
Wilsonova choroba
Wilson’s disease
Wilson’s disease is an inherited disorder leading to accumulation of copper in tissues, mainly in the liver and brain. Genetic defect is in the gene coding ATPase type P (ATP7B). The inheritance is autosomal recessive. Up to now, more then 500 mutations causing Wilson’s disease were described. The most frequent mutation in Central Europe is mutation H1069Q.
The manifestation of Wilson’s disease is usually hepatic or neurologic. Hepatic form is manifested by acute or chronic hepatitis, steatosis or cirrhosis. Neurologic involvement is manifested usually after 20 year of age by motor disturbances (tremor, disturbed speech, problems with writing), which could progress into severe extrapyramidal syndrome with tremor, rigidity, dysartria, dysfagia and muscle contracture.
Diagnosis is based on clinical and laboratory examinations (neurologic symptoms, liver disease, low serum ceruloplasmin levels, elevated free copper concentration in serum, high urine copper excretion, and presence of Kayser-Fleischer rings). Confirmation of diagnosis is done by hepatic copper concentration in liver biopsy or by genetic examination.
Untreated disease leads to the death of a patient. Treatment is based on chelating agents decreasing the copper content by excretion into urine (D-penicillamine, trientine) or on agents preventing absorption of copper from food (zinc, ammonium-tetrahiomolybdene). Patients with asymptomatic Wilson’s disease have to be treated as well. In Czech Republic either penicillamine or zinc are used. Liver transplantation is indicated in patients with fulminant liver failure or decompensated cirrhosis.
Screening in families of affected patients (all siblings) is obvious.
Key words:
Wilson’s disease, liver cirrhosis, extrapyramidal syndrome, copper metabolism, penicillamine, zinc.
Autoři:
R. Brůha 1; Z. Mareček 4; P. Martásek 2; S. Nevšímalová 3; J. Petrtýl 1; P. Urbánek 4; H. Kalistová 3; L. Pospíšilová 2
Působiště autorů:
Univerzita Karlova v Praze, 1. lékařská fakulta, IV. interní klinika VFN
1; Univerzita Karlova v Praze, 1. lékařská fakulta, Klinika dětského a dorostového lékařství VFN
2; Univerzita Karlova v Praze, 1. lékařská fakulta, Neurologická klinika VFN
3; Univerzita Karlova v Praze, 1. lékařská fakulta, Interní klinika ÚVN
4
Vyšlo v časopise:
Čas. Lék. čes. 2009; 148: 544-548
Kategorie:
Přehledový článek
Souhrn
Wilsonova choroba je vrozené onemocnění vedoucí k akumulaci mědi ve tkáních, především játrech a mozku. Genetický defekt je v genu kódujícím ATPázu typu P (ATP7B). Dědičnost je autozomálně recesivní. Je známo přes 500 mutací způsobujících Wilsonovu chorobu. Ve střední Evropě je nejčastější mutací H1069Q.
Wilsonova choroba se nejčastěji projevuje jaterním či neurologickým postižením. Jaterní forma se manifestuje jako akutní či chronická hepatitida, steatóza nebo cirhóza. Neurologické postižení se projeví nejčastěji po 20. roce věku poruchami motoriky (třes, poruchy řeči a písma), které mohou progredovat do těžkého extrapyramidového syndromu s třesem a rigiditou, dysartrií, dysfagií a svalovými kontrakturami.
Diagnózu lze stanovit na základě klinických a laboratorních vyšetření (neurologické projevy, jaterní léze, nízký ceruloplazmin, vyšší volná měď v séru, vysoké odpady Cu močí, Kayser-Fleischerův prstenec). Potvrzení diagnózy přinese vyšetření Cu v jaterní tkáni nebo genetické vyšetření.
Neléčená Wilsonova choroba vede ke smrti nemocného. Léčba je založena buď na odstranění mědi z organismu chelačními látkami, které se vylučují do moči (penicilamin, trientin), nebo na omezení vstřebávání mědi ze střeva a snížení toxicity mědi (zinek, amonium-tetrathiomolybden). V našich podmínkách se používá penicilamin nebo zinek. Jaterní transplantace je indikována u pacientů s fulminantním jaterním selháním nebo s dekompenzovanou jaterní cirhózou.
V rodině je nezbytné provést screening u všech sourozenců postiženého jedince a případné asymptomatické nemocné také léčit.
Klíčová slova:
Wilsonova choroba, jaterní cirhóza, extrapyramidový syndrom, metabolismus mědi, penicilamin, zinek.
Úvod
Wilsonova choroba je svou prevalencí tří nemocných na 100 000 obyvatel jednou z nejčastějších vrozených poruch metabolismu. Vyskytuje se na celém světě bez větších rasových rozdílů. Podkladem choroby je vrozená porucha vylučování mědi do žluče s následnou akumulací mědi v játrech i jiných orgánech. Nadměrné množství mědi pak vede k poškození především jater a mozku. Choroba byla popsána před necelými 100 lety jako familiární „progresivní lentikulární degenerace“ – smrtelná neurologická choroba provázená jaterní cirhózou (1). V následujících desetiletích byla postupně objevena souvislost Wilsonovy choroby s mědí a byl stanoven autozomálně recesivní typ dědičnosti.
Měď je nezbytnou součástí proteinů, které jsou odpovědné za mnoho enzymatických pochodů v živém organismu – například v respiračním řetězci mitochondrií, při biosyntéze melaninu, v metabolismu dopaminu, při udržení homeostázy železa, pro antioxidační ochranné mechanismy, při aminaci peptidů a při metabolismu pojivových tkání (2).
Měď přijatá v potravě se vstřebává neomezeně v duodenu a jejunu a je přednostně vychytávána jaterními buňkami – několik hodin po perorálním podání radioizotopu mědi je 95 % podaného množství akumulováno v játrech (3). Denní potřeba mědi se pohybuje kolem 0,9 mg. V běžné potravě je přitom obsaženo 2–5 mg/den a je zřejmé, že většina vstřebané mědi musí být z organismu vyloučena. Jedinou přirozenou cestou eliminace mědi je exkrece do žluče.
Za transport mědi v hepatocytu je odpovědno několik proteinů: na plazmatické membráně jaterních buněk je umístěn tzv. přenašeč mědi 1 (copper transporter 1 – Ctr1), který má vysokou afinitu pro měď (4). Intracelulárně se měď váže na metalothiony (MT I a II; mohou vázat i jiné kovy jako kadmium a zinek a hrají významnou úlohu v ochraně buňky před poškozením těmito kovy) a metalochaperony, které transportují měď především do Golgiho aparátu, kde probíhá vlastní inkorporace mědi do proteinů (5, 6). Klíčovou roli v přenosu mědi přes nitrobuněčné membrány i v přenosu přes membránu na žlučovém pólu hepatocytu, a tím v exkreci do žluče hraje enzym ATPáza 7ß (ATP7B) lokalizovaný v Golgiho aparátu (7). Další funkcí tohoto proteinu je inkorporace mědi do apoceruloplazminu. Uvolňování apoceruloplazminu z jaterních buněk bez navázané mědi pak vede k typickému snížení samotného ceruloplazminu v séru, neboť apoceruloplazmin má podstatně kratší poločas. Ceruloplazmin je plazmatická bílkovina, glykoprotein obsahující 8 atomů mědi, který je nezbytný pro hematopoézu a umožňuje uvolňování železa z buněk. Katalyzuje oxidaci dvojmocného železa, proto se někdy též nazývá „ferrooxidáza“. Donátorem mědi pro tkáně se stává teprve při svém katabolismu. Jeho snížená hladina u pacientů s Wilsonovou chorobu je druhotná a ve vývoji choroby se neuplatňuje, jeho význam je u této choroby při diagnostice.
Gen pro ATP7B je umístěn na 13. chromozomu. Mutace tohoto genu je podkladem Wilsonovy choroby, neboť vede k poruše funkce ATP7B, následně k narušení exkrece mědi do žluče, a tím k akumulaci mědi v organismu (8, 9). V současnosti je známo již více než 500 mutací ATP7B, není však jisté, zda všechny chorobu způsobí.
Stále nevyjasněný zůstává vztah mezi genetickým defektem a fenotypickým projevem. Někteří autoři popisují přímý vztah mezi jednou z mutací (H1069Q) a neurologickou formou Wilsonovy choroby (10), na jiných souborech však tento vztah tak jasný není. Je pravděpodobné, že přítomnost mutace H1069Q, která se u nás vyskytuje nejčastěji, může vést jak k jaterní, tak neurologické či smíšené formě (11). Lze předpokládat existenci dalších endogenních či exogenních vlivů, které způsobí rozdílnou klinickou manifestaci. Je například známo, že genetické variace apolipoproteinu E (ApoE) mohou hrát roli ve vývoji choroby a zdá se, že některé izoformy ApoE mohou mít protektivní vliv především u pacientů s neurologickou formou Wilsonovy choroby (12).
Klinická manifestace
Wilsonova choroba se nejčastěji manifestuje jaterním postižením nebo neuropsychiatrickými příznaky (jaterní nebo neurologická forma). Může se projevit jen jaterním postižením, samostatným neurologickým postižením či oběma společně. Neurologická forma se objevuje v průměru až o 10 let později než forma jaterní. V době neurologické manifestace je u většiny pacientů s Wilsonovou chorobou přítomno též jaterní poškození, pokud se jako první diagnostikuje jaterní forma, pak se většinou neurologické postižení nevyvine. Vzácně se choroba manifestuje izolovanými psychiatrickými projevy. V rámci rodinného screeningu můžeme chorobu zachytit ve zcela asymptomatické fázi.
Jaterní forma
Akutní hepatitida. Wilsonova choroba je chronické onemocnění, které se však může manifestovat akutní hepatitidou. Ta se nejčastěji objeví v dětství nebo dospívání a klinicky se podobá akutní hepatitidě jiné etiologie (infekční, autoimunitní) s elevací transamináz, mírným ikterem, únavou a případně dyspepsií. U většiny pacientů akutní fáze odezní, většinou pak přetrvávají vyšší jaterní testy i jiné známky chronické jaterní choroby (hepatomegalie). U části pacientů však může progredovat do fulminantního jaterního selhání. V takovém případě je typická hemolýza (13) a velmi vysoké odpady Cu močí; hodnota ceruloplazminu nemusí být v této situaci spolehlivým vodítkem. Důležitá je též těžká koagulopatie jako projev jaterního selhání.
Chronická hepatitida, cirhóza. Choroba se může manifestovat jaterní cirhózou v mladém věku (i před 20. rokem života) nebo chronickou hepatitidou s různým stupněm fibrózy. Klinicky se nemusí lišit od chronických hepatitid jiného původu. Stejně tak se může projevit steatózou či steatohepatitidou.
Neurologická forma
Tato forma se objevuje většinou kolem 20. roku života a později, vzácně až po 30. roce věku. Prvními projevy bývá třes rukou, obtíže s psaním a dalšími jemnými činnostmi, jako je například zapínání knoflíků, poruchy řeči, nestabilita chůze. Neléčená choroba pak většinou progreduje do plného parkinsonského syndromu s dysatrií, dysfagií a rigiditou, která vede se svalovým kontrakturám a následným deformitám končetin.
Téměř všichni pacienti s neurologickou formou mají jaterní postižení (u více než poloviny je přítomna již jaterní cirhóza) a v anamnéze mají často přechodnou a řádně nevyšetřenou elevaci jaterních testů v dětství.
V současné době na našem pracovišti sledujeme 114 pacientů s Wilsonovou chorobou. Klinická manifestace i další údaje jsou uvedeny v tabulce 1.

Diagnóza
Diagnóza je založena na kombinaci klinických a laboratorních parametrů; výjimečně může být založena jen na molekulárně biologickém vyšetření.
Při fyzikálním vyšetření lze nalézt výše popsané neurologické projevy, případně projevy akutní či chronické jaterní choroby. Žádný z těchto příznaků však není specifický pro Wilsonovu chorobu. Takovým specifickým příznakem je Kayser-Fleischerův prstenec. Bývá přítomen u většiny pacientů s neurologickou formou, ale jen u méně než poloviny pacientů s jaterní formou. Kombinace typických neurologických příznaků, nízkého ceruloplazminu a Kayser-Fleischerova prstence je dostačující ke správné diagnóze, většinou jsou však nezbytná další vyšetření včetně jaterní biopsie.
Ceruloplazmin bývá výrazně snížen (pod 0,1 g/l). Jedná se však o typický protein akutní fáze, jehož koncentrace může být při jakémkoliv zánětlivém procesu v organismu zvýšena.
Měď v séru. Celková měď nemá pro stanovení diagnostiky význam, při Wilsonově chorobě bývá zvýšena volná měď v séru. Tento parametr není příliš spolehlivý.
Odpad mědi do moči. Zvýšený nativní odpad Cu do moči (na více než dvojnásobek normy) je pro Wilsonovu chorobu velmi specifický. Normální nativní odpad mědi močí je 0,6–0,7 μmol za 24 hodin. V poslední době se už neklade takový důraz na „zátěžový“ penicilaminový test, který spočívá v indukci vylučování mědi do moči penicilaminem. Test není validován pro dospělé osoby, jediný prokázaný přínos penicilaminového testu v diagnostice Wilsonovy choroby je u dětí, kdy se podá 500 mg penicilaminu na začátku sběru, dalších 500 mg po 12 hodinách a celkový odpad za 24 hodin by měl přesáhnout 25 μmol (21). Vzhledem k dostupnosti penicilaminu v České republice lze připustit podání 600 mg namísto původně popisovaných 500 mg. U dospělých se používají různé dávky penicilaminu i různý interval sběru (až 3 dny, 1200 mg penicilaminu denně), při kterém by měly hodnoty Cu v moči dosáhnout alespoň pětinásobku původní nativní hodnoty. Není však zřejmé, jak pozitivní výsledek testu zpřesní diagnózu. Zásadní podmínkou správně provedeného sběru moči na odpady mědi je použití sběrných nádob, které jsou zbaveny případné kontaminace (tj. řádné vymytí destilovanou vodou).
V diagnostice Wilsonovy choroby může být někdy zavádějící pozitivita autoprotilátek, která se může někdy u pacientů s Wilsonovou chorobou vyskytnout.
Jaterní biopsie slouží jak k diagnostice (zvýšená hodnota mědi na 250 μg/g sušiny jaterní tkáně), tak ke stagingu jaterního postižení. Histologický obraz může být velmi různý – od prosté steatózy až po jaterní cirhózu, ale není specifický. I při speciálním barvení mědi se granula mědi podaří málokdy mikroskopicky prokázat. Staging jaterní choroby je tak indikací k jaterní biopsii i u pacientů s geneticky prokázanou chorobou.
Genetické vyšetření přineslo velký pokrok v diagnostice Wilsonovy choroby. Problémem je skutečnost, že v dnešní době je známo více než 500 různých mutací, které mohou chorobu způsobit, a stále se popisují nové. Aktuální údaje o známých mutacích lze nalézt na internetové adrese: http://www.wilsondisease.med.ualberta.ca/database.asp. V daném geografickém regionu by většina pacientů měla mít jen několik nejčastěji se opakujících mutací, ale stále existuje až 20 % pacientů, u kterých se nepodaří genetický defekt jednoduchými metodami nalézt. Prevalentní mutací ve střední Evropě je H1069Q (15–17).
Nedávno byl vypracován skórovací systém (18), který zahrnuje jak klinické nálezy, tak laboratorní vyšetření (tab. 2). U pacientů s bodovým hodnocením 4 a více je diagnóza Wilsonovy choroby vysoce pravděpodobná. Všichni pacienti sledovaní na našem pracovišti pro Wilsonovu chorobu splňují tato diagnostická kritéria.
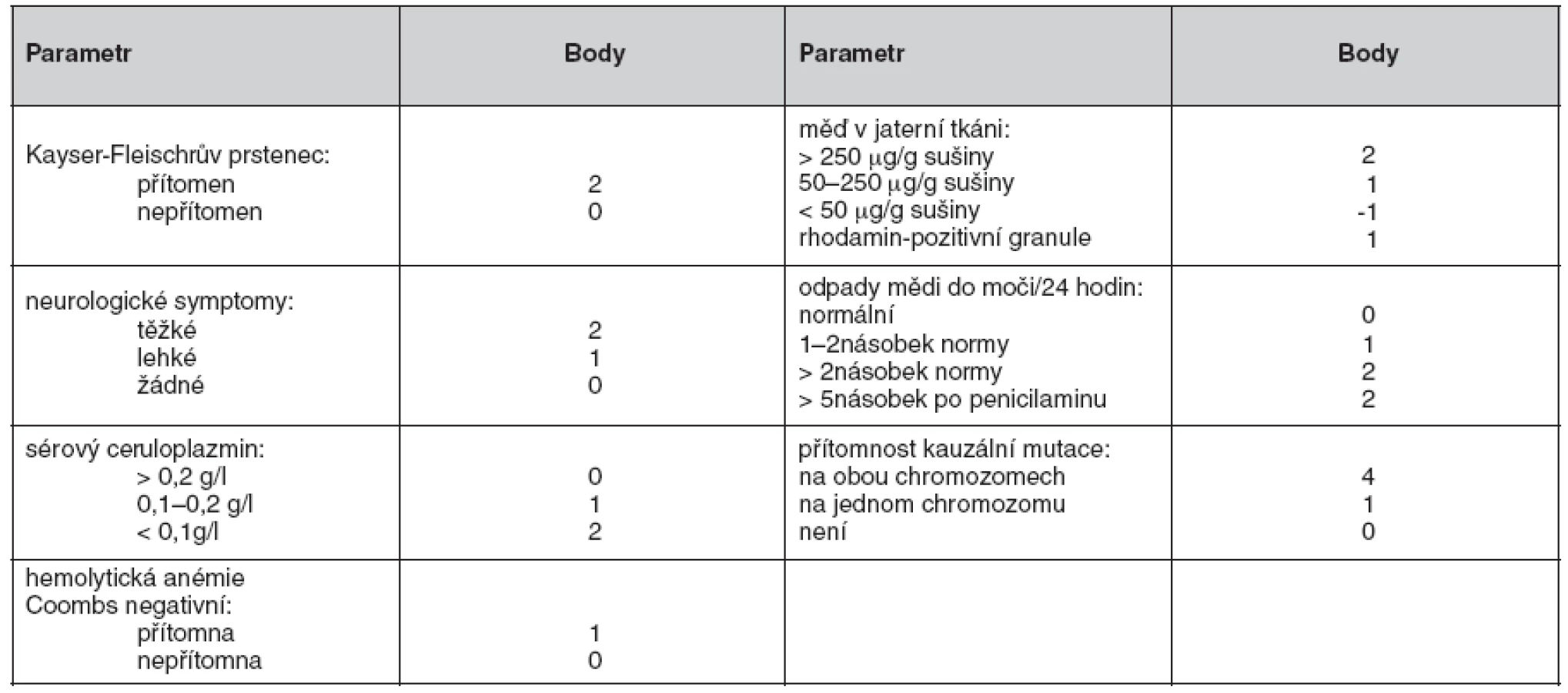
Metodou screeningu je buď molekulárně genetické vyšetření u známé mutace, nebo bazální vyšetření metabolismu mědi (ceruloplazmin, volná měď v séru, nativní odpady Cu močí), vyšetření jaterních testů, sonografie jater, případně neurologické vyšetření tam, kde není genetická diagnostika jednoznačná. Vyšetřeni musí být všichni sourozenci postiženého jedince. V rámci genetického poradenství většinou vyšetřujeme i děti nemocného s Wilsonovou chorobou.
Jednotlivá diagnostická kritéria pacientů sledovaných na našem pracovišti jsou uvedena v tabulce 1. Pro praxi je důležité pozorování týkající se spolehlivosti jednotlivých parametrů při diagnostice Wilsonovy choroby v české populaci (graf 1). Spolehlivým se jeví stanovení Cu v jaterní tkání – 79 pacientů z 81 dostupných biopsií (98,75 %) mělo hodnotu Cu v sušině jaterní tkáně vyšší než 250 μg/g (oba pacienti s hodnotou Cu nižší než 250 μg/g sušiny jaterní tkáně měli Wilsonovu chorobu potvrzenou genetickým vyšetřením). Velmi spolehlivým parametrem je nižší hodnota ceruloplazminu (přítomna u 90,5 % pacientů). Nativní odpady Cu do moči dosahovaly dvojnásobku normy u 78 % pacientů v našem souboru. Naopak velmi málo spolehlivým parametrem je Kayser-Fleischerův prstenec – byl popsán jen u 47 % našich pacientů. Genetické vyšetření bylo pozitivní u 80 % pacientů s Wilsonovou chorobou.

Léčba
Léčba se liší podle manifestace a stadia choroby (19). Neléčená Wilsonova choroba vede ke smrti nemocného. Při řádné léčbě nemusí být život pacientů výrazně zkrácen.Léčba je založena buď na odstranění mědi z organismu chelačními látkami, které se vylučují do moči (penicilamin, trientin), nebo na omezení vstřebávání mědi ze střeva a snížení toxicity mědi v organismu (zinek, amonium tetrathiomolybden). V našich podmínkách se používá penicilamin nebo zinek (Zn-sulfát, či Zn-acetát).
Při fulminantním jaterním selhání na podkladě Wilsonovy choroby je jedinou účinnou léčbou jaterní transplantace (20, 21). Jinak vede tato forma manifestace téměř ve 100 % ke smrti postiženého jedince.
Penicilamin je indikován v léčbě jaterní formy Wilsonovy choroby (krom fulminantního jaterního selhání – viz výše). Naprosto nezbytné je zahájit léčbu nízkou dávkou penicilaminu (150 mg/den) a k cílové denní dávce 900–1200 mg/den dospět v průběhu 3–6 měsíců. Při okamžitém nasazení plné dávky hrozí vznik ireverzibilního neurologického postižení či jaterního poškození (14). Dále je nezbytné léčbu penicilaminem doplňovat podáváním pyridoxinu 2 tbl/den. Jaterní chorobu lze léčbou téměř kompletně stabilizovat, většinou dojde k poklesu transamináz, u pacientů s cirhózou dojde k dlouhodobé stabilizaci. Léčbu kontrolujeme pravidelným vyšetřováním odpadů mědi do moči. Pokud pacienti nemají hodnoty Cu v moči zvýšeny, je velmi pravděpodobné, že penicilamin neužívají. Ve fázi stabilizace choroby je možno přejít na podávání preparátů zinku. Symptomatická léčba pacientů s jaterní formou spočívá v prevenci komplikací portální hypertenze, tj. betablokátory v prevenci krvácení, případně antibiotika v prevenci spontánní bakteriální peritonitidy při rizikovém ascitu. Účinek různých hepatoprotektiv býval v minulosti velmi přeceňován.
U pacientů s neurologickou symptomatologií není výběr léčby tak jednoznačný (22). Řada autorů se v poslední době přiklání k léčbě zinkem (23) (či trientinem). Dle našich zkušeností jsou však negativní důsledky penicilaminu v této indikaci (tj. zhoršení neurologické symptomatolgie) dány především neadekvátně rychlým zvyšováním dávky. Denní dávka esenciálního zinku musí být nejméně 150 mg (tj. přibližně 600 mg Zn sulphuricum). Potravinové doplňky obsahující řádově miligramy zinku jsou naprosto nedostatečné. Donedávna jsme používali Zincteral® (Polfa), při nedostupnosti preparátu nyní podáváme magistraliter připravovaný Zn suplhuricum či při jeho intoleranci komerčně dostupný Zn aceticum (Wilzin®, Orphan). Plná dávka zinku se podává od zahájení léčby, nástup jeho účinku je však pomalejší než u penicilaminu. Plné účinnosti při neurologickém postižení lze dosáhnout i po několika letech (24). Neurologické příznaky u většiny pacientů mohou vymizet či z velké míry ustoupit. Léčbu můžeme kontrolovat vyšetřením odpadů Zn do moči – ty by měly být vyšší než 35 μg/den.
Velkou roli při neurologickém postižení má i symptomatická léčba extrapyramidového syndromu.
Asymptomatické jedince zachycené screeningem léčíme v dnešní době většinou zinkem (25).
Dietní opatření spočívající v omezení potravin s vysokým obsahem zinku mají snad význam v úvodních fázích léčby, při stabilizaci choroby již přísné dietní restrikce není třeba.
V tabulce 1 jsou uvedeny jednotlivé léčebné metody v naší skupině pacientů. Úvodní léčbou byl u 86 % pacientů penicilamin. V průběhu sledování se počet pacientů léčených penicilaminem snížil na 63 %. Nejčastějšími důvody ke změně na zinek byly nežádoucí účinky nebo neúčinnost penicilaminu, případně stabilizace choroby. Vysoký počet asymptomatických pacientů léčených penicilaminem je dán skutečností, že část pacientů byla diagnostikována v době, kdy se penicilamin jevil jako ideální lék pro všechny formy choroby (průměrná doba sledování této skupiny pacientů je 24,4 roku).
Prognóza
Neléčená choroba má velmi špatnou prognózu a téměř jistě vede ke smrti nemocného. Při správné (celoživotní) léčbě nemusí být život pacientů s Wilsonovou chorobou výrazněji zkrácen (26). Za neúspěchem léčby může být i špatná adherence pacienta. Vysazení léčby může vést k nevratnému neurologickému poškození či k dekompenzaci jaterní choroby. Je třeba pacienty pravidelně kontrolovat, dostatečně motivovat a provádět laboratorní kontroly léčby (tj. odpady Cu do moči při léčbě penicilaminem nebo odpady Zn do moči při léčbě zinkem). U stabilní choroby zveme pacienty ke kontrolám 2–3× do roka, při známkách aktivity choroby častěji. Nezbytná je spolupráce s neurologickým pracovištěm, které se dané problematice věnuje.
Závěr
Wilsonova choroba je závažné onemocnění, které má při správné léčbě vynikající prognózu. Je na ní třeba myslet u jakékoliv jaterní choroby dětí a mladých pacientů, pokud není známá jasná příčina (tj. například infekční hepatitida, mononukleóza apod.). Dále je nutné na Wilsonovu chorobu pamatovat u mladších pacientů s neurologickými extrapyramidovými projevy a nevysvětlitelnými psychickými poruchami. Základní metodou screeningu je vyšetření ceruloplazminu a volné mědi v séru, jaterních testů, nativních odpadů mědi do moči za 24 hodin a oční vyšetření na přítomnost Kayser-Fleischerova prstence. Při negativitě uvedených testů je diagnóza Wilsonovy choroby velmi nepravděpodobná.
Zkratky
ApoE – apolipoprotein E
ATP7B – ATPáza 7ß
Ctr1 – přenašeč mědi 1 (copper transporter 1)
Cu – měď
K-F prstenec – Kayser-Fleischerův prstenec
MT – metalothion
PNC – penicilamin
SD – směrodatná odchylka
Zn – zinek
Práce byla podpořena grantem IGA MZ ČR NR-9406/3.
Adresa pro korespondenci:
doc. MUDr. Radan Brůha, CSc.
IV. interní klinika 1. LF UK a VFN
U Nemocnice 2, 128 08 Praha 2
e-mail: bruha@cesnet.cz
Zdroje
1. Wilson SAK. Progressive lenticular degeneration: a familial nervous disease associated with cirrhosis of the liver. Lancet 1912; 1 : 1115–1119.
2. Culota VC, Gitlin JD. Disorders of copper transport. In: Scriver CS. TheMolecular and Metabolic Basis of Inherited Disease. New York: McGraw-Hill 2001; 3105–3126.
3. Hellman N, Gitlin JD. Ceruloplasmin metabolism and function. Ann Rev Nutr 2002; 22 : 439–458.
4. Klomp AE, Tops BB, van Denberg IE, Berger R, Klomp LW. Biochemical characterisation and subcellular localization of human copper transporter 1 (hCTR1). Biochem J 2002; 364 : 497–505.
5. Kelley EJ, Palmiter RJ. A murine model of Menkes disease reveals a physiological function of metallothionein. Nat Genet 1996; 13 : 219–222.
6. Rae T, Schmidt P, Pufahl R, Culotta VC, O’Halloran TV. Undetectable intracellular free copper: The requirement of a copper chaperone for superoxide dismutase. Science 1999; 284 : 805–808.
7. Schaefer M, Hopkins R, Failla M, Gitlin JD. Hepatocyte-specific localisation and copper-dependent trafficking of the Wilson’s disease protein in the liver. Am J Physiol 1999; 276: G639–G646.
8. Tanzi RE, Petrukhin K, Chernov I, Pellequer UL, Wasco W, et al. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat Genet 1993; 5 : 344–350.
9. Bull AI, Thomas GR, Rommens JM, Forbes JR, Cox DW. The Wilson disease gene is a putative copper transporting P-type ATPase similar to Menkes gene. Nat Genet 1993; 5 : 327–337.
10. Stapelbroek JM, Bollen CW, Ploos van Amstel JK, Erpecum KJ, et al. The H1069Q mutation in ATP7B is associated with late and neurologic presentation in Wilson disease: results of a meta-analysis. J Hepatol 2004; 41 : 758–763.
11. Mareček Z. Wilsonova choroba. Praha: Galén 1996; 143 s.
12. Schiefermeier M, Kollegger H, Madl C, Polli C, Oder W, et al. The impact of apolipoprotein E genotypes on age at onset of symptom and phenotypic expression in Wilson’s disease. Brain 2000; 123 : 585–590.
13. Janda J, Kotalová R, Nevoral J, Šuláková T, Smíšek P. Akutní hemolytická krize se selháním jater jako první manifestace morbus Wilson u dětí. Čs Pediat 1996; 51 : 509–514.
14. Roberts EA, Schilsky ML. A practice guideline on Wilson disease. Hematology 2003; 37 : 1475–1492.
15. Vrábelová S, Váňová P, Kopečková L, Trunečka P, Smolka V, et al. Molekulární analýza Wilsonovy choroby. Čas Lék čes 2002; 141 : 642–645.
16. Vrábelová S, Letocha O, Borský M, Kozák L. Mutation analysis of the ATP7B gene and genotype/phenotype correlation in 227 patients with Wilson disease. Mol Genet Metab 2005; 86 : 277–285.
17. Gojová L, Jansová E, Külm M, Pouchlá S, Kozák L. Genotyping microarray as a novel approach for the detection of ATP7B gene mutations in patients with Wilson disease. Clin Genet 2008; 73 : 441–452.
18. Ferenci P, Caca K, Loudianos G, et al. Diagnosis and phenotypic classification of Wilson disease. Liver 2003; 23 : 139–142.
19. Wiggelinkhuizen M, Tilanus MEC, Bollen CW, Houwen RHJ. Systematic review: clinical efficacy of chelator agents and zinc in the initial treatment of Wilson disease. Aliment Pharmacol Ther 2009; 29 : 947–958.
20. Petrášek J, Jirsa M, Šperl J, Kozák L, Taimr P, Špičák J, Filip K, Trunečka P. Revisited Kings’s College score for liver transplantation in adult patients with Wilson’s disease. Liver Transpl 2007; 13 : 55–61.
21. Geissler I, Heinemann K, Rohm S, et al. Liver transplantation for hepatic and neurological Wilson’s disease. Transplant Proc 2003; 35 : 1445–1446.
22. Wiggelinkhuizen M, Tilanus MEC, Bollen CW, Houwen RHJ. Systematic review: clinical efficacy of chelator agents and zinc in the initial treatment of Wilson disease. Aliment Pharmacol Ther 2009; 29 : 947–958.
23. Hoogenraad TU. Zinc treatment of Wilson’s disease. J Lab Clin Med 1998; 132 : 240–241.
24. Brewer GJ, Dick RD, JohnsonVD, Brunberg JA, Kluin KJ, Fink JK. Treatment of Wilson’s disease with zinc: XV Long-term follow-up studies. J Lab Clin Med 1998; 132 : 264–278.
25. Huster D, Leonhardt K, Mossner J. Wilson disease – update on pathophysiology and management. Ces a Slov Gastroent a Hepatol 2008; 62 : 220–228.
26. Merle U, Schaefer M, Ferenci P, Stremmel W. Clinical presentation, diagnosis and long-term outcome of Wilson’s disease: a cohort study. Gut 2007; 56 : 115–120.
Štítky
Adiktologie Alergologie a imunologie Angiologie Audiologie a foniatrie Biochemie Dermatologie Dětská gastroenterologie Dětská chirurgie Dětská kardiologie Dětská neurologie Dětská otorinolaryngologie Dětská psychiatrie Dětská revmatologie Diabetologie Farmacie Chirurgie cévní Algeziologie Dentální hygienistkaČlánek vyšel v časopise
Časopis lékařů českých

- Jak a kdy u celiakie začíná reakce na lepek? Možnou odpověď poodkryla čerstvá kanadská studie
- Jaké zdravotní benefity může mít popíjení kávy nebo čaje?
- Doc. Jitka Fricová: V USA nasazovali fentanyl poměrně nekriticky, v Česku je situace jiná
- Efektivita kartáčku Sonicare For Kids u dětí předškolního věku
- Když léky přinášejí jinou chuť Vánoc...
Nejčtenější v tomto čísle
- Wilsonova choroba
- Posuzování zdravotního stavu a pracovní schopnosti u onkologicky nemocných
- Případ akutní virové hepatitidy E získané v České republice
- BEZKREVNÍ MEDICÍNA V KLINICKÉ PRAXI
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy