
Rothmundův-Thomsonův syndrom sdružený s anaplastickým velkobuněčným T lymfomem – popis případu
Rothmund-Thomson Syndrome Associated with Anaplastic Large Cell T-lymphoma: Case Report
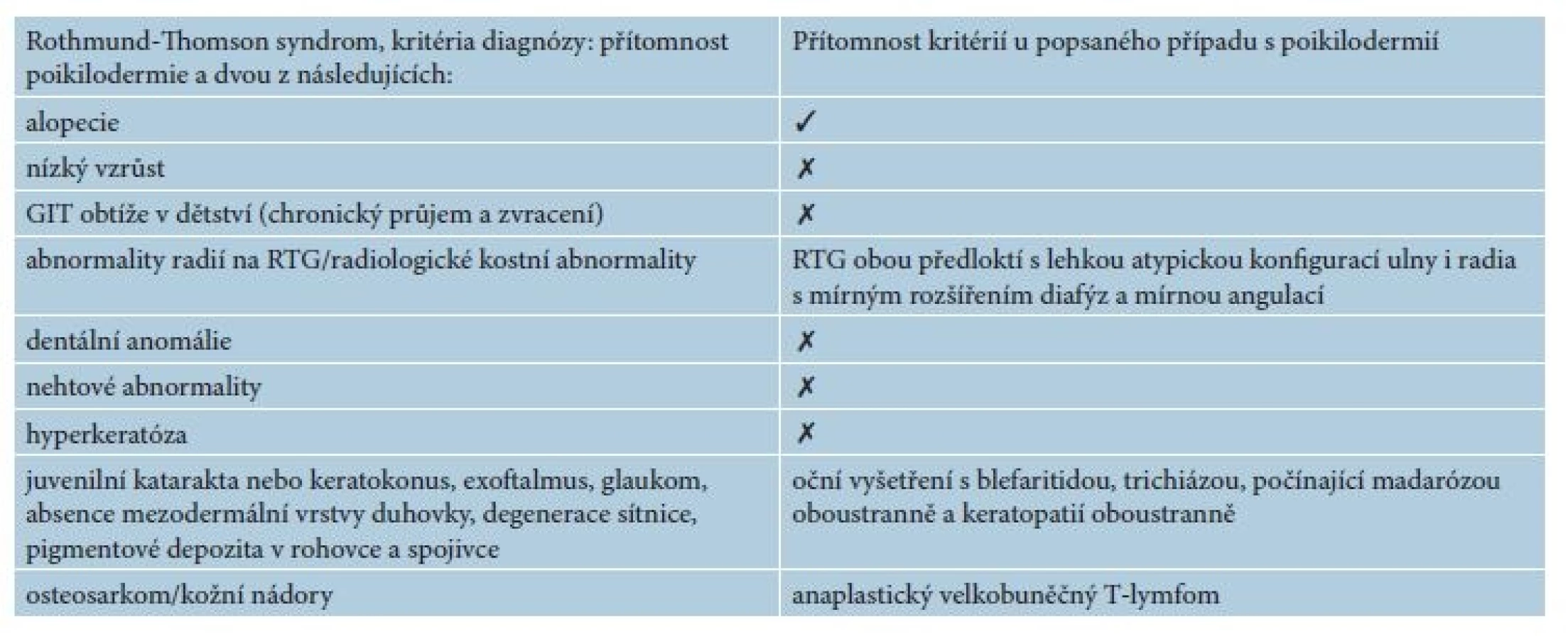
Rothmund-Thomson syndrome (RTS) is characterized by poikiloderma, sparse hair, eyelashes, and/or eyebrows, small stature, skeletal and dental abnormalities, cataracts and an increased risk of cancer, especially osteosarcoma. Authors describe a case of 35 year old man with poikiloderma, universal alopecia and anaplastic large T-cell lymphoma (ALCL). The skin manifestation first appeared at age of 23 years of the patient. Diagnosis of RTS was based on the clinical manifestation and genetic examination, that showed RECQ4 mutations presented at multiple levels. Sequencing of fibroblasts by Western blot analysis confirmed Gln253His mutation typical for RTS. Despite combined chemotherapy of ALCL the patient died 17 days after its initiation due to cardiac arrest. RTS is a rare genodermatosis. Because of increased risk of skin cancer, follow-up of patients with RTS is mandatory.
Key words:
poikiloderma – Rothmund-Thomson syndrome – anaplastic large T-cell lymphoma
Autoři:
S. Vachatová 1; M. Salavec 1; L. Krejčí 2
Působiště autorů:
Klinika nemocí kožních a pohlavních, Fakultní nemocnice Hradec Králové, Univerzita Karlova v Praze, Lékařská fakulta v Hradci Králové, přednosta doc. MUDr. Miloslav Salavec, CSc.
1; Národní centrum pro výzkum biomolekul, Přírodovědecká fakulta, Masarykova univerzita v Brně, ředitel ústavu prof. RNDr. Jaroslav Koča, DrSc.
2
Vyšlo v časopise:
Čes-slov Derm, 93, 2018, No. 1, p. 20-26
Kategorie:
Kazuistika
Souhrn
Rothmund-Thomson syndrom (RTS) je charakterizován poikilodermií, prořídlými vlasy, řasami a/nebo obočím, nízkým vzrůstem, kosterními a dentálními abnormalitami, kataraktou a zvýšeným rizikem malignit, a to zejména osteosarkomem. V článku popisujeme případ 35letého muže s poikilodermií, univerzální alopecií a anaplastickým velkobuněčným T lymfomem (Anaplastic Large Cell Lymphoma, ALCL), se vznikem kožních projevů ve 23 letech věku. Diagnóza RTS byla stanovena na základě klinických projevů a genetického vyšetření, které prokázalo mutace genu RECQ4 přítomné na více úrovních. Sekvenace fibroblastů westernblotem odhalila mutaci Gln253His svědčící pro RTS. I přes terapii ALCL kombinací chemoterapeutik pacient zemřel na srdeční zástavu 17. den po zahájení léčby. RTS je vzácná genodermatóza. Nemocné, kteří trpí RTS, je nutné dispenzarizovat pro jejich zvýšené riziko výskytu kožních nádorů.
Klíčové slova:
poikilodermie – Rothmund-Thomson syndrom – anaplastický velkobuněčný T lymfom
POPIS PŘÍPADU
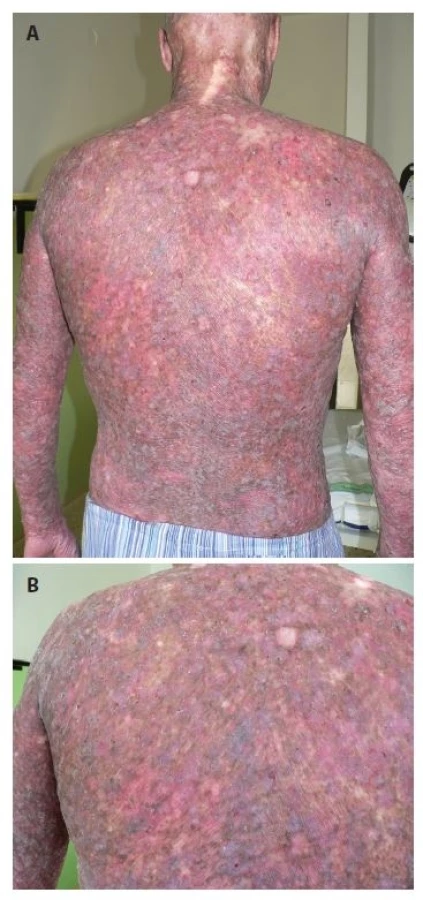
Pacientem byl 35letý muž, který se kromě kožních potíží s ničím neléčil. V rodině se otec léčil s trombofilií, jinak matka, sestra, bratr a dcera byli zdrávi. Trvale neužíval žádnou farmakoterapii. Od věku 23 let pozoroval postupně se rozšiřující erytematózní ložiska s pruritem, která se objevila nejdříve kolem kotníků a ve flexurách. Ve věku 29 let, byl v roce 2010 hospitalizován na kožní klinice s klinickým obrazem rozsáhlých mapovitých ložisek, pokrývajících kůži celého těla. V ložiscích byl nález živě červených erozí, střídajících se s hyperpigmentovanými makulami a se suchou kůží. Výsledný vzhled kůže tvořila síťovitá hyper - a hypopigmentace. Alopetické ložisko bylo nalezeno nad levým uchem a ve kštici. V dubnu 2010 byly provedeny celkem tři průbojníkové biopsie, které vykázaly fokálně nevýraznou hyperkeratózu epidermis, zcela ojediněle i s drobnými ložisky parakeratózy. Na rozhraní epidermis a dermis byl nalezen souvislý pruh tvořený lymfoidními elementy, v těchto místech byly patrná rovněž četnější depozita pigmentu. Bazální vrstvy epidermis byly ložiskově narušené s vakuolární degenerací. Histologie byla uzavřena jako obraz chronické superficiální dermatitidy s inkontinencí pigmentu, nález byl slučitelný s diagnózou poikilodermie. Laboratorní vyšetření – myoglobin 3, antinukleární autoprotilátky, ENA protilátky a protilátky proti kardiolipinu – bylo negativní. Následně v období let 2010–2015 nebyl sledován v žádné z kožních ambulancí. Kožní nález od roku 2010 s tendencí k výrazné progresi. V březnu 2016 byl přijat k další léčbě za hospitalizace pro asi měsíc trvající febrilie, společně s impetiginizovanou dermatitidou a lymfadenopatií. Objektivní vyšetření prokázalo generalizovanou ztenčelou kůži se síťovitou hyper - a hypopigmentací, místy s erytematózními makulózními ložisky, četnými plošnými erozemi místy se serózní sekrecí, místy pokryté hemoragickoserózní krustou (obr. 1a, 1b), byla přítomna univerzální alopecie (obr. 2). V krevním obrazu byla nalezena mikrocytární anémie (hemoglobin 126 g/l; norma 135–175 g/l), trombofílie (trombocyty 492 x 109/l; norma 150–400 x 109/l) s přítomností metamyelocytů a myelocytů. Biochemicky byly zaznamenány vyšší hodnoty gama-glutamyltransferázy (GMT, 2,72 μkat/l, norma: 0–1 μkat/l), alkalické fosfatázy (ALP, 2, 86 μkat/l, norma 0,67–2,17 μkat/l), cholesterolu (5,7 mmol/l, norma 0–5,2 mmol/l), C-reaktivního proteinu (CRP, 169 mg/l; norma 0–5 mg/l), laktátdehydrogenázy (LDH, 6,46 μkat/l, norma 2,25–3,75 μkat/l). Bakteriální stěr z kůže s kultivací prokázal přítomnost Staphylococcus aureus. Nález na rentgenu (RTG) plic byl normální. Scintigrafie skeletu poskytla nález atypicky homogenně zvýšeného metabolismu v páteři, žebrech a pánve nejasné etiologie. Bylo vysloveno podezření na Rothmundův-Thomsonův syndrom (RTS), jehož kritéria shrnuje tabulka 1, protože náš pacient splňoval dvě ze tří nutných kritérií pro stanovení diagnózy Rothmundova-Thomsonova syndromu: alopecii a poikilodermii (tab. 1). Genetické vyšetření prokázalo mutace v genu RECQ4 a Gln253His svědčící pro Rotmundův-Thomsonův syndrom. Biopsie provedená z projevů v oblasti levé lopatky potvrdila atypickou T-lymfoidní proliferaci s výrazným epidermotropismem – s vytvářením objemnějších lymfoepitelových lézí. Invazivní elementy měly středně velká okrouhlá jádra s drobnými jadérky a bohatší světlou cytoplazmu (obr. 3a). Imunofenotypizace byla netypická pro mycosis fungoides. Imunohistochemicky byly invazivní elementy pozitivní při průkazu antigenu CD30, dále koexprimovaly antigen CD3, CD7, a CD8. CD4 byl pozitivní na zbytku menších elementů (obr. 3b). Vzhledem k imunofenotypu se jevilo postižení kůže jako sekundární v rámci agresivní systémové T lymfoproliferace. Sternální punkce byla bez patologického nálezu. Uroporfyrin v moči nebyl nalezen – talkový test byl negativní.
Obr. 1b. Detail kožního postižení levé lopatky
Přítomná deskvamace, poikilodermie (síťovitá hyper- a hypopigmentace,
teleangiektázie, atrofie kůže).



Lokální terapie spočívala ve vysychavých obkladech superoxidovaným roztokem, dále byl aplikován hydrofilní metakrylátový gel, 2% chloramfenikol v 0,05% dexametason synderman ung., mřížky s jodem. Systémová léčba zahrnovala antibiotika perorálně (amoxicilinem v kombinaci s kyselinou klavulanovou, azitromycinem, penicilinem). Terapie vedla ke zmírnění lokálních obtíží. Postupně se hojily plošné eroze, ložiska již byly bez serózní sekrece, hemoragickoserózní krusty se odhojily. Po 24 dnech hospitalizace na kožní klinice je pacient pro nález suspektní agresivní systémové T lymfoproliferace přeložen na hematoonkologickou kliniku, kde dále podstoupil vyšetření CT/PET s výsledkem generalizované lymfadenopatie s mnohočetným postižením skeletu, sleziny. Pro lymfadenopatii byla exstirpována krční uzlina. Histopatologický nález prokázal infiltraci uzliny agresivním T-non Hodgkinovým lymfomem (genotyp CD30+, CD8+). Lymfatická uzlina byla prostoupena difuzně rostoucí proliferací značně pleomorfní populací elementů, dominovaly buňky středně velké, velké a anaplastické s vezikulárními jádry (obr. 4, barvení hematoxylin-eosinem). Na prvním místě byl diferenciálně diagnosticky anaplastický velkobuněčný T lymfom, CD 8+, CD 30+, anaplastická lymfomová kináza (ALK) negativní.

Genetické vyšetření bylo s nálezem mutace v genu RECQ4 přítomné na více úrovních. Sekvenace fibroblastů metodou westernblot odhalila mutaci Gln253His svědčící pro Rotmund-Thomson syndrom. Byla zahájena léčba kortikoidy, poté snaha o zahájení studijní terapie Echolon (Brentuximab, konjugát protilátky anti CD30), ale pro ultrasonografický nález asteatotického ložiska jater při žlučníku o velikosti 2 cm nejasného původu, nebylo možné pacienta zařadit do studie a terapií brentuximabem léčen nebyl. Byl proto léčen CHOP chemoterapií ve složení: cyklofosfamid, doxorubicin, vinkristin, methylprednisolon. V důsledku léčby došlo k řadě nežádoucích účinků včetně respiračního selhání a k febrilní neutropenii. Po 20 dnech hospitalizace na hematologické klinice v důsledku náhlé asystolie pacient zemřel.
DISKUSE
RTS byl původně stanoven v roce 1868 německým oftalmologem Augustem von Rothmundem, který popsal poikilodermii společně s retardací růstu a juvenilní kataraktu u 10 dětí v bavorské vesnici. V roce 1936 anglický dermatolog Matthew Sydney Thomson hlásil tři podobné pacienty s „kongenitální poikilodermií (atrofií kůže spojenou s hyperpigmentacemi, depigmentacemi a teleangiektaziemi)“ a růstovou retardací. Tito pacienti však netrpěli juvenilní kataraktou, ale disponovali skeletálními abnormalitami. Eponym Rothmund-Thomson syndrom byl vytvořen Williamem B. Taylorem v roce 1957, který stanovil stejnou patofyziologii u obou dříve popsaných skupin a sledoval další pacienty s výše uvedeným postižením [7].
Etiologie tohoto syndromu je předmětem mnoha spekulací. V roku 1999 byla objevena kauzativní mutace v RECQL4 genu [4]. Tato mutace byla detekována asi u 66 % pacientů [1]. Gen RECQL4 je lokalizován na chromozomu 8q24.3 zahrnujícím 21 exonů a patří do rodiny proteinů DNA helikáz. DNA helikázy jsou enzymy zúčastňující se různých typů oprav DNA. Jsou důležité při genové reparaci, při opravě nukleotidů excisí a při přímých opravách nukleotidů [2]. Na základě ne/přítomnosti mutace v genu RECQL4 se pacienti s RTS dělí na 2 typy. Typ 1 – pacienti bez RECQL4 genové mutace, u kterých se předpokládá mutace v jiném genu, který kóduje protein v rodině helikázy RecQ. Typ 2 pak představuje pacienty s mutací v genu RECQL4.
Pacienti trpící RTS sice mají při narození kůži normálního vzhledu, ale již kolem třetího a šestého měsíce se u pacientů vyvíjejí eflorescence typu erytému, edému a puchýřů. Projevy jsou zpočátku lokalizovány v obličeji a poté se šíří do oblasti hýždí a končetin. V průběhu měsíců až let mají eflorescence chronický charakter se síťovou hypo - a hyperpigmentací, ložiskovou atrofií a telangiektaziemi. Hyperkeratotické léze dlaní a plosek se vyskytují přibližně u jedné třetiny jedinců [13]. Náš pacient uváděl kožní potíže až od svých 23 let, na rozdíl od literárních údajů, které uvádí vznik nemoci od kojeneckého věku. V úvahu připadá, že nemocný trpěl mírnými projevy, které nevnímal jako nemoc, již dříve nebo by se mohlo jednat o dosud nepopsanou formu nemoci, která se projevuje až v pozdějším věku. Klinicky náš pacient jasně splňoval diagnostická kritéria (tab. 1) poikilodermie, alopecie a lehké atypické konfigurace ulny a radia s mírným rozšířením diafýz a mírnou angulací. Dále je splněno i kritérium sdružení s maligním onemocněním. Podle Wanga et al. je možné stanovit diagnózu RTS jen na základě klinického obrazu, protože žádná laboratorní metoda není spolehlivě diagnostická. Kožní biopsie může ukázat poikilodermní změny: hyperkeratózu, atrofii epidermis, inkontinenci pigmentu, teleangiektazie, lymfohistocytární infiltrát horní dermis, které jsou nespecifické, ale v souladu s kritérii RTS [13]. Genetická mutace v RECQL4 genu je prokazatelná jen u zhruba 66 % pacientů. U našeho pacienta byly mutace v genu RECQL4 přítomné na vícero úrovních. Doposud, ale není jasné, která z mutací byla dominantní a způsobila klinický obraz poikilodermie. Je potřeba důsledně geneticky vyšetřit oba rodiče a potomka pacienta. Simon et al. popsali podobný případ chlapce s RTS se současně přítomným anaplastickým velkobuňečným T lymfomem. Jednalo se o chlapce, u kterého byla zaznamenána během raného dětství retardace růstu a byly detekovány kostní abnormality s následným vznikem poikilodermie, prořídnutím vlasů, ztráty obočí a řas. Genová analýza byla u pacienta provedena v jeho 21 letech a prokázala mutaci v genu RECQL4. Ve věku 9 let byla u chlapce zjištěna lymfadenopatie uzlin krku, axil a paraaortálních lymfatických uzlin. Histologicky se jednalo o anaplastický velkobuněčný T lymfom. U pacienta se lymfom podařilo vyléčit na základě střídajících se šesti cyklů chemoterapie A (dexametazon, metotrexát, ifosfamid, etoposid a cytarabin) s chemoterapií B (dexametazon, metotrexát, cyklofosfamid a doxorubicin) zakončené COPP cyklem (cyklofosfamid, vinkristin, prokarbazin a prednison). Další osud pacienta popsaného z literatury byl následovný: ve věku 14 let mu byla diagnostikována centroblastická varianta velkobuněčného maligního B lymfomu. Po exstirpaci postižené krční uzliny pacient dosáhl remise, bez nutnosti podstoupit chemoterapii. Následně v jeho 14,5 roku byl u něj detekován osteosarkom levé tibie. Po deseti týdnech adjuvantní chemoterapie pacient podstoupil kompletní odstranění nádoru. Akutní leukémie mu byla diagnostikována ve věku 21 let, v tom samém věku nakonec pro progresi leukémie zemřel [10].
Pacienti s RTS vykazují vyšší riziko onemocnět maligním nádorem a zároveň mají vyšší riziko vzniku druhého nádorového onemocnění než ostatní populace. Sekundární maligní onemocnění bylo hlášeno u 11 z 61 RTS pacientů, kteří podstoupili léčbu osteosarkomu, sarkomu měkkých tkání nebo trpících hematologickou malignitou [11].
Náš pacient trpěl anaplastickým velkobuněčným T lymfomem (ALCL), který tvoří 2–8 % non-Hodgkinských lymfomů u dospělých a až 10–15 % u dětí. Vyznačuje se silnou pozitivitou exprese Ki-1 antigenu (CD30). Původně byl nazýván maligní histiocytózou nebo anaplastickým karcinomem. Biologie ALCL byla pochopena popisem opakované strukturální cytogenetické abnormality t (2; 5) (p23; q35). Tato translokace prokázala vznik nového fuzního proteinu, který zahrnuje novou kinázu pojmenovanou jako anaplastická lymfomová kináza (ALK), jehož gen je lokalizován na chromozomu 2 [9]. Podle exprese ALK proteinu klasifikujeme ALCL na subtypy – ALCL CD30+ALK+ a ALCL CD30+ ALK-. ALK protein pozitivní ALCL se vyskytuje převážně ve věkových kategoriích do třetího decenia, ALK protein negativní případy postihují starší osoby s věkovým průměrem 60 let. Obě skupiny lymfomů mají agresivní průběh, avšak ALK+ lymfomy reagují dobře na systémovou léčbu a mají příznivější prognózu. Primárně kožní ALCL spadají do rámce T lymfoproliferativních onemocnění kůže a mají ve většině případů příznivý průběh bez generalizace [5]. Primárně kožní ALCL může přejít do systémové formy ALCL, ale toto riziko je nízké [3]. ALCL postihuje nejčastěji lymfatické uzliny a kůži, postižené mohou být také plíce, gastrointestinální trakt, kosti nebo CNS [8].
Nejčastěji indikovaná forma léčby ALCL je chemoterapie CHOP ve složení cyklofosfamid, doxorubicin, vinkristin, prednison. Pětileté přežití při této kombinaci chemoterapeutik je udáváno u ALK + pacientů 58%, u ALK - skupiny 34%. Riziko recidivy bylo stejně vysoké pro obě skupiny. Dalšími možnostmi terapie ALCL je kombinace chemoterapeutik EPOCH (ve složení etoposid, prednison, vinkristin, cyklofosfamid, doxorubicin), transplantace kostní dřeně, radioterapie (volba této terapie dominuje u kožní formy ALCL). Novými formami terapie ALCL je užití Crizotinibu, který je inhibitorem ALK, inhibitoru Alisertib-Aurora Kinázy - Alisertibu; Bortezomibu, který inhibuje proteosomy; Brentuximabu vedotinu, který je konjugátem monoklonální protilátky anti CD30; Pralatrexatu - antifolátu a Vorinostatu - inhibitoru histondeacetylázy [8]. Náš pacient byl léčen základní formou CHOP terapie.
ZÁVĚR
Pokud je u pacienta RTS diagnostikován, je nutné provádět každoroční kontroly dermatologem a oftalmologem. Důležitá je fotoprotekce s aplikací krémů s vysokým ochranným faktorem proti UVA i UVB. Oční lékař dispenzarizuje pacienty k vyloučení katarakty. Vhodné je i pravidelné rentgenologické sledování pacienta pro možný výskyt skeletálních dysplazií. Doporučuje se každoroční zhotovení rentgenových snímků dlouhých kostí od 5 let věku života u všech pacientů s RTS [13]. Dermatolog hraje důležitou roli při detekci vzácných genodermatóz, včetně Rothmund-Thomsonova syndromu a v dispenzarizaci osob se zvýšenou predispozicí k výskytu kožních nádorů.
Poděkování
Speciální poděkování patří paní doktorce Markétě Nové z Fingerlandůvho ústavu patologie FNHK za její pomoc při odečtu histopatologie.
Do redakce došlo dne 20. 7. 2017
Adresa pro korespondenci
MUDr. Simona Vachatová
Klinika nemocí kožních a pohlavních,
Fakultní nemocnice Hradec Králové
Sokolská tř. 581
500 05 Hradec Králové
e-mail: vachatova.simona@gmail.com
Zdroje
1. CABRAL, R. E., QUEILLE, S., BODEMER, C. et al. Identification of new RECQL4 mutations in Caucasian Rothmund‐Thomson patients and analysis of sensitivity to a wide range of genotoxic agents. Mutat. Res, 2008, 643, 1-2, p. 41–47.
2. CROTEAU, D. L., SINGH, D. K., HOH FERRARELLI, L. et al. RECQL4 in genomic instability and aging. Trends Genet, 2012, 28, 12, p. 624–631.
3. HAPGOOD, G., PICKLES, T., SEHN, L. H. et al. Outcome of primary cutaneous anaplastic large cell lymphoma: a 20-year British Columbia Cancer Agency experience. Br. J. Haematol., 2016. DOI: 10.1111/bjh.14404. ISSN 00071048.
4. KITAO, S., LINDOR, N. M., SHIRATORI, M. et al. Rothmund–Thomson syndrome responsible gene, RECQL4: Genomic structure and products. Genomics, 1999, 61, 3, p. 268–276.
5. KODET, R., MRHALOVÁ, M., KRSKOVÁ, L., STEJSKALOVÁ, E. Anaplastický velkobuněčný lymfom: přehled problematiky. Čes.-slov. Patol., 2003, 3, p. 102–114.
6. LARIZZA, L., ROVERSI, G., VEROLOES, A. Clinical utility gene card for: Rothmund‐Thomson syndrome. Eur. J. Hum. Genet, 2013, 21, 7. DOI:10.1038/ejhg.2012.260.
7. LARIZZA, L., ROVERSI, G., VOLPI, L. Rothmund-Thomson syndrome. Orphanet J. Rare Dis., 2010, 5. DOI: 10.1186/1750-1172-5-2. ISSN 17501172.
8. MA, H., ABDUL-HAY, M. T-cell lymphomas, a challenging disease: types, treatments, and future. Int. J. Clin. Oncol., 2016. DOI: 10.1007/s10147-016-1045-2. ISSN 13419625.
9. MIHÁL, V., MICHÁLKOVÁ, K., JAROŠOVÁ, M. a kol. Anaplastický velkobuněčný lymfom jako příčina horečky neznámého původu s uzlinovým syndromem. Pediatr. pro Praxi, 2009, 10, 6, p. 412–413.
10. SIMON, T., KOHLHASE, J., WILHEM, C. et al. Multiple malignant diseases in a patient with Rothmund-Thomson syndrome with RECQL4 mutations: Case report and literature review. Am. J. Med. Genet A, 2010, 152A, 6, p. 1575–1579.
11. STINCO, G., GOVERNATORI, G., MATTIGHELLO, P. et al. Multiple cutaneous neoplasms in a patient with Rothmund-Thomson syndrome: case report and published work review. J. Dermatol., 2008, 35, 3, p. 154–161.
12. WANG, L. L., PLON, S. E. Rothmund-Thomson Syndrome. SourceGeneReviews, 1999 [updated 2016] In: PAGON, R. A., ADAM, M. P., ARDINGER, H. H. et al. GeneReviews, 1993–2016.
13. WANG, L. L., LEVY, M. L., LEWIS, R. A. et al. Clinical manifestations in a cohort of 41 Rothmund-Thomson syndrome patients. Am. J. Med. Genet., 2001, 102, 1, p. 11–17.
Štítky
Dermatologie Dětská dermatologieČlánek vyšel v časopise
Česko-slovenská dermatologie

2018 Číslo 1
- Nástroje k hodnocení závažnosti psoriázy v klinických studiích
- Systémová léčba atopické dermatitidy konečně i u dětí
- Inovace v hojení ran: krytí Zetuvit Plus Silicone Border pro optimální management exsudátu z ran
Nejčtenější v tomto čísle
- PITYRIASIS VERSICOLOR(syn. Tinea versicolor)
- Diagnostika melanomu a současná doporučení pro léčbu a sledování
- IMPETIGO
- Kožní leiomyosarkom – popis případu
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy