
Soubor pacientů s diagnózou primárního lymfedému sledovaných v Lymfologickém centru Dermatovenerologické kliniky UK 2. LF a FN Na Bulovce v letech 2000–2006
Patiens with Primary Lymphoedema Followed in the Lymphology Center of the Department of Dermatology of the 2nd Medical School and Bulovka University Hospital in the Years 2000–2006
Primary lymphoedema is a chronic disease caused by abnormalities of lymphatics, mostly involving lower legs. According to the age of onset it is divided into lymphoedema congenitum, preacox and tardum. From pathophysiological point of view it can be divided into the hypoplastic and hyperplastic hereditary type (Nonne-Milroy syndrome and Meige syndrome, respectively), and idiopathic type. In diagnostics history and physical examination helps to exclude other causes of oedema, in more difficult cases lymphoscintigraphy is performed. The therapy is based on the complex decongestion therapy using combination of manual and instrumental lymphodrainage and compression bandage, in some cases surgical and pharmacological treatment is indicated. In the Lymphology Center of the Department of Dermatology of the 2nd Medical School and Bulovka University Hospital 44 patients with diagnosis of primary lymphoedema were newly treated from January 2000 to December 2006. Diagnosis was made on the basis of history, clinical picture and lymphoscintigraphy. Most patiens underwent the complex decongestion therapy in the phase of oedema reduction. Maintenance therapy is always compressive (bandage, compressive stockings or leggings of II – IV class) including home instrumental therapy in a number of patients. The therapeutical effect is asessed by a regular measurement of extremity perimeter according to the established scheme. Each patient with primary lymphoedema is regularly followed up.
Key words:
primary lymphoedema – lymphoscintigraphy – complex decongestion therapy
Autoři:
N. Vojáčková; L. Šipulová; R. Schmiedbergerová; J. Hercogová
Působiště autorů:
Dermatovenerologická klinika UK 2. LF a FN Na Bulovce
přednostka prof. MUDr. Jana Hercogová CSc.
Vyšlo v časopise:
Čes-slov Derm, 83, 2008, No. 4, p. 187-190
Kategorie:
Klinické a laboratorní práce
Souhrn
Soubor pacientů s diagnózou primárního lymfedému sledovaných v Lymfologickém centru Dermatovenerologické kliniky UK 2. LF a FN Na Bulovce v letech 2000–2006
Primární lymfedém je chronické onemocnění postihující především končetiny, které vzniká na podkladě abnormality lymfatického systému. Podle doby vzniku prvních projevů se dělí na lymphoedema congenitum, praecox a tardum. Podle patofyziologie vzniku se dělí na typ hypoplastický a hyperplastický, s dědičným výskytem (Nonneho-Milroyův syndrom a Meigeův syndrom) a idiopatický. V diagnostice se uplatňuje anamnéza, fyzikální vyšetření, vyloučení jiné příčiny otoku a v nejasných případech se využívá lymfoscintigrafie. Základem léčby je tzv. komplexní dekongestivní terapie, využívající kombinaci manuální a přístrojové lymfodrenáže a komprese bandáží, v indikovaných případech chirurgická a farmakologická terapie.
V Lymfologickém centru Dermatovenerologické kliniky UK 2. LF a FN Na Bulovce bylo v období od ledna 2000 do prosince 2006 zařazeno do péče 44 pacientů s diagnózou primárního lymfedému. Diagnóza byla stanovena na podkladě anamnézy, lokálního nálezu a lymfoscintigrafie. Většina pacientů podstoupila v rámci fáze redukce otoku komplexní léčbu. Udržovací terapie je vždy kompresí (bandáží, kompresivními punčochami nebo návleky II. - IV. KT) a u řady pacientů domácí přístrojovou léčbou. Efekt léčby je hodnocen pravidelným měřením obvodu končetiny podle zavedeného schématu. Všichni pacienti s primárním lymfedémem jsou trvale sledováni.
Klíčová slova:
primární lymfedém – lymfoscintigrafie – komplexní dekongestivní terapie
Úvod
Lymfedém je vysokoproteinový otok, který vzniká na podkladě patologie lymfatického systému. Nedostatečnost lymfatického systému může mít různé příčiny, jejich rozpoznání pouze na základě klinického obrazu je obtížné, protože objektivní nález bývá u různých typů lymfedému shodný.
Základy klasifikace lymfedému stanovil v roce 1934 Allen, který rozdělil lymfedém na dva základní typy: zánětlivý a nezánětlivý a do dvou skupin: primární a sekundární. Primární lymfedém vzniká bez zjevné příčiny, sekundární vzniká druhotně následkem zánětu, operačního výkonu, radioterapie, úrazu nebo nádorové infiltrace.
Příčinou primárního lymfedému je patologický stav lymfatického systému, který vzniká na podkladě vývojové poruchy (4). Lymfografické studie, provedené Kinmonthem a Taylorem v roce 1954, prokázaly, že primární lymfangioadenopatie se nemusí projevit hned po narození. Primární lymfedém se proto dělí na lymphoedema congenitum (projevy po narození do dvou let), lymphoedema praecox (projevy do 35. let věku), lymphoedema tardum (projevy po 35. roce věku). Z hlediska patofyziologie rozdělil primární lymfedém Foldi v roce 1969. Toto dělení je přesné a podrobné, ale příliš složité, proto se nerozšířilo. Zjednodušené dělení z klinického pohledu je do dvou skupin na lymfedém hypoplastický, podmíněný hypoplazií, a lymfedém hyperplastický, podmíněný lymfektazií.
Hypoplastický lymfedém dále dělíme na proximální typ, kdy jsou dominantně postiženy lymfatické uzliny, a distální typ, kdy jsou dominantně postiženy lymfatické cévy. Dále dělíme primární lymfedém na lymfedém s dědičným výskytem, kam řadíme Nonneho-Milroyův syndrom (11, 12) a Meigeův syndrom, a lymfedém idiopatický, který vzniká izolovaně nebo v náhodné koincidenci s jinými vrozenými vadami a je nejčastější (10).
Nonneho-Milroyův syndrom je vzácné autosomálně dominantně dědičné onemocnění, které se projevuje při nebo krátce po narození (6, 13). Otok postihuje častěji dolní končetiny, vzácně horní končetiny, většinou symetricky (3). Meigeův syndrom je charakterizován familiárním výskytem otoku končetin, který je podmíněn dědičným faktorem Mendelova typu, jenž se uplatňuje recesivně. Postihuje častěji ženy, projevuje se v období dospívání. K této skupině lymfedému řadíme též Turnerův syndrom, pro který jsou charakteristické deformity skeletu, dysplazie gonád a ve 30 % lymfedém dolních končetin.
Klinický obraz jednotlivých typů primárního lymfedému je shodný. Otok postihuje převážně dolní končetiny, vzniká distálně a postupně se šíří proximálním směrem, častěji jsou postiženy ženy, projevy vznikají v první polovině života a může se vyskytovat dědičně. Rozlišení jednotlivých typů lymfedému z hlediska klinické praxe má malý význam, protože terapeutický přístup je stejný, mohou se lišit prognosticky (proximální typ hypoplastického lymfedému mívá horší prognózu než typ distální).
Při vyšetření pacienta s otokem zpravidla vystačíme s anamnézou a fyzikálním vyšetřením, v některých nejasných případech potom doplňujeme přístrojová vyšetření a v případě primárního lymfedému je též vhodné genetické vyšetření. V rodinné anamnéze se zaměříme na výskyt otoků v rodině, při podezření na dědičný původ otoků je vhodné vyšetřit rodinné příslušníky, v osobní anamnéze pátráme po vrozených vadách a onemocněních, zjišťujeme operace a úrazy, choroby v dětství a před vznikem otoku. V gynekologické anamnéze jsou důležité údaje o menstruačním cyklu, počtu porodů a potratů. V rámci nynějšího onemocnění klademe důraz na získání údajů o době vzniku otoku, jeho lokalizaci a rozsahu, charakteru (konzistence, barva kůže), směru šíření, důležitý je časový údaj prvních známek otoku. Primární lymfedém začíná v distálních částech končetin a odtud se šíří proximálně.
Fyzikální vyšetření je důležité k vyloučení jiných příčin otoku. Provedeme celkové orientační somatické vyšetření, poté se zaměříme na oblast postiženou otokem. Zjišťujeme rozsah a kvalitu otoku, přítomnost fibrózních změn, barvu a teplotu kůže, přítomnost pigmentací a trofických změn. Nezbytné je též vyšetření regionálních lymfatických uzlin. Primární lymfedém postihuje zpravidla obě končetiny, někdy bývá jedna postižena v časovém předstihu, stupeň postižení může být rozdílný (1, 2).
Z přístrojových vyšetření je na prvním místě lymfoscintigrafie, od dříve používané rentgenkontrastní lymfografie se upustilo pro možnou retikuloendotelovou reakci s následným zhoršením otoku (1). V některých případech je možno využít ultrasonografické vyšetření a CT.
Diferenciálně diagnosticky je třeba odlišit sekundární lymfedém, flebedém a lipedém, otoky z interních příčin, otoky na podkladě endokrinní poruchy a poruchy hospodaření s elektrolyty a vodou, otoky způsobené léky (5).
Nejčastější komplikace lymfedému jsou infekce (erysipel, tinea pedum, onychomycosis) a kožní změny (verrucosis lymphostatica, ekzém, hypo - a hyperpigmentace, unguis incarnatus), lymphorhea, vzácné jsou ulcus lymphaticum a lymfosarkom (14).
Základem léčby všech typů lymfedému je komplexní dekongestivní terapie, která zahrnuje manuální lymfodrenáž, přístrojovou lymfodrenáž a kompresi. Tato intenzivní fáze léčby, někdy označovaná jako fáze redukce otoku, může probíhat ambulantně nebo za hospitalizace, zpravidla zahrnuje 4–6 týdenní cyklus, kdy léčba probíhá 5krát týdně, pokud pacient dochází ambulantně a 7krát týdně, pokud je hospitalizován. Následuje udržovací fáze léčby, která zahrnuje autolymfodrenáž, ve které je zaučen pacient nebo rodinný příslušník, kompresi elastickými obinadly nebo kompresivními pomůckami II.–IV. kompresivní třídy, cvičení, polohování a v některých případech též domácí přístrojovou léčbu.
Materiál a metodika
Jde o retrospektivní analýzu souboru pacientů s diagnózou primárního lymfedému, sledovaných v lymfologické ambulanci Dermatovenerologické kliniky UK 2. LF a FN Na Bulovce v letech 2000–2006. Do souboru byli zařazeni všichni pacienti s primárním lymfedémem, u nichž byla diagnóza stanovena na podkladě anamnézy, klinického vyšetření a ve většině případů též lymfoscintigraficky. V souboru jsme sledovali rozložení podle pohlaví, lokalizaci lymfedému, dobu vzniku prvních příznaků, typ lymfedému z různých hledisek. V souboru jsme posuzovali zvolená léčebná schémata a hodnotili efekt terapie.
Výsledky
Do souboru bylo zařazeno 44 pacientů s diagnózou primárního lymfedému, z toho bylo 14 mužů a 30 žen. Lymfedém byl lokalizován ve 24 případech na obou dolních končetinách, v 15 případech byla dosud postižena pouze jedna dolní končetina, obě horní končetiny byly postiženy u 4 pacientů a v jednom případě jsme zaznamenali lymfedém jedné horní a jedné dolní končetiny stejné strany. Věk v době prvních příznaků zachycuje tabulka 1, nejvíce pacientů bylo ve věku 11–15 let.

Typ lymfedému jsme hodnotili z různých hledisek. Podle věku nástupu otoku byl kongenitální typ ve 3 případech, typ praecox ve 31 případech a tardum u 10 pacientů. Na podkladě provedeného lymfoscintigrafického vyšetření jsme rozdělili soubor na typ hypoplastický proximální (3 případy), hypoplastický distální (36 případů), hyperplastický (1 pacient), kongenitálně chybné založení lymfatické drenáže smíšeného typu (1 pacient), ve 3 případech nebyla lymfoscintigrafie provedena.
Dědičný výskyt lymfedému jsme prokázali v 13 případech, z toho u 11 pacientů byla stanovena diagnóza Meigeova syndromu a ve 2 případech jsme se přiklonili k diagnóze Nonneho-Milroyova syndromu. Ve zbývajících 31 případech se jednalo o idiopatický hypoplastický lymfedém a v 1 případě o idiopatický hyperplastický lymfedém.
Léčba u všech pacientů probíhala ve dvou fázích: fázi redukce otoku a fázi udržovací. Fázi redukce otoku absolvovali pacienti ambulantně nebo za hospitalizace v délce 4–6 týdnů, v některých případech bylo nutné tuto léčebnou intenzivní kúru opakovat. Komplexní léčbu lymfedému podstoupilo 38 nemocných. Tři pacienti byli v minulosti pro lymfedém operováni, tzv. radikální Bařinkovou operací. Pouze kompresí byli léčeni 3 pacienti. U všech pacientů následovala udržovací terapie, která probíhá dosud a předpokládá se, že bude zapotřebí léčby trvalé. Přístroj pro domácí léčbu má k dispozici 25 pacientů a stav udržují stabilizován spolu s autolymfodrenáží a kompresí. Autolymfodrenáž a komprese je dostačující pro udržení stabilizovaného stavu u 15 pacientů a pouze autolymfodrenáž u 4 pacientů.
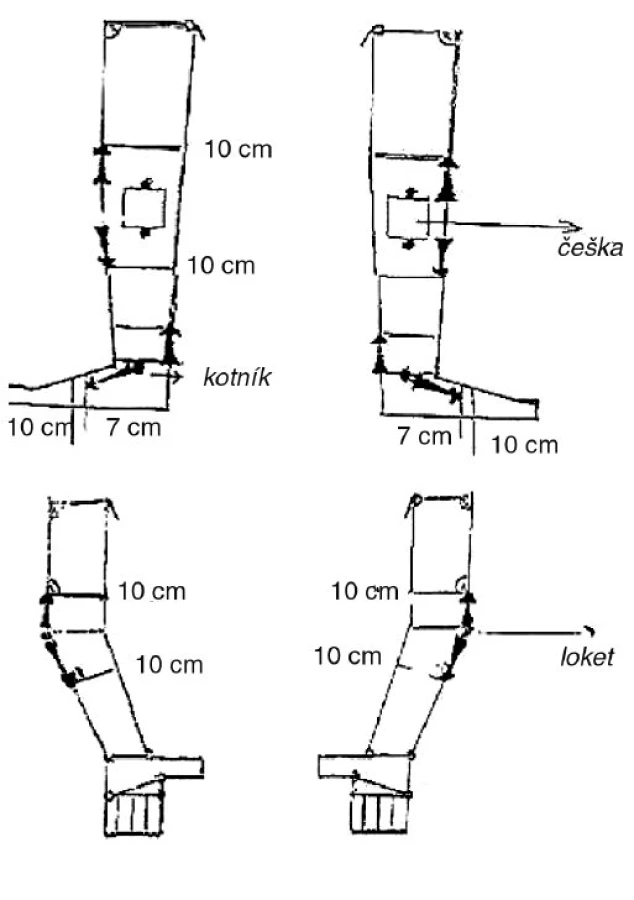
Efekt terapie jsme hodnotili ze dvou hledisek, subjektivního – spokojenost pacienta, všichni pacienti udávali úlevu tíhy postižené končetiny a zlepšení mobility. Objektivně jsme sledovali redukci v měřených obvodech, měření se provádí v pravidelných intervalech a zapisuje se schematicky (obr. 1). U všech pacientů došlo ke zmenšení v měřených obvodech, v průměru o 1–10 cm. Bylo provedeno měření obou dolních končetin u 24 pacientů a jedné dolní končetiny u 16 pacientů, celkem 64 dolních končetin (tab. 2), měření obou horních končetin ve 4 případech a jedné horní končetiny v 1 případě, celkem 5 horních končetin (tab. 3). Pacienti nadále zůstávají v péči naší lymfologické ambulance.


Diskuse
Primární lymfedém je chronické onemocnění, které zatěžuje nemocné fyzicky i psychicky a pokud není včas a adekvátně léčen, vede k ireverzibilním změnám a komplikacím. Všichni pacienti s otokem, který zpravidla vzniká plíživě, zhoršuje se večer a po námaze, ale bez zjevné příčiny, by měli být vyšetřeni se zaměřením na podezření na otok lymfatického původu (15).
V souboru pacientů s primárním lymfedémem je více žen, údaj o vyšší incidenci primárního lymfedému u žen v literatuře jsme však nenašli. Lokalizace primárního lymfedému je převážně na dolních končetinách, horní končetiny jsou postiženy vzácně (7).
Klasifikace primárního lymfedému je možná z různých hledisek. Podle věku v době prvních příznaků bylo nejvíce pacientů ve skupině lymphoedema praecox – 31 případů, nejméně ve skupině lymphoedema congenitum – 3 pacienti. U 10 pacientů se první příznaky objevily po 35. roce věku (lymphoedema tardum). Z hlediska dědičnosti se podařilo dědičný původ lymfedému prokázat u 13 pacientů, jako idiopatický se ukázal v 31 případech.
Většina pacientů podstoupila komplexní terapii lymfedému ve fázi redukce otoku, 3 pacienti byli v minulosti pro lymfedém operováni (8). Od radikálních Bařinkových operací se ustoupilo vzhledem ke komplikacím v pooperační péči za vzniku četných jizev a následně možného rozvoje sekundárního lymfedému. U všech pacientů došlo po léčbě ke stabilizaci stavu, který je nadále s různým úspěchem zachován v rámci udržovací léčby. Všichni pacienti nebo jejich rodinní příslušníci si osvojili autolymfodrenáž, kterou je nutno provádět denně. Denně je také potřebná komprese postižené končetiny obinadly nebo kompresivními pomůckami. U 25 pacientů byl stav indikován k následné domácí přístrojové léčbě.
Efekt terapie je možné hodnotit různými způsoby (9). U našich pacientů jsme jej hodnotili pomocí redukce v měřených obvodech. Měření podle předem stanoveného schématu provádí lymfoterapeut, končetina je měřena před léčbou, v průběhu léčby a po léčbě. Porovnáním rozměrů před léčbou a po léčbě došlo k redukci v měřených obvodech o minimálně 1 cm, maximálně 10 cm.
Pacienti s primárním lymfedémem by měli být trvale sledováni lymfologem, udržovací léčba by měla být též trvalá. Nedílnou součástí léčby jsou i režimová opatření, o kterých jsou pacienti poučeni (15).
Došlo do redakce: 14. 4. 2008
MUDr. Naděžda Vojáčková
Dermatovenerologická klinika UK 2. LF a
FN Na Bulovce
Budínova 2, 180 00 Praha 8
E-mail: nadavojackova@seznam.cz
Zdroje
1. Bechyně, M. Mízní otok-lymfedém. Praha. Phlebomedica, 1997.
2. Benda, K., Bařinka, L. Lymfedém končetin. Avicenum, 1981.
3. Brice, G., Child, AH., Bell, R. et al. Milroy disease and the VEGFR-3 mutation phenotype. J Med Genet, 2005, 42, p. 98-102.
4. Byung-Boong, LEE. Lymphoedema-angiodysplasia syndrome: a prodigal form of lymphatic malformation. Lymphology, 2004, 47, p. 324-332.
5. Ely, JW., Osberoff, JE., Chambliss, ML. et al. Approach to leg edema of unclear etiology. J Am Board Fam Med, 2006, 19, p. 148-160.
6. Ghalamkarpour, A., Morlot, S., Raas-Rotschild A. et al. Hereditary lymphedema type I associated with VEGFR3 mutation: the first de novo case atypical presentations. Clin Genet, 2006, 70, p. 330-335.
7. Harel, L., Amir, J., Nussinowitch, M. et al. Lymphedema praecox seen as isolated unilateral arm involvement: Case report and review of the literature. J Ped, 1997, 130, 3, p. 492-494.
8. Heinig, B., Wolina, U. Surgrery in congenital lymphedema: a follow-up of 50 years. Low Extr Wounds, 2003, 2, p. 173-176.
9. Lerner, DS., Klose, G., Cosimi, AB. Effective treatment of lymphedema of the extremities. Arch Surg, 1998, 133, p. 452-458.
10. Meige, H. Dystrophie oedemateuse hereditaire. Presse Méd, 1898, 6, p. 341-343.
11. Milroy, WF. An undescribed variety of hereditary oedema. NY Med J, 1892, 56, p. 505-508.
12. Milroy, WF. Chronic hereditary edema. Milroy’s disease. JAMA, 1928, 91, p. 1172-1175.
13. Štelcl, J., Hudeček, R. Primární lymfedém: kazuistika. Prak Gyn, 2004, s. 16-17.
14. Vignes, S., Dupuy, A. Recurrence of lymphedema-associated cellulitis under prophylactic antibiotherapy: a retrospective cohort study. JEADV, 2006, 20, p. 818-822.
15. Vojáčková, N., Šebková, M., Schmiedbergerová, R. et al. Soubor nemocných s lymfedémem sledovaný v Lymfologickém centru Dermatovenerologické kliniky UK 2. LF a FN Na Bulovce v letech 2000-2005. Retrospektivní analýza. Čas Lék Čes, 2007, 146, s. 57-61.
Štítky
Dermatologie Dětská dermatologieČlánek vyšel v časopise
Česko-slovenská dermatologie

2008 Číslo 4
- Nástroje k hodnocení závažnosti psoriázy v klinických studiích
- Systémová léčba atopické dermatitidy konečně i u dětí
- Inovace v hojení ran: krytí Zetuvit Plus Silicone Border pro optimální management exsudátu z ran
Nejčtenější v tomto čísle
- Novšie vírusové exantémy detského veku – komplexný pohľad na problematiku
- Kazuistický prípad pacienta s blastickým NK-bunkovým lymfómom, syn. CD4+/CD56+ hematodermická neoplázia, syn. včasná plazmocytoidná leukémia/lymfóm z dendritických buniek
- Klinický prípad: Stopkatá polypoidná lézia na stehne
- Novinky v léčbě psoriázy biologiky a standardními systémovými léky
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy