, je spektrum chronickej lymfocytovej leukémie (CLL) a malobunkového B-lymfocytového lymfómu (SLL). Je známe, že CLL by nemala byť bioptickou diagnózou, s čím však nekorešponduje klinická prax, kedy je stále veľká časť prípadov CLL diagnostikovaná na základe bioptického vyšetrenia tkaniva LU alebo KD (27). Diagnostika CLL je postavená na tzv. IWCLL kritériách z roku 2008, z ktorých nepriamo vyplýva, že diagnózu CLL je možné stanoviť aj v prípadoch s nádorovou lymfocytózou pod 5.109/l, ak sú súčasne prítomné cytopénie a/alebo symptómy asociované s CLL (28). Podľa revidovanej WHO klasifikácie 2016 by to už nemalo byť možné a prípady s lymfocytózou pod 5.109/l by mali byť klasifikované ako monoklonová B-lymfocytóza (MBL) (3). Aby sa však zohľadnila heterogenita prípadov MBL vo vzťahu k podielu klonálnych lymfocytov v PK, mala by sa rozlišovať MBL s nízkym („low count“) a vysokým počtom buniek („high count“) (29). MBL s nízkym počtom buniek, definovaná podielom CLL lymfocytov pod 0,5.109/l, je stav s extrémne nízkym rizikom progresie do CLL, preto títo pacienti nevyžadujú sledovanie. MBL s vysokým počtom buniek, definovaná podielom CLL lymfocytov nad 0,5.109/l, je ochorenie s vlastnosťami podobnými nízkemu štádiu CLL (Rai 0), od čoho sa odvíja aj klinický manažment a pravidelné kontroly pacientov, pričom klinicky významnou je spravidla až lymfocytóza nad 2.109/l. Dnes je známe, že MBL virtuálne predchádza všetky prípady CLL/SLL. Okrem MBL s typickým CD5+/CD23+ fenotypom existujú aj MBL s fenotypom atypickej CLL či raritne iným než CLL, napr. podobným lymfómom z B-buniek marginálnej zóny (MZBL) či MCL.
WHO klasifikácia 2016 venuje pozornosť aj pseudofolikulom (PSF) a tiež sa vyjadruje k novým molekulovým abnormalitám CLL. PSF predstavujú štruktúry, v ktorých dochádza k proliferácii nádorových buniek a k interakciám s bunkami mikroprostredia. PSF tak predstavujú štruktúry s veľkým významom pre patogenézu ochorenia, proliferáciu a prežívanie nádorových buniek a evolúciu rezistencie. Expandované PSF sú považované za nepriaznivý prognostický znak odrážajúci aktivitu ochorenia (30). Bunky v PSF môžu koexprimovať cyklín-D1 a c-myc, ale bez prestavby príslušných génov (31). Nové genetické abnormality ako sú mutácie génov NOTCH1, SF3B1 a BIRC3, boli identifikované pomocou sekvenačných analýz. Ani jedna z nich pravdepodobne nie je spúšťajúcou mutáciou CLL, ale podľa štúdií sú asociované s nepriaznivou prognózou (32,33). Keďže ich individuálna frekvencia je pomerne nízka, vyšetrovanie náročné a výsledok neovplyvňuje samotnú diagnózu ani liečbu, WHO neodporúča ich analýzu v rutinnej praxi (3). Najvýznamnejšími molekulovými abnomalitami, ktoré majú vplyv na terapeutický manažment pacientov s CLL, tak aj naďalej ostávajú len abnomality TP53/17p13.
Zmeny zasiahli aj kategóriu najčastejšieho indolentného B-bunkového lymfómu, ktorým je folikulový lymfóm (FL). Pôvodné úvahy, že blastický FL G3B bude odčlenený od FL G1-3A sa nenaplnili a spoločným menovateľom týchto lymfómov aj naďalej ostáva nodulárny rast a pôvod v bunkách zárodočných centier lymfatických folikulov. Klasický nodálny FL pravdepodobne ostáva bez významnejších zmien (3). Zmeny, ktoré sa pri FL udiali, sa preto týkajú predovšetkým existencie zriedkavých foriem. V roku 2008 bola zavedená provizórna jednotka s názvom pediatrický FL (2). Keďže v praxi sa jej zavedenie ukázalo ako opodstanené, po novom sa mení na definitívnu a súčasne sa mení jej názov na FL pediatrického typu (PTFL) (3). Tieto zmeny zohľadňujú fakt, že prípady s podobnými vlastnosťami sa nevyskytujú len u detí, ale raritne aj u adolescentov či dospelých. Postihujú najčastejšie LU v oblasti hlavy a krku, morfologicky pripomínajú explozívnu reaktívnu folikulovú hyperpláziu, majú vysokú proliferačnú aktivitu a zvyčajne blastoidnú morfológiu, ktorá nezriedka vedie k problémom s gradingom. Môžu exprimovať bcl-2, ale spravidla nevykazujú prestavbu génu BCL2 ani BCL6 či MYC. Ide o prípady s veľmi dobrou prognózou, kde častokrát postačuje chirurgická excízia, preto sú niektorými autormi považované za benígne klonálne lymfoproliferácie s nízkym malígnym potenciálom (34). Najmä u dospelých pacientov treba dávať pozor na ich možnú zámenu s blastickým FL G3 a odlíšiť ich treba aj od prípadov veľkobunkových B-lymfómov s prestavbou IRF4 génu, ktoré boli spomenuté vyššie.
Implementované zmeny ďalej akcentujú význam miesta vzniku FL. Ten je väčšinou lymfóm nodálny, vznikajúci v LU, no existujú aj prípady primárne extranodálne, niektoré s unikátnymi vlastnosťami. Do spektra primárne extranodálnych FL sa okrem už dobre známeho primárneho kožného FL (PCFCL), vyčleneného už v minulosti, po novom pridáva aj tzv. duodenálny typ FL tenkého čreva (3). Ide o veľmi zriedkavé ochorenie, spravidla postihujúce duodenum, jejunum, raritne i žalúdok, ktoré je odlišné od iných FL postihujúcich tráviaci trakt. Ide o sliznicovú lymfoproliferáciu, ktorá je spravidla bcl-2+ a má prestavbu BCL2 génu, no jej správanie je neagresívne, s veľmi nízkym rizikom diseminácie (35).
WHO 2016 venuje pozornosť aj nízkomalígnym FL s črtami difúneho rastu, ktoré sú väčšinou diagnosticky problematické. Nakoľko v časti týchto prípadov bola dokázaná delécia 1p36, táto sa považuje za možnú príčinu uvedeného fenoménu. To sa premietlo do zavedenia novej provizórnej jednotky, tzv. predominantne difúzneho FL s deléciou 1p36. Tieto lymfómy sa najčastejšie vyskytujú v inguinálnej oblasti, kde zvyčajne tvoria rozsiahlu lokalizovanú tumoróznu masu. Nevykazujú prestavbu BCL2 génu (36). Uvedený difúzny rast treba odlíšiť od difúzne rastúcich blastických FL, kde táto črta znamená transformáciu do blastického lymfómu, najčastejšie DLBCL.
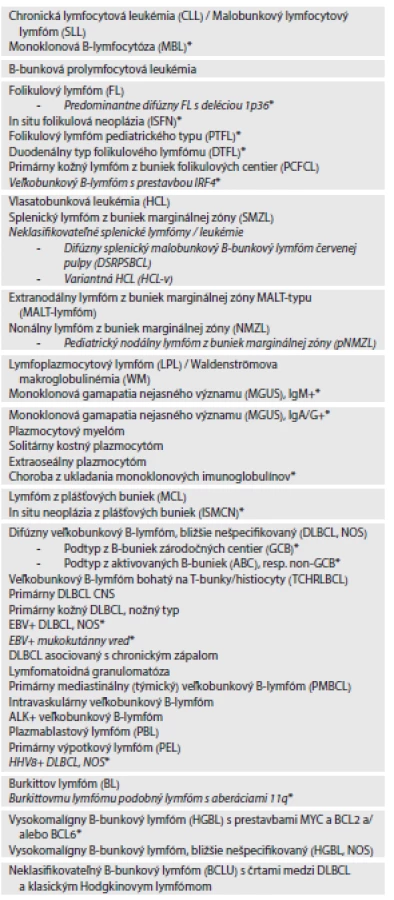

Nakoniec treba ešte spomenúť včasné B-bunkové lézie. Okrem už opísanej MBL sa WHO klasifikácia 2016 venuje aj iným včasným „prelymfomatóznym“ léziám ako sú FL „in situ“ (FLIS) a MCL „in situ“ (MCLIS), ktoré boli uvedené v roku 2008 (2). V rámci dnešného konzervatívnejšieho prístupu k týmto léziám sa mení ich názov na „in situ“ folikulovú neopláziu (ISFN) a „in situ“ neopláziu z plášťových buniek (ISMCN), čím sa z názvu vypúšťa označenie lymfóm (3). Praktická využiteľnosť týchto jednotiek je v skutočnosti nízka, nakoľko v oboch prípadoch ide o extrémne raritné, incidentálne lézie, definované v prvom prípade nálezom bcl-2+ buniek s prestavbou BCL2 génu v izolovaných či ojedinelých zárodočných centrách a v druhom prípade cyklín-D1+ buniek s prestavbou CCND1 génu vo vnútornej vrstve lymfatických folikulov inak reaktívne vyzerajúcich LU. Ide o lézie s veľmi nízkym rizikom progresie do rozvinutého FL či MCL, ktoré treba odlíšiť od parciálnej infiltrácie LU (37,38). MCLIS môže byť diseminovaná lézia a môže sa vyskytovať aj v asociácii s inými lymfómami.
MZBL, splenické lymfómy iné než HCL a plazmocytový myelóm či iné plazmocytové neoplázie doteraz publikovaná verzia WHO klasifikácie 2016 nekomentuje, čiže prípadné zmeny v týchto kategóriách zatiaľ nie sú známe (3).
ZRELÉ (PERIFÉRNE) T-BUNKOVÉ NEOPLÁZIE
T-bunkové neoplázie v našej populácii síce patria k pomerne zriedkavým (cca 10-15 %), no fakt, že často ide o lézie diagnosticky problematické a prognosticky zlé, zvyšuje nielen záujem, ale aj potrebu reorganizácie tejto extrémne heterogénnej skupiny lymfómov. Žiaľ mnoho avizovaných, resp. skôr navrhovaných zmien je postavených na analýze nových genetických abnormalít, pochádzajúcich z GEP alebo sekvenačných analýz (39), ktorých vyšetrovanie je náročné a pre rutinnú prax nedostupné, čo limituje až znemožňuje ich praktické využitie, ako to priznáva aj samotná WHO (3). Nepodarilo sa dotiahnuť ani zvažovanú klasifikáciu T-bunkových neoplázií na základe ich bunkového pôvodu. V konečnom dôsledku sa tak revízia klasifikácie T-bunkových neoplázií javí ako dosť neprehľadná, najmä dovtedy, pokým nebude známe jej kompletné znenie v podobe monografie.
K najdôležitejším zmenám v skupine T-bunkových neoplázií určite patrí vyjasnenie postavenia kategórie ALK-negatívneho anaplastického veľkobunkového lymfómu (ALCL), ktorý bol v roku 2008 zavedený ako provizórna jednotka na jeho odlíšenie od ostatných CD30+ bližšie nešpecifikovaných periférnych T-bunkových lymfómov (PTCL, NOS) (2). Po revízii z roku 2016 sa jeho postavenie zmenilo na definitívne, pretože jeho odseparovanie od ALK-pozitívneho (ALK+) ALCL sa ukázalo ako správny krok (3). Ide totiž o prognosticky horšiu formu ALCL, i keď samotné prežívanie ALK-negatívneho ALCL je veľmi variabilné. Po novom sem svetlo vnáša opäť genetika, keďže ALK-negatívny ALCL je geneticky heterogénna entita (40). Prípady s abnormalitami v oblasti 6p25 (DUSP22, IRF4) sú monomorfnejšie a svojim správaním podobné prognosticky lepšiemu ALK+ ALCL. Naopak prípady s abnormalitami TP63 sú prognostiky najhoršie. Prípady ALK-negatívneho ALCL bez uvedených abnormalít, označované aj ako „triple“ negatívne, sú prognosticky intermediárne (41). Abnormality 6p25 boli identifikované aj v časti prípadov primárneho kožného ALCL (cALCL) a lymfomatoidnej papulózy (LyP).Spektrum osobitných foriem ALCL sa rozrástlo o tzv. ALCL asociovaný s prsníkovými implantátmi, označovaný aj ako ALK-negatívny ALCL asociovaný s výpotkom či serómom (3). Ide o extrémne raritné ochorenie, opísané v prípadoch priemerne 10 rokov po aplikovaní prsníkových implantátov, ktoré v prípade, že je lokalizované a neinvaduje kapsulu vytvorenú okolo implantátu, má excelentnú prognózu (42).
Významnou zmenou je reorganizácia kategórie a spresnenie diagnostických kritérií intestinálnych T-lymfómov, najmä pôvodného T-bunkového lymfómu asociovaného s enteropatiou (EATL), ktorý bol v minulosti rozdeľovaný na typ I a II (2,3). Po novom sa typ I, tzv. klasický polymorfný, ktorý je asociovaný s celiakiou a častejší v Severnej Európe, označuje proste ako T-bunkový lymfóm asociovaný s enteropatiou (EATL). Typ II, tzv. monomorfný, sa po novom označuje ako monomorfný epiteliotropný intestinálny T-bunkový lymfóm (MEITL). Tento je častejší u Aziatov a Hispáncov, nie je asociovaný s enteropatiou, je často CD8+ a CD56+ a na rozdiel od EATL častejšie vykazuje klonálnu prestavbu γδ reťazcov T-bunkového receptora (TCR) (43,44). Spektrum črevných T-lymfoproliferácií sa ďalej rozrástlo o indolentné T a NK-bunkové lymfoproliferatívne ochorenia GIT-u, čo je provizórna kategória, u ktorej došlo k zámernému vypusteniu označenia lymfóm. Jedná sa totiž o raritné indolentné proliferácie z T alebo NK-buniek. Tie tvoria blandne vyzerajúce povrchové infiltráty z malých lymfocytov s nízkou proliferačnou aktivitou. Sú však klonálne a niektoré môžu časom progredovať, ich ideálny klinický manažment však nie je určený (45).
Skutočnou výzvou pre autorov WHO klasifikácie bola snaha o reorganizáciu najväčšej kategórie T-bunkových neoplázií, ktorými sú bližšie nešpecifikované PTCL, NOS. Tie sú spravidla vnímané ako veľký odpadkový kôš, kam sa zaraďuje veľká časť (>60 %) PTCL, či už z objektívnych dôvodov, kedy nespĺňajú kritériá iných definovaných jednotiek, alebo z dôvodu nemožnosti vykonania všetkých fenotypových či genetických analýz. V rámci snahy vyčleniť z tejto skupiny niektoré entity bol napríklad vytvorený koncept nodálnych T-lymfómov s fenotypom pomocných folikulových (TFH) buniek (3). Keďže tieto lymfómy vykazujú podobný fenotyp a genetické abnormality ako angioimunoblastový T-lymfóm (AITL), výsledkom bolo ich zoskupenie do kategórie non-kutánnych PTCL s TFH-fenotypom, ktorý je definovaný expresiou prinajmenej 2 znakov zahŕňajúcich PD-1, CD10, bcl-6, CXCL3, ICOS, SAP a CCR5 (46). Do takto vytvoreného spektra sa zaraďujú okrem AITL po novom aj prípady folikulového T-bunkového lymfómu (FTCL) a ostatných nodálnych PTCL s TFH-fenotypom, pričom dve posledne menované kategórie boli zavedené ako provizórne (3). AITL a FTCL sú okrem neoplastických TFH-buniek známe aj prítomnosťou EBV+ B-blastov pripomínajúcich Hodgkinove alebo Sternbergove-Reedovej bunky. FTCL sa častejšie vyskytujú ako lokalizované ochorenie (47).
Skupinu PTCL, NOS možno stratifikovať na základe GEP na 3 skupiny podľa expresie génov skupiny GATA3, TBX21 a cytotoxických génov, čo by sa malo dať identifikovať aj imunohistochemickou detekciou príslušných proteínov. Prípady s expresiou génov TBX21 klastra sú považované za prognosticky lepšie, avšak realizovateľnosť tejto stratifikácie v praxi je sporná (39).
Z ďalších zmien v kategórii T-bunkových lymfoproliferácií možno spomenúť spresnené kritériá primárneho kožného γδ+ T-bunkového lymfómu, ktoré by mali napomôcť jeho ľahšiemu odlíšeniu od iných γδ+ T-bunkových neoplázií ako sú mycosis fungoides (MF) či LyP (48). Okrem primárneho kožného CD8+ agresívneho epidermotropického cytotoxického T-lymfómu bola zavedená nová provizórna jednotka tzv. primárneho kožného akrálneho CD8+ T-bunkového lymfómu, ktorý predstavuje spravidla lokalizované ochorenie kože, najčastejšie v oblasti ucha, ktoré sa správa indolentne a na jeho liečbu postačuje excízia (49). V rámci spektra kožných T-lymfoproliferácií sa ďalej zmenil názov primárneho kožného CD4+ T-lymfómu z malých/stredných T-buniek na primárne kožné CD4+ T-lymfoproliferatívne ochorenie z malých/stredných T-buniek, čo vyplýva z už spomenutého konzervatívnejšieho prístupu k niektorým indolentným procesom (50). Indolentné T-lymfoproliferatívne ochorenia GIT-u, primárny kožný akrálny CD8+ T-bunkový lymfóm, ako aj ALCL asociovaný s prsníkovými implantátmi, sa zaraďujú do skupiny T-lymfómov z cytotoxických CD8+ T-buniek, ktoré majú indolentný klinický priebeh.
ZÁVER
Záverečné konštatovanie uvedené v prehľade myeloidných neoplázií v zásade platí aj pre tie lymfoidné. Uvedený text je len zostručneným prehľadom informácií, ktoré boli doposiaľ zverejnené a publikované autormi WHO klasifikácie v súvislosti s jej revíziou. Plné znenie klasifikácie a teda všetky zavedené zmeny tak budú známe až po jej knižnom vydaní. Dovtedy sa treba spoliehať na údaje publikované v jednotlivých originálnych článkoch, ktorých formálna záväznosť je otázna, avšak hematopatológmi všeobecne akceptovaná. Ostáva len veriť, že naša séria článkov prispeje k pochopeniu aktuálnej situácie a pomôže zorientovať sa v nastupujúcich zmenách, a to nielen patológom, ale aj klinikom, ktorí možu byť frustrovaní častými zmenami klasifikácie.
PREHLASENIE
Autor práce prehlasuje, že v súvislosti s témou, vznikom a publikácií tohto článku nie v konflikte záujmov a vznik ani publikácia článku neboli podporené žiadnou farmaceutickou firmou. Toto prehlasenie sa týka i všetkých spoluautorov.
Adresa pre korešpondenciu:
MUDr. Tomáš Balhárek
ÚPA JLF UK a UNM
Kollárova 2,
03659 Martin,
Slovensko
tel.: +421 43 4133002,
fax: +421 43 4203370
e-mail: balharek@jfmed.uniba.sk
Zdroje
1. Cazzola M. Introduction to a review series: The 2016 revision of the WHO classification of tumors of hematopoietic and lymphoid tissues. Blood 2016; 127(20): 2361-2364.
2. Swerdlow SH, Campo E, Harris NL et al, eds. WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues. IARC Press: Lyon, 2008.
3. Swerdlow SH, Campo E, Pileri SA et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016; 127(20): 2375-2390.
4. Scott DW, Wright GW, Williams PM et al. Determining cell-of-origin subtypes of diffuse large B-cell lymphoma using gene expression in formalin-fixed paraffin-embedded tissue. Blood 2014; 123(8): 1214-1217.
5. Hans CP, Weisenburger DD, Greiner TC et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood 2004; 103(1): 275-282.
6. Roschewski M, Staudt LM, Wilson WH. Diffuse large B-cell lymphoma-treatment approaches in the molecular era. Nat Rev Clin Oncol 2014; 11(1): 12-23.
7. Nicolae A, Pittaluga S, Abdullah S et al. EBV-positive large B-cell lymphomas in young patients: a nodal lymphoma with evidence for a tolerogenic immune environment. Blood 2015; 126(7): 863-872.
8. Dojcinov SD, Venkataraman G, Raffeld M et al. EBV positive mucocutaneous ulcer - a study of 26 cases associated with various sources of immunosuppression. Am J Surg Pathol 2010; 34(3): 405-417.
9. Swerdlow SH. Diagnosis of „double hit“ diffuse large B-cell lymphoma and B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and Burkitt lymphoma: when and how, FISH versus IHC. Hematology Am Soc Hematol Educ Program 2014; 2014(1): 90-99.
10. Johnson NA, Savage KJ, Ludkovski O et al. Lymphomas with concurrent BCL2 and MYC translocations: the critical factors associated with survival. Blood 2009; 114(11): 2273-2279.
11. Campo E. MYC in DLBCL: partners matter. Blood 2015; 126(22): 2439-2440.
12. Johnson NA, Slack GW, Savage KJ et al. Concurrent expression of MYC and BCL2 in diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine and prednisone. J Clin Oncol 2012; 30(28): 3452-3459.
13. Horn H, Ziepert M, Becher C et al. MYC status in concert with BCL2 and BCL6 expression predicts outcome in diffuse large B-cell lymphoma. Blood 2013; 121(12): 2253-2263.
14. Green TM, Young KH, Visco C et al. Immunohistochemical double-hit score is a strong predictor of outcome in patients with diffuse large B-cell lymphoma treated with Rituximab plus Cyclophosphamide, Doxorubicin, Vincristine and Prednisone. J Clin Oncol 2012; 30(28): 3460-3467.
15. Schmitz R, Young RM, Ceribelli M et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 2012; 490(7418): 116-120.
16. Ferreiro JF, Morscio J, Dierickx D et al. Post-transplant molecularly defined Burkitt lymphomas are frequently MYC-negative and characterized by the 11q-gain/loss pattern. Haematologica 2015; 100(7): 275-279.
17. Salaverria I, Philipp C, Oschlies I et al. Molecular mechanisms in Malignant lymphomas network project of the Deutsche Krebshilfe; German high-grade lymphoma study group; Berlin-Frankfurt-Munster-NHL Trial Group. Translocations activating IRF4 identify a subtype of germinal center-derived B-cell lymphoma affecting predominantly children and young adults. Blood 2011; 118(1): 139-147.
18. Tiacci E, Trifonov V, Schiavoni G et al. BRAF mutations in hairy-cell leukemia. N Engl J Med 2011; 364(24): 2305-2315.
19. Waterfall JJ, Arons E, Walker RL et al. High prevalence of MAP2K1 mutations in variant and IGHV4-34-expressing hairy-cell leukemias. Nat Genet 2014; 46(1): 8-10.
20. Treon SP, Xu L, Yang G et al. MYD88 L265P somatic mutation in Waldenström’s macroglobulinemia. N Engl J Med 2012; 367(9): 826-833.
21. Swerdlow SH, Kuzu I, Dogan A et al. The many faces of small B cell lymphomas with plasmacytic differentiation and the contribution of MYD88 testing. Virchows Arch 2016; 468(3): 259-275.
22. Roccaro AM, Sacco A, Jimenez C et al. C1013G/CXCR4 acts as a driver mutation of tumor progression and modulator of drug resistance in lymphoplasmacytic lymphoma. Blood 2014; 123(26): 4120-4131.
23. Furtado M, Rule S. Indolent mantle cell lymphoma. Haematologica 2011; 96(8): 1086–1088.
25. Jares P, Colomer D, Campo E. Molecular pathogenesis of mantle cell lymphoma. J Clin Invest 2012; 122(10): 3416-3423.
26. Bea S, Valdes-Mas R, Navarro A et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci 2013; 110(45): 18250-18255.
27. Nodit L, Bahler DW, Jacobs SA, et al. Indolent mantle cell lymphoma with nodal involvement and mutated immunoglobulin heavy chain genes. Hum Pathol 2003; 34(10): 1030-1034.
28. Balhárek T, Plank L. Úloha patológa v manažmente chronickej lymfocytovej leukémie. Onkológia 2014; 9(6): 365–370.
29. Hallek, M., Cheson, B.D., Catovsky, D. et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute – Working Group 1996 guidelines. Blood 2008; 111(12): 5446-5456.
30. Rawstron AC, Shanafelt T, Lanasa MC et al. Different biology and clinical outcome according to the absolute numbers of clonal B-cells in monoclonal B-cell lymphocytosis (MBL). Cytometry B Clin Cytom 2010; 78(suppl 1): S19-S23.
31. Gine E, Martinez A, Villamor N et al. Expanded and highly active proliferation centers identify a histological subtype of chronic lymphocytic leukemia (“accelerated” chronic lymphocytic leukemia) with aggressive clinical behavior. Haematologica 2010; 95(9): 1526-1533.
32. Gradowski JF, Sargent RL, Craig FE et al. Chronic lymphocytic leukemia/small lymphocytic lymphoma with cyclin D1 positive proliferation centers do not have CCND1 translocations or gains and lack SOX11 expression. Am J Clin Pathol 2012; 138(1): 132-139.
33. Rossi D, Rasi S, Spina V et al. Integrated mutational and cytogenetic analysis identifies new prognostic subgroups in chronic lymphocytic leukemia. Blood 2013; 121(8): 1403-1412.
34. Baliakas P, Hadzidimitriou A, Sutton LA et al. European Research Initiative on CLL (ERIC). Recurrent mutations refine prognosis in chronic lymphocytic leukemia. Leukemia 2015; 29(2): 329-336.
35. Louissaint A, Ackerman AM, Dias-Santagata D et al. Pediatric-type nodal follicular lymphoma: an indolent clonal proliferation in children and adults with high proliferation index and no BCL2 rearrangement. Blood 2012; 120(12): 2395-2404.
36. Schmatz AI, Streubel B, Kretschmer-Chott E et al. Primary follicular lymphoma of the duodenum is a distinct mucosal/submucosal variant of follicular lymphoma: a retrospective study of 63 cases. J Clin Oncol 2011; 29(11): 1445-1451.
37. Katzenberger T, Kalla J, Leich E et al. A distinctive subtype of t(14;18)-negative nodal follicular non-Hodgkin lymphoma characterized by a predominantly diffuse growth pattern and deletions in the chromosomal region 1p36. Blood 2009; 113(5): 1053-1061.
38. Jegalian AG, Eberle FC, Pack SD et al. Follicular lymphoma in situ: clinical implications and comparisons with partial involvement by follicular lymphoma. Blood 2011; 118(11): 2976-2984.
39. Carvajal-Cuenca A, Sua LF, Silva NM et al. In situ mantle cell lymphoma: clinical implications of an incidental finding with indolent clinical behavior. Haematologica 2012; 97(2): 270-278.
40. Iqbal J, Wright G, Wang C et al. Lymphoma Leukemia Molecular Profiling Project and the International Peripheral T-cell Lymphoma Project. Gene expression signatures delineate biological and prognostic subgroups in peripheral T-cell lymphoma. Blood 2014; 123(19): 2915-2923.
41. Parilla Castellar ER, Jaffe ES, Said JW et al. ALK-negative anaplastic large cell lymphoma is a genetically heterogeneous disease with widely disparate clinical outcomes. Blood 2014; 124(9): 1473-1480.
42. Agnelli L, Mereu E, Pellegrino E et al. European T-Cell Lymphoma Study Group. Identification of a 3-gene model as a powerful diagnostic tool for the recognition of ALK-negative anaplastic large cell lymphoma. Blood 2012; 120(6): 1274-1281.
43. Miranda RN, Aladily TN, Prince HM et al. Breast implant-associated anaplastic large-cell lymphoma: long-term follow-up of 60 patients. J Clin Oncol 2014; 32(2): 114-120.
44. Deleeuw RJ, Zettl A, Klinker E et al. Whole genome analysis and HLA genotyping of enteropathy-type T-cell lymphoma reveals 2 distinct lymphoma subtypes. Gastroenterology 2007; 132(5): 1902-1911.
45. Chan JK, Chan AC, Cheuk W et al. Type II enteropathy-associated T-cell lymphoma: a distinct aggressive lymphoma with frequent γδ T-cell receptor expression. Am J Surg Pathol 2011; 35(10): 1557-1569.
46. Perry AM, Warnke RA, Hu Q et al. Indolent T-cell lymphoproliferative disease of the gastrointestinal tract. Blood 2013; 122(22): 3599-3606.
47. Lemonnier F, Couronne L, Parrens M et al. Recurrent TET2 mutations in peripheral T-cell lymphomas correlate with TFH-like features and adverse clinical parameters. Blood 2012; 120(7): 1466-1469.
48. Nicolae A, Pittaluga S, Venkataraman G et al. Peripheral T-cell lymphomas of follicular T-helper cell derivation with Hodgkin/Reed-Sternberg cells of B-cell lineage: both EBVpositive and EBV-negative variants exist. Am J Surg Pathol 2013; 37(6): 816-826.
49. Guitart J, Weisenburger DD, Subtil A et al. Cutaneous gd T-cell lymphomas: a spectrum of presentations with overlap with other cytotoxic lymphomas. Am J Surg Pathol 2012; 36(11): 1656-1665.
50. Petrella T, Maubec E, Cornillet-Lefebvre P et al. Indolent CD8-positive lymphoid proliferation of the ear: a distinct primary cutaneous T-cell lymphoma? Am J Surg Pathol 2007; 31(12): 1887-1892.
51. Garcia-Herrera A, Colomo L, Camos M et al. Primary cutaneous small/medium CD4+ T-cell lymphomas: a heterogeneous group of tumors with different clinicopathologic features and outcome. J Clin Oncol 2008; 26(20): 3364-3371.
Štítky
Patologie Soudní lékařství ToxikologieČlánek vyšel v časopise
Česko-slovenská patologie

2017 Číslo 3
Nejčtenější v tomto čísle
- Uzlinová metastáza karcinomu z Merkelových buněk bez kožního primárního ložiska – kazuistické sdělení
- Zmeny WHO klasifikácie lymfoidných neoplázií v kontexte revízie z roku 2016
- JAN JESENSKÝ - JESSENIUS (1566 - 1621)
- Izolovaná infekční endokarditida pulmonální chlopně: kazuistika
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy