
Hereditární ichtyózy
Hereditary Ichthyoses
Hereditary ichthyoses form a very heterogeneous group of disorders manifested by dry, rough and scaly skin. An impairment of cutaneous barrier, to various degrees, is present in all forms of ichthyoses. Hereditary ichthyoses are caused by different gene mutations. Clinical manifestations of individual types of ichthyosis change during patient’s life. The authors are currently engaged in the diagnosis and classification of congenital ichthyosis based on molecular diagnostics and they emphasize the importance of comprehensive genetic counseling and genetic prevention. Patients should be followed up in specialized centers. Recently developing patient organization represents a great benefit for the patients.
Key words:
ichthyosis – diagnostics – DNA molecular analysis
Autoři:
B. Pinková 1; H. Bučková 1; R. Borská 2; L. Fajkusová 2
Působiště autorů:
Dětské kožní oddělení Pediatrické kliniky, LF MU a FN Brno
primář MUDr. Hana Bučková, Ph. D.
1; Centrum molekulární biologie a genové terapie IHOK FN Brno a LF MU v Brně
vedoucí pracoviště doc. RNDr. Lenka Fajkusová, Ph. D.
2
Vyšlo v časopise:
Čes-slov Derm, 91, 2016, No. 1, p. 3-12
Kategorie:
Souborné referáty (doškolování lékařů)
Souhrn
Hereditární (dědičné) ichtyózy jsou velmi heterogenní skupinou onemocnění projevující se suchou, hrubou a šupící kůží. U všech forem ichtyóz je v různém rozsahu porušena kožní bariéra. Dědičné ichtyózy jsou způsobeny mutacemi různých genů. Klinické projevy jednotlivých typů ichtyóz se v průběhu věku pacienta mění. Autoři se věnují současné diagnostice a klasifikaci vrozených ichtyóz založené na molekulární diagnostice, zdůraňují význam komplexního genetického poradenství a genetické prevence. Pacienti by měli být dispenzarizováni ve specializovaných centrech. Velkým přínosem pro pacienty je v poslední době se rozvíjející pacientská organizace.
Klíčová slova:
ichtyóza – diagnostika – DNA molekulární analýza
ÚVOD
Ichtyózy jsou velmi heterogenní skupinou onemocnění projevující se suchou, různě hrubou a šupící kůží. Dělí se na hereditární (dědičné) a získané.
Získané ichtyózy mohou být paraneoplastickým projevem některých nádorových onemocnění, např. morbus Hodgkin, nehodgkinských lymfomů a vzácně karcinomů vnitřních orgánů.
Dědičné ichtyózy patří do skupiny vzácných onemocnění – keratodermií.
U všech dědičných forem ichtyóz je v různém rozsahu porušena kožní bariéra, tím dochází k vyšší transepidermální ztrátě vody a snížení schopnosti udržet dostatečné množství vody v epidermis. Epidermální bariéra se skládá z nahromaděných korneocytů ve stratum corneum. Tyto buňky jsou obklopeny velmi hydrofobními molekulami lipidové vrstvy, které jsou vytvořeny sekrecí lamelárních či Odlandových tělísek na přechodu stratum granulosum a corneum. U ichtyóz je sekrece těchto lipidů pozměněna, či snížena a vede ke zvýšené transepidermální ztrátě vody. Hyperproliferace a zánět v kůži je považován za kompenzační mechanismus, který vede k obnovení této bariéry.
Deskvamace je konečný výsledek proteolytické degradace korneodesmosomů (intercelulárních spojů ve stratum corneum) potencovaný třením a buněčnou hydratací. Deskvamace vyžaduje normální epidermální diferenciaci a závisí na gradientu pH, přítomnosti proteázovoých inhibitorů a vzniku hygroskopických molekul v buňkách stratum corneum. Abnormality v deskvamaci vyústí v nahromadění korneocytů (retenční hyperkeratóza) nebo ve zvýšenou proliferaci (epidermální hyperplazie).
Etiologie
Příčinou ichtyóz jsou různé poruchy v terminální diferenciaci epidermis. Jde o defekty strukturálních proteinů cytoskeletu – tj. keratinu, neadhezivních a transmembránových proteinů, jako jsou např. konexiny či proteiny tzv. „cornified cell envelope“ – kornifikované obálky jako jsou filagrin, loricrin, popř. enzymy k jejímu vytvoření, jako je transglutamináza 1 (TGM 1).
Možné jsou také defekty výměny lipidů kůže. Jsou známy změny v transportu v keratinosomech a hepoxillinu. Tyto defekty odhalí testy na molekulární úrovni.
Diagnostika
Zařazení dědičné ichtyózy vychází z klinického obrazu, histologického nálezu, histochemických výsledků, ale největším přínosem k přesnému určení typu ichtyózy je diagnostika na molekulární úrovni [10, 13]. Klinické projevy jednotlivých typů ichtyóz jsou pestré, mění se s věkem pacienta i s ročním obdobím. Některé vrozené ichtyózy se projeví u novorozence, u jiných typů se příznaky objeví později. Klinická diagnostika vyžaduje dermatologa s bohatými zkušenostmi. Přesto zařadit typ ichtyózy podle klinického obrazu a histologického vyšetření je v dnešní době nedostačující.
Histologický obraz
Běžné histopatologické změny nejsou pro ichtyózy nijak specifické, často nacházíme pouze epidermální hyperplazii a různé stupně ortohyperkeratózy. Důležitou roli hraje elektronová mikroskopie, která je vodítkem pro diagnostiku ichtyóz a jejich ultrastrukturálních markerů. U jednotlivých typů ichtyóz nacházíme změny v množství lamelárních tělísek nebo změny ve stratum corneum. Hustota lamelárních tělísek je snížena, jejich tvar je menší a často mají také vakuolární tvar a tím neplní zcela normálně svoji funkci. Právě toto abnormální uspořádání lamelárních tělísek vede ke snížené bariérové funkci kůže. Současně také u některých typů ichtyóz dochází k chybnému zpracování profilagrinu a změněnému metabolismu ceramidů. Tím dochází ke změně kornifikované obálky [4]. Kornifikovaná obálka je zvláštní formou zrohovatělých buněk. Skládá se z 10 nm tlusté vrstvy nerozpustných proteinů, s kovalentně vázanými ceramidy. Toto uspořádání extracelulárních lipidů do lamel je nezbytné pro účinnou bariérovou funkci kůže.
Molekulární diagnostika
Molekulární diagnostika je v současné době základem pro přesné určení typu vrozené ichtyózy. Má velký význam v genetické prevenci, umožňuje rodinám, kde se onemocnění již vyskytlo, komplexní genetické poradenství, prenatální nebo preimplantační diagnostiku. Základní typy ichtyóz a jejich molekulární diagnostika jsou uvedeny v tabulce 1.

Terapie
U dědičných ichtyóz je symptomatická, převažuje lokální terapie. Výjimečně se volí systémová terapie retionoidy. Kauzální genová terapie je stále ve stadiu výzkumu.
Klasifikace
Aktuální klasifikace ichtyóz z roku 2009 je přehledná. Docent Vinzenz Oji dělí hereditární ichtyózy na vrozené – kongenitální, jejichž manifestace je zřejmá při porodu, a ichtyózy s pozdějším nástupem (Ichtyosis vulgaris, X-recesivně vázaná ichtyóza), Ichthyoses of a delayed onset [10].
A) HEREDITÁRNÍ ICHTYÓZY S POZDĚJŠÍM NÁSTUPEM PROJEVŮ (Ichthyoses of a delayed onset)
1. Ichthyosis vulgaris (IV)
Prevalence 1 : 250, jedná se o nejčastější typ ichtyózy.
Etiologie: IV je onemocnění AD, či semidominantní, to znamená, že při postižení obou rodičů je klinický stav horší než při postižení jednoho z rodičů. Onemocnění se zakládá na inaktivující mutaci genu pro filagrin /loss-of function-mutation/ (chromosom 1q21), který patří ke komplexu diferenciace kůže. Filagrin je hlavní součástí keratohyalinových proteinů a podílí se na propojení keratinových filament. Mutace v genu pro filagrin zvyšují i riziko atopické dermatitidy [17].
Klinický obraz: ichthyosis vulgaris (IV) – obrázek 1, se může již u novorozenců projevit zvýšenou suchostí kůže a jemným, bělavým olupováním, ale klinické příznaky se převážně objeví až ve věku čtyř až šesti měsíců. Predilekční místa jsou extenzorové strany končetin, především bérců a břicha. Obličej a ohybové strany končetin jsou ušetřeny. U 80–90 % pacientů nacházíme hyperlinearitu dlaní. Suchost kůže je výraznější v zimě. Pacienti neudávají jiné subjektivní potíže [8].

Asi ve 40 % případů je ichhtyosis vulgaris asociována s keratosis pillaris a atopickou dermatitidou.
Histologické vyšetření: je přítomna epidermální hyperplazie a různé stupně ortohyperkeratózy, chybí nebo je zúžené stratum granulosum. V elektronovém mikroskopu jsou zmenšená keratohyalinní granula [4, 10, 11].
Diagnostika: pozitivní rodinná anamnéza, typické kožní projevy, známky atopie přispějí k správné diagnóze, kterou je vhodné potvrdit DNA molekulární analýzou.
Terapie: nejdůležitější je terapie emoliencii. V lokální terapii používáme většinou lehčí – hydrofilní základy tak, aby nedošlo k okluzivnímu efektu extern. Volíme preparáty s glycerolem, při větším zašupení i preparáty s ureou, v koncetraci 5%, vyjma kojenců. Celková terapie retinoidy u ichthyosis vulgaris indikována není.
2. X chromozomálně recesivně vázaná ichtyóza (XLI)
Prevalence: s incidencí 1 : 2000 až 1 : 4000 chlapců se jedná o druhý nejčastější typ ichtyózy. Vyskytuje se jen u chlapců, ženy jsou pouze přenašečky.
Etiologie: XLI vzniká na základě defektu enzymu steroid sulfatázy, defekt STS genu je kódován na chromozomu Xp 22.32.
Klinický obraz: obrázek 2, XLI se někdy manifestuje při porodu, velmi zřídka se narodí collodion baby. Častěji se projeví v prvních dvou, třech měsících života dítěte, kdy zpočátku dominuje generalizované olupování drobnými bělavými šupinkami, které typicky vynechávají kubitální a popliteální jamky. Ve věku čtyř až šesti měsíců jsou šupiny polygonální, hrubší, šedohnědé barvy. Šupiny pevně lpí, ve starším věku jsou zejména v oblasti šíje a retroaurikulárně, ale také na extenzorových stranách končetin a bocích trupu. Flexury jsou většinou ušetřeny. Někdy bývá postižen i obličej, zde spíše laterální části a vlasatá část hlavy. Pacienti nemají hyperlinearitu dlaní a projevy atopické dermatitidy. Kožní projevy se typicky zlepšují v létě.

Onemocnění je u 50 % nemocných a 25 % přenašeček asociováno se ztluštěním rohovky v důsledku ukládání cholesterolsulfátu na zadní membráně, většinou však bez funkčního postižení [5]. Asi u 24 % pacientů nacházíme gonadální dysfunkce (kryptorchismus a infertilitu). Pro matky dítěte s XLI jsou typické slabé porodní bolesti, protrahovaný porod, který je indikací k provedení císařského řezu.
Histologie: typické změny pro XLI nejsou, nacházíme pouze mírnou hyperkeratózu s akantózou a zachované stratum granulosum.
Diagnostika: typické kožní projevy, histologické vyšetření potvrzení XLI DNA molekulární analýzou. Anamnestické údaje o protrahovaném porodu přispějí k diagnóze XLI u dítěte.
Terapie: u pacientů s XLI je volbou pouze lokální terapie, hydrofilní preparáty, na hyperkeratózy na šíji volíme preparáty s ureou, koncentrace může být na tyto menší plochy 5–10%.
XLI bývá kromě výše uvedných extrakutánních změn take asociována s níže uvedenými syndromy:
Kallmanův syndrom: (KAL 1, Xp 22.3) je charakterizován hypogonadotropním hypogonadismem a anosmií.
Bývá také asociace XLI a chondrodysplasia punctata = Conradiův-Hünermannův syndrom. Jedná se o kombinaci ichtyózy a změn na kostech, které se v rentgenovém obraze projevují jako tečkovitá projasnění epifýz – chondrodysplasia punctata.
B) HEREDITÁRNÍ ICHTYÓZY S PROJEVY JIŽ PŘI NAROZENÍ – KONGENITÁLNÍ
1. Autosomálně recesivní kongenitální ichtyózy (ARCI)
K těmto onemocněním řadíme celé spektrum ichtyóz, které jsou AR dědičné. Jejich akronymem je proto zkratka ARCI (autosomal recesive congenital ichthyoses). Incidence se odhaduje mezi 1 : 100.000 až 1 : 500.000. V populacích, kde dochází k příbuzenským svazkům, je incidence vyšší.
Etiologie: je velmi různorodá, molekulárně geneticky lze rozlišit cca 7 mutací genů. Nejčastějšími mutacemi jsou mutace v ALOXE3 a ALOX 12B (viz tabulka 1). Tyto enzymy štěpí deriváty kyseliny arachidonové – leukotrieny v další metabolity. Mutace těchto enzymů pak vede k výrazně porušené bariérové funkci kůže a tím ke ztrátám vody kůží.
Dalším nejčastěji mutovaným enzymem je transglutamináza 1 (TGM 1), enzym potřebný k vytvoření kornifikované obálky „cornified cell envelope”. Je zodpovědný za tvorbu „cross-links“ proteinů stratum corneum, tj. udržuje buňky stratum corneum při sobě.
Klinický obraz: nezávisle na rozdílné etiologii vykazují děti s těžkými formami ichtyóz celou řadu společných klinických znaků:
- Collodion baby (obr. 3) u 80–90 % pacientů s lamelární ichtyózou, či kongenitální ichtyosiformí erytrodermií je povrch těla novorozence pokryt koloidní membránou, kterou lze přirovnat k tlustému celofánu. Nejde o specifický projev určitého typu ichtyózy. Ectropium a eclabium vznikají v důsledku napětí kůže pokryté koloidní membránou, popř. pak následně při vzniku hyperkeratóz tahem na perorální a periokulární oblast. V průběhu dvou až tří týdnů po porodu se membrána zcela uvolní a sloupne. Stejnou membránu najdeme vzácně i u jiných typů ichtyóz (např. někdy u XLI, IV), u střádavých lysomálních chorob – m. Gaucher typu 2, kde je příčinou defekt glucosylceramid – beta-glucosidázy. U 5–6 % novorozenců se následně z collodion baby nevyvíjí žádná forma ichtyózy (např. Self healing collodion baby, nyní se užívá název Self improving congenital ichthyosis /SICI/).
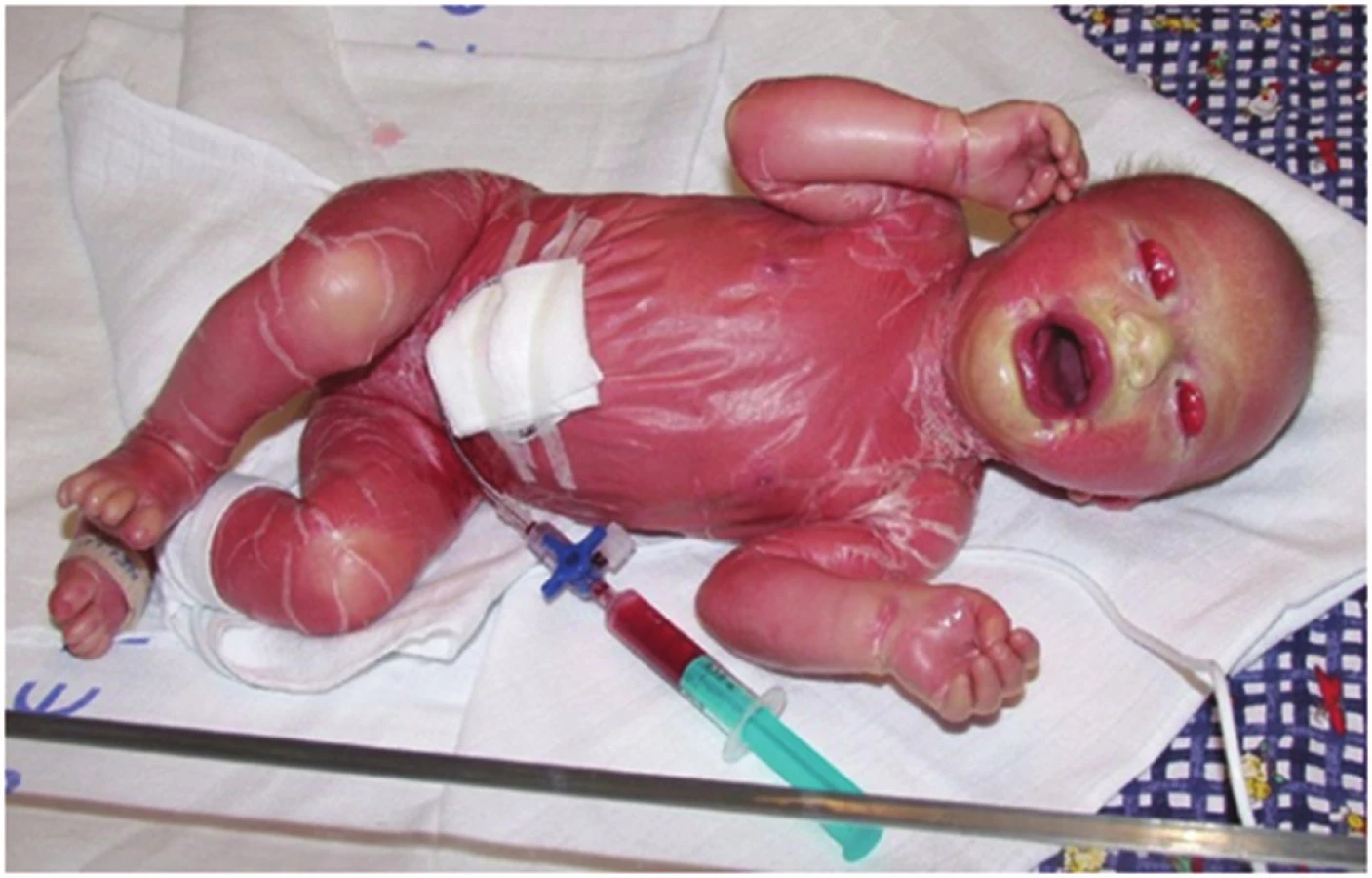
U novorozence s collodion baby snadno dochází k hypotermii a dehydrataci, které mohou vyústit v křeče, hypotenzi a šokový stav dítěte. Ragády v koloidní membráně mohou být sekudárně infikovány.
Terapie: dítě se ukládá do inkubátoru se zvýšenou vlhkostí (70–80%), kůže se ošetřuje hydrofilními krémy, ragády se obkládají dezinfekčními roztoky a ošetřují externy, která podporují epitelizaci, kombinují se se zinkovými preparáty. Zevní zvukovod se zvláčňuje hydrofilními externy nebo olivovým olejem, pravidelně se ošetřují oči, aby nedocházelo k poškození rohovky.
Dalšími společnými znaky u ARCI jsou palmoplantární hyperkeratózy, někdy změny na nehtech, alopecie a v neposlední řadě hypohidróza [4].
a) Lamelární ichtyóza (LI)
Incidence LI je asi okolo 1 : 100.000. Dědičnost je AR, ale v některých případech může být AD.
Etiologie: LI mají rozdílný genotyp, ale podobný fenotyp. Nejčastějšími mutacemi jsou: v 32 % TGM 1, v 16 % ichthyin /NIPAL 4/, ve 12 % ALOX 12B, v 8 % CYP4F22, v 5 % ABCA12, v 5 % ALOXE 3 a v 22 % neprokázána zatím žádná mutace (viz tab. 1). U LI je nejčastěji defektní enzym TGM1 v keratinocytech. Je zodpovědný za tvorbu „cross-links“ proteinů stratum corneum, tj. udržuje buňky stratum corneum při sobě. Druhou nejčastější mutaci je Ichthyin gen (NIPAL 4): je to transmembránový protein, který je důležitý pro transport lipidů do lamelárních tělísek. Ostatní ARCI geny, jako jsou ALOX 12B, ALOXE 3 a CGI -58, kódují rozdílné transportní proteiny a enzymy, které syntetizují speciální lipidové komponenty stratum corneum [15].
Klinický obraz: obrázek 3, kožní projevy u LI jsou velmi pestré. 80–90 % dětí se rodí jako collodion baby. Po sloupnutí membrány je kůže na rozdíl od CIE méně zarudlá a vykazuje větší, hrubější šupiny. Rozsah postižení je rozdílný. Asi 70 % pacientů má palmoplantární hyperkeratózu. K dalším projevům patří ragády či fisury, ectropia, eclabia a deformity nehtů. Děti mohou mít v raném věku opožděnou růstovou křivku, která se později srovná. Pacienti nemají systémové onemocnění, nejsou mentálně opoždění.
U mutace TGM1 se může rozvinout tzv. bathing suit ichthyosis, kdy drobné šupiny jsou v oblasti zapářek (vyšší tělesné teploty), tj. plavková lokalizace a axily. Je to dáno termosenzitvní mutací TGM 1.
Histologické vyšetření: u pacientů s ARCI byly pozorovány tyto společné histologické změny – nepřítomnost lamel, abnormální rozestup a přerušení lamelových spojů a intracelulárních lipidových kapének, a to jak v rámci korneocytů, tak buněk stratum granulosum. Následně také výrazná ortohyperkeratóza.
Diagnostika: kožní projevy, histologické vyšetření kůže, imunohistochemie pro aktivitu TGM 1, molekulární genetika.
b) Kongenitální ichtyosiformní erytrodermie (CIE)
Etiologie: toto onemocnění je převážně AR. Molekulárně gentické vyšetření je pro CIE stejně jako pro LI překryvné a vykazuje mutace v genech pro stejné enzymy, viz tabulka 2. U CIE mírně převažují mutace v genech pro ALOXE3 a ALOX12B [1].

Klinický obraz: obrázek 4, klinický obraz CIE je klinicky a geneticky heterogenní. 80–90 % dětí se rodí jako collodion baby. Hlavním symptomem je erytrodermie, s generalizovaným šupením, které je zpočátku bělavé a spíše jemné. V průběhu jsou pak šupiny hrubší a tmavší. Vyjádření erytrodermie a ektropia je variabilní. Často je doprovodným znakem palmoplantární hyperkeratóza, jejíž rozsah se také u jednotlivých pacientů liší. U některých dětí může dojít až k jizvící alopecii a eventuálně také k poruchám růstu nehtů. Pacienti nemají systémové onemocnění, nejsou mentálně opožděni. Děti se i po stránce intelektu vyvíjejí zcela normálně.

Histologické vyšetření: změny podobné jako u LI (výraznější hyperkeratóza, parakeratóza, akantóza, stratum granulosum bez odchylek nebo vakuolizace ve stratum granulosum).
Diagnostika: klinický obraz, histologické vyšetření kůže, DNA molekulární diagnostika, eventuálně imunohistochemie pro aktivitu TGM 1.
Terapie: projevy těchto forem ichtyóz patří k spíše závažnějším, proto je péče multioborová. Velmi důležitá je správná péče o kůži, která se volí podle stupně závažnosti klinických projevů LI nebo CIE. Nutné jsou pravidelné kontroly laboratorních ukazatelů (stěry z kůže, ionty, CRP a FW) správná výživa, rehydratace, zejména v novorozeneckém věku a poté podle stavu cca do 3 let věku [6].
Lokální ošetření kůže: děti jsou zejména v prvních 3–4 týdnech života velmi náchylné k infekcím, termolabilitě, a vyžadují proto velmi specifickou, intenzivní péči. Po porodu je vhodné zajistit i. v. přístup pupeční žilou. Novorozence je nutné po porodu zabalit do aluminiové fólie a transportovat na novoreneckou JIP. Novorozenci s těžkou formou vrozené ichtyózy by měli být první dny života umístěni v inkubátoru s vlhkostí 60–80 %, podle klinického obrazu, kůže musí být pravidelně ošetřována 6–8krát denně, musí se dbát na rehydrataci.
Terapie mastmi se řídí podle klinického obrazu kůže pacienta. Čím zarudlejší je kůže, tím jemnější a hydratující masti musíme použít. V prvních týdnech života je novorozenec v inkubátoru, proto se na kůži používají hydrofilní základy. Postupně se vlhkost snižuje a novorozenec se ukládá do postýlky. Vedle hydrofilních základů lze užít i krémové základy. Masťové základy jsou pro možnou poruchu termoregulace na základě hyperkeratózy kontraindikovány. Tato stejná pravidla platí i při horkém letním počasí. Aplikace hydrofilních základů je optimální cca 6–8krát denně, u mastnějších základů se doporučuje pouze asi 3–4krát denně.
Přídavky do mastí: 5–10% glycerol, u starších pak možno až na 8%, urea od 2. do 4. roku 3–5%, na plosky 10%. Kůže hlavy: keratolytické šampóny, ponechat působit alespoň 5–10 minut.
Koupele: Přístup ke koupelím je individuální, řídí se kožním nálezem a celkovým klinickým stavem a věkem dítěte. Převážně jsou efektní každodenní koupele, částečně s přídavkem olejů, později olejové koupele střídavě s natriumhydrogenkarbonatem – jedlou sodou, v dávce 50–150 g/koupel, která působí keratolyticky tím, že zvyšuje pH. Používá se převážně u LI, neboť blokuje LEKTI, inhibitor serinových proteáz, a tak dochází k zvýšení aktivity proteáz, což vede ke snadnějšímu odšupení [13 ].
U některých pacientů je prospěšná mechanická keratolýza: po 10–20 minutách změkčení hyperkeratóz ve vaně je možno jemným mechanickým třením podle závažnosti ichtyózy 1–7krát týdně tyto šupiny odstranit. Odstranění by mělo být prováděno stále ve vlhku, při namočené kůži. K užití je možno užít různé brusné kameny, či houby, které jsou volně k dostání. U těžce postižených pacientů může tento proces trvat 60–90 minut, provádí se denně [13 ].
Systémová terapie: U těžkých forem LI, CIE lze nasadit celkovou terapii retinoidy /Oral retinoids and retinoic acid metabolism blocking agents (RAMBAs)/. Retinoidy mají keratolytické účinky, které usnadňují odlupování šupin z povrchu a zabraňují nadměrné hyperkeratóze, což vede k normální tloušťce a lepší funkci rohové vrstvy. U LI lze použít oba typy retinoidů, isotretinoin a aromatické retinoidy (etretinate, acitretin). Aromatické retinoidy jsou účinější u palmoplantarních hyperkeratóz. Acitretin má delší poločas eliminace než isotretinoin, v organismu může setrvávat po měsíce po vysazení léčiva. U těžkých forem ichtyóz se proto doporučuje isotretinoin [15].
Retinoidy se výjimečně nasazují již u novorozenců nebo kojenců. Větší zkušenosti jsou s celkovou léčbou u pacientů školního věku, dávka retinoidů je 0,3–0,7 mg/kg/den. Podání je perorální, ale přináší s sebou celou řadu nežádoucích účinků. Retinoidy velmi zlepší nález hyperkeratózy a ektropií, ale ne erytrodermie. Účinky a vedlejší účinky terapie jsou ale reverzibilní [2, 9]. Před zahájením terapie je nutno provést tato vyšetření: RTG dlouhých kostí a páteře, KO, JT, TAG a cholesterol, urea, kreatinin, ionty a moč. V průběhu terapie pak vyšetření viz výše nejprve á 4 týdny v prvních dvou měsících terapie, poté každé tři měsíce [12]. Nežádoucími účinky mohou být: suchost kůže a sliznic, panaricia, pyogenní granulomy, hyperostózy skeletu, kalcifikace vazů, která však může být přítomna i u ostatních mladistvých, proto je nutno provádět RTG i před zahájením terapie. Předčasný uzávěr epifyzárních štěrbin se vyskytuje jen velmi zřídka při velmi vysokých dávkách retinoidů [2]. Asi u 25 % pacientů nacházíme hypertriacylglycerolémii a asi u 15 % elevaci transamináz. Tyto hodnoty jsou však závislé na dávce a jsou reverzibilní.
Teratogenita: ženy v produktivním věku při systémovém užívání retinoidů musí být zajištěny antikoncepcí a pravidelně každý měsíc musí být prováděn těhotenský test, který se provádí i před zahájením terapie. Rodiče pacientů podepisují informovaný souhlas s terapií retiniody.
Pacienti se závažnými formami ichtyózy jsou v péči specialistů různých odborností podle příznaků:
- Oční: kontroly rohovky se provádí pravidelně, výrazná ectropia se řeší ve spolupráci s plastickým chirurgem.
- ORL: nánosy šupin se šetrně odstraňují ze zvukovodu. Ve školním věku se řeší přirostlý boltec chirurgickou cestou.
- Ortopedie a rehabilitace: v důsledku ztluštění rohoviny se vytváří krunýř in utero, který způsobí kontraktury a omezenou hybnost u novorozence. Postnatálně prsty, které jsou v semiflekčním postavení, vyžadují rehabilitaci a vyvazování.
- Výživa: v důsledku zvýšené metabolické výměny u těžkých ichtyóz se dbá na zvýšený metabolický příjem, sleduje se hladina bílkovin, železa a iontů.
- Dětský psycholog: kvalita života pacientů je snížena. Děti se cítí sociálně izolovány, trpí svým vzhledem. Psychoterapie je nezbytnou součástí komplexní péče o pacienty.
Fungující pacientská organizace je oporou rodinám a samotným pacientům se závažnými formami ichtyózy, organizace spolupracuje s týmem specialistů.
c) Harlequin ichthyosis (HI)
Etiologie: jedná se vlastně o velmi závažnou vystupňovanou formu lamelární ichtyózy.
Vzniká na základě recesivně získané mutace genu pro enzym ABCA 12 /ATP-binding cassette transporter 12/. Tento enzym je důležitý pro transport lipidů a je kódován na chromozomu 2q34. V důsledku této mutace dochází k poruše transportu nově syntetizovaného glucosylceramidu, namísto do lamelárních tělísek do intersticiálních prostor stratum corneum, což vede k výraznému snížení bariérové funkce kůže.
Klinický obraz: děti se rodí většinou předčasně s kůží, která připomíná oblek šaška Harlekýna z „Commedia dell´arte“. Šupiny jsou velké, silné a s četnými hlubokými ragádami. Je silně vyjádřeno eclabium, ectropium a k hlavě přirostlé boltce, rty mají tvar O a připomínají ústa ryby. Vzhledem ke kožnímu nálezu se rozvíjí ohybové kontraktury a prsty rukou a nohou splývají pod nánosem šupin. Pohyblivost celého tělíčka je vzhledem k nánosům až pancéřovitých šupin omezena.
Onemocnění však není asociováno s jinými interními či neurologickými onemocněními. Oproti dříve uváděným údajům je pozdější život dítěte relativně normální a i neonatální mortalita poklesla z dřívějších téměř 100 % na nynějších cca 10 %. Vzhledem k předčasnému narození a kožnímu nálezu jsou děti ohroženy hlavně vznikem infekcí, dále pak hypertermií a sekundárními poruchami elektrolytové rovnováhy. Při intenzivní terapii se šupiny během prvních několika týdnů postupně odloučí, avšak během doby se vyvíjí v poměrně těžkou formu lamelární ichtyózy.
Histologické vyšetření: v elektronové mikroskopii nacházíme tvarově vysoce abnormální lamelární tělíska. Dalšími změnami jsou vezikulární organely ve stratum granulosum a vysoce amorfní materiál v intersticiálních prostorech stratum corneum.
Diagnostika: klinický obraz, histologické vyšetření, DNA molekulární diagnostika.
Terapie: se řídí doporučením, která platí pro závažné typy ichtyóz. Péče se koncentruje na stabilizování tělesné teploty, elektrolytové rovnováhy a hydratace. Používají se externa s obsahem 5–10 % glycerolu, nikdy pak vazelinové základy pro svůj okluzivní charakter. Dále se doporučují dezinfekční koupele, např. s obsahem octenidinu. Hluboké ragády se dezinfikují Cytealem (ředění 1 : 10). Aplikují se epitelizační a dezinfekční gely do ragád, např. Hemagel, eventuálně lokální antibiotika.
2. Keratinopatické ichtyózy
K bulózním ichtyózám se počítají epidermolytická ichtyóza, superficiální epidermolytická ichtyóza a ichthyosis Curth-Macklin. Jsou známy mutace genů, které zapříčiňují vakuolární degeneraci stratum granulosum a horních vrstev stratum spinosum, a vedou tak k tvorbě intraepidermálních puchýřků. Současně dochází k poruše uvolňování lipidů z lamelárních tělísek a tyto lipidy pak nejsou uvolňovány do mezibuněčných prostor. Stejně jako u ARCI, vykazuje kůže výraznou orthohyperkeratózu, která v průběhu prvního roku života ještě dále sílí (epidermolytická hyperkeratóza). S postupným ztluštěním epidermis klesá tvorba puchýřů.
a) Epidermolytická ichtyóza (EI)/dříve kongenitální bullózní ichtyosiformní erytrodermie BCIE = morbus Brocq/
Incidence je asi 1 : 200.000 až 1 : 300.000.
Etiologie: toto AD dědičné onemocnění je založeno na mutaci genu pro keratin 1 (CK 1 : 12q13), popř. keratin 10 (CK 10: chromosom 17q21-22), které vedou k charakteristickému obrazu epidermolytické hyperkeratózy. Pacienti s mutací pro CK 1 mají současně i palmoplantární hyperkeratózu, to je dáno rozložením CK1 v organismu.
Při postzygotické, somatické mutaci CK 1 nebo CK10 v brzké embryogenezi dochází k tvorbě epidermálního névu, který je uložen v Blaschkeho liniích. Pokud somatický mozaicismus zasáhne také somatické buňky, může se u další generace vyskytnout EI. Měli by být proto vyšetřeni rodiče dětí s EI, zda nemají epidermální névy a pokud ano, pak musí být provedena histologie, která může vykazovat známky epidermolytické hyperkeratózy.
Klinický obraz: ihned po porodu se u dětí vyskytuje erytrodermie a plošné eroze, které mohou připomínat epidermolysis bullosa congenita nebo stafylokokový SSS syndrom. Během 2.–3. roku života postupně dochází ke zmírnění tvorby puchýřů a postupně se tvoří lamelární hyperkeratózy, které jsou postupně hřebínkové, nahnědlého odstínu (obr. 5). Stále však přetrvává výrazný sklon k bakterielním infekcím kůže. Klinicky se rozlišuje 6 podtypů EI, hlavním rozlišovacím kritériem je palmoplantární keratóza, která je přítomna u typu I–III, nebo není přítomna u typu IV–V [3]. Hlavními komplikacemi u kojenců s EI je dehydratace, bakteriální superinfekce, která hrozí pacientům po celý život. Častým patogenem na kůži je Streptococcus pyogenes. Infekce jsou u kojenců nebezpečné, mohou vést k sepsi, popř. až ke smrti dítěte. U starších dětí jsou infekce kůže spojeny s nepříjemným zápachem, který je stigmatizuje a vyčleňuje ze společnosti. Nutné jsou proto pravidelné stěry z kůže a sledování hodnot ASLO, dodržovat zásady dezinfekce kůže.


Histologické vyšetření: u EI je kornifikovaná obálka a rozložení lipidů v keratinocytech zcela normální. Dochází však ke změnám ve vláknech keratinu, která jsou v buňkách a tvoří drobné shluky, což vede k abnormální funkci cytoskeletu, a lamelární tělíska tak zůstávají “uvězněna “ v keratinocytech a neplní tak svoji normální funkci
Diagnostika: klinický obraz, histologické vyšetření, DNA molekulární diagnostika.
Terapie: terapie závisí na věku a rozsahu projevů.
Raný kojenecký věk: na základě ohrožení masivní infekcí a plošnými erozemi, musí být tyto děti uloženy na jednotkách intenzivní péče. Celkovou terapii antibiotiky je nutné uvážit podle stavu.
Raný dětský věk: kvůli sklonům k tvorbě puchýřů a erozí, musí být keratolytická terapie, zvláště v prvním roce života, prováděna velmi opatrně. Preparáty s vysokým obsahem urey se nesmí užívat. Vhodné jsou dezinfekční koupele v hypermanganu, naředěném Cytealu. Ke zmírnění nánosů hyperkeratóz eventuálně vedou koupele s přídavkem NaHC03, jedlé sody, viz výše. Nesmí se však používat u akutně zanícených, zarudlých ploch, pouze u nánosů hyperkeratóz. Kůže se zvláčňuje hydrofilními krémy, lze kombinovat se zinkovými nebo taninovými preparáty. Při superinfekci se dále užívají antibiotika, podle nálezu ve stěru, podle citlivosti nejčastěji penicilin, či amoxicilin.
Systémová terapie: Terapie s acitretinem může být užita pouze u hyperkeratóz velmi na terapii rezistentních, ale smí být zahájena teprve až po odeznění erozivní fáze, a to v dávce 0,3 mg/kg, ne více! Orální retinoidy lze užít ve velmi nízkým dávkách, 0,3–0,5 mg/kg, tj. asi 10 mg obden u dospělých. Mutace CK1 vede převážně k projevům na dlaních a ploskách, a proto je zde terapie acitretinem nutná jen velmi uváženě, může naopak vést ke zhoršení kožních bulózních projevů. Mutace v CK10 vede k projevům na celém těle a končetinách, naopak dlaně a plosky vynechává, stejně tak obličej – jedná se o rozložení CK10 v těle. Zde je terapie retinoidy v nižších dávkách možná, ale nemusí být efektní.
b) Ichthyosis Curth–Macklin (dříve ichthyosis hystrix)
Tato ichtyóza je podmíněna mutací CK1. Stejně jako EI je AD dědičná. První projevy jsou již v raném dětství a s velmi rychle se zhoršujícími klinickými projevy jako jsou difuzní či pruhovitá palmoplantární keratóza až s následnými flexulárními kontrakturami. Zejména na rukou a nohou a nad velkými klouby se vyvíjejí velké žlutohnědé šupiny špičatého, ostnitého tvaru.
c) Superficiální epidermolytická ichtyóza (dříve ichthyosis bullosa typ Siemens)
Tento typu ichtyózy je nejlehčím typem z bulózních ichtyóz. Je podmíněn mutací pro CK 2.
Klinický obraz: hyperkeratózy jsou přítomny jen v mírné formě a palmoplantární keratózy chybí zcela. Místo štěpení epidermis a puchýře jsou umístěny velmi povrchově, proto záhy praskají a slupují se v četné povrchové eroze.
Histologické vyšetření: epidermolýza se vyskytuje pouze ve vrchních vrstvách epidermis.
Kongenitální ichtyózy asociované se syndromy
a) Sjögrenův-Larssonův syndrom
Incidence: asi 1 : 250.000.
Etiologie: tento syndrom je AR dědičný, jeho základem je mutace, která deaktivuje fetaldehyd-dehydrogenázu (FALDH – chromozom 17p). Dochází tak ke kumulaci toxických metabolitů kyseliny arachidonové nejen v kůži, ale i v mozku.
Klinický obraz: u dětí je nejprve nápadná erytrodermie a různé šupení (od jemných šupin a až po velké hrubé), které se kolem 2. roku života stává nápadnějším, šupiny jsou tmavší, bývá palmoplantární keratóza. V důsledku ukládání metabolitů je typickým doprovázejícím znakem výrazný pruritus. Teprve sekundárně se kolem 2.–3. roku života vyvíjejí neurologické komplikace: nejistá chůze, poškození pyramidové dráhy, di - až tetraplegie, stejně tak jako motorická až mentální retardace.
Diagnostika: klinické příznaky, molekulárně genetické vyšetření s nálezem mutace genu pro FALDH, která pak může být nápomocna i při prenatální diagnostice. Při vyšetření oka se nacházejí na uvee charakteristické bělavé skvrny.
Terapie: pokud se u pacientů dodrží dieta s omezením tuků, která je založena pouze na rostlinných tucích, popř. jen na TAG se středně dlouhým řetězcem, může se progrese onemocnění výrazně zpomalit. Velmi důležitá je také rehabilitace ke zmírnění progrese motorické retardace. Kožní příznaky reagují velmi dobře na terapii retinoidy, ať už celkovými, či lokálními. Svědění lze omezit antagonisty leukotrienů, jako jsou např. montelukast.
b) Chanarinův-Dorfmanův syndrom = neutrální lipidy střádající choroba
Výskyt: tato relativně málo častá choroba je AR dědičná a vyskytuje se převážně u národů žijících kolem Středozemního moře.
Etiologie: syndrom je charakterizován akumulací triacylglycerolů v různých tkáních. Onemocnění je kódováno na chromozomu 3p21 (NCIE2, respektive CGI-58) a postihuje gen esterázy /lipázy/ thioesterázy.
Klinický obraz: klinicky dominuje kongenitální, jemně šupící ichtyóza na erytematózní kůži. U dětí se vyvíjí hepatosplenomegalie. V adolescentním věku se vyvíjí katarakta, dochází k postižení sluchu a myopatii, která při dalším zhoršení vývoje může vést až k ataxii a neurologickým komplikacím. U některých dětí se vyvíjí mikrocefalie.
Diagnostika: klinické projevy, průkaz vakuolizovaných (díky lipidovým kapkám) granulocytů a monocytů v periferní krvi.
Diferenciální diagnostika: Sjögrenův-Larssonův syndrom.
Terapie: ichtyóza dobře odpovídá na terapii retinoidy, ale zda dieta s omezením tuků může ovlivnit základ této choroby, není ještě zcela známo.
c) Nethertonův syndrom
Výskyt: incidence asi 1 : 200 000.
Etiologie: základem tohoto syndromu je AR dědičný defekt Serin-Protease-Inhibitor-Kazal typ 5 (SPINK5, chromozom 5q32), který vede ke zvýšené hydrolytické aktivitě ve stratum corneum, snížení desmosomálních cadherinů a zvýšené deskvamaci s následkem poruchy kožní bariéry [7]. Je zajímavé, že také mnoho atopiků vykazuje polymorfismus tohoto SPINK5 genu [16].
Klinický obraz: klinicky nacházíme jak příznaky atopického ekzému, tak také zpočátku generalizované, později jen lokalizované formy ichtyózy.
V prvním roce dominuje exfoliativní dermatitida, která může progredovat až do erytrodermie, stejně tak jako poruchy prospívání, které jsou hlavně zapříčiněny častými průjmy a infekcemi. Kožní nález připomíná těžký průběh atopické dermatitidy – děti mají vysoké IgE a eozinofilii v krevním obraze. Sklon k infekcím je založen na snížené syntéze antipolysacharidových protilátek [14]. V průběhu prvních 2 let života se pak také přidává zvýšená lomivost vlasů, které tak rostou jen velmi málo a zůstávají krátké. Toto se zakládá na strukturální anomálii vlasů, která se nazývá trichorrhexis invaginata „bambusové vlasy“. Tyto změny nejsou prokazatelné v každém vlasu, ale jsou viditelné i v řasách a obočích. Později se vyvíjí ichthyosis lienaris circumflexa – plochá erytematoskvamózní ložiska, v jejichž centru jsou nahromaděné dvojité šupiny.
Histologické vyšetření: vykazuje psoriasiformní epidermis, tj. hyperplazii, ale minimální hyperkeratózu. Z toho pohledu by se nejednalo o „pravou“ ichtyózu. Jsou však velmi změněna lamelární tělíska, a dochází tak poruše bariérové funkce kůže.
Diagnostika: Klinické příznaky, anomálie vlasů – trichorrhexis invaginata. Imunohistochemicky, DNA molekulární analýza, průkaz SPINK5.
Terapie: v 1. roce života intenzivní terapie, která je zaměřena na ošetření kůže hydrofilními krémy, na prevenci kvasinkových infekcí v zapářkové lokalizaci. Později je péče o kůži podobná pacientům s atopickým ekzémem. V prvním roce života mohou dětí trápit průjmovité stolice.
Infekčním projevům na kůži dítěte se předchází obklady s dezinfekčními roztoky a gely. Při větším, plošném postižení se podávají celkově antibiotika proti stafylokokům a streptokokům, např. Cefuroxim.
Pokud se vyskytují při terapii rezistentní hyperkeratózy, pak je možno, stejně jako u keratinopatické ichtyózy ve velmi nízkých dávkách užití acitretinu, tj. 0,1–0,15 mg/kg/den.
ZÁVĚR
Ačkoliv jsou ichtyózy velmi heterogenní skupinou onemocnění s pestrým klinickým obrazem, metody molekulární biologie výrazně zpřesnily jejich diagnostiku, umožnily komplexní genetické poradenství v rodinách, kde se již ichtyóza vyskytla, a také genetickou prevenci. Naproti tomu stále zůstávají velmi omezené možnosti lokální terapie i systémové terapie, která je vzhledem k častým nežádoucím účinkům problematická. Dosud neexistuje kauzální terapie u ichtyóz, je ve stadiu výzkumu.
U závažných typů ichtyóz je nutná multioborová spolupráce, nejlépe ve specializovaných centrech s návazností na neonatologická pracoviště. Tato specializovaná centra se sdružují v mezinárodní týmy expertů.
Do redakce došlo dne 16. 12. 2015.
Adresa pro korespondenci:
MUDr. Blanka Pinková
Dětské kožní oddělení
Pediatrické kliniky FN Brno
Černopolní 9, 613 00 Brno
e-mail: b.pinkova@seznam.cz
Zdroje
1. AKIYAMA, M., SHIMIZU, H. An update on molecular aspects of the non-syndromic ichthyoses. Experimental Dermatology, 2008, 17, p. 371–382.
2. BRECHER, A. R., ORLOW, S. I. Oral retinoid therapy for dermatologic conditions in children and adolescents. Journal of American Academy of Dermatology, 2003, 49, p. 171–182.
3. DI GIOVANNA, J. J., BALE, S. J. Clinical heterogenity in epidermolytic hyperkeratosis. Archives of Dermatology, 1994, 130, p. 1026–1035.
4. ELIAS, P. M., WILLIAMS, M. L., CRUMRINE, D., SCHMUTH, M. Ichthyoses, clinical, biochemical, pathogenic and diagnostic assessment. Current problems in dermatology, 2010, 39.
5. FERNANDES, N. F., JANNINER, C. K., SCHWARTZ, R. A. X-linked ichthyosis: an occulocutaneous genodermatosis. Journal of American Academy of Dermatology, 2010, 62, p. 480–485.
6. HÖGER, P. Kindermatologie – differenzialdiagnostik und Therapie bei Kindern und Jugendlichen. 3. vollständing überarbeitete und erweiterte Auflage, 2011, ISBN 978-3-7945-2730-4.
7. KOMATSU, N., TAKATA, M., OTSUKI, N., OHKA, R. Elevated stratum corneum hydrolytic activity in Nethertone syndrome suggests an inhibitory regulation of desquamation by SPINK 5 derived peptides. Journal of Investigative Dermatology, 2002, 118, p. 436–443.
8. KRUG, M., OJI, V., TRAUPE, H., BERNEBURG, M. Ichthyosen – Teil 1: Differentialdiagnose Vulgärer Ichthyosen und therapeutische Erwägungen. Journal Deutscher Dermatologischer Gesellschaft, 2009, 7, p. 511–520.
9. LACOUR, M., MEHTA - NIKHAR, B., ATHERTON, D., HARPER, J. I. An appraisal of acitretin therapy in children with inherited disorders of keratinization. British Journal of Dermatology, 1996, 134, p. 1023–1029.
10. OJI, V., TADINI, G., AKIYMA, M., BLANCHET BARDON, C., BODEMER, C., BOURRAT, E., COUDIERE, P., DIGIOVANNA, J. J., ELIAS, P., FISCHER, J., FLECKMAN, P., GINA, M., HARPER, J., HASHIMOTO, T., HAUSSER, I., HENNIES, H. C., HOLS, D., HOVNANIAN, A., ISHIDA – YAMAMOTO, A., JACYK, W. K., LEACHMAN, S., LEIGH, I., MAZEREEUW-HAUTIER, J., MILSTONE, L., MORICE-PICARD, F., PALLER, A. S., RICHARD, G., SCHMUTH, M., SHIMIZU, H., SPRECHER, E., VAN STEENSEL, M., TAIEB, A., TORO, J. R., VABRES, P., VAHLQUIST, A., WILLIAMS, M., TRAUPE, H. Revised nomenclature and classification of inherited ichthyoses: results of the First Ichthyosis Consensus Conference in Sorèze 2009. Journal of American Academy of Dermatology, 2010, 63, 4, p. 607–641.
11. OJI, V., TRAUPE, H. Ichthyoses: differential diagnosis and molecular genetics. European Journal of Dermatology, 2006, 16, p. 349–359.
12. ORMEROD, A. D., CAMPALANI, E., GOODFIELD, M. I. BAD Clinical standards Unit British Association of dermatologist guidelines on efficacy and use of acitretin in dermatology. British Journal of Dermatology, 2010, 162, p. 952–963.
13. PREIL, M. L., TRAUPE, H. Diagnostik und Therapie der Ichthyosen. AWMF - Leitlinie , 2008, 9, Nr.13/043.
13. STRYK, S., SIEGFRIED, E. C., KNUSTON, A. P. Selective antibody deficiency to bacterial polysacharide antigens in patients with Netherton syndrome. Pediatric Dermatology, 1999, 16, p. 19–22.
14. VAHLQUIST, A., GANEMO, A., VIRTANEN, M. Congenital ichthyoses: an overview of current and emerging therapies. Acta Dermato – Venereology, 2008, 88, p. 4–14.
15. WALLEY, A. J., CHAVANAS, S., MOFFATT, M. F., ESROUF, R. M., WONG, K., HARPER, J. I. Gene polymorfismus in Nethertone syndrome and common atopic diseases. Nature Genetics, 2001, 29, p. 175–178.
16. WEIDINGER, S., O´SULLIVAN, M., ILLIG, T., BARNETT, H., DEPNER, M., RODRIGUEZ, E., RUETHER, A., KLOPP, N., VOGELBERG, C., WEILAND, S. K. Filaggrin mutation, atopic eczema, hay fever and asthma in children. Journal of Allergology and Clinical Immunology, 2009, 121, p. 1203–1209.
Štítky
Dermatologie Dětská dermatologieČlánek vyšel v časopise
Česko-slovenská dermatologie

2016 Číslo 1
- Nástroje k hodnocení závažnosti psoriázy v klinických studiích
- Systémová léčba atopické dermatitidy konečně i u dětí
- Inovace v hojení ran: krytí Zetuvit Plus Silicone Border pro optimální management exsudátu z ran
Nejčtenější v tomto čísle
- Hereditární ichtyózy
-
Dermatoskopie nepigmentovaných kožních nádorů.
Cévní nádory a krvácivé projevy - Kongenitální forma dermatofibrosarkoma protuberans
- Psoriáza, psoriatická artritida a tofózní dna
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy